

We have previously shown that Kaposi’s sarcoma-associated herpesvirus (KSHV)-transformed cells depend on glutamine rather than glucose for energy production and amino acid and nucleotide syntheses. In this study, we have further examined how the KSHV-reprogramed metabolic pathways are regulated and discovered that KSHV hijacks the citrulline-nitric oxide (NO) cycle to promote growth proliferation and transformation. Multiple KSHV-encoded microRNAs upregulate argininosuccinate synthase 1 (ASS1), a key enzyme in the citrulline-NO cycle. ASS1 is required for KSHV-induced proliferation, colony formation in soft agar, and NO generation of KSHV-transformed cells, which also depends on inducible nitric oxide synthase. By maintaining intracellular NO levels, ASS1 mediates KSHV activation of the STAT3 pathway, which is essential for KSHV-induced abnormal cell proliferation and transformation. These results illustrate a novel mechanism by which an oncogenic virus hijacks a key metabolic pathway to promote growth transformation and reveal a potential novel therapeutic target for KSHV-induced malignancies.
KEYWORDS: argininosuccinate synthase 1, ASS1, citrulline-nitric oxide cycle, inducible nitric oxide synthase, iNOS, KSHV, Kaposi’s sarcoma, nitric oxide, NO, STAT3, microRNA
ABSTRACT
Cancer cells are required to rewire existing metabolic pathways to support their abnormal proliferation. We have previously shown that, unlike glucose-addicted cancers, Kaposi’s sarcoma-associated herpesvirus (KSHV)-transformed cells depend on glutamine rather than glucose for energy production and amino acid and nucleotide syntheses. High-level consumption of glutamine is tightly regulated and often coupled with the citrulline-nitric oxide (NO) cycle. We have found that KSHV infection accelerates nitrogen efflux by upregulating the expression of argininosuccinate synthase 1 (ASS1), a key enzyme in the citrulline-NO cycle. KSHV utilizes multiple microRNAs to upregulate ASS1 expression. Depletion of either ASS1 or inducible nitric oxide synthase (iNOS) in KSHV-transformed cells suppresses growth proliferation, abolishes colony formation in soft agar, and decreases NO generation. Furthermore, by maintaining intracellular NO levels, ASS1 expression facilitates KSHV-mediated activation of the STAT3 pathway, which is critical for virus-induced transformation. These results illustrate a novel mechanism by which an oncogenic virus hijacks a key metabolic pathway to promote growth transformation and reveal a potential novel therapeutic target for KSHV-induced malignancies.
IMPORTANCE We have previously shown that Kaposi’s sarcoma-associated herpesvirus (KSHV)-transformed cells depend on glutamine rather than glucose for energy production and amino acid and nucleotide syntheses. In this study, we have further examined how the KSHV-reprogramed metabolic pathways are regulated and discovered that KSHV hijacks the citrulline-nitric oxide (NO) cycle to promote growth proliferation and transformation. Multiple KSHV-encoded microRNAs upregulate argininosuccinate synthase 1 (ASS1), a key enzyme in the citrulline-NO cycle. ASS1 is required for KSHV-induced proliferation, colony formation in soft agar, and NO generation of KSHV-transformed cells, which also depends on inducible nitric oxide synthase. By maintaining intracellular NO levels, ASS1 mediates KSHV activation of the STAT3 pathway, which is essential for KSHV-induced abnormal cell proliferation and transformation. These results illustrate a novel mechanism by which an oncogenic virus hijacks a key metabolic pathway to promote growth transformation and reveal a potential novel therapeutic target for KSHV-induced malignancies.
INTRODUCTION
Metabolism is a process that occurs in cells that utilize nutrients to generate macromolecules and energy for anabolic proliferation or homeostatic maintenance (1). Numerous intermediates, such as nitric oxide (NO) and reactive oxygen species (ROS), produced during metabolic processes can act as messengers to regulate cellular signaling and physiological functions (2, 3). In cancer cells, metabolism is commonly dysregulated as a result of aberrant expression of oncogenes and/or tumor suppressors (1).
Kaposi’s sarcoma-associated herpesvirus (KSHV) is an oncogenic gammaherpesvirus associated with four malignancies: Kaposi’s sarcoma (KS), primary effusion lymphoma (PEL), a subset of multicentric Castleman’s disease (MCD), and KSHV inflammatory cytokine syndrome (KICS) (4). KSHV latent infection is essential for the development of KSHV-induced malignancies. During KSHV latency, a small subset of KSHV genes is expressed, including vFLIP (ORF71), vCyclin (ORF72), LANA (ORF73), and a cluster of 12 precursor microRNAs (pre-miRNAs) (5). These latent genes play essential roles in KSHV-induced cellular transformation and tumorigenesis (5).
Viruses often induce metabolic alterations in host cells to facilitate their persistence and spread. KSHV infection alone is sufficient to trigger cellular metabolic reprogramming. KSHV infection or expression of KSHV-encoded miRNAs increases glucose consumption and the Warburg effect (6–9). Both KSHV-infected endothelial cells and PEL cells have increased lipogenesis (9, 10). In addition, KSHV-infected endothelial cells have increased levels of glutamine consumption and require glutaminolysis for survival (11, 12). However, in KSHV-transformed cells, the alterations of metabolic pathways are different from those in untransformed cells. KSHV-transformed cells consume glutamine and depend on glutamine but not glucose for proliferation and transformation (12, 13). In fact, there are reduced levels of glucose consumption and production of lactate in KSHV-transformed cells (13).
It has been reported that high-level consumption of glutamine is tightly regulated and often coupled with the citrulline-nitric oxide (citrulline-NO) cycle (14). In this study, we have shown that the citrulline-NO cycle is indeed reprogramed in KSHV-transformed cells. Two indispensable enzymes in the citrulline-NO cycle, argininosuccinate synthase 1 (ASS1) and inducible nitric oxide synthase (iNOS), are upregulated in KSHV-transformed cells. The coordinated expression of ASS1, iNOS, and argininosuccinate lyase (ASL) ensures the flow of the citrulline-NO cycle and NO production. iNOS converts arginine into citrulline, which is recycled back to replenish intracellular arginine for NO production through ASS1 and ASL (15–17).
Elevated expression levels of ASS1 have been reported in primary epithelial, ovarian, gastric, colorectal, and lung cancers (18–21). However, the consequences of ASS1 upregulation in these cancers are largely unknown. In contrast, ASS1 silencing through promoter methylation supports the proliferation of osteosarcoma by fostering de novo pyrimidine synthesis (22, 23). Loss of ASS1 causes genetic diseases, such as type I citrullinemia, as a result of decreased nitrogen flux, indicating a critical role of ASS1 in nitrogen metabolism, the citrulline-NO cycle, and NO production (17, 24).
Endogenous NO is an important gas signal transmitter synthesized from arginine by a family of NOSs and regulates diverse physiological processes (2, 3). Mammalian cells express three types of NOS: neuronal NOS (nNOS), iNOS, and endothelial NOS (eNOS) (25). nNOS and eNOS are constitutively and predominantly expressed in neuronal and vascular endothelial cells, respectively, whereas iNOS is often expressed in tumor cells. ASS1 is the rate-limiting enzyme for de novo arginine synthesis, and arginine is the unique substrate for NO production. Thus, ASS1-driven recycling of citrulline, which produces arginine, appears to be a prerequisite for all NO-producing cells (15, 26, 27). NO is known to exert its functions by activating the soluble guanylyl cyclase-cGMP pathways and causing direct nitrosylation or nitration of proteins, which can be either cytotoxic or tumorigenic (28–31).
STAT3, a transcription factor often activated in cancer cells, is important for the proliferation and survival of tumor cells and tumor angiogenesis, immunosuppression, and invasion (32, 33). Latently KSHV-infected cells have an increased phosphorylation level of STAT3 as a result of complement and Toll-like receptor 4 (TLR4) activation, which promotes the proliferation and survival of KSHV-infected cells (34, 35). Moreover, both NO donors and ASS1 have been shown to regulate STAT3 to either promote or inhibit cell proliferation and survival (21, 36, 37).
In this study, we have discovered that multiple KSHV-encoded miRNAs upregulate ASS1 expression to promote cell proliferation and transformation by inducing NO and STAT3 activation. Both ASS1 and iNOS are required for maintaining the intracellular NO level despite excessive extracellular arginine, a process which seems to be at odds with the arginine paradox (38). Additionally, our data indicated that NO production is required for STAT3 phosphorylation. These findings have demonstrated that the concomitant expression of iNOS and ASS1 is indispensable for NO generation and STAT3 activation.
