

Abstract
For decades, symptoms of depression have been treated primarily with medications that directly target the monoaminergic brain systems, which typically take weeks to exert measurable effects and months to exert remission of symptoms. Low, subanesthetic doses of (R,S)-ketamine (ketamine) result in the rapid improvement of core depressive symptoms, including mood, anhedonia, and suicidal ideation, occurring within hours following a single administration, with relief from symptoms typically lasting up to a week. The discovery of these actions of ketamine has resulted in a reconceptualization of how depression could be more effectively treated in the future. In this review, we discuss clinical data pertaining to ketamine and other rapid-acting antidepressant drugs, as well as the current state of pharmacological knowledge regarding their mechanism of action. Additionally, we discuss the neurobiological circuits that are engaged by this drug class and that may be targeted by a future generation of medications, for example, hydroxynorketamine; metabotropic glutamate receptor 2/3 antagonists; and N-methyl-D-aspartate, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, and γ-aminobutyric acid receptor modulators.
Keywords: depression, antidepressant, ketamine, hydroxynorketamine, glutamate, AMPA receptor, NMDA receptor
INTRODUCTION
Depression is a chronic, severe, and often life-threatening affliction. A diverse combination of symptoms can characterize depression, including changes in mood (affect), sleep, appetite, cognition, and motivation, as well as a decreased capacity to experience pleasure (anhedonia) or thoughts of suicide. Major depressive disorder (MDD) is operationally, but not biologically, defined as having a sufficient number of these symptoms, and significant distress or impairment in functioning, for at least two weeks. This diagnosis occurs in approximately 16% of the population over the course of a lifetime (1). For decades, depressive symptoms have been primarily treated with medications that target the serotonin and norepinephrine brain systems. These drugs include monoamine oxidase inhibitors and tricyclic antidepressants, as well as the subsequently developed (in the 1980s) selective serotonin and serotonin/norepinephrine reuptake inhibitors (SSRIs and SNRIs), all of which result in increased synaptic levels of serotonin and/or norepinephrine. It is generally believed that the increase in synaptic monoamines initiates a slowly evolving cascade of changes in synaptic strength within mood-regulating brain circuits, resulting in antidepressant actions (2–5).
These traditional treatments for depression typically take weeks to exert measurable effects in depressed individuals and months to achieve full remission of symptoms. In addition to this lag time for treatment effectiveness, up to 30% of depressed patients remain depressed despite treatment with multiple structurally distinct medications (6), which is an outcome that defines treatment-resistant depression (TRD). The lack of response when subsequent medications are used is not altogether surprising considering that the available medications all target the same monoamine neurotransmitter systems. In essence, a noneffective medication is typically replaced by another medication that exerts similar pharmacological effects. A nonpharmacological treatment, electroconvulsive therapy (ECT), is traditionally viewed as having the greatest efficacy in TRD, although a significant number of patients remain refractory even to ECT (7).
While waiting and hoping for current treatments to take effect, individuals continue to experience suicidal thoughts or actions. The potential severe consequences of this lag time, as well as the high nonresponse rate, emphasize the need for better, faster-acting medications to treat these serious and life-threatening psychiatric manifestations of depression. A new class of antidepressant medication that can act rapidly and effectively would offer significant advantages. Ideally, a rapid-acting antidepressant drug would exert its effects within hours or days, as opposed to weeks or months. Historically, ECT, when efficacious, has a faster onset than monoamine-targeting antidepressants (8). Sleep deprivation is another treatment with a rapid onset of antidepressant action. One complete night of sleep deprivation results in alleviation of depressive symptoms in greater than 50% of patients with TRD (9). Thus, the concept that individuals with TRD can experience rapid remission of symptoms is not entirely new. However, practical application of sleep deprivation is limited by the procedure itself and, even more so, by the temporary nature of the recovery, as the effect is lost with sleep recovery (9). ECT is similarly limited by the time-consuming nature of the procedure; requirements for anesthesia, physiological monitoring, and, typically, a specialized clinic; cognitive deficits that are a common side effect; the percentage of patients that remain refractory after treatment; and the fact that a typical course requires multiple treatments to be effective (10).
The discovery of the rapid antidepressant efficacy of (R,S)-ketamine (referred to as ketamine below) has resulted in a reconceptualization of how depression could be treated and studied in the future. In this review, we discuss clinical data pertaining to rapid-acting antidepressant drugs, including ketamine, as well as the current state of pharmacological knowledge regarding their mechanism of action. Additionally, we discuss the neurobiological circuits that are engaged by rapid-acting antidepressants and that may be targeted by a future generation of medications.
