

RSV is an important cause of LRTI in infants and young children for which there are no suitable antiviral drugs offered. We evaluated the efficacy of KPT-335 as an anti-RSV drug and show that KPT-335 inhibits XPO1-mediated nuclear export, leading to nuclear accumulation of RSV M protein and reduction in RSV levels. KPT-335 treatment also resulted in inhibition of proinflammatory pathways, which has important implications for its effectiveness in vivo.
KEYWORDS: exportin 1, KPT-335, M protein, RSV, Verdinexor, XPO1, nuclear export, respiratory syncytial virus
ABSTRACT
Respiratory syncytial virus (RSV) is a leading cause of hospitalization of infants and young children, causing considerable respiratory disease and repeat infections that may lead to chronic respiratory conditions such as asthma, wheezing, and bronchitis. RSV causes ∼34 million new episodes of lower respiratory tract illness (LRTI) in children younger than 5 years of age, with >3 million hospitalizations due to severe RSV-associated LRTI. The standard of care is limited to symptomatic relief as there are no approved vaccines and few effective antiviral drugs; thus, a safe and efficacious RSV therapeutic is needed. Therapeutic targeting of host proteins hijacked by RSV to facilitate replication is a promising antiviral strategy as targeting the host reduces the likelihood of developing drug resistance. The nuclear export of the RSV M protein, mediated by the nuclear export protein exportin 1 (XPO1), is crucial for RSV assembly and budding. Inhibition of RSV M protein export by leptomycin B correlated with reduced RSV replication in vitro. In this study, we evaluated the anti-RSV efficacy of Verdinexor (KPT-335), a small molecule designed to reversibly inhibit XPO1-mediated nuclear export. KPT-335 inhibited XPO1-mediated transport and reduced RSV replication in vitro. KPT-335 was effective against RSV A and B strains and reduced viral replication following prophylactic or therapeutic administration. Inhibition of RSV replication by KPT-335 was due to a combined effect of reduced XPO1 expression, disruption of the nuclear export of RSV M protein, and inactivation of the NF-κB signaling pathway.
IMPORTANCE RSV is an important cause of LRTI in infants and young children for which there are no suitable antiviral drugs offered. We evaluated the efficacy of KPT-335 as an anti-RSV drug and show that KPT-335 inhibits XPO1-mediated nuclear export, leading to nuclear accumulation of RSV M protein and reduction in RSV levels. KPT-335 treatment also resulted in inhibition of proinflammatory pathways, which has important implications for its effectiveness in vivo.
INTRODUCTION
Respiratory syncytial virus (RSV) causes acute respiratory tract infections in infants and young children, including serious lower respiratory tract disease with bronchiolitis and pneumonia (1). Most children are infected with RSV by age 3. Recurrent RSV infections are common throughout life, and exposure does not induce robust immunity against subsequent infections; thus, the elderly and immunocompromised are at risk for developing complications associated with RSV infection (2, 3).
RSV is a member of the Pneumoviridae family (4). It has an outer envelope derived from the host plasma membrane and a negative-sense RNA genome. The viral envelope contains an attachment (G) protein, a fusion (F) protein, and a small hydrophobic (SH) protein. The matrix (M) protein occurs under the viral envelope and surrounds a nucleocapsid core composed of a complex of genomic viral RNA, the nucleocapsid protein (N), the phosphoprotein (P), the large polymerase subunit (L), and the M2-1/M2-2 proteins (5).
RSV infection is initiated when the G protein attaches to a cell surface receptor followed by F protein-mediated fusion (5). The nucleocapsid is released into the cell cytoplasm where the L and P polymerase complex directs the transcription of the RSV genome to generate the primary mRNA transcripts, which are translated into viral nonstructural and structural proteins (5, 6). The genome is replicated into a full-length complementary copy, the antigenome, which is used as a template to direct the synthesis of genomic RNA (5). The nascent genome associates with the N, P, and L proteins to form an active viral ribonucleoprotein (vRNP) complex within characteristic cytoplasmic inclusion bodies (7, 8). The M2-1 protein associates with the vRNP complex to promote transcription of the genome. The F, G, and SH proteins associate with each other to form a glycoprotein complex (9). The vRNP assembles with the envelope glycoprotein complex, and the virus buds from the apical surface within lipid rafts, facilitated by the interaction of M protein with the vRNP, envelope proteins, and the cellular membrane (7, 10–12).
RSV M protein modulates virus assembly and egress from the respiratory epithelium (13). It has been shown to localize to the nucleus of infected cells early in the viral life cycle (14), moving to cytoplasmic inclusion bodies at later time points and associating with the vRNP complex (7). Studies have shown that nuclear uptake of M protein is mediated by importin β1 (a nuclear import receptor) while exportin 1 (XPO1) shuttles the M protein from the nucleus to the cytoplasm (15, 16), and inhibition of XPO1-mediated nuclear export by leptomycin B (LMB; a prototypical inhibitor of XPO1 produced by Streptomyces) results in reduced virus production and M protein nuclear accumulation (15). Additionally, modification of the M protein nuclear export signal (NES) leads to its nuclear accumulation, and RSV containing an M protein with a mutated NES is not viable (15). Thus, it remains unclear why M protein translocates to the nucleus, but findings suggest a critical role for nuclear M protein in RSV replication and a likely role in RSV pathogenesis (14).
Substantial research has converged on identifying compounds that target RSV to inhibit replication and reduce disease severity, but none has translated into approved anti-RSV drugs for clinical use. One of the major challenges has been the development of drug resistance. Currently, one prophylactic, palivizumab (Synagis), a humanized monoclonal antibody specific to the RSV F protein, is administered to high-risk patients, typically pediatric cases with underlying congenital heart disease, pulmonary disease, or immune deficiencies (17, 18). Treatment has been shown to be partly effective at preventing RSV disease. Another FDA-approved treatment for RSV is inhaled ribavirin; its effectiveness is debatable, and it is difficult to administer (19, 20). Accordingly, there is a need for new RSV preventative and therapeutic strategies to prevent or decrease the severity of RSV infections in the young, elderly, and immunocompromised.
A novel class of selective inhibitors of nuclear export (SINE) compounds that target XPO1-mediated export have shown success as antitumor and antiviral agents in preclinical and multiple clinical studies (21–25). These orally bioavailable compounds are slowly reversible inhibitors that interfere with XPO1-mediated nuclear export of proteins. Karyopharm has a drug candidate, Selinexor (KPT-330), which is being evaluated in late-stage clinical trials in patients with relapsed and/or refractory hematological and solid tumor malignancies. KPT-335 (Verdinexor) is being investigated as a potential broad-spectrum treatment for viral diseases. Previously, KPT-335 has exhibited broad-spectrum antiviral activity against various influenza A and B viruses in the mouse and ferret model systems (26, 27). In these studies, KPT-335 inhibited nuclear export of the vRNP, resulting in reduced influenza virus replication and decreased disease pathogenesis (26, 27). KPT-335 has also proven effective against Venezuelan equine encephalitis virus (VEEV) in vitro by inhibiting the nuclear export of the capsid protein (28). In a previous study conducted as a randomized, double-blind, placebo-controlled, dose-escalating phase 1 clinical trial in healthy human volunteers, KPT-335 was found to be generally safe and well tolerated, with adverse events occurring in similar numbers and grades as placebo (ClinicalTrials.gov registration number NCT02431364).
In the current study, we have evaluated the antiviral efficacy of KPT-335 against RSV in vitro. We show that KPT-335 inhibits XPO1-mediated nuclear export, leading to nuclear accumulation of RSV M protein and a reduction in RSV titers. KPT-335 treatment also results in inhibition of proinflammatory pathways, which has important implications for the effectiveness of KPT-335 in vivo.
RESULTS
XPO1 expression is required for RSV replication.
We examined the requirement of exportin 1 (XPO1) expression for RSV replication. A549 cells were treated with a SMARTpool of small interfering RNAs (siRNAs) designed to target XPO1 mRNA (siXPO1) and inhibit its expression. At 48 h post-siXPO1 treatment, A549 cells were infected (multiplicity of infection [MOI] of 0.5) with RSV A2, and the infection was monitored by anti-F protein immunostaining and plaque assay. Compared to siRNA nontargeting control (siNTC)-treated or nontreated (NT) A549 cells, XPO1 knockdown significantly (P < 0.05) inhibited RSV replication, as confirmed by anti-RSV F immunostaining (Fig. 1A) and quantification of RSV infectious particles by plaque assay (Fig. 1B). As shown in Fig. 1C, siXPO1 treatment inhibited XPO1 expression compared to the level in A549 cells treated with siNTC or in nontreated (NT) cells as early as 24 h posttreatment (Fig. 1C), with sustained inhibition up to 96 h post-siXPO1 transfection (48 h postinfection [p.i.]) (Fig. 1C, R48). These data confirm that XPO1 is a host factor essential for RSV replication in A549 respiratory epithelial cells.