RESULTS
ASS1 is upregulated in KSHV-infected and -transformed cells.
We have previously shown that KSHV-transformed cells depend on glutamine for anabolic proliferation (12). Because active glutamine consumption is tightly regulated, we hypothesized that KSHV-transformed cells would have an active citrulline-NO cycle. ASS1 has a critical role in the citrulline-NO cycle, and its expression is upregulated in several types of human cancer by an unknown mechanism (18). Thus, we examined ASS1 expression in KSHV-infected and -transformed cells. We have recently shown that KSHV can efficiently infect and transform primary rat embryonic metanephric mesenchymal (MM) cells, and KSHV-transformed MM (KMM) cells efficiently induce KS-like tumors in nude mice (39). Since this is the first efficient system of KSHV-induced cellular transformation of primary cells, which is useful for delineating the mechanism of KSHV-induced growth transformation, we examined the expression of ASS1 in this system. The expression of the ASS1 transcript was elevated by 108-fold in KMM cells compared to MM cells (Fig. 1A). To confirm upregulated ASS1 expression in KMM cells at the protein level, we tested several commercial ASS1 antibodies. However, none of them worked except one, which detected the ASS1 band and an additional nonspecific lower band. Using MM and KMM cells with and without overexpression of ASS1, we confirmed that the top band was indeed the ASS1 protein (Fig. 1B). As expected, the expression level of the ASS1 protein was much higher in KMM than in MM cells (Fig. 1C). We further examined ASS1 expression in other types of KSHV-infected cells. KSHV infection of a renal carcinoma cell line stably expressing doxycycline-inducible RTA (iSLK) upregulated ASS1 expression at both the transcript and protein levels albeit to a lesser extent, which was possibly due to the relatively high expression level of ASS1 in the uninfected iSLK cells (Fig. 1D). PEL is an aggressive B-cell non-Hodgkin’s lymphoma associated with KSHV infection (4). Several KSHV-infected PEL cell lines, including BCBL1, BC3, and BCP1, expressed high levels of ASS1 with an increase of transcript levels ranging from 1.6- to 7.0-fold compared to BJAB, an Epstein-Barr virus (EBV)- and KSHV-negative Burkitt’s lymphoma cell line. Interestingly, KSHV infection of BJAB cells increased the expression of the ASS1 transcript by 11.5-fold (Fig. 1E). These results were further confirmed at the protein level (Fig. 1E). Hence, latent KSHV infection upregulated ASS1 expression in different cell types. However, the extent of upregulation varied according to the individual types of cells.
FIG 1.

Multiple KSHV-encoded miRNAs mediate upregulation of ASS1 expression. (A) The ASS1 transcript is upregulated in KSHV-transformed cells. RT-qPCR was used to examine the level of the ASS1 transcript. (B) Validation of the specificity of ASS1 antibody. Western blotting was used to examine MM and KMM cells stably expressing ASS1 or a vector control. The intensity of the upper band was dramatically increased in cells expressing ASS1 but not the vector control, indicating that the upper band is specific for the ASS1 protein. (C) ASS1 protein is upregulated in KSHV-transformed cells. Western blotting was used to examine the level of ASS1 protein. (D and E) ASS1 transcript and protein are upregulated in KSHV-infected cells. RT-qPCR and Western blot results show that the expression of ASS1 was upregulated at both the mRNA and protein levels in iSLK-KSHV cells (D) and BJAB-KSHV cells (E) compared to uninfected cells and in PEL cells compared to BJAB cells (E). (F and G) The KSHV miRNA cluster is required for the upregulation of ASS1 expression. Shown are data from analysis of ASS1 expression in cells infected by different recombinant viruses, including wild-type KSHV (KMM) and a mutant with a deletion of a cluster of 10 pre-miRNAs (ΔmiRs), vFLIP (ΔvFLIP), or vCyclin (ΔvCyclin), by RT-qPCR (F) and Western blotting (G). (H and I) Expression of the KSHV miRNA cluster is sufficient to upregulate ASS1 expression. ASS1 expression was detected by Western blotting (H) and RT-qPCR (I) in MM cells and ΔmiRs cells overexpressing the KSHV miRNA cluster or the control empty vector pITA (MM+pITA). (J) Expression of multiple individual miRNAs is sufficient to upregulate ASS1 expression. ASS1 protein was detected by Western blotting in ΔmiRs cells overexpressing individual miRNAs. β-Actin and β-tubulin were used as internal controls for RT-qPCR and Western blotting, respectively. ** and *** represent P values of <0.01 and <0.001, respectively, while NS indicates not significant.
Multiple KSHV-encoded miRNAs upregulate ASS1 expression.
Most KSHV-transformed cells are latently infected by KSHV and mainly express viral latent genes, including LANA, vCyclin, vFLIP, and a cluster of 12 pre-miRNAs (39). To identify which viral product is responsible for ASS1 upregulation in KMM cells, we examined MM cells latently infected by a KSHV mutant with a deletion of either vFLIP, vCyclin, or a cluster of 10 of the 12 pre-miRNAs (miR-K1 to -9 and -11), named ΔvFLIP, ΔvCyclin, and ΔmiRs, respectively (13, 40–42). We were not able to obtain MM cells latently infected by the KSHV mutant with LANA deleted or mutated because of LANA’s essential role in viral genome persistence (43, 44). Deletion of vFLIP or vCyclin had a minimal effect on ASS1 expression (Fig. 1F and G). In contrast, deletion of the miRNA cluster completely abolished ASS1 expression at both the transcript and protein levels (Fig. 1F and G). Expression of the miRNA cluster in ΔmiRs cells partially rescued ASS1 expression (Fig. 1H and I). Interestingly, expression of the miRNA cluster in MM cells was sufficient to upregulate ASS1 expression although to a lesser extent than in KMM cells (Fig. 1H and I). These results indicated that KSHV miRNAs were required for KSHV-induced ASS1 upregulation. However, other KSHV genes might also contribute to the maximal induction of ASS1 expression. To identify the individual miRNA(s) that might mediate KSHV-induced ASS1 upregulation, we examined ASS1 expression in ΔmiRs cells expressing individual miRNAs (miR-K1 to -12) (45, 46). Western blot results showed that numerous miRNAs, including miR-K2, -K6, -K7, -K8, -K9, -K10, -K11, and -K12, partially upregulated ASS1 expression in ΔmiRs cells (Fig. 1J).
Suppression of ASS1 inhibits KSHV-induced cell proliferation and cellular transformation.
To explore the role of ASS1 in KSHV-transformed cells, we used 3 different short hairpin RNAs (shRNAs) to deplete ASS1 expression. The shRNAs achieved approximately 80% knockdown efficiencies at the RNA level, which were confirmed at the protein level (Fig. 2A and B). Depletion of ASS1 expression with the 3 shRNAs reduced cell proliferation by 45%, 54%, and 85% in KMM cells, respectively, and by 33%, 41%, and 58% in MM cells, respectively (Fig. 2C). Depletion of ASS1 expression also significantly reduced the efficiencies of colony formation of KMM cells in a soft agar assay as shown by the reduced sizes and numbers of colonies (Fig. 2D). As expected, MM cells with and without depletion of ASS1 expression failed to form any colonies in soft agar assays (Fig. 2D). Taken together, these results demonstrated that ASS1 was essential for KSHV-induced cell proliferation and cellular transformation.
FIG 2.

ASS1 is essential for KSHV-induced cell proliferation and cellular transformation. (A and B) Analysis of ASS1 expression in MM and KMM cells following depletion of ASS1 expression by RT-qPCR (A) and Western blotting (B). (C and D) ASS1 knockdown inhibits cell proliferation and cellular transformation of KMM cells. Cell proliferation (C) and colony formation in soft agar (D) were examined following knockdown of ASS1 with 3 shRNAs (sh1, sh2, or sh3) or a scrambled shRNA (control [ctl]). Representative pictures at a ×40 magnification from soft agar assays are shown (D, left). Colonies with diameters of >50 μm were counted, and the relative number of colonies per field was quantified (D, right). (E and F) ASS1 knockdown induces cell cycle arrest and apoptosis. (E) The cell cycle was analyzed by flow cytometry 48 h following transduction with ASS1 shRNAs (shASS1). (F) Apoptotic cells were detected by annexin V staining 72 h following transduction with ASS1 shRNAs. *, **, and *** represent P values of <0.05, <0.01, and <0.001, respectively, while NS indicates “not significant.”