CLINICAL STUDIES WITH RAPID-ACTING ANTIDEPRESSANTS
For more than 50 years, ketamine has been safely used to induce anesthesia (11) and for procedural sedation in children in emergency medical settings (12). Over the past two decades, multiple controlled studies have described rapid, robust, and relatively sustained antidepressant responses following a single infusion of subanesthetic ketamine to patients with treatment-refractory MDD (13–19). For instance, individuals with TRD were relieved of their symptoms following slow intravenous administration of 0.5 mg/kg ketamine; at 4, 24, and 72 hours, response rates were 50%, 70%, and 35%, respectively (14, 17). Similar effects have been noted in individuals with bipolar depression treated concomitantly with mood stabilizers (lithium and valproate) (20, 21). A meta-analysis of seven studies encompassing a total of 147 patients treated with ketamine found that a single ketamine infusion had rapid antidepressant effects. Odds ratios for response and the remission of symptoms at 24 hours were 9.87 [95%, confidence interval (CI): 4.37–22.29] and 14.47 (95%, CI: 2.67–78.49), respectively (22). No other treatment for depression—especially for TRD—has demonstrated comparable effects in terms of the magnitude of response following a single administration.
Notable from a public health perspective, ketamine has also been found to have rapid and significant antisuicidal effects in clinical trials (23–27). Ketamine’s therapeutic action for the rapid reduction in suicidal thoughts remained significant when controlling for the effects of ketamine on other depressive symptoms (28). Ketamine also rapidly improves anhedonia in depressed patients (29, 30). The reduction in thoughts of suicide associated with ketamine treatment is correlated with the antianhedonic response, independent of depression improvement (31). These data have led to ketamine’s off-label use and the emergence of specialized ketamine clinics and emergency room protocols to treat severely depressed and suicidal individuals (32). We do note that, to date, most studies with ketamine have only documented these rapid effects occurring in TRD. This does not mean that ketamine is not an effective antidepressant in non-TRD, but rather reflects the fact that this exploratory treatment has not been thoroughly assessed in non-TRD populations. However, independent of efficacy in non-TRD, ketamine may be reserved in its use for those who have failed to respond to existing treatments, considering the fact that its side effect profile typically limits use to specialized clinics (33). Ketamine, even when administered at antidepressant subanesthetic doses, leads to dissociative and psychotomimetic effects, likely mediated by its inhibitory actions at the N-methyl-D-aspartate receptor (NMDAR) (34).
In addition to the studies conducted with racemic ketamine [(R,S)-ketamine], several phase II clinical trials examining the efficacy of intravenous or intranasal (S)-ketamine (sometimes referred to as esketamine) have been completed. One study found that an infusion of either 0.20 mg/kg or 0.40 mg/kg of (S)-ketamine led to robust, rapid (<2 hours), and sustained (>4 days) antidepressant effects when compared to placebo (35). In another study, 67 subjects with TRD received 28 mg, 56 mg, or 84 mg of intranasal (S)-ketamine. On day 8 following treatment, changes in Montgomery Asberg Depression Rating Scale (MADRS) total scores were statistically superior to placebo in all three (S)-ketamine treatment groups, with a significant ascending dose–response relationship (36).
While ketamine’s antidepressant effects have been consistently replicated over the past decade (14), efforts to develop novel or more effective agents that mimic ketamine’s mechanism of action have proven difficult. As discussed in the next section, ketamine exerts it greatest replicated potency as an NMDAR antagonist, and the drug appears to exert its anesthetic, dissociative, psychotomimetic, and rewarding properties at least in part due to inhibition of the NMDAR (37). It has been assumed that NMDAR inhibition is the most likely antidepressant mechanism of ketamine. As a result, several alternative NMDAR antagonists have been studied for antidepressant efficacy.