FIG 1.
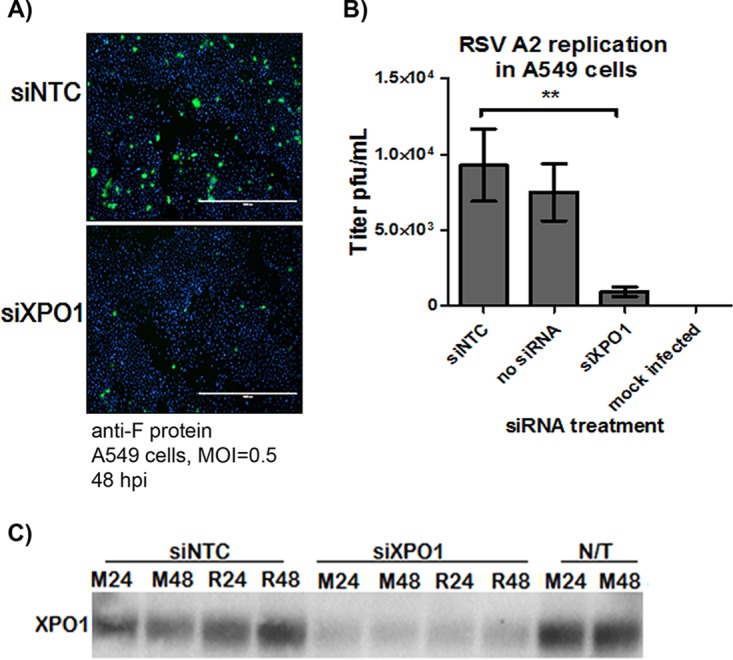
XPO1 expression is required for RSV infection. A549 cells were transfected with a SMARTpool siRNA designed to target XPO1 mRNA (siXPO1) or with an siRNA nontargeting control (siNTC). At 48 h post-siRNA transfection, cells were infected with RSV A2 (MOI of 0.5), and the infection was monitored at 48 h p.i. by F protein immunostaining (A) and RSV plaque assay (B). Inhibition of XPO1 expression was confirmed by Western blotting (C) in A549 cells infected with RSV (R) or mock infected (M) at 24 or 48 h p.i. (48 h and 96 h post-siRNA transfection, respectively). A Mann-Whitney nonparametric, two-tailed test was used to determine differences between groups, and significant differences are indicated. **, P < 0.001.
KPT-335, a selective XPO1 inhibitor, inhibits RSV replication.
We examined the activity of KPT-335 (reversible inhibitor of XPO1) against RSV replication. A549 cells were infected (MOI of 0.5) with RSV for 2 h and treated with increasing concentrations of KPT-335, and at 72 h p.i. RSV titers were quantified by plaque assay (Fig. 2). At KPT-335 concentrations of ≥10 μM, RSV replication was inhibited by nearly 100% with ∼20% cytotoxicity (50% cytotoxic concentration [CC50] of ∼38 μM). At 1 μM, KPT-335 inhibited RSV A2 replication by ≥50% (50% inhibitory concentration [IC50] of 0.96 μM) without affecting cell viability; thus, this concentration was selected for all subsequent experiments.
FIG 2.

KPT-335 inhibits RSV replication. (A) A549 cells were treated with DMSO or increasing concentrations of KPT-335 for 72 h. At 70 h posttreatment, 20 μl of CellTiter Blue reagent was added to each well and incubated for 2 h at 37°C. The plate was shaken for 10 s, and fluorescence emission was measured at 560/590 nm. The average fluorescence value of the culture medium background (wells containing no cells) was subtracted from each experimental well. The mean fluorescence emission values of the wells containing DMSO only were considered 100% viability and were used to calculate percent relative cell viability of the wells containing KPT-335. The percent viability versus the log10 concentration of KPT-335 was plotted using GraphPad Prism, and the values were fitted to a nonlinear regression curve to determine the CC50. The data shown are from triplicate samples from one experiment and are representative of three independent experiments. (B) A549 cells were infected with RSV A2 (MOI of 0.5) and treated with increasing amounts of KPT-335 from 2 h p.i. Samples were collected at 72 h p.i., and titers of infectious RSV were determined by immuno-plaque assay as described in Materials and Methods. Average RSV titer in infected samples treated with DMSO was taken as 100% and served as the control; this value was used to calculate percent infection of the samples treated with KPT-335. The percent infection versus the log10 concentration of KPT-335 was plotted using GraphPad Prism, and the values were fitted to a nonlinear regression curve to determine the IC50. The data shown are from triplicate samples from one experiment and are representative of three independent experiments.
To understand the temporal requirement, XPO1-dependent nuclear export was inhibited with KPT-335 at different times post-RSV infection. A549 cells were infected with RSV A2 (MOI of 0.5) for 2 h, the inoculum was removed, and KPT-335 was added as a single dose at 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, or 22 h p.i. At 24 h p.i. (approximately one round of replication), RSV titers were quantified by plaque assay. As shown in Fig. 3A, compared to infection levels in cells treated with dimethyl sulfoxide (DMSO) (100% infection), addition of 1 μM KPT-335 earlier than 10 h p.i. reduced infection between 70 and 90%. When KPT-335 was added later than 10 h p.i., no inhibition was detected (Fig. 3A). These findings correlate with our previous study showing that most of RSV M protein is exported to the cytoplasm at 8 to 12 h p.i. (15) and that inhibition of XPO1-dependent nuclear export by KPT-335 at time points beyond 10 h p.i. has little or no impact on RSV replication (15).
FIG 3.

KPT-335 is effective against RSV when provided therapeutically or prophylactically. (A) A549 cells were infected with RSV A2 for 2 h (MOI of 0.5), inoculum was removed, and KPT-335 was added as a single dose (1 μM) at 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, or 22 h p.i. At 24 h p.i., samples were collected, and percent RSV infection was determined by immunostaining plaque assay. Average RSV titer at 24 h p.i. in infected samples treated with DMSO was taken as 100% and served as a control; this value was used to calculate percent infection of the samples treated with KPT-335. The means ± standard errors of the means of percent infection versus time of addition of KPT-335 are shown. (B) A549 cells were treated with 1 μM KPT-335 at 72, 48, 24, or 2 h prior to infection (prophylactic treatment) or at 2 or 24 h p.i. (therapeutic treatment). Samples were collected at 72 h p.i., and virus production was quantified by plaque assay. Average RSV titers at 72 h p.i. in infected samples treated with DMSO was taken as 100% and served as control; this value was used to calculate percent infection of the samples treated with KPT-335. The mean ± SEM of percent infection versus time of addition of KPT-335 is shown. The data shown are from three independent experiments. The experimental timeline is depicted above each graph.
The therapeutic window of 2 to 10 h p.i. between adverse and desired effects is potentially narrow in terms of antiviral treatment; therefore, we examined KPT-335 prophylactic or therapeutic treatment at earlier and later time points. KPT-335 was added to A549 cells as a single dose at 2 h, 24 h, 48 h, or 72 h prior to infection (prophylactic treatment) or at 2 h or 24 h p.i. (therapeutic treatment). Infection progressed for 72 h (approximately 3 replication cycles), at which time virus production was quantified by plaque assay. Prophylactic treatment with KPT-335 at 72 h, 48 h, or 24 h prior to infection completely inhibited RSV replication in A549 cells, whereas addition of KPT-335 between 2 h and 24 h p.i. inhibited RSV replication by ∼60% (Fig. 3B). These data show that KPT-335 is efficacious when provided prophylactically or therapeutically, but it was more effective when administered at 24 to 72 h prior to infection. This indicates that prophylactic addition of KPT-335 favors interaction with XPO1, possibly making XPO1 unavailable for interacting with the RSV M protein. Notably, addition of KPT-335 at 24 h p.i. inhibited RSV replication by 60%, demonstrating that addition of this drug when viral replication has occurred can inhibit subsequent rounds of replication and reduce the viral load.