To further investigate how ASS1 might regulate KSHV-induced cell proliferation and cellular transformation, we examined cell cycle progression and apoptosis. Depletion of ASS1 expression with 3 shRNAs induced cell cycle arrest in KMM cells by increasing the percentage of G1-phase cells from 50.6% to 55.3 to 70.5% and decreasing the percentage of S-phase cells from 36.5% to 10.4 to 20.9% (Fig. 2E). For MM cells, the percentage of G1-phase cells was increased from 51.1% to 57.9 to 69.7%, while the percentage of S-phase cells was decreased from 35.5% to 7.3 to 27.2% (Fig. 2E). Depletion of ASS1 expression also significantly increased the numbers of dead cells from 5.3% to 15.4 to 52.7% in KMM cells and from 9.1% to 16 to 27.3% in MM cells (Fig. 2F). Overall, depletion of ASS1 expression suppressed the proliferation of KSHV-transformed cells by inducing both cell cycle arrest and apoptosis, while there was a lesser effect on untransformed cells.
Inhibition of iNOS induces cell cycle arrest and apoptosis of KSHV-transformed cells.
ASS1 is a metabolic enzyme involved in the citrulline-NO cycle, arginine metabolism, and NO synthesis (16, 17, 38). To delineate the mechanism by which ASS1 regulates KSHV-induced growth proliferation, we complemented medium with various ASS1 downstream metabolites to determine which of them could rescue the effect of ASS1 knockdown. Altogether, we tested polyamines, arginine, glutamine, asparagine, fumarate, proline, α-ketoglutarate, and their different combinations, but none of them showed any rescue effect on KMM cells with ASS1 knockdown (data not shown). Another metabolite related to ASS1 is NO. However, due to the short half-life of commercially available NO donors, we could not perform NO rescue experiments.
Alternatively, if ASS1 impacts the proliferation of KSHV-transformed cells by regulating NO production, knockdown of the enzymes catalyzing NO production should induce a phenotype similar to that with ASS1 depletion. There are three different NOSs in mammalian cells: iNOS, nNOS, and eNOS (2, 3). We first examined the expression levels of these three NOSs in KMM cells. Surprisingly, KSHV infection had no effect on the expression of eNOS and increased the expression of nNOS by only 2-fold (Fig. 3A). In contrast, KSHV infection increased the expression of iNOS by 7-fold, suggesting that iNOS might play a role in KSHV-transformed cells (Fig. 3A). We then performed knockdown of iNOS to examine its role in KSHV-induced growth transformation (Fig. 3B and C). Depletion of iNOS expression suppressed the growth proliferation of both MM and KMM cells (Fig. 3D). Depletion of iNOS expression significantly inhibited the colony formation efficiencies of KMM cells in a soft agar assay (Fig. 3E). Upon iNOS depletion, the percentages of G1-phase cells increased from 51.9% to 61 to 73.1% and from 48.1% to 70.1 to 72.5% in MM and KMM cells, respectively. Accordingly, the percentages of S-phase cells were reduced from 31.35% to 9.5 to 12.9% and from 35.6% to 8.6 to 9.7% in both MM and KMM cells, respectively (Fig. 3F). The results of apoptosis assays showed that the percentages of dead cells were increased from 11.0% to 33.0 to 40.0% in MM cells and from 6.23% to 24.3 to 37.3% in KMM cells (Fig. 3G). Taken together, these data showed that depletion of iNOS expression gave phenotypes similar to those with depletion of ASS1 expression, indicating that ASS1 and iNOS might function through the same downstream effector, NO.
FIG 3.

iNOS is essential for KSHV-induced cell proliferation and cellular transformation. (A) Examination of eNOS, iNOS, or nNOS expression by RT-qPCR in MM and KMM cells. (B and C) Analysis of iNOS expression in MM and KMM cells following iNOS knockdown by RT-qPCR (B) and Western blotting (C). (D and E) iNOS knockdown inhibits cell proliferation and cellular transformation of KMM cells. Cell proliferation (D) and colony formation in soft agar (E) were determined following depletion of iNOS expression with 3 shRNAs (sh1, sh2, or sh3) or a scrambled shRNA (ctl). Representative pictures at a ×40 magnification from soft agar assays are shown (E, left). Colonies with diameters of >50 μm were counted, and the results were graphed (E, right). (F and G) iNOS knockdown induces cell cycle arrest and apoptosis. (F) The cell cycle was analyzed by flow cytometry 48 h following transduction with iNOS shRNAs. (G) Apoptotic cells were detected by annexin V staining 72 h following transduction with ASS1 shRNAs. *, **, and *** represent P values of <0.05, <0.01, and <0.001, respectively, while NS indicates “not significant.”
Knockdown of ASS1 or iNOS reduces the intracellular NO level.
ASS1 is the rate-limiting enzyme for de novo arginine synthesis, providing an essential substrate for NO production (16, 17, 38). The reduced expression of ASS1 or iNOS can decrease NO production despite an excessive extracellular arginine supply (15, 26, 47). To evaluate the roles of ASS1 and iNOS in NO production in KSHV-transformed cells, the endogenous NO level was measured in MM and KMM cells with or without depletion of ASS1 or iNOS expression using DAR-4M AM (DAR), which is a cell-permeable diaminorhodamine-based dye emitting red fluorescence upon reacting with NO in the presence of ROS (48–50) (Fig. 4A). Since the presence of ROS is a prerequisite for DAR to emit fluorescence when reacting with NO, we first compared ROS between MM and KMM cells. Surprisingly, MM cells had much a higher intracellular ROS level than that of KMM cells (Fig. 4B). However, treatment with S-nitroso-N-acetyl-d,l-penicillamine (SNAP), a known NO donor, had no effect on endogenous ROS in both MM and KMM cells (Fig. 4C), indicating that DAR could be used as a reliable sensor for endogenous NO detection after depletion of ASS1 expression in these cells. Depletion of either iNOS or ASS1 expression indeed led to a dramatic decrease in intracellular NO production in both MM and KMM cells (Fig. 5A to D). Nω-nitro-l-arginine methyl ester (L-NAME) inhibits the generation of NO by inhibiting iNOS function but not iNOS expression (51). Treatment with L-NAME inhibited NO production in both MM and KMM cells (Fig. 6A and B). As expected, L-NAME also reduced cell proliferation of both MM and KMM cells in a dose-dependent manner (Fig. 6C), which was consistent with the results of iNOS knockdown (Fig. 3B to D). Collectively, our results demonstrated that ASS1 and iNOS were necessary for endogenous NO generation, which was essential for the proliferation of KSHV-transformed cells.
FIG 4.

Detection of intracellular NO levels with DAR is not altered by different intracellular ROS levels. (A) Chemical equation showing that the reaction of DAR with NO requires ROS. (B) MM cells have higher intracellular ROS levels than KMM cells. (C) Flow cytometry analysis showing that the NO donor SNAP does not alter intracellular ROS levels in MM and KMM cells. APC-A indicates allophycocyanin (APC) fluorescent intensity.
FIG 5.

Silencing of ASS1 or iNOS expression reduces the intracellular production of NO. (A and B) Detection of NO with DAR in MM and KMM cells following knockdown of ASS1 with 3 shRNAs (sh1, sh2, or sh3) or a scrambled shRNA (ctl). The intracellular NO level was examined with a fluorescence microscope (A) and quantified for relative intensity with ImageJ (B). (C and D) Detection of NO with DAR in MM and KMM cells following knockdown of iNOS with 3 shRNAs (sh1, sh2, or sh3) or a scrambled shRNA (ctl). The intracellular NO level was detected with a fluorescence microscope (C), and the relative intensity was quantified with ImageJ (D). CTCF represents the corrected total cell fluorescence by ImageJ. * and ** represent P values of <0.05 and <0.01, respectively, while NS indicates “not significant.”
FIG 6.