Memantine is an NMDAR antagonist that, similar to ketamine, acts by blocking the NMDAR channel. Memantine was the first alternative NMDAR antagonist studied for the antidepressant activities discovered for ketamine. However, memantine has failed to exert antidepressant actions in multiple placebo-controlled trials for the treatment of MDD (38–41). The antidepressant potential of another NMDAR channel blocker, AZD6765 (lanicemine), was assessed in a study of 22 unmedicated subjects with treatment-resistant MDD. A single infusion of AZD6765 (150 mg) resulted in significant improvements in MADRS scores at 80 and 110 minutes after administration when compared to placebo but was not antidepressant at later time points (42). AZD6765 resulted in no psychotic or dissociative side effects, likely due to its relatively rapid dissociation rate, when compared to ketamine, once glutamate is no longer bound to the NMDAR (i.e., low trapping within the channel pore). However, the response rate was low and was not as robust or sustained as that typically observed with ketamine (42). Another placebo-controlled study of repeated adjunctive AZD6765 infusions administered daily over three weeks to subjects with treatment-resistant MDD found that AZD6765 (given at doses of either 100 mg or 150 mg) had antidepressant effects without dissociative side effects but was not rapid acting, as measured at early time points (43). However, a larger 12-week course, adjunctive, repeated-dose study of AZD6765 (50 mg or 150 mg) found that this agent did not separate from placebo (44). While this result was confounded by a large placebo response (39% at trial end point) the development of AZD6765 for MDD was discontinued.
In addition, as an approach to increasing antidepressant specificity (see the section titled Pharmacology of Rapid-Acting Antidepressants), antagonists selective for the NMDAR GluN2B subunit have also been tested as proof-of-concept agents for TRD. CP-101,606 (traxoprodil) is an GluN2B subtype–selective NMDAR antagonist whose antidepressant efficacy as an adjunctive treatment to paroxetine in treatment-resistant MDD was assessed in a placebo-controlled, randomized, double-blind study (n = 15/group) (45). A single CP-101,606 infusion did not result in rapid effects within 2 days, but it did improve MADRS scores at 5 and 8 days post-treatment. Overall, MDD subjects had a 60% response rate to CP-101,606, compared to 20% in the placebo group (45). Despite these results, development of CP-101,606 was stopped because of potential cardiovascular toxicity. The antidepressant efficacy of the oral GluN2B antagonist, CERC-301 (MK-0657), was tested in a 12-day, randomized, double-blind, placebo-controlled, crossover pilot study of five patients with TRD (46). The study, which administered CERC-301 daily as monotherapy (4–8 mg/day), found no improvement in depressive symptoms over placebo as assessed by the MADRS, but some improvement was seen using the clinician-administered HAM-D and the self-reported BDI. However, these effects were not observed until 5 days post–treatment initiation and were not consistently observed at time points thereafter. No serious or dissociative adverse effects were noted. A subsequent small placebo-controlled trial of CERC-301 (12 mg or 20 mg) found that CERC-301 exerted no significant antidepressant effects compared to placebo, although significant improvements in depressive symptoms were observed at day 2 with the 20-mg dose (47). CERC-301 was also tested as a daily adjunctive treatment in patients with severe MDD and recent active suicidal ideation, but administration for 28 days had no effect on primary end-point measures (change in HAM-D17) (48).
To date, there are limited human data regarding rapid antidepressant actions of medications proposed to act independently of NMDAR inhibition. Glyxin 13 (GLYX-13; rapastinel) is reported to act as a functional partial agonist of the NMDAR (for discussion, see Reference 49). To date, the observed human clinical effects of GLYX-13 have been ambiguous and have yet to be fully reported, but they do suggest potential rapid and sustained antidepressant effects (49, 50). Phase III trials are ongoing (51). In addition to drugs that act directly on the NMDAR, a few human studies have identified relatively rapid antidepressant actions of the muscarinic receptor antagonist scopolamine. An early study found that intramuscular administration of scopolamine resulted in an antidepressant effect following the second of three administrations (52). More recent controlled studies observed antidepressant actions of intravenous scopolamine 3–5 days following a single treatment (53, 54). It was subsequently reported that oral, twice-daily scopolamine had a small but superior antidepressant effect, compared to placebo, within 4 days of treatment initiation (55). However, scopolamine did not significantly relieve more severe or treatment-refractory depression in a more recent randomized, placebo-controlled, crossover trial of 23 subjects with MDD (56).