The M protein nuclear export signal (NES; amino acids 194 to 206) is conserved among RSV strains (Fig. 4). We examined whether KPT-335 would inhibit RSV A and B. As shown in Fig. 5, 1 μM KPT-335 inhibited RSV A2, RSV Long, and RSV B1 replication in A549 by 50 to 75% compared to levels in the DMSO control. LMB had a stronger inhibitory activity than KPT-335, but this effect was largely due to the cytotoxicity of LMB. The antiviral effect of KPT-335 was also confirmed in normal bronchial epithelium cells derived from a noncancerous individual (BEAS-2B cells). These data demonstrate that KPT-335 is effective against both RSV A and B strains.
FIG 4.

M protein is conserved across RSV A and B strains. The amino acid sequence of the RSV M protein from the indicated strains is shown. The nuclear export signal (NES) sequence is shown within a black frame. NES are 8 to 15 residues long and conform to the consensus Φ1-X2,3-Φ2-X2,3-Φ3-X-Φ4 where Φn represents Leu, Val, Ile, Phe, or Met, and X can be any amino acid (39). GenBank accession numbers are indicated at the end of the sequence. #, viruses tested in this study.
FIG 5.

KPT-335 inhibits replication of RSV A and B strains independent of the host cell line. A549 or BEAS-2B cells were infected with RSV A2, A/Long, or B1 strain at an MOI of 0.5. At 2 h p.i., medium was replaced with fresh medium containing 1 μM KPT-335 or 750 nm LMB or DMSO as a control. Samples were collected at 24 h p.i., and virus production was quantified by plaque assay. Average RSV titers at 24 h p.i. in infected samples treated with DMSO was taken as 100% and served as control; this value was used to calculate percent infection of the samples treated with KPT-335 or LMB. The means ± standard errors of the means of percent infection versus time of addition of KPT-335 are shown. The data shown are from three independent experiments. The cell lines and virus strains are indicated on the graphs. The Kruskal-Wallis one-way analysis of variance (ANOVA) was used to determine differences between groups, and significant differences are indicated. *, P < 0.05; ns, nonsignificant (P > 0.05).
KPT-335 results in limited nuclear accumulation of RSV M protein.
RSV M protein is involved in the XPO1-mediated nuclear export pathway. The M protein is essential for RSV assembly in the cytoplasm, and its restriction to the nucleus results in reduced RSV infectious titers (15). To determine if KPT-335 mediated inhibition of RSV replication via retention of M protein in the nucleus, we examined M protein subcellular distribution. A549 cells were infected with RSV, followed by treatment with 1 μM KPT-335 from 2 h p.i., and subcellular distribution of M protein was analyzed at 24 h p.i. (22 h of KPT-335 treatment) by immunofluorescence confocal microscopy (Fig. 6A and B). DMSO treatment did not result in detectable nuclear retention of M protein (Fig. 6B) (nuclear- to cytoplasmic-fluorescence ratio [Fn/c] of <1); however, there was a significant (P = 0.0003) change in the relative amount of M protein in the nucleus compared to that in the cytoplasm when infected cells were treated with KPT-335. The immunofluorescence data were confirmed by Western blot analysis of nuclear and cytoplasmic fractions of KPT-335-treated RSV-infected cells (Fig. 6C and D). Treatment with KPT-335 from 2 to 24 h p.i. increased M protein in the nucleus compared to the level with DMSO treatment, similar to the effect of treatment with LMB. Interestingly, cells treated with KPT-335, with or without RSV infection, had lower XPO1 expression than DMSO-treated controls (Fig. 6C), suggesting that KPT-335 negatively affects XPO1 expression.
FIG 6.

KPT-335 treatment leads to nuclear accumulation of M protein and XPO1. (A) Monolayers of A549 cells propagated to 80% confluence in 12-well plates containing coverslips were infected with RSV A2 followed by treatment with 1 μM KPT-335 during the early (6 to 18 h p.i.) or late stages (18 to 30 h p.i.) of infection and analyzed at 48 h p.i. by immunofluorescence confocal microscopy for localization of M protein. Cells treated with DMSO were taken as a control. Digital images of 0.5-μm sections were captured with a Nikon Ti-Eclipse confocal system and NIS AR Elements software. (B) Image J was used to determine the nuclear/cytoplasmic ratio (Fn/c) of RSV-M [Fn/c = (Fn − Fb)/(Fc − Fb)], where Fn is the nuclear fluorescence, Fc is the cytoplasmic fluorescence, and Fb is the background or autofluorescence in images such as those shown in panel A. Data shown are means ± standard errors of the means (n ≥ 30) and representative of three independent experiments. Two-way ANOVA followed by Fisher’s least significant difference test was used to determine differences between groups, and statistically significant differences are indicated. **, P < 0.01. (C) RSV A2 infected (R) or mock-infected (M) A549 cells were treated with 1 μM KPT-335, DMSO, or 750 nM LMB from 2 h p.i. Fractions were collected at 24 h p.i., utilizing an NE-PER nuclear and cytoplasmic purification kit. Equivalent protein concentrations of the 24-h p.i. cytoplasmic and nuclear fractions were resolved by SDS-polyacrylamide gel electrophoresis, followed by immunoblotting. Blots were probed for XPO1 and RSV M protein. GAPDH and lamin B1 were used as markers for cytoplasmic and nuclear fractions, respectively. The primary antibodies used are indicated on the left. Images are single blots from one experiment representative of three different experiments; the white line observed in the lamin B1 blot is an artifact produced during exposure and image capture. (D) The relative amount of RSV M protein in the nucleus compared to that in the cytoplasm was estimated by densitometric scanning of the protein bands using ImageJ. Data shown are relative to value for the DMSO-treated samples, taken as 1. (E) A549 cells were transfected using Lipofectamine. After 18 h of incubation, the cells were treated with DMSO or 1 μM KPT-335 for 6 or 24 h. Digital images of 0.5-μm sections were captured with a Nikon Ti-Eclipse confocal system and NIS AR Elements software and representative images are shown. Image J was used to analyze the digital images and determine the Fn/c as described in the legend of Fig. 5. Data are shown in histograms for GFP-Rev after 6 h of treatment with KPT-335 or DMSO (control) (F) and GFP and GFP-M after 6 h and 24 h of treatment with KPT-335 or DMSO (control) (G). Data shown are means ± standard errors of the means (n ≥ 30) and are representative of three independent experiments. Two-way ANOVA followed by Fisher’s least significant difference test was used to determine differences between groups, and statistically significant differences are indicated. *, P < 0.05; **, P < 0.001.
To determine if subcellular localization of M protein was specific to KPT-335, M protein localization in the presence of KPT-335 was investigated using a transfected cell system (Fig. 6E to G). A549 cells were transfected with a plasmid expressing green fluorescent protein-fused M protein (GFP-M), and 1 μM KPT-335 (or DMSO control) was added 18 h later for 6 h or 24 h, followed by collection of digital images using live-cell confocal laser scanning microscopy (CLSM) (Fig. 6E). Plasmids expressing GFP alone or GFP fused to the HIV Rev protein NES (GFP-Rev) were used as negative and positive controls, respectively. KPT-335 treatment for 6 h resulted in increased GFP-M in the nucleus relative to the level with DMSO treatment, and this increased with longer treatment (images labeled GFP-M). However, GFP-M did not accumulate in the nucleus more than in the cytoplasm (Fig. 6G, Fn/c ≤ 1). This was not due to lack of KPT-335 inhibition of the XPO1 pathway, as shown by the nuclear retention of GFP-Rev (Fig. 6F, Fn/c = 21) after 6 h treatment. There was no detectable change in the subcellular localization of GFP alone (Fig. 6G), showing that the effect of KPT-335 on GFP-M localization was specific to M. Figure 6 shows that treatment with KPT-335 induces a limited but significant (P < 0.001) nuclear accumulation of M protein correlating to reduced viral titers, as shown in Fig. 3. The data showing that KPT-335 treatment resulted in a reduction of XPO1 suggest that this is likely the molecular mechanism underlying the nuclear retention of M protein and not a direct interaction or effect of KPT-335 on M protein. Transient inhibition of XPO1-mediated transport would also disrupt other pathways that are regulated by XPO1.
KPT-335 reduces XPO1 expression while increasing p53 expression.