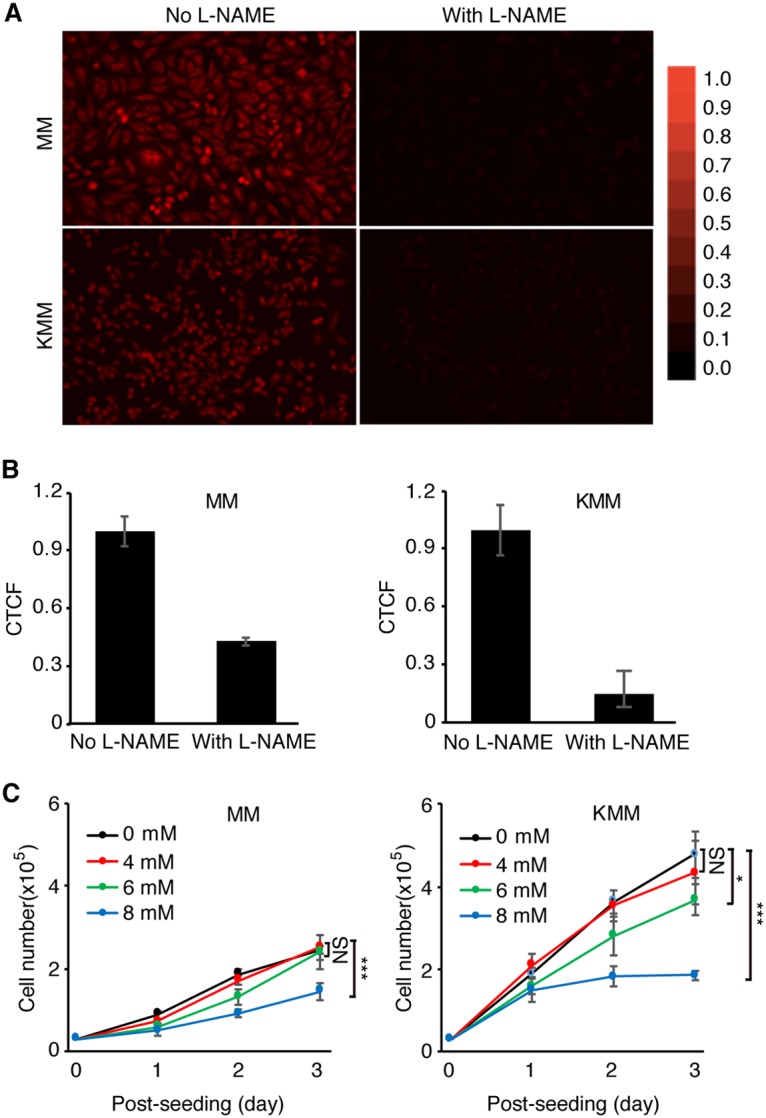
The iNOS inhibitor L-NAME reduces intracellular NO levels and inhibits cell proliferation of MM and KMM cells. (A) Detection of intracellular NO levels with DAR in MM and KMM cells with or without treatment with the NO donor L-NAME. Representative images were captured at a ×20 magnification with a fluorescence microscope. (B) Quantification of intracellular NO levels in MM and KMM cells with ImageJ. CTCF represents the corrected total cell fluorescence by ImageJ. (C) Inhibition of cell proliferation of MM and KMM cells by L-NAME. Numbers of MM and KMM cells treated with 0, 4, 6, or 8 mM L-NAME were counted over time. * and *** represent P values of <0.05 and <0.001, respectively, while NS indicates “not significant.”
NO produced through the ASS1-iNOS cycle activates STAT3 to promote the proliferation of KSHV-transformed cells.
ASS1 expression has been reported to correlate with STAT3 protein expression in human gastric cancer cells by an unknown mechanism (21). We observed a marginal increase of STAT3 protein expression in KMM cells compared to MM cells (Fig. 7A). STAT3 plays a crucial role in promoting tumor progression and thus is a favorable target for cancer therapy (52, 53). In latently KSHV-infected and -transformed cells, it has been shown that STAT3 is activated with increased phosphorylation at Tyr705 through the activation of alternative complement and TLR4 pathways and that STAT3 activation is essential for the survival of KSHV-transformed cells (34, 35). Since NO has been shown to activate STAT3 in ovarian cancer cells (37), we hypothesized that STAT3 might be a potential common downstream target of NO, ASS1, and iNOS. Depletion of ASS1 expression reduced the phosphorylation of STAT3 (Tyr705) in KMM cells, suggesting that STAT3 was regulated by ASS1 in KSHV-transformed cells (Fig. 7A).
FIG 7.

NO mediates ASS1 and iNOS induction of STAT3 activation. (A) Depletion of ASS1 expression inhibits STAT3 tyrosine phosphorylation. MM and KMM cells were transduced with 3 ASS1 shRNAs (sh1, sh2, or sh3) or a scrambled shRNA (ctl) and examined for the levels of total and phosphorylated STAT3 (Y705) by Western blotting. The ASS1 protein level was also examined to monitor knockdown efficiencies, while β-tubulin was used as a loading control. (B and C) The NO donor SNAP increases intracellular NO levels. MM and KMM cells were treated with 0.5 mM SNAP for 1 h and examined for intracellular NO by DAR staining. The intracellular NO level was examined with a fluorescence microscope (B), and the relative intensity was quantified with ImageJ (C). (D) ASS1 knockdown has no effect on ROS production. MM and KMM cells transduced with 3 ASS1 shRNAs (sh1, sh2, or sh3) or a scrambled shRNA (ctl) for 2 days were examined for intracellular ROS levels. (E) The NO donor SNAP partially rescues STAT3 activation following ASS1 knockdown. MM and KMM cells transduced with an ASS1 shRNA (shRNA3) or a scrambled shRNA (ctl) for 2 days were treated with 0.5 mM SNAP for 0.5 h and then examined for the levels of total and phosphorylated STAT3 (Y705) by Western blotting. β-Tubulin was used as a loading control. APC-A indicates allophycocyanin (APC) fluorescent intensity.
To determine if NO mediated ASS1-induced STAT3 activation, we searched for a method to increase intracellular NO levels. Since SNAP is a known NO donor, we determined whether SNAP could increase intracellular NO levels. We treated MM and KMM cells with SNAP and then measured NO by fluorescence live-cell imaging following treatment with DAR. As expected, stronger fluorescent DAR signals were observed in the SNAP-treated cells than in untreated cells (Fig. 7B and C). To rule out the possibility that SNAP might influence ROS and thereby give rise to stronger fluorescence, we checked the intracellular ROS level by flow cytometry after SNAP administration. The results showed that there was no change of intracellular ROS levels before or after SNAP treatment (Fig. 7D), indicating that SNAP is an effective donor for NO in MM and KMM cells.
Finally, we examined if NO mediated ASS1-induced STAT3 activation. We treated MM and KMM cells with SNAP after depletion of ASS1 expression and then examined STAT3 phosphorylation. As expected, SNAP partially rescued the decreased phosphorylation of STAT3 caused by depletion of ASS1 expression, which implied that NO mediated STAT3 activation in KSHV-transformed cells (Fig. 7E). However, SNAP failed to rescue the reduced cell proliferation rate caused by depletion of ASS1 expression (results not shown), which was possibly due to its short half-life of 4 h. Taken together, these results indicated that STAT3 activation in KSHV-transformed cells was at least partially dependent on ASS1-mediated NO production.
Glucose metabolism does not regulate ASS1 and intracellular NO and vice versa.
We have previously shown that KSHV inhibits aerobic glycolysis to promote cell survival in KSHV-transformed cells by downregulating the expression levels of GLUT1 and GLUT3 (13). Our results so far showed the importance of ASS1 upregulation and the citrulline-NO cycle in KSHV-transformed cells. Thus, we further examined the role of ASS1 in the expression of GLUT1 and GLUT3 in KSHV-transformed cells. Knockdown of ASS1, which also led to a reduced level of intracellular NO (Fig. 5C and D), did not alter the levels of GLUT1 and GLUT3 (Fig. 8A). Overexpression of ASS1 also did not alter the levels of GLUT1 and GLUT3 (Fig. 8B). Hence, it is unlikely that ASS1 and the citrulline-NO cycle regulate the expression of GLUT1 and GLUT3 and glucose metabolism. Similarly, glucose deprivation, which increases glutamine uptake (12), did not alter ASS1 expression and intracellular NO levels (Fig. 8C and D). Therefore, glucose metabolism does not regulate ASS1 expression and the citrulline-NO cycle.
FIG 8.