Thus, ketamine is, by far, the most-studied rapid-acting antidepressant treatment. To date, it has also proven to be the most effective. Overall, while some modest antidepressant efficacy is observed with alternative NMDAR antagonists, these drugs lack the full therapeutic profile seen with ketamine (Table 1). Specifically, none of these drugs appear to possess ketamine’s combination of rapid onset, robust differentiation from placebo, and sustained antidepressant effects—particularly in subjects with TRD (Table 1)—or its antisuicidal properties and antianhedonic effects. While many studies are ongoing, and the many results of some studies have yet to be fully reported (51), the available data suggest the distinct possibility of additional alternative mechanism(s) for ketamine’s action beyond NMDAR inhibition, which has not been sufficiently considered to date. Thus, we focus on the pharmacology of ketamine as the prototypical and established antidepressant that exerts rapid antidepressant effects in humans.
Table 1.
Comparison of the human clinical effects of ketamine to other selective NMDAR antagonists tested for antidepressant actions
| NMDAR antagonist | Rapid onset (within hours) | Robust separation from placebo | Sustained action following a single administration |
|---|---|---|---|
| Ketamine/(S)-ketamine | + | + | + |
| Memantine (namenda) | − | − | − |
| AZD6765 (lanicemine) | +/− | − | − |
| CP-101,606 (traxoprodil) | − | − | + |
| MK-0657/CERC-301 (rislenemdaz) | − | − | − |
+ indicates consistently positive human clinical effects; − indicates a lack of human clinical effects; +/− indicates ambiguous effects between studies. Abbreviation: NMDAR, N-methyl-D-aspartate receptor.
PHARMACOLOGY OF RAPID-ACTING ANTIDEPRESSANTS
Ketamine was initially synthesized as an alternative dissociative anesthetic to phencyclidine (PCP) (57). Compared to PCP, ketamine has less potent dissociative and psychotomimetic effects, a broader anesthetic safety window, and a shorter half-life (57). As a result, ketamine has become widely accepted and used worldwide as an anesthetic, in contrast to PCP, which was removed from the market in 1965 because of these issues.
Ketamine is recognized as an activity-dependent blocker of the NMDAR (58, 59). A functional NMDAR in brain cortical regions is most typically composed of two GluN1 (historically referred to as NR1) subunits and two GluN2 (historically referred to as NR2) subunits (60). The two GluN2 subunits may be either GluN2A, GluN2B, GluN2C, or GluN2D, or they may, in some cases, be a combination of two of these. In addition to binding glutamate, activation of the NMDAR requires binding of glycine to the obligatory glycine coagonist site (glycineB receptor) on the GluN1 subunit (60).
Ketamine’s affinity for the NMDAR is in the low micromolar range [Ki between 0.35 and 3.1 μM; IC50 in brain slices typically approximately 10 μM (37)]. One study reports that ketamine exerts greater inhibition of GluN2C- and GluN2D-containing NMDARs than of GluN2A- and GluN2B-containing subunits (61). However, other studies suggest no such selectivity (62, 63). Ketamine is generally believed to exert anesthetic, dissociative, and psychotomimetic actions, largely via its inhibition of the NMDAR (37). Indeed, intraperitoneal administration of NMDA to mice impairs the anesthetic actions of ketamine (64). However, there is evidence that other targets contribute to anesthetic actions (65), such as ketamine blockade of the hyperpolarization-activated cyclic nucleotide-gated (HCN) channel (66). Ketamine is abused because of its dissociative and psychotomimetic effects (34), and rodents will self-administer ketamine (67, 68).
Multiple possible mechanisms could account for an antidepressant action of ketamine that results from NMDAR inhibition (Figure 1). One putative mechanism involves preferential ketamine-mediated inhibition of NMDARs localized to inhibitory γ-aminobutyric acid (GABA)ergic interneurons (69, 70). This is proposed to result in the disinhibition of glutamatergic pyramidal neurons, thereby increasing these neurons’ evoked release of glutamate and triggering resultant changes in the postsynaptic activation of mechanistic target of rapamycin (mTOR) complex 1 (mTORC1), brain-derived neurotrophic factor (BDNF) release, and increases in synaptic strength (71, 72). A second hypothesis proposes that brief inhibition of postsynaptic NMDAR activity elicited by spontaneous (nonevoked) release of glutamate leads to a cascade of intracellular changes [involving eukaryotic elongation factor 2 (eEF2)] that result in an increase in translation and release of BDNF and subsequent synaptic potentiation (73, 74). A third hypothesis proposes a role for extrasynaptic NMDARs composed primarily of GluN2B that are tonically activated by ambient glutamate and, when inhibited, lead to activation of cellular plasticity cascades (75, 76). These three hypotheses are proposed to converge on activation of cellular plasticity cascades, resulting in the strengthening of excitatory synapses in the brain’s positive valence (reward) circuitry (Figure 1), specifically in circuits weakened by the pathophysiology underlying depression (Figure 2a) (5, 77). A fourth, more recent, putative NMDAR-dependent mechanism proposes ketamine inhibition of NMDAR-dependent high-frequency burst firing of a specific neuronal population in the lateral habenula, which will result in a decreased activity of negative valence brain circuits that are proposed to be abnormally enhanced in depression (Figure 2b) (78, 79).