A previous study showed that KPT-185, a drug analogue of KPT-335, induces cell death of a primary effusion lymphoma cell line by causing nuclear accumulation of p53 tumor suppressor protein, as well as inhibition of NF-κB activity, resulting in cell cycle arrest and apoptosis induction (24). In addition, the XPO1 gene promoter has a p53 binding site, and p53 binding to this promoter represses XPO1 expression (29). To assess the effect of KPT-335 treatment on p53 expression, RSV-infected or mock-infected A549 cells were treated with KPT-335, and total cell lysate was analyzed by Western blotting. KPT-335 treatment of mock- and RSV-infected cells reduced XPO1 expression, while increasing p53 expression compared to that with DMSO treatment (Fig. 7A). Interestingly, although LMB binds and inhibits XPO1-dependent nuclear export in a fashion similar to that of KPT-335, LMB treatment did not reduce XPO1 expression. These results suggest that KPT-335 and LMB have similar mechanisms of XPO1 inhibition but that the downstream activation pathways are likely different. KPT-335 was synthesized to bind to the same pocket within the XPO1 structure as LMB, and this is the mechanism behind the inhibition of XPO1 activity by both drugs. However, the downstream pathways that are affected appear to be different, as evidenced by the difference in the effects on XPO1 and p53 expression levels.
FIG 7.

KPT-335 reduces XPO1 expression and increases p53 expression without inducing apoptosis. (A) RSV-infected (R) or mock-infected (M) A549 cells were treated with 1 μM KPT-335, DMSO, or 750 nM LMB from 2 h p.i. Total cell lysate was collected at 24 h p.i. and analyzed by Western blotting as described in the legend of Fig. 5. Blots were probed for XPO1 and p53, with GAPDH used as a loading control. The primary antibodies used are indicated on the left. XPO1 and p53 expression was quantified using ImageJ, normalized to the level of GAPDH. Data shown are relative to values for DMSO-treated samples, taken as 1. (B) A549 cells were infected with RSV at an MOI of 0.5 or 2.0. KPT-335, DMSO, or LMB was added at 2 h p.i., and samples were collected at 24 h p.i. Assay controls included wells without cells (background), mock-infected cells (negative control), and cells treated with 1 μM staurosporine (STP) for 4 h (positive control). At 24 h p.i., plates were equilibrated at room temperature for 30 min, and 100 μl of Caspase-Glo 3/7 reagent was added to each well and incubated for 3 h at room temperature. Luminescence was measured using a Tecan microplate reader, and the background RLU count (wells without cells) was subtracted from each well. A Kruskal-Wallis one-way ANOVA was used to determine differences between groups, and statistically significant differences are indicated. *, P < 0.05; ns, nonsignificant (P > 0.05). (C) A549 cells were infected with RSV at an MOI of 0.5 or 2.0. KPT-335, DMSO, or LMB was added at 2 h p.i., and samples were collected at 6, 24, or 48 h p.i. At the indicated times plates were equilibrated at room temperature for 30 min, and 100 μl of Caspase-Glo 3/7 reagent was added to each well and incubated for 3 h at room temperature. Luminescence was measured using a Tecan microplate reader, and the background RLU count (wells without cells) was subtracted from each well.
Although treatment of A549 cells with 1 μM KPT-335 did not induce substantial cytotoxicity or detectable cytopathic effects (Fig. 2), alteration of p53 expression was examined as this could contribute to induction of apoptosis. Activation of caspase-3 and caspase-7 was measured in A549 cells infected with RSV (MOI of 0.5 or 2.0) for 24 h and treated with KPT-335 (from 2 h to 24 h p.i.). As shown in Fig. 7B, KPT-335 treatment of mock- or RSV-infected cells did not substantially affect caspase-3 and -7 activation compared to treatment with the DMSO control. Staurosporine treatment, as expected, resulted in substantial apoptosis. Short (6 h) or long (48 h) incubation of staurosporine did not affect the outcome (Fig. 7C); however, treatment of mock- or RSV-infected cells with LMB resulted in caspase-3 and caspase-7 activation at 24 h and 48 h p.i., with general cell death occurring by 48 h p.i. (Fig. 7C). These data show that the effect of KPT-335 on p53 expression does not result in the cleavage of procaspase-3 and -7; thus, it is unlikely that the reduction in RSV replication is due to induction of apoptosis.
KPT-335 induces NF-κB nuclear accumulation and reduces cytokine expression.
It is known that several highly inducible cytokine genes are modulated by NF-κB following RSV infection, where NF-κB-dependent genes include members of the interferon (IFN) regulatory family along with suppressors of cytokine signaling (SOCS) and cytokine-inducible SH2 (CIS) protein families. Further, KPT-185 was shown to induce inhibition of NF-κB activity (30). As NF-κB-regulated genes are important for the innate immune response to RSV infection, we examined the effect of KPT-335 on NF-κB-regulated genes following RSV infection. As shown in Fig. 8A, inhibition of XPO1-dependent nuclear export by KPT-335 or LMB induced nuclear retention of NF-κB p65. The evaluation of the effect of KPT-335 on the expression of several proinflammatory cytokines and chemokines was examined (Fig. 8B). Two important chemokines, RANTES and macrophage inflammatory protein 1α (MIP-1α), have been associated with increased RSV pathogenesis and were examined along with 10 other chemokines. KPT-335 treatment (from 2 to 24 h p.i.) was associated with a decreased level of RANTES compared to the level in DMSO-treated cells, but this reduction was not significant (P < 0.05). Growth-regulated oncogene alpha (GRO-α), monocyte chemoattractant protein 1 (MCP-1), and interleukin-8 (IL-8) were detected, but no significant change was observed in the presence of KPT-335 compared to levels with treatment with the DMSO control although there was a trend toward reduction of MCP-1 and IL-8. The cytokine/chemokine response to KPT-335 treatment was similar to the response to LMB treatment (Fig. 8B). No change was observed for IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, IL-12, IL-17α, IFN-γ, tumor necrosis factor alpha (TNF-α), or granulocyte-macrophage colony-stimulating factor (GM-CSF) (data not shown). Taken together, these results indicate that KPT-335 inhibition of XPO1-dependent nuclear export leads to NF-κB nuclear accumulation; however, this change resulted in a nonsignificant reduction in cytokine expression levels compared to those of the control.
FIG 8.

KPT-335 induces NF-κB nuclear accumulation and varied cytokine expression. RSV A2-infected A549 cells were treated with 1 μM KPT-335, DMSO, or 750 nM LMB from 2 h p.i. Fractions were collected at 24 h p.i. utilizing an NE-PER nuclear and cytoplasmic purification kit, and analyzed by Western blotting as described in the legend of Fig. 6. Blots were probed for p65, and GAPDH and lamin B1 were used as controls. The relative amount of p65 in the nucleus compared to that in the cytoplasm was estimated by densitometric scanning of the protein bands using ImageJ. Data shown are relative to values for DMSO-treated samples, taken as 1. (B) RSV-infected A549 cells were treated with DMSO, KPT-335, or LMB from 2 h p.i.; mock-infected cells served as controls. Supernatants were collected at 24 h p.i. Culture supernatants were clarified by centrifugation before proinflammatory cytokine and chemokine protein expression was examined with a Multi-Analyte ELISArray kit. Fold change was calculated relative to values in mock-infected samples.
DISCUSSION
XPO1-mediated transport has an essential role for RNA viral replication, and disruption of this pathway by LMB results in reduced replication (31). SINE compounds were designed to reversibly bind to XPO1 and disrupt nuclear export based on the same mechanism of action as LMB but without the cytotoxic side effects (32, 33). SINE compounds are small-molecule drugs designed to reversibly interact with XPO1 at the NES binding groove solely through hydrophobic interactions and without being hydrolyzed by XPO1, enabling a stable interaction (32, 33). In this study, we show that KPT-335 reduces RSV replication, induces partial nuclear accumulation of M protein, and increases the nuclear retention of p53 and NF-κB-p65.
Previous studies have demonstrated that the nuclear import of M protein by importin β1 occurs during the early stages of infection, followed by XPO1-mediated shuttling into the cytoplasm in the late stages of infection (16). We have previously shown that RSV replication is negatively affected by XPO1-mediated nuclear transport inhibition using LMB at nanomolar concentrations (15). Targeting XPO1 with KPT-335 has also proven effective both in vitro and in vivo against several strains of the influenza virus (26, 27) and against the Venezuelan equine encephalitis virus (VEEV) in vitro (28). siRNAs were used to inhibit expression of XPO1 in A549 cells, followed by infection with RSV A2, and this was associated with substantial reduction in RSV replication in human epithelial cells.