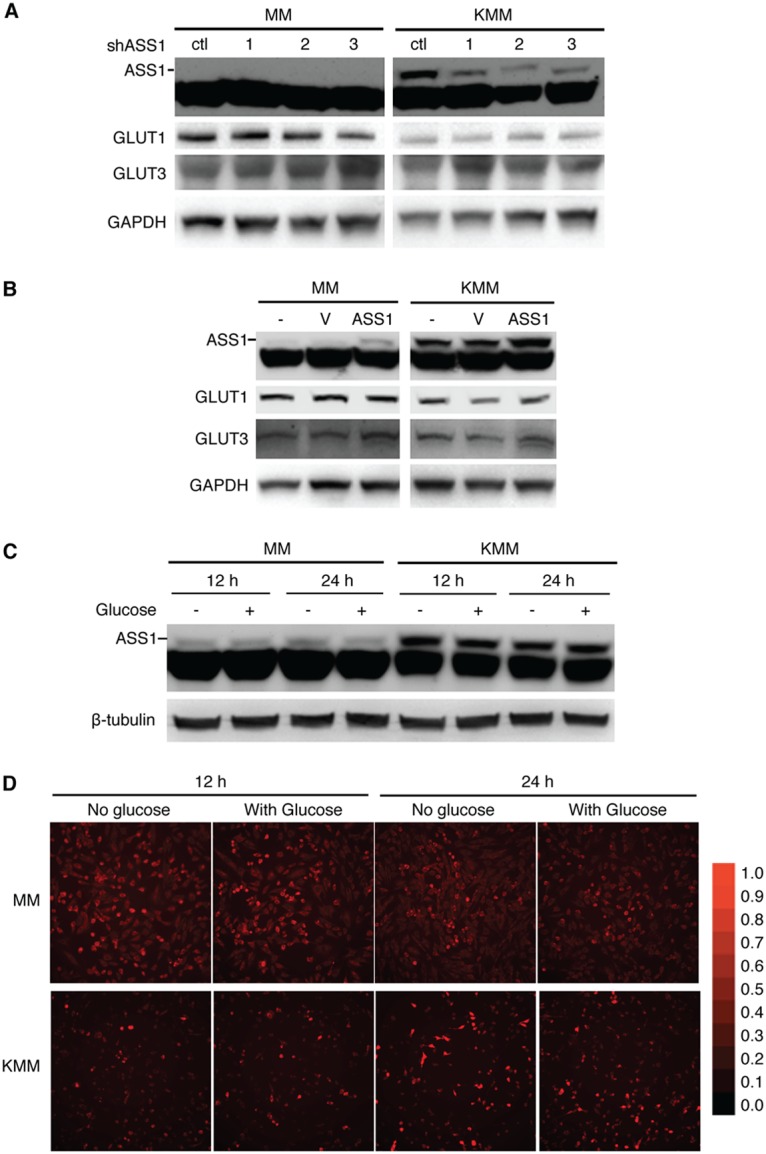
ASS1 does not regulate the expression of GLUT1 and GLUT3, and glucose deprivation does not affect ASS1 protein and intracellular NO levels. (A) ASS1 knockdown has no effect on the expression of GLUT1 and GLUT3 proteins. MM and KMM cells transduced with 3 ASS1 shRNAs (shRNA1, -2, or -3) or a scrambled shRNA (ctl) for 2 days were examined for the levels of GLUT1 and GLUT3 proteins. (B) Overexpression of ASS1 has no effect on the expression of GLUT1 and GLUT3 proteins. MM and KMM cells overexpressing ASS1 or a vector control (V) for 2 days were examined for the levels of GLUT1 and GLUT3 proteins. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (C and D) Glucose deprivation does not affect ASS1 protein and intracellular NO levels. MM and KMM cells were seeded overnight in full medium, cultured in medium with and without glucose for 12 and 24 h, and examined for the level of ASS1 protein (C) or intracellular NO (D).
DISCUSSION
We have previously shown that KSHV suppresses glucose uptake and aerobic glycolysis but upregulates glutamine metabolism to promote cell survival and growth proliferation in KSHV-transformed cells (12, 13). In this report, we have demonstrated that KSHV regulates ASS1 expression to maintain NO production, which is essential for the survival and proliferation of KSHV-transformed cells. Depletion of ASS1 expression significantly reduced intracellular NO levels, and knockdown of iNOS gave a phenotype similar to that of depletion of ASS1 expression. Additionally, ASS1 silencing decreased STAT3 activation, which was partially rescued by NO. Taken together, these results reveal a novel mechanism by which an oncogenic virus rewires metabolic pathways to support the growth proliferation and survival of KSHV-transformed cells by sustaining NO generation and STAT3 activation (Fig. 9).
FIG 9.
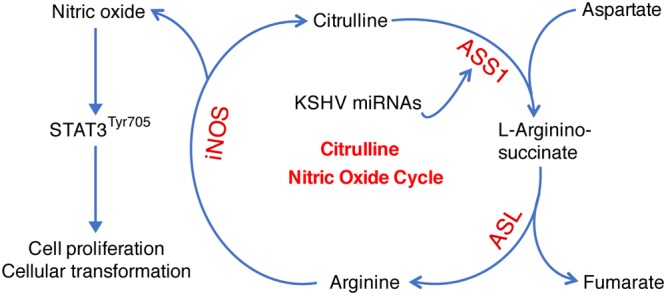
Working model illustrating that KSHV hijacks the citrulline-NO cycle to promote KSHV-induced cell proliferation and cellular transformation. Multiple KSHV miRNAs upregulate ASS1 expression to maintain intracellular NO levels and STAT3 activation, which is essential for KSHV-induced cell proliferation and cellular transformation.
ASS1 is a rate-limiting enzyme in the citrulline-NO cycle, arginine synthesis, and NO production (16, 26, 54). Previous studies have shown that ASS1 expression is upregulated in several types of cancer, but the role of ASS1 in cancer as well as the molecular basis mediating ASS1 upregulation remain unclear (18–21). In KSHV-transformed cells, we found that numerous KSHV-encoded miRNAs were sufficient for upregulating ASS1 expression but to much lesser extents than the whole virus (Fig. 1). The contributions of these miRNAs to ASS1 upregulation also vary with individual miRNAs. Whether they have any synergistic effects remains to be further investigated. Significantly, we showed that ASS1 was required for the proliferation and colony formation of KSHV-transformed cells in soft agar (Fig. 2). These findings are consistent with the results of another study showing that inhibition of ASS1 results in decreased proliferation and tumorigenicity of colorectal cancer (55). Interestingly, ASS1 expression is downregulated in several types of cancer cells, leading to the increased dependence of tumor cells on exogenous arginine and, hence, enhanced sensitivity to arginine deprivation (56, 57). However, the significance of ASS1 loss in cancer is currently unclear.
By far, the only known function of ASS1 is to recycle citrulline to synthesize argininosuccinate that is further converted into arginine by ASL to provide the substrate for NO production. Although the extracellular arginine concentration is much higher than the reported Km of arginine for iNOS, NO generation still depends on the availability of intracellular arginine in multiple cell lines (26, 54, 58, 59). Results of a previous microarray study showed that ASS1 upregulation is positively correlated with NO production, suggesting a possible role of ASS1 in NO generation (60). Our results demonstrated that ASS1 was required for NO generation in KSHV-transformed cells. Suppressing either ASS1 or iNOS expression significantly reduced the NO concentration in vitro (Fig. 5). Interestingly, we did not detect a higher concentration of NO in KMM cells than in MM cells despite higher expression levels of both ASS1 and iNOS in KMM cells (Fig. 6A) (50). We speculate that higher levels of ASS1 and the citrulline-NO cycle are required for maintaining the intracellular NO level, which is essential for sustaining the proliferation of KSHV-transformed cells. Indeed, knockdown of iNOS reduced the intracellular NO level as well as cell proliferation (Fig. 3B to D and Fig. 5A and B), indicating that the normal flow of the citrulline-NO cycle carried out by iNOS and ASS1 is indispensable for the proliferation of KSHV-transformed cells. Paradoxically, overexpression of ASS1 did not further increase the intracellular NO level (results not shown), suggesting that ASS1 needs to cooperate with other components of the citrulline-NO cycle. As excess NO might be toxic to the cells (2, 3), maintaining the homeostasis of NO could maximize the survival of KSHV-transformed cells.
NO is a multifunctional regulator implicated in diverse physiological and pathological processes (30, 61, 62). A known NO donor, SNAP can both activate and inactivate STAT3 (36, 37, 51), but there is no report that has linked ASS1 to STAT3 activation. Our results showed that STAT3 was inactivated following depletion of ASS1 expression, which was partially rescued by SNAP (Fig. 7E). These results linked ASS1 to STAT3 activation, which was mediated by NO. Our previous studies have shown that STAT3 activation is essential for the survival and proliferation of KSHV-transformed cells (35). Hence, the effect of ASS1 depletion on reduced KSHV-induced growth proliferation and transformation was likely due to the inactivation of STAT3 resulting from the decreased intracellular NO level. Our results, for the first time, demonstrated that STAT3 activation was closely regulated by ASS1, NO, and hence the citrulline-NO cycle. Further mechanistic studies are required to delineate the mechanism by which NO mediates STAT3 activation.