Figure 1.
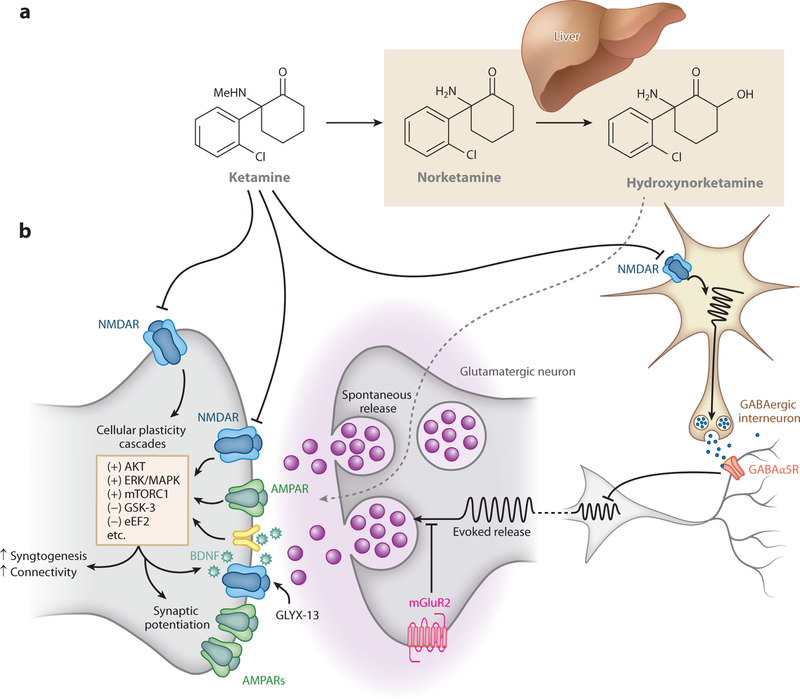
Pharmacology of rapid-acting antidepressants in corticomesolimbic brain circuits. Preclinical data suggest that ketamine and other rapid-acting antidepressants potentiate excitatory synapses in mood-relevant brain regions, potentially reversing disease-induced deficits. New treatments may potentiate these connections while leaving synapses primarily involved in other brain functions unaltered. (a) The principle ketamine metabolic pathway occurs in the liver, resulting first in demethylation of ketamine to norketamine, followed by metabolism to the HNKs. The most prominent HNK metabolites are (2R,6R)- and (2S,6S)-HNK. (b) Rapid-acting antidepressants potentiate glutamatergic synapses. Ketamine, acting as an NMDAR antagonist, may act by preferentially blocking NMDARs localized to GABAergic interneurons, resulting in disinhibition of glutamatergic neurons and an increase in these neurons’ evoked release of glutamate. Ketamine may also act by inhibiting synaptic NMDARs that are activated by spontaneously released glutamate or via inhibition of extrasynaptic NMDARs. Alternatively (or in addition), ketamine conversion to (2R,6R; 2S,6S)-HNK may result in actions on glutamatergic neurons to promote glutamate release and AMPA receptor (AMPAR)-mediated excitation. All hypotheses converge on synaptic processes that strengthen excitatory synapses, activate cellular plasticity cascades, increase activity of AMPARs, and increase connectivity within circuits previously weakened by the pathophysiology underlying depression. Alternative approaches that may have the same net effect include inhibition of GABAα5Rs, resulting in disinhibition of glutamatergic neurons; inhibition of muscarinic acetylcholine receptors (not shown), resulting in inhibition of GABAergic neurons and disinhibition of glutamatergic neurons; inhibition of mGluR2s, resulting in increased probability of glutamate release; and direct activation of synaptic AMPARs and NMDARs (via AMPAkines or NMDAR modulators such as GLYX-13). Abbreviations: AKT, protein kinase B; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; BDNF, brain-derived neurotrophic factor; eEF2, eukaryotic elongation factor 2; ERK/MAPK, extracellular signal-regulated kinase/mitogen-activated kinase; GABAα5R, γ-aminobutyric acid α 5 receptor; GLYX-13, glyxins 13; GSK-3, glycogen synthase kinase-3; HNK, hydroxynorketamine; mGluR2, metabotropic glutamate receptor 2; NMDAR, N-methyl-D-aspartate receptor; mTORC1, mechanistic target of rapamycin complex 1.