SINE compounds have been shown to inhibit replication of HIV, influenza A virus, and hepatitis C virus (25, 26, 34). KPT-335 reduced RSV replication at a 1 μM concentration with low cytotoxicity, an important factor for therapeutic applications. We show that treatment using a 1 μM dose during the early stages of replication (2 to 10 h p.i.) reduces RSV titers by 60 to 90% compared to titers in DMSO control-treated cells. For influenza A virus, treatment with 1 μM KPT-335 for 2 h preinfection increased the nuclear retention of vRNP (26). The same prophylactic treatment in A549 cells with 2.5 μM KPT-335 prior to infection with VEEV resulted in nuclear accumulation of the viral capsid at 16 h p.i. (28). Minimal impact on RSV replication was observed on treatment after 10 h p.i., most likely due to the export of M protein to the cytoplasm after 8 to 12 h p.i. (15). Longer periods of prophylactic treatment of A549 cells with KPT-335 (24 to 72 h prior to infection) were more effective than short periods of treatment (2 h prior to infection), leading to 100% inhibition of RSV replication. In comparison, therapeutic treatment between 2 and 24 h p.i. reduced viral load to 60%. These findings indicate that KPT-335 is effective as both a prophylactic and a therapeutic antiviral. It has been shown that the mechanism of action of KPT-335 treatment in influenza virus and VEEV infections is associated with disruption of viral assembly or budding, leading to reduced viral replication (26, 28).
In this study, we show that treatment with 1 μM KPT-335 reduced replication of RSV strains A2, Long, and B in A549 cells and BEAS-2B cells. The data show that KPT-335 inhibits the replication of different RSV strains and suggests that the mechanism of inhibition is likely similar. Importantly, a mutated strain of RSV A2 lacking an active NES in the M protein could not be recovered in vitro, highlighting the importance of the nuclear export process of this viral protein (15). The M protein is largely conserved among both A and B strains (35). This implies that targeting the host factor XPO1 using KPT-335 is a broadly effective strategy against RSV.
The subcellular location of M protein is linked to the viral life cycle during which M protein enters the nucleus by interacting with importin-β and probably reduces the transcriptional activity in the host cell (14, 16). In the later stages of infection, M protein shuttles back into the cytoplasm to mediate inhibition of viral replication and initiate viral assembly at the plasma membrane, filament formation, and budding (7, 10, 36, 37). In this study, nuclear accumulation of M protein was observed in A549 cells treated with KPT-335 using both immunofluorescence and Western blotting. Interestingly, the decrease in RSV titers did not correlate with a reduction in the amount of cytoplasmic M protein. It is possible that translocation of M protein through the nucleus is required for virus assembly and that the majority of M protein in the cytoplasm has not traversed through the nucleus. In this regard, we have shown previously that M protein is phosphorylated at the threonine at position 205 and that inhibition of CK2 phosphorylation leads to reduced RSV titers (37). Taking these findings together, it is possible that movement of M protein through the nucleus results in its phosphorylation by CK2 and that phosphorylated M protein is required for its assembly functions; however, this remains to be confirmed. Unlike influenza A virus and VEEV, which replicate within the nucleus, RSV replication is confined to the cytoplasm, synthesizing structural glycoproteins and nucleocapsid proteins at the Golgi compartment and within inclusion bodies (5). The inhibition of M protein nuclear export does not completely inhibit virus production but is important for efficient assembly and maturation of viral filaments, which is reflected by reduced viral titers (15). This suggests that nuclear retention of RSV M protein occurs during KPT-335 treatment which can disrupt the later stages of viral infection and result in reduced replication.
As M protein is in the cytoplasm by 18 h p.i. (7), inhibition of XPO1 after that time and assaying for virus at 24 h p.i. (∼1 cycle) will not show any effect. Indeed, treatment with KPT-335 at 22 h p.i. followed by plaque assay at 24 h p.i. will not show any reduction in RSV titers. However, treatment at 24 h p.i. and assaying for virus at 72 h p.i. (∼3 cycles) will exhibit an effect due to inhibition of XPO1 activity in newly infected cells that were in the early stages of infection at 24 h p.i.
The inhibitory effect of KPT-335 treatment of A549 cells transfected to express GFP-Rev or GFP-M was similar to the inhibitory effect of LMB, where increased nuclear localization of NES carrying proteins occurred in the presence of the compound (15). It has been shown that the NES signal of p53 (FXXLXXXLXL), which uses XPO1 as the sole nuclear exporter, is weaker than that of Rev (LPXLXXLXL) (38, 39). Similarly, the lesser degree of nuclear accumulation of GFP-M than of GFP-Rev was most likely due to the differences in strength of the interaction with XPO1 and ease of accessibility for binding to the nuclear transporter.
XPO1 expression was significantly (P < 0.05) lower in KPT-335-treated A549 cells than in DMSO-treated A549 cells, which suggests that alterations of downstream activation pathways dependent on XPO1-mediated transport may occur. This includes anti-apoptotic pathways, cell cycle progression, and inflammatory pathways. SINE compounds, including KPT-335, induce the nuclear accumulation of tumor suppressor proteins such as FOXO, p53, and p21 in cancer cells that overexpress XPO1 in order to evade apoptosis (40). For example, the nuclear accumulation of tumor suppressor proteins (including p53) in the presence of SINE compounds has been shown to result in apoptosis and reduced malignancy in different hematological and solid tumors (32). KPT-185 and KPT-330 have been shown to cause cell cycle arrest in Jurkat, MOLT-4, and T-ALL cancer cell lines, resulting in a dramatic decrease in tumor size (22). Treatment with KPT-127, KPT-185, KPT-205, or KPT-225 targeting pancreatic cancer cell lines increased the nuclear retention of several tumor suppressor proteins, including FOXO and p53, inducing cell cycle arrest and apoptosis (23). The reduction in XPO1 expression in the presence of KPT-335 leads to disruption of the nuclear export of nearly ∼220 proteins (39) and several RNAs, including mediators of proliferative and prosurvival signaling pathways that are known to be required for the survival of cancer cells and for effective infection and release by RSV (15). Thus, the mechanism of action of KPT-335 to reduce RSV replication is linked to a combined effect of inhibition of M nuclear export and reduced XPO1 expression.
We observed decreased levels of XPO1 and increased levels of p53 in both infected and noninfected A549 cells treated with 1 μM KPT-335. However, activation of procaspases-3 and -7 was absent in both short- and long-term treatments with KPT-335 while treatment with LMB resulted in their activation at both 24 and 48 h posttreatment. This suggests that reduction in XPO1 expression in SINE-treated A549 cells prevents the induction of caspase-dependent apoptosis by RSV, and reduction in RSV replication is not likely due to induction of apoptosis. It has been shown that KPT-335 treatment of influenza virus-infected cells reduces the expression of TNF-α, IL-1β, and IL-6 by inhibiting NF-κB signaling pathway (26). The NF-κB inhibitor IkB utilizes XPO1 to repress NF-κB signaling. IκB binds to NF-κB (composed of the subunits p50 and p65) in the cytoplasm and renders it inactive by masking the nuclear localization signal (NLS) of NF-κB, preventing its trafficking to the nucleus and the transcriptional activation of several genes (41, 42). IκB kinase (IKK) phosphorylates the IκB regulatory domain, initiating the signal-induced degradation of IκB and releasing the NF-κB complex to enter the nucleus, where it can induce the expression of specific genes (41, 42). In the nucleus, NF-κB also induces the expression of the repressor IκB, and the newly synthesized IκB then reinhibits NF-κB, forming an auto-feedback loop (41, 42). XPO1 is required for the replenishment of cytoplasmic pools of NF-κB/IκB complexes and the subsequent regulation of NF-κB activation (43). We show in this study that KPT-335 treatment of mock- or RSV-infected A549 cells results in the nuclear retention of NF-κB subunit p65, thus likely leading to inactivation of NF-κB signaling. It has been shown that RSV infection induces the expression of several NF-κB-dependent genes and mediates proinflammatory cytokine expression of IL-8, IL-6, RANTES, and MIP-1α among several other cytokines (44). KPT-335 treatment of RSV-infected cells led to a minor reduction in RANTES expression, and it did not affect other cytokines and chemokines measured in this study.
Conclusion.
XPO1-mediated transport has a crucial role in RSV replication, and targeted inhibition of XPO1 likely has therapeutic applications as an antiviral approach. KPT-335 reduces RSV replication for both prophylactic and therapeutic treatments. In the absence of inhibition, RSV M protein is translocated from the nucleus to the cytoplasm in the later stages of infection to mediate viral assembly and budding. Treatment with KPT-335 induces limited nuclear accumulation of M protein. KPT-335 increases the nuclear retention of p5 but does not activate caspase-dependent apoptosis, and it has limited effects on the proinflammatory cytokines induced by RSV. Prospective studies will examine the in vivo efficacy of KPT-335 on RSV disease pathogenesis and will explore other host gene pathways that are regulated by XPO1-mediated transport and may be crucial for RSV infection and disease intervention.
MATERIALS AND METHODS
Cell culture and RSV.