Interestingly, we did not find a role of ASS1 or the citrulline-NO cycle in regulating the expression of GLUT1 and GLUT3 even though they are downregulated in KSHV-transformed cells (12). Glucose deprivation also did not alter ASS1 expression and intracellular NO levels. These results are expected, as the glutamine pathway is not reprogrammed in MM cells, while KMM cells are already reprogrammed by KSHV to utilize glutamine with or without glucose deprivation (13). Hence, these are the distinct properties of MM and KMM cells that define their primary and transformed features, respectively.
In summary, we have shown that KSHV miRNAs upregulate ASS1 expression in KSHV-transformed cells, resulting in enhanced cell proliferation and cellular transformation by regulating NO-mediated STAT3 activation (Fig. 9). These findings might also be relevant for other types of cancer that have dysregulated ASS1 expression.
MATERIALS AND METHODS
Cell culture and reagents.
Rat primary embryonic metanephric mesenchymal (MM) cells; KSHV-transformed MM (KMM) cells (39); MM cells infected by a KSHV mutant with a cluster of 10 precursor miRNAs deleted (ΔmiRs) (41), vFLIP deleted (ΔvFLIP) (42), or vCyclin deleted (ΔvCyclin) (40); MM cells infected by ΔmiRs complemented with individual miRNAs (45, 46); and HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich St. Louis, MO) and antibiotics (100 μg/ml penicillin and 100 μg/ml streptomycin). SNAP (NO donor) was purchased from Cayman Chemical. ROS detection dye was purchased from Thermo Scientific (catalog number C10422).
Knockdown of cellular genes with lentiviral shRNAs.
The ASS1 shRNAs were prepared as previously described (12). The shRNA DNA fragments for ASS1 and iNOS were ligated into the pLKO1 lentiviral vector. The target sequences for ASS1 included GCTCGCAAACAAGTGGAAATT for shRNA1, GCACATCCTTGGACCTCTTCA for shRNA2, and GCATGGATGAGAACCTTATGC for shRNA3. The target sequences for iNOS included GCACAGAATGTTCCAGAATCC for shRNA1, GCATATCTGCAGACACATACT for shRNA2, and GCTGAAATCCCTCCAGAATCT for shRNA3. The sequence for a scrambled shRNA is TTGTACTACACAAAAGTACTG. To package the lentiviruses, a vector expressing shRNA of the scrambled control, ASS1, or NOS2 was cotransfected with pMDLg/pRRE, pRSVRev, and pMD2.G packaging plasmids into exponential-phase HEK293T cells using Lipofectamine 2000 transfection reagent. At day 3 posttransfection, the supernatant of HEK293T cells was collected and then filtered. Targeted cells were transduced with the lentiviruses by spinning infection at 1,500 rpm for 1 h in the presence of 10 μg/ml Polybrene. The knockdown efficiency was confirmed by both real-time reverse transcription-quantitative qPCR (RT-qPCR) and Western blotting after day 3 posttransduction.
Soft agar colony assay.
A soft agar colony assay was performed as previously described (39). Generally, a total of 2 × 104 cells suspended in 1 ml of 0.3% top agar (catalog number A5431; Sigma-Aldrich) were plated onto 1 well of 0.5% base agar in 6-well plates. After 2 to 3 weeks, colonies with diameters of >50 μm were counted and photographed with a microscope.
RT-qPCR.
Total RNA was isolated with Tri reagent (catalog number T9424; Sigma) according to the instructions of the manufacturer. Reverse transcription was performed with total RNA using a Maxima H Minus first-strand cDNA synthesis kit (catalog number K1652; Thermo Fisher Scientific, Waltham, MA). qPCR analysis was performed on an Eppendorf Real Plex system using Kapa SYBR fast qPCR kits (catalog number KK4602; Kapa Biosystems, Wilmington, MA). The relative expression levels of genes were normalized with the internal control β-actin, which yielded a 2−ΔΔCT value. All reactions were run in triplicates. The primers for rat ASS1 were rat ASS1-F (CTGGAGGATGCCCGAGTTTT) and rat ASS1-R (TCCAGGATTCGAGCCTGGTA), the primers for rat iNOS were rat iNOS-F (CACCTTGGAGTTCACCCAGT) and rat iNOS-R (ACCACTCGTACTTGGGATGC), and the primers for rat β-actin were rat β-actin-F (CCATGTACCCAGGCATTGCT) and rat β-actin-R (AGCCACCAATCCACACAGAG).
Western blot analysis.
Total cell lysates were separated in SDS-polyacrylamide gels and electrophoretically transferred to nitrocellulose membranes (GE Healthcare, Pasadena, CA). The membranes were then blocked with 5% nonfat milk for 1 h and sequentially incubated with primary and secondary antibodies. The signal was revealed with the Luminiata Crescendo Western HRP substrate (catalog number WBLUR0500; EMD Millipore, San Diego, CA) and imaged with a UVP BioSpectrum imaging system (UVP, LLC, Upland, CA). The antibodies used for Western blotting included mouse monoclonal antibodies (mAbs) for ASS1 (catalog number ab124465; Abcam), STAT3 (catalog number 9139; Cell Signaling Technology), and β-tubulin (clone 7B9; Sigma); a rabbit polyclonal antibody to iNOS (catalog number sc-651; Santa Cruz); and a rabbit monoclonal antibody to phospho-STAT3 (catalog number 9145; Cell Signaling Technology).
Cell cycle analysis, apoptosis assay, and bromodeoxyuridine incorporation.
Cell cycle analysis was performed by propidium iodide (PI) staining as previously described (13, 63). Apoptotic cells were detected by eFluor 660 fixable viability dye staining (catalog number 650864; eBioscience, San Diego, CA) and with a phycoerythrin (PE)-Cy7 annexin V apoptosis detection set (catalog number 88810374; eBioscience) according to the instructions of the manufacturer. Samples were examined with a FACSCanto system (BD Biosciences, San Jose, CA) and analyzed with FlowJo (FlowJo, LLC, Ashland, OR).
Live-cell imaging.
Live-cell imaging was performed to detect intracellular NO as previously described (48). Cells grown on a 24-well plate at 37°C in 5% CO2 were treated with 5 μM DAR-4M AM for 1 h at 37°C, followed by washing with phosphate-buffered saline (PBS) four times for 5 min each time at room temperature. Plates were then examined with a Nikon Eclipse Ti-S fluorescence microscope (Nikon instruments Inc., Melville, NY, USA). Imaging was performed with a 20× objective. ImageJ was used for fluorescence quantification.
ROS detection.
CellROX deep red reagent (catalog number C10422; Thermo Fisher) is a fluorogenic probe designed to measure ROS in living cells. The cell-permeable CellROX deep red dye is a nonfluorescent dye in a reduced state but exhibits fluorescence upon oxidation with maximal excitation/emission wavelengths at 640/665 nm. Cells were incubated with CellROX deep red reagent at a final concentration of 5 μM for 30 min at 37°C. The cells were washed with PBS three times and examined with a FACSCanto II flow cytometer.
Statistical analysis.
A two-tailed t test or F-test of equality of variances was performed on results, and a P value of <0.05 was considered statistically significant.
ACKNOWLEDGMENTS
We thank members of the Gao laboratory for technical assistance and helpful discussions.
This work was in part supported by grants from the NIH to S.-J. Gao (CA096512, CA124332, CA132637, CA177377, CA213275, DE025465, and CA197153) and J. U. Jung (CA180779, CA082057, HL110609, DE023926, AI073099, and AI116585), grants from the National Natural Science Foundation of China (81730062 and 81761128003) and Nanjing Medical University (KY101RC1710) to C. Lu, and a grant from the National Natural Science Foundation of China (31701256) to Y. Zhu.