A549 cells, a human type II respiratory epithelial cell line (ATCC CCL-185TM), Vero cells, an African green monkey kidney epithelial cell line (ATCC CCL-81), and BEAS-2B cells, an immortalized, human lung epithelial cell line (ATCC CRL-9609) were grown in Dulbecco's modified Eagle's medium (DMEM) (Sigma) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (FBS) (Bovogen or Atlanta Biologics). Cells were maintained in a humidified 37°C incubator supplied with 5% CO2. RSV strains (15) were grown as described below and stored at −80°C or in liquid N2 vapor until use.
Virus production.
RSV A and B strains were propagated in Vero cells. Briefly, 85% to 90% confluent T75 flasks of Vero cells were infected with RSV at an MOI of 0.1 in 5 ml of DMEM. Cells were infected for 1 h at 37°C and 5% CO2. After infection, 7 ml of DMEM with 2% fetal bovine serum was added to the flask. Flasks were incubated for 2 days or until extensive syncytium formation was observed. On the day of harvest, all except 3 ml of supernatant was removed and placed on ice in 50-ml centrifuge tubes. Three milliliters of sucrose-phosphate-glutamate-albumin (SPGA) medium was utilized for the extraction of cell-free viruses. SPGA (218 mM sucrose, 7.1 mM K2HPO4, 4.9 mM sodium glutamate, and 1% bovine serum albumin) was added, and the flask was stored overnight at −80°C. The flask was thawed, and cells were scraped from the flask, added into a 15-ml centrifuge tube, and centrifuged at 4,000 rpm for 15 min at 4°C to separate the cell debris. The same was done for the virus supernatant collected. Uninfected flasks were treated identically to generate control samples.
siRNA transfection.
A549 cells were seeded onto 96-well plates to a confluence of ∼104 cells/well in antibiotic-free DMEM containing 5% FBS and incubated at 37°C with 5% CO2 overnight. On the day of the assay, a SMARTpool ON-TARGETplus XPO1 siRNA (siXPO1) (Dharmacon L-003030-00-0005) or a control SMARTpool ON-TARGETplus nontargeting control pool siRNA (siNTC) (Dharmacon D-001810-10-20) was diluted to 5 μM in serum-free (SF) DMEM. Briefly, for each well to be transfected, 10 μl of diluted siRNA was prepared by mixing 0.5 μl of siRNA with 9.5 μl of SF-DMEM. Separately, 0.2 μl of Dharmafect-1 transfection reagent (Lafayette, CO) was mixed with 9.8 μl of SF-DMEM and incubated at room temperature for 5 min. The diluted siRNA and transfection reagent were mixed and incubated for 20 min at room temperature. Eighty microliters of antibiotic-free DMEM containing 5% FBS was added to the mix to achieve a final volume of 100 μl of transfection medium and a final siRNA concentration of 25 nM. The cell culture medium was removed from the 96-well plate containing the A549 cells and replaced by 100 μl of transfection medium. Finally, the cells were incubated for 24 to 48 h at 37°C in 5% CO2, at which point XPO1 expression was measured by immunostaining and Western blot assay as described below.
Immunostaining plaque assay.
Viral titers were determined as previously described (45). Briefly, 10-fold serial dilutions of whole-cell extracts of RSV-infected A549 cells were prepared in serum-free DMEM and added to confluent Vero cell monolayers in 24-well plates. After adsorption for 2 h at 37°C, 2 ml of overlay medium containing DMEM, 5% FBS, and 0.5% methylcellulose (Sigma St. Louis, MO, USA) was added to each well and incubated at 37°C for 3 days. At day 3 postinfection, overlay was carefully removed, and cells were fixed with ice-cold acetone-methanol (60/40%). RSV plaques were immunostained with a mouse monoclonal antibody directed against the RSV F protein (clone 131-2A) and a secondary antibody conjugated to alkaline phosphatase. Finally, plaques were visualized by addition of 1-Step nitroblue tetrazolium/5-bromo-4-chloro-3-indolylphosphate (NBT/BCIP) substrate solution (ThermoFisher Scientific) and enumerated using a dissecting microscope.
Western blotting.
(i) XPO1/p53-expression. Whole-cell extracts of RSV-infected A549 cells were collected at 24 h p.i. using lysis buffer (150 mM sodium chloride, 1.0% Triton X-100, 0.5% sodium deoxycholate, 10% sodium dodecyl sulfate [SDS], and 50 mM Tris, pH 8.0) treatment. The lysates were mixed with reducing SDS sample buffer (4× buffer contained 40% glycerol, 240 mM Tris-HCl, pH 6.8, 8% SDS, 0.04% bromophenol blue, and 5% β-mercaptoethanol), boiled, and electrophoresed on a 12% polyacrylamide gel; 10 μl of each sample was loaded per lane. XPO1 or p53 expression was detected by mouse anti-XPO1 (611832; BD Biosciences) or mouse anti-p53 (ab28; Abcam) diluted 1:1,000 in 1% skim milk in phosphate-buffered saline (pH 7.2) containing 0.1% Tween 20 (PBST). Bound primary antibody was detected with horseradish peroxidase-conjugated secondary antibodies diluted 1:5,000 in 1% skim milk in PBST. Bound antibodies were visualized by addition of SuperSignal West Dura Extended Duration Substrate (Pierce/Thermo Scientific) with a FluorChem-E Western imaging system (ProteinSimple).
(ii) Nuclear accumulation of proteins. A549 cells were infected with RSV A2 at an MOI of 0.5 for 2 h prior to being treated with 1 μM KPT-335, DMSO (diluent), or 750 nM LMB. Fractions were collected at 24 h p.i., utilizing an NE-PER nuclear and cytoplasmic purification kit (Thermo Scientific). Equivalent protein concentrations of cytoplasmic and nuclear fractions at 24 h p.i. were diluted in SDS sample buffer, boiled, and resolved by SDS-polyacrylamide gel electrophoresis, followed by immunoblotting. Antibodies against XPO1 (611832; BD Biosciences), RSV M protein (produced in-house), NF-κB subunit p65 (ab32536; Abcam), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (ab8245; Abcam), and lamin B1 (ab16048; Abcam) were used as primary antibodies. Horseradish peroxidase-conjugated species-specific antibodies were used as secondary antibody. Protein bands were visualized as for XPO1 and p53 above.
Cell viability.
A CellTiter Blue (Promega) cell viability assay was used for monitoring cell viability according to the manufacturer’s instructions. Briefly, 96-well plates containing A549 cells or BEAS-2B cells were treated with DMSO or increasing concentrations of KPT-335 for 72 h. At 70 h posttreatment, 20 μl of CellTiter Blue reagent was added to each well and incubated for 2 h at 37°C. The plate was mixed for 10 s, and fluorescence emission was measured at 560/590 nm. The average fluorescence value of the culture medium background (wells containing no cells) was subtracted from the value for each experimental well. The mean fluorescence emission values of the wells containing DMSO only was taken as 100% viability and used to calculate percent relative cell viability of KPT-335-treated wells. The percent viability versus the log10 concentration of KPT-335 was plotted using GraphPad Prism, and the values were fitted to a nonlinear regression curve to determine the 50% cytotoxic concentration (CC50).
Infection and immunofluorescence.
A549 cells were grown to 80% confluence on glass coverslips and infected (MOI of 0.5) with RSV A2. The cells were treated with KPT-335 (1 μM) for the time interval specified in the legend to Fig. 6. At the indicated times (Fig. 6) postinfection, cells were fixed with 4% formaldehyde, permeabilized with 0.1% Triton X-100, and immunostained with a monoclonal antibody specific for M protein (46) followed by Alexa Fluor 488-conjugated secondary antibody (Invitrogen). Hoechst 33342 (ThermoFisher) diluted in PBS was used for nuclear staining. The coverslips were washed twice in PBS and mounted using fluorescence mounting medium (Dako), followed by confocal laser scanning microscopy (CLSM; described below). Cells in 96-well plates that were transfected with siRNA and infected with RSV (described above) were fixed, permeabilized, and immunostained with a monoclonal antibody specific for F protein followed by Alexa Fluor 488-conjugated secondary antibody and Hoechst 33342 counterstaining. Cells were imaged by epifluorescence microscopy using a 10× objective.
Transfection.
Transfection of A549 cells was performed using Lipofectamine (Invitrogen) mixed 1:1 with DNA and reagent, and cells were cultured for 24 h before analysis by CLSM. The cells were transfected to express GFP (pGFP-DESTC), GFP-M (pEPI-GFP-M), or GFP-Rev [pEPI-GFP-REV(2-116)] (15). KPT-335 (1 μM) was added 18 h posttransfection for 6 or 24 h prior to live-cell CLSM and image analysis with cells treated identically with DMSO, taken as the control.