REFERENCES
- 1.Cairns RA, Harris IS, Mak TW. 2011. Regulation of cancer cell metabolism. Nat Rev Cancer 11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 2.Somasundaram V, Nadhan R, Hemalatha SK, Kumar Sengodan S, Srinivas P. 2016. Nitric oxide and reactive oxygen species: clues to target oxidative damage repair defective breast cancers. Crit Rev Oncol Hematol 101:184–192. doi: 10.1016/j.critrevonc.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Keshet R, Erez A. 2018. Arginine and the metabolic regulation of nitric oxide synthesis in cancer. Dis Model Mech 11:dmm033332. doi: 10.1242/dmm.033332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhutani M, Polizzotto MN, Uldrick TS, Yarchoan R. 2015. Kaposi’s sarcoma-associated herpesvirus-associated malignancies: epidemiology, pathogenesis, and advances in treatment. Semin Oncol 42:223–246. doi: 10.1053/j.seminoncol.2014.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ye F, Lei X, Gao SJ. 2011. Mechanisms of Kaposi’s sarcoma-associated herpesvirus latency and reactivation. Adv Virol 2011:193860. doi: 10.1155/2011/193860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delgado T, Carroll PA, Punjabi AS, Margineantu D, Hockenbery DM, Lagunoff M. 2010. Induction of the Warburg effect by Kaposi’s sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proc Natl Acad Sci U S A 107:10696–10701. doi: 10.1073/pnas.1004882107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma T, Patel H, Babapoor-Farrokhran S, Franklin R, Semenza GL, Sodhi A, Montaner S. 2015. KSHV induces aerobic glycolysis and angiogenesis through HIF-1-dependent upregulation of pyruvate kinase 2 in Kaposi’s sarcoma. Angiogenesis 18:477–488. doi: 10.1007/s10456-015-9475-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yogev O, Lagos D, Enver T, Boshoff C. 2014. Kaposi’s sarcoma herpesvirus microRNAs induce metabolic transformation of infected cells. PLoS Pathog 10:e1004400. doi: 10.1371/journal.ppat.1004400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhatt AP, Jacobs SR, Freemerman AJ, Makowski L, Rathmell JC, Dittmer DP, Damania B. 2012. Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma. Proc Natl Acad Sci U S A 109:11818–11823. doi: 10.1073/pnas.1205995109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delgado T, Sanchez EL, Camarda R, Lagunoff M. 2012. Global metabolic profiling of infection by an oncogenic virus: KSHV induces and requires lipogenesis for survival of latent infection. PLoS Pathog 8:e1002866. doi: 10.1371/journal.ppat.1002866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanchez EL, Carroll PA, Thalhofer AB, Lagunoff M. 2015. Latent KSHV infected endothelial cells are glutamine addicted and require glutaminolysis for survival. PLoS Pathog 11:e1005052. doi: 10.1371/journal.ppat.1005052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Y, Li T, Ramos da Silva S, Lee JJ, Lu C, Eoh H, Jung JU, Gao SJ. 2017. A critical role of glutamine and asparagine gamma-nitrogen in nucleotide biosynthesis in cancer cells hijacked by an oncogenic virus. mBio 8:e01179-17. doi: 10.1128/mBio.01179-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu Y, Ramos da Silva S, He M, Liang Q, Lu C, Feng P, Jung JU, Gao SJ. 2016. An oncogenic virus promotes cell survival and cellular transformation by suppressing glycolysis. PLoS Pathog 12:e1005648. doi: 10.1371/journal.ppat.1005648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maestri NE, McGowan KD, Brusilow SW. 1992. Plasma glutamine concentration: a guide in the management of urea cycle disorders. J Pediatr 121:259–261. doi: 10.1016/S0022-3476(05)81200-4. [DOI] [PubMed] [Google Scholar]
- 15.Qualls JE, Subramanian C, Rafi W, Smith AM, Balouzian L, DeFreitas AA, Shirey KA, Reutterer B, Kernbauer E, Stockinger S, Decker T, Miyairi I, Vogel SN, Salgame P, Rock CO, Murray PJ. 2012. Sustained generation of nitric oxide and control of mycobacterial infection requires argininosuccinate synthase 1. Cell Host Microbe 12:313–323. doi: 10.1016/j.chom.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haines RJ, Pendleton LC, Eichler DC. 2011. Argininosuccinate synthase: at the center of arginine metabolism. Int J Biochem Mol Biol 2:8–23. [PMC free article] [PubMed] [Google Scholar]
- 17.Husson A, Brasse-Lagnel C, Fairand A, Renouf S, Lavoinne A. 2003. Argininosuccinate synthetase from the urea cycle to the citrulline-NO cycle. Eur J Biochem 270:1887–1899. doi: 10.1046/j.1432-1033.2003.03559.x. [DOI] [PubMed] [Google Scholar]
- 18.Szlosarek PW, Grimshaw MJ, Wilbanks GD, Hagemann T, Wilson JL, Burke F, Stamp G, Balkwill FR. 2007. Aberrant regulation of argininosuccinate synthetase by TNF-alpha in human epithelial ovarian cancer. Int J Cancer 121:6–11. doi: 10.1002/ijc.22666. [DOI] [PubMed] [Google Scholar]
- 19.Wu MS, Lin YS, Chang YT, Shun CT, Lin MT, Lin JT. 2005. Gene expression profiling of gastric cancer by microarray combined with laser capture microdissection. World J Gastroenterol 11:7405–7412. doi: 10.3748/wjg.v11.i47.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delage B, Fennell DA, Nicholson L, McNeish I, Lemoine NR, Crook T, Szlosarek PW. 2010. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int J Cancer 126:2762–2772. doi: 10.1002/ijc.25202. [DOI] [PubMed] [Google Scholar]
- 21.Shan YS, Hsu HP, Lai MD, Yen MC, Chen WC, Fang JH, Weng TY, Chen YL. 2015. Argininosuccinate synthetase 1 suppression and arginine restriction inhibit cell migration in gastric cancer cell lines. Sci Rep 5:9783. doi: 10.1038/srep09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rabinovich S, Adler L, Yizhak K, Sarver A, Silberman A, Agron S, Stettner N, Sun Q, Brandis A, Helbling D, Korman S, Itzkovitz S, Dimmock D, Ulitsky I, Nagamani SC, Ruppin E, Erez A. 2015. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 527:379–383. doi: 10.1038/nature15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Syed N, Langer J, Janczar K, Singh P, Lo Nigro C, Lattanzio L, Coley HM, Hatzimichael E, Bomalaski J, Szlosarek P, Awad M, O’Neil K, Roncaroli F, Crook T. 2013. Epigenetic status of argininosuccinate synthetase and argininosuccinate lyase modulates autophagy and cell death in glioblastoma. Cell Death Dis 4:e458. doi: 10.1038/cddis.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beaudet AL, O’Brien WE, Bock HG, Freytag SO, Su TS. 1986. The human argininosuccinate synthetase locus and citrullinemia. Adv Hum Genet 15:161–196, 291–292. [DOI] [PubMed] [Google Scholar]
- 25.Fukumura D, Kashiwagi S, Jain RK. 2006. The role of nitric oxide in tumour progression. Nat Rev Cancer 6:521–534. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 26.Goodwin BL, Solomonson LP, Eichler DC. 2004. Argininosuccinate synthase expression is required to maintain nitric oxide production and cell viability in aortic endothelial cells. J Biol Chem 279:18353–18360. doi: 10.1074/jbc.M308160200. [DOI] [PubMed] [Google Scholar]
- 27.Flam BR, Eichler DC, Solomonson LP. 2007. Endothelial nitric oxide production is tightly coupled to the citrulline-NO cycle. Nitric Oxide 17:115–121. doi: 10.1016/j.niox.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 28.Friebe A, Koesling D. 2003. Regulation of nitric oxide-sensitive guanylyl cyclase. Circ Res 93:96–105. doi: 10.1161/01.RES.0000082524.34487.31. [DOI] [PubMed] [Google Scholar]
- 29.Stamler JS, Lamas S, Fang FC. 2001. Nitrosylation. The prototypic redox-based signaling mechanism. Cell 106:675–683. doi: 10.1016/S0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 30.Lala PK, Chakraborty C. 2001. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol 2:149–156. doi: 10.1016/S1470-2045(00)00256-4. [DOI] [PubMed] [Google Scholar]
- 31.Ekmekcioglu S, Tang CH, Grimm EA. 2005. NO news is not necessarily good news in cancer. Curr Cancer Drug Targets 5:103–115. doi: 10.2174/1568009053202072. [DOI] [PubMed] [Google Scholar]
- 32.Yu H, Jove R. 2004. The STATs of cancer—new molecular targets come of age. Nat Rev Cancer 4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 33.Buettner R, Mora LB, Jove R. 2002. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res 8:945–954. [PubMed] [Google Scholar]