CLSM and image analysis.
Fixed or live (transfected)-cell samples were imaged as described previously (15, 47). Digitized fluorescent cell images were collected using a Nikon Ti Eclipse CLSM with a Nikon 60×/1.40 numeric aperture oil immersion lens (Plan Apo VC OFN25 DIC N2; optical section of 0.5 μm) and the NIS Elements AR software; data from four individual scans were averaged to obtain the final images. Digital images were analyzed as described previously using ImageJ software to determine the fluorescence intensity above background (Fb) in the nucleus (Fn) compared to that in the cytoplasm (c) and to determine the nuclear-to-cytoplasmic fluorescence ratio (Fn/c) (47).
Caspase-3/7 activity.
Caspase-3/7 activation was measured using a Capsase-Glo 3/7 assay (Promega) according to the manufacturer’s recommendations. Briefly, white-walled 96-well plates were seeded with 100 μl/well of A549 cells (105 cells/ml) and incubated at 37°C overnight. Cells were infected (MOI of 0.5) with RSV A2 for 24 h. KPT-335 and LMB treatment was initiated at 2 h p.i. and maintained until the end of the infection. Assay controls included wells without cells (background), mock-infected cells (negative control), and cells treated with 1 μM staurosporine for 4 h (positive control). At 24 h p.i., plates were equilibrated at room temperature for 30 min; 100 μl of Caspase-Glo 3/7 reagent was added to each well and incubated for 3 h at room temperature. Luminescence (in relative light units [RLU]) was measured using a Tecan microplate reader, and background RLU count (wells without cells) was subtracted from each well.
ELISA.
To assess the proinflammatory cytokine and common chemokine expression, supernatants were collected from A549 cells infected (MOI of 0.5) with RSV A2 or medium only (mock) for 2 h and then treated with 1 μM KPT-335, 750 nM LMB, or DMSO until 24 h p.i. Culture supernatants were collected and clarified by centrifugation before proinflammatory cytokine and chemokine protein expression was examined with a Multi-Analyte ELISArray kit (Qiagen). Fold-change was calculated relative to levels in mock-infected cell and compared to the levels in DMSO-treated cells.
Statistical analysis.
GraphPad Prism, version 7.0, was used for all statistical analysis. The statistical tests used to determine differences between groups are as indicated in the legends to respective figures, and significance was accepted at a P value of <0.05.
ACKNOWLEDGMENTS
We thank Karyopharm Therapeutics for kindly providing KPT-335 for the study.
This research was supported by an NIAID grant (R21-AI119903) and the Georgia Research Alliance.
REFERENCES
- 1.Iwane MK, Edwards KM, Szilagyi PG, Walker FJ, Griffin MR, Weinberg GA, Coulen C, Poehling KA, Shone LP, Balter S, Hall CB, Erdman DD, Wooten K, Schwartz B, New Vaccine SN. 2004. Population-based surveillance for hospitalizations associated with respiratory syncytial virus, influenza virus, and parainfluenza viruses among young children. Pediatrics 113:1758–1764. doi: 10.1542/peds.113.6.1758. [DOI] [PubMed] [Google Scholar]
- 2.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. 2005. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med 352:1749–1759. doi: 10.1056/NEJMoa043951. [DOI] [PubMed] [Google Scholar]
- 3.Falsey AR, Walsh EE, Looney RJ, Kolassa JE, Formica MA, Criddle MC, Hall WJ. 1999. Comparison of respiratory syncytial virus humoral immunity and response to infection in young and elderly adults. J Med Virol 59:221–226. doi:. [DOI] [PubMed] [Google Scholar]
- 4.Rima B, Collins P, Easton A, Fouchier R, Kurath G, Lamb RA, Lee B, Maisner A, Rota P, Wang L, ICTV Report Consortium . 2017. ICTV virus taxonomy profile: Pneumoviridae. J Gen Virol 98:2912–2913. doi: 10.1099/jgv.0.000959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collins PL. 2011. Human respiratory syncytial virus, p 341–410. In Samal SK. (ed), The biology of paramyxoviruses. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 6.Lamb RA, Parks GD. 2006. Paramyxoviridae: the viruses and their replication, p 1449–1496. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 7.Ghildyal R, Mills J, Murray M, Vardaxis N, Meanger J. 2002. Respiratory syncytial virus matrix protein associates with nucleocapsids in infected cells. J Gen Virol 83:753–757. doi: 10.1099/0022-1317-83-4-753. [DOI] [PubMed] [Google Scholar]
- 8.Lindquist ME, Lifland AW, Utley TJ, Santangelo PJ, Crowe JE. Jr, 2010. Respiratory syncytial virus induces host RNA stress granules to facilitate viral replication. J Virol 84:12274–12284. doi: 10.1128/JVI.00260-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghildyal R, Jans DA, Bardin PG, Mills J. 2012. Protein-protein interactions in RSV assembly: potential targets for attenuating RSV strains. Infect Disord Drug Targets 12:103–109. [DOI] [PubMed] [Google Scholar]
- 10.Marty A, Meanger J, Mills J, Shields B, Ghildyal R. 2004. Association of matrix protein of respiratory syncytial virus with the host cell membrane of infected cells. Arch Virol 149:199–210. doi: 10.1007/s00705-003-0183-9. [DOI] [PubMed] [Google Scholar]
- 11.Ghildyal R, Li D, Peroulis I, Shields B, Bardin PG, Teng MN, Collins PL, Meanger J, Mills J. 2005. Interaction between the respiratory syncytial virus G glycoprotein cytoplasmic domain and the matrix protein. J Gen Virol 86:1879–1884. doi: 10.1099/vir.0.80829-0. [DOI] [PubMed] [Google Scholar]
- 12.Li D, Jans DA, Bardin PG, Meanger J, Mills J, Ghildyal R. 2008. Association of respiratory syncytial virus M protein with viral nucleocapsids is mediated by the M2-1 protein. J Virol 82:8863–8870. doi: 10.1128/JVI.00343-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meshram CD, Baviskar PS, Ognibene CM, Oomens AG. 2016. The respiratory syncytial virus phosphoprotein, matrix protein, and fusion protein carboxy-terminal domain drive efficient filamentous virus-like particle formation. J Virol 90:10612–10628. doi: 10.1128/JVI.01193-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghildyal R, Baulch-Brown C, Mills J, Meanger J. 2003. The matrix protein of human respiratory syncytial virus localises to the nucleus of infected cells and inhibits transcription. Arch Virol 148:1419–1429. doi: 10.1007/s00705-003-0112-y. [DOI] [PubMed] [Google Scholar]
- 15.Ghildyal R, Ho A, Dias M, Soegiyono L, Bardin PG, Tran KC, Teng MN, Jans DA. 2009. The respiratory syncytial virus matrix protein possesses a Crm1-mediated nuclear export mechanism. J Virol 83:5353–5362. doi: 10.1128/JVI.02374-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghildyal R, Ho A, Wagstaff KM, Dias MM, Barton CL, Jans P, Bardin P, Jans DA. 2005. Nuclear import of the respiratory syncytial virus matrix protein is mediated by importin β1 independent of importin α. Biochemistry 44:12887–12895. doi: 10.1021/bi050701e. [DOI] [PubMed] [Google Scholar]
- 17.Aujard Y, Fauroux B. 2002. Risk factors for severe respiratory syncytial virus infection in infants. Respir Med 96(Suppl B):S9–S14. [PubMed] [Google Scholar]
- 18.Hussman JM, Lanctot KL, Paes B. 2013. The cost effectiveness of palivizumab in congenital heart disease: a review of the current evidence. J Med Econ 16:115–124. doi: 10.3111/13696998.2012.734886. [DOI] [PubMed] [Google Scholar]
- 19.Jorquera PA, Tripp RA. 2017. Respiratory syncytial virus: prospects for new and emerging therapeutics. Expert Rev Respir Med 11:609–615. doi: 10.1080/17476348.2017.1338567. [DOI] [PubMed] [Google Scholar]
- 20.Sullivan BJ, Treuhaft MW. 1987. Ribavirin for respiratory syncytial virus infection. Pediatr Infect Dis J 6:697–698. [DOI] [PubMed] [Google Scholar]
- 21.Breit MN, Kisseberth WC, Bear MD, Landesman Y, Kashyap T, McCauley D, Kauffman MG, Shacham S, London CA. 2014. Biologic activity of the novel orally bioavailable selective inhibitor of nuclear export (SINE) KPT-335 against canine melanoma cell lines. BMC Vet Res 10:160. doi: 10.1186/1746-6148-10-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Etchin J, Sanda T, Mansour MR, Kentsis A, Montero J, Le BT, Christie AL, McCauley D, Rodig SJ, Kauffman M, Shacham S, Stone R, Letai A, Kung AL, Thomas Look A. 2013. KPT-330 inhibitor of CRM1 (XPO1)-mediated nuclear export has selective anti-leukaemic activity in preclinical models of T-cell acute lymphoblastic leukaemia and acute myeloid leukaemia. Br J Haematol 161:117–127. doi: 10.1111/bjh.12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gravina GL, Mancini A, Sanita P, Vitale F, Marampon F, Ventura L, Landesman Y, McCauley D, Kauffman M, Shacham S, Festuccia C. 2015. KPT-330, a potent and selective exportin-1 (XPO-1) inhibitor, shows antitumor effects modulating the expression of cyclin D1 and survivin [corrected] in prostate cancer models. BMC Cancer 15:941. doi: 10.1186/s12885-015-1936-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.London CA, Bernabe LF, Barnard S, Kisseberth WC, Borgatti A, Henson M, Wilson H, Jensen K, Ito D, Modiano JF, Bear MD, Pennell ML, Saint-Martin JR, McCauley D, Kauffman M, Shacham S. 2014. Preclinical evaluation of the novel, orally bioavailable selective inhibitor of nuclear export (SINE) KPT-335 in spontaneous canine cancer: results of a phase I study. PLoS One 9:e87585. doi: 10.1371/journal.pone.0087585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng Y, Gery S, Sun H, Shacham S, Kauffman M, Koeffler HP. 2014. KPT-330 inhibitor of XPO1-mediated nuclear export has anti-proliferative activity in hepatocellular carcinoma. Cancer Chemother Pharmacol 74:487–495. doi: 10.1007/s00280-014-2495-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perwitasari O, Johnson S, Yan X, Howerth E, Shacham S, Landesman Y, Baloglu E, McCauley D, Tamir S, Tompkins SM, Tripp RA. 2014. Verdinexor, a novel selective inhibitor of nuclear export, reduces influenza a virus replication in vitro and in vivo. J Virol 88:10228–10243. doi: 10.1128/JVI.01774-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perwitasari O, Johnson S, Yan X, Register E, Crabtree J, Gabbard J, Howerth E, Shacham S, Carlson R, Tamir S, Tripp RA. 2016. Antiviral efficacy of Verdinexor in vivo in two animal models of influenza A virus infection. PLoS One 11:e0167221. doi: 10.1371/journal.pone.0167221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lundberg L, Pinkham C, de la Fuente C, Brahms A, Shafagati N, Wagstaff KM, Jans DA, Tamir S, Kehn-Hall K. 2016. Selective inhibitor of nuclear export (SINE) compounds alter new world alphavirus capsid localization and reduce viral replication in mammalian cells. PLoS Negl Trop Dis 10:e0005122. doi: 10.1371/journal.pntd.0005122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Golomb L, Bublik DR, Wilder S, Nevo R, Kiss V, Grabusic K, Volarevic S, Oren M. 2012. Importin 7 and exportin 1 link c-Myc and p53 to regulation of ribosomal biogenesis. Mol Cell 45:222–232. doi: 10.1016/j.molcel.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ranganathan P, Yu X, Na C, Santhanam R, Shacham S, Kauffman M, Walker A, Klisovic R, Blum W, Caligiuri M, Croce CM, Marcucci G, Garzon R. 2012. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood 120:1765–1773. doi: 10.1182/blood-2012-04-423160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mathew C, Ghildyal R. 2017. CRM1 inhibitors for antiviral therapy. Front Microbiol 8:1171. doi: 10.3389/fmicb.2017.01171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun Q, Carrasco YP, Hu Y, Guo X, Mirzaei H, Macmillan J, Chook YM. 2013. Nuclear export inhibition through covalent conjugation and hydrolysis of leptomycin B by CRM1. Proc Natl Acad Sci U S A 110:1303–1308. doi: 10.1073/pnas.1217203110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang K, Wang M, Tamayo AT, Shacham S, Kauffman M, Lee J, Zhang L, Ou Z, Li C, Sun L, Ford RJ, Pham LV. 2013. Novel selective inhibitors of nuclear export CRM1 antagonists for therapy in mantle cell lymphoma. Exp Hematol 41:67–78.e4. doi: 10.1016/j.exphem.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 34.Boons E, Vanstreels E, Jacquemyn M, Nogueira TC, Neggers JE, Vercruysse T, van den Oord J, Tamir S, Shacham S, Landesman Y, Snoeck R, Pannecouque C, Andrei G, Daelemans D. 2015. Human exportin-1 is a target for combined therapy of HIV and AIDS related lymphoma. EBioMedicine 2:1102–1113. doi: 10.1016/j.ebiom.2015.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan L, Coenjaerts FE, Houspie L, Viveen MC, van Bleek GM, Wiertz EJ, Martin DP, Lemey P. 2013. The comparative genomics of human respiratory syncytial virus subgroups A and B: genetic variability and molecular evolutionary dynamics. J Virol 87:8213–8226. doi: 10.1128/JVI.03278-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Henderson G, Murray J, Yeo RP. 2002. Sorting of the respiratory syncytial virus matrix protein into detergent-resistant structures is dependent on cell-surface expression of the glycoproteins. Virology 300:244–254. [DOI] [PubMed] [Google Scholar]
- 37.Bajorek M, Caly L, Tran KC, Maertens GN, Tripp RA, Bacharach E, Teng MN, Ghildyal R, Jans DA. 2014. The Thr205 phosphorylation site within respiratory syncytial virus matrix (M) protein modulates M oligomerization and virus production. J Virol 88:6380–6393. doi: 10.1128/JVI.03856-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henderson BR, Eleftheriou A. 2000. A comparison of the activity, sequence specificity, and CRM1-dependence of different nuclear export signals. Exp Cell Res 256:213–224. doi: 10.1006/excr.2000.4825. [DOI] [PubMed] [Google Scholar]
- 39.Xu D, Grishin NV, Chook YM. 2012. NESdb: a database of NES-containing CRM1 cargoes. Mol Biol Cell 23:3673–3676. doi: 10.1091/mbc.E12-01-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gravina GL, Tortoreto M, Mancini A, Addis A, Di Cesare E, Lenzi A, Landesman Y, McCauley D, Kauffman M, Shacham S, Zaffaroni N, Festuccia C. 2014. XPO1/CRM1-selective inhibitors of nuclear export (SINE) reduce tumor spreading and improve overall survival in preclinical models of prostate cancer (PCa). J Hematol Oncol 7:46. doi: 10.1186/1756-8722-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Christian F, Smith EL, Carmody RJ. 2016. The regulation of NF-kappaB subunits by phosphorylation. Cells 5:E12. doi: 10.3390/cells5010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guijarro C, Egido J. 2001. Transcription factor-kappa B (NF-kappa B) and renal disease. Kidney Int 59:415–424. doi: 10.1046/j.1523-1755.2001.059002415.x. [DOI] [PubMed] [Google Scholar]
- 43.Pickens JA, Tripp RA. 2018. Verdinexor targeting of CRM1 is a promising therapeutic approach against RSV and influenza viruses. Viruses 10:E48. doi: 10.3390/v10010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mohapatra SS, Boyapalle S. 2008. Epidemiologic, experimental, and clinical links between respiratory syncytial virus infection and asthma. Clin Microbiol Rev 21:495–504. doi: 10.1128/CMR.00054-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jorquera PA, Tripp RA. 2016. Quantification of RSV infectious particles by plaque assay and immunostaining assay. Methods Mol Biol 1442:33–40. doi: 10.1007/978-1-4939-3687-8_3. [DOI] [PubMed] [Google Scholar]
- 46.Orvell C, Norrby E, Mufson MA. 1987. Preparation and characterization of monoclonal antibodies directed against five structural components of human respiratory syncytial virus subgroup B. J Gen Virol 68:3125–3135. doi: 10.1099/0022-1317-68-12-3125. [DOI] [PubMed] [Google Scholar]
- 47.Walker E, Jensen L, Croft S, Wei K, Fulcher AJ, Jans DA, Ghildyal R. 2016. Rhinovirus 16 2A protease affects nuclear localization of 3CD during infection. J Virol 90:11032–11042. doi: 10.1128/JVI.00974-16. [DOI] [PMC free article] [PubMed] [Google Scholar]