- 34.Gruffaz M, Vasan K, Tan B, Ramos da Silva S, Gao SJ. 2017. TLR4-mediated inflammation promotes KSHV-induced cellular transformation and tumorigenesis by activating the STAT3 pathway. Cancer Res 77:7094–7108. doi: 10.1158/0008-5472.CAN-17-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee MS, Jones T, Song DY, Jang JH, Jung JU, Gao SJ. 2014. Exploitation of the complement system by oncogenic Kaposi’s sarcoma-associated herpesvirus for cell survival and persistent infection. PLoS Pathog 10:e1004412. doi: 10.1371/journal.ppat.1004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kielbik M, Klink M, Brzezinska M, Szulc I, Sulowska Z. 2013. Nitric oxide donors: spermine/NO and diethylenetriamine/NO induce ovarian cancer cell death and affect STAT3 and AKT signaling proteins. Nitric Oxide 35:93–109. doi: 10.1016/j.niox.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 37.Huang K, Li LA, Meng YG, You YQ, Fu XY, Song L. 2014. Arctigenin promotes apoptosis in ovarian cancer cells via the iNOS/NO/STAT3/survivin signalling. Basic Clin Pharmacol Toxicol 115:507–511. doi: 10.1111/bcpt.12270. [DOI] [PubMed] [Google Scholar]
- 38.Morris SM., Jr. 2007. Arginine metabolism: boundaries of our knowledge. J Nutr 137:1602S–1609S. doi: 10.1093/jn/137.6.1602S. [DOI] [PubMed] [Google Scholar]
- 39.Jones T, Ye F, Bedolla R, Huang Y, Meng J, Qian L, Pan H, Zhou F, Moody R, Wagner B, Arar M, Gao SJ. 2012. Direct and efficient cellular transformation of primary rat mesenchymal precursor cells by KSHV. J Clin Invest 122:1076–1081. doi: 10.1172/JCI58530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones T, Ramos da Silva S, Bedolla R, Ye F, Zhou F, Gao SJ. 2014. Viral cyclin promotes KSHV-induced cellular transformation and tumorigenesis by overriding contact inhibition. Cell Cycle 13:845–858. doi: 10.4161/cc.27758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moody R, Zhu Y, Huang Y, Cui X, Jones T, Bedolla R, Lei X, Bai Z, Gao SJ. 2013. KSHV microRNAs mediate cellular transformation and tumorigenesis by redundantly targeting cell growth and survival pathways. PLoS Pathog 9:e1003857. doi: 10.1371/journal.ppat.1003857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ye FC, Zhou FC, Xie JP, Kang T, Greene W, Kuhne K, Lei XF, Li QH, Gao SJ. 2008. Kaposi’s sarcoma-associated herpesvirus latent gene vFLIP inhibits viral lytic replication through NF-kappaB-mediated suppression of the AP-1 pathway: a novel mechanism of virus control of latency. J Virol 82:4235–4249. doi: 10.1128/JVI.02370-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Q, Zhou F, Ye F, Gao SJ. 2008. Genetic disruption of KSHV major latent nuclear antigen LANA enhances viral lytic transcriptional program. Virology 379:234–244. doi: 10.1016/j.virol.2008.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ye FC, Zhou FC, Yoo SM, Xie JP, Browning PJ, Gao SJ. 2004. Disruption of Kaposi’s sarcoma-associated herpesvirus latent nuclear antigen leads to abortive episome persistence. J Virol 78:11121–11129. doi: 10.1128/JVI.78.20.11121-11129.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou FC, Zhang YJ, Deng JH, Wang XP, Pan HY, Hettler E, Gao SJ. 2002. Efficient infection by a recombinant Kaposi’s sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J Virol 76:6185–6196. doi: 10.1128/JVI.76.12.6185-6196.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lei X, Bai Z, Ye F, Xie J, Kim CG, Huang Y, Gao SJ. 2010. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat Cell Biol 12:193–199. doi: 10.1038/ncb2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mun GI, Kim IS, Lee BH, Boo YC. 2011. Endothelial argininosuccinate synthetase 1 regulates nitric oxide production and monocyte adhesion under static and laminar shear stress conditions. J Biol Chem 286:2536–2542. doi: 10.1074/jbc.M110.180489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pandit L, Kolodziejska KE, Zeng S, Eissa NT. 2009. The physiologic aggresome mediates cellular inactivation of iNOS. Proc Natl Acad Sci U S A 106:1211–1215. doi: 10.1073/pnas.0810968106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steinert JR, Kopp-Scheinpflug C, Baker C, Challiss RA, Mistry R, Haustein MD, Griffin SJ, Tong H, Graham BP, Forsythe ID. 2008. Nitric oxide is a volume transmitter regulating postsynaptic excitability at a glutamatergic synapse. Neuron 60:642–656. doi: 10.1016/j.neuron.2008.08.025. [DOI] [PubMed] [Google Scholar]
- 50.Lacza Z, Horvath EM, Pankotai E, Csordas A, Kollai M, Szabo C, Busija DW. 2005. The novel red-fluorescent probe DAR-4M measures reactive nitrogen species rather than NO. J Pharmacol Toxicol Methods 52:335–340. doi: 10.1016/j.vascn.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 51.Pan X, Wang X, Lei W, Min L, Yang Y, Wang X, Song J. 2009. Nitric oxide suppresses transforming growth factor-beta1-induced epithelial-to-mesenchymal transition and apoptosis in mouse hepatocytes. Hepatology 50:1577–1587. doi: 10.1002/hep.23156. [DOI] [PubMed] [Google Scholar]
- 52.Herrmann A, Kortylewski M, Kujawski M, Zhang C, Reckamp K, Armstrong B, Wang L, Kowolik C, Deng J, Figlin R, Yu H. 2010. Targeting Stat3 in the myeloid compartment drastically improves the in vivo antitumor functions of adoptively transferred T cells. Cancer Res 70:7455–7464. doi: 10.1158/0008-5472.CAN-10-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. 2008. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest 118:3367–3377. doi: 10.1172/JCI35213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Solomonson LP, Flam BR, Pendleton LC, Goodwin BL, Eichler DC. 2003. The caveolar nitric oxide synthase/arginine regeneration system for NO production in endothelial cells. J Exp Biol 206:2083–2087. doi: 10.1242/jeb.00361. [DOI] [PubMed] [Google Scholar]
- 55.Bateman LA, Ku WM, Heslin MJ, Contreras CM, Skibola CF, Nomura DK. 2017. Argininosuccinate synthase 1 is a metabolic regulator of colorectal cancer pathogenicity. ACS Chem Biol 12:905–911. doi: 10.1021/acschembio.6b01158. [DOI] [PubMed] [Google Scholar]
- 56.Feun LG, Marini A, Walker G, Elgart G, Moffat F, Rodgers SE, Wu CJ, You M, Wangpaichitr M, Kuo MT, Sisson W, Jungbluth AA, Bomalaski J, Savaraj N. 2012. Negative argininosuccinate synthetase expression in melanoma tumours may predict clinical benefit from arginine-depleting therapy with pegylated arginine deiminase. Br J Cancer 106:1481–1485. doi: 10.1038/bjc.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Szlosarek PW, Klabatsa A, Pallaska A, Sheaff M, Smith P, Crook T, Grimshaw MJ, Steele JP, Rudd RM, Balkwill FR, Fennell DA. 2006. In vivo loss of expression of argininosuccinate synthetase in malignant pleural mesothelioma is a biomarker for susceptibility to arginine depletion. Clin Cancer Res 12:7126–7131. doi: 10.1158/1078-0432.CCR-06-1101. [DOI] [PubMed] [Google Scholar]
- 58.Wu G, Morris SM Jr.. 1998. Arginine metabolism: nitric oxide and beyond. Biochem J 336:1–17. doi: 10.1042/bj3360001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xie L, Gross SS. 1997. Argininosuccinate synthetase overexpression in vascular smooth muscle cells potentiates immunostimulant-induced NO production. J Biol Chem 272:16624–16630. doi: 10.1074/jbc.272.26.16624. [DOI] [PubMed] [Google Scholar]
- 60.McCormick SM, Eskin SG, McIntire LV, Teng CL, Lu CM, Russell CG, Chittur KK. 2001. DNA microarray reveals changes in gene expression of shear stressed human umbilical vein endothelial cells. Proc Natl Acad Sci U S A 98:8955–8960. doi: 10.1073/pnas.171259298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bogdan C. 2015. Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol 36:161–178. doi: 10.1016/j.it.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 62.Serio R, Zizzo MG, Mule F. 2003. Nitric oxide induces muscular relaxation via cyclic GMP-dependent and -independent mechanisms in the longitudinal muscle of the mouse duodenum. Nitric Oxide 8:48–52. doi: 10.1016/S1089-8603(02)00144-1. [DOI] [PubMed] [Google Scholar]
- 63.Li Y, Qin J, Wei X, Liang G, Shi L, Jiang M, Xia T, Liang X, He M, Zhang Z. 2016. Association of SIRT6 gene polymorphisms with human longevity. Iran J Public Health 45:1420–1426. [PMC free article] [PubMed] [Google Scholar]