Figure 2.
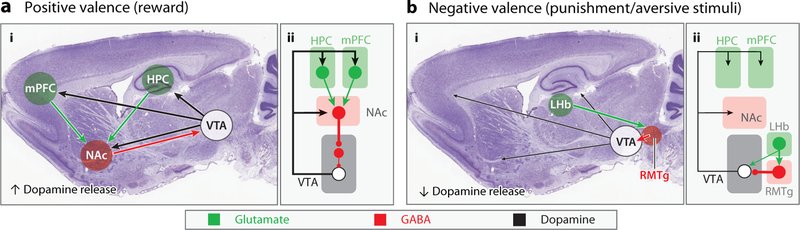
Circuits implicated in rapid antidepressant action. Simplified diagram of circuits promoting reward behavior in response to rewarding stimuli (a) and limiting reward behavior in response to aversive stimuli (b). Locations of key nodes in the reward circuitry are shown (i) superimposed on a parasagittal section of the mouse brain and (ii) in schematic form. Excitatory glutamatergic neurons and projections (green), inhibitory GABAergic neurons and projections (red), and dopaminergic neurons and projections (black) are shown. (a) Critical features of rewarding stimuli are perceived in the HPC and PFC and relayed by excitatory projections to the NAc. GABAergic medium spiny neurons in the NAc synapse onto VTA GABAergic interneurons reducing their inhibition of dopamine back to the telencephalon, overall promoting behaviors that combine to signal reward via dopaminergic connections. (b) Aversive stimuli decrease dopamine output by the VTA as the result of projections from the LHb to the RMTg. As shown in Figure 1, ketamine and hydroxynorketamines may act by strengthening excitatory synapses weakened in depression (5, 122). Ketamine may also act as an NMDAR antagonist to reduce depression-induced high-frequency burst firing of LHb neurons (78). Abbreviations: GABA, γ-aminobutyric acid; HPC, hippocampus; LHb, lateral habenula; mPFC, medial prefrontal cortex; NAc, nucleus accumbens; PFC, prefrontal cortex; RMTg, rostromedial tegmentum; VTA, ventral tegmental area.
Several lines of evidence indicate that ketamine’s antidepressant actions may be independent of NMDAR inhibition. Ketamine exists as equal portions of two stereoisomers, (R)- and (S)-ketamine. (S)-ketamine has an approximately fourfold higher affinity to, and functional inhibition of, the NMDAR compared to (R)-ketamine (37). (S)-ketamine is the more potent anesthetic (37). An NMDAR inhibition hypothesis predicts that the antidepressant potency of ketamine’s enantiomers correlates with NMDAR potency. However, rodent studies from several groups indicate superior antidepressant potency of (R)-ketamine compared to (S)-ketamine (68, 80–82). These data suggest that the antidepressant actions of ketamine in rodents are due to a unique target in lieu of, or in addition to, the NMDAR. It is critically important for future human clinical trials to assess the antidepressant efficacy of (R)-ketamine and to compare the potency of (R)-ketamine to that of (S)-ketamine (83).
Consistent with the limited clinical efficacy of memantine and AZD6765 (Table 1), NMDAR channel blockers, such as MK801, have typically not shown the sustained antidepressant actions of ketamine in preclinical models (68, 74, 84). Similarly, although NMDAR GluN2B–selective compounds consistently exert ketamine-like antidepressant actions in preclinical models (71, 84–87), clinical studies with selective GluN2B antagonists, while not definitive, do not indicate that they exert complete ketamine-like rapid and sustained antidepressant actions (Table 1). The NMDAR glycine site antagonist brain-penetrant prodrug 4-chlorokynurenine has been shown to exert antidepressant activity in preclinical models and is in human phase II clinical trials with results not yet reported (88). Overall, these data add to the evidence that ketamine may exert antidepressant actions, at least in part, via a mechanism distinct from NMDAR inhibition. Over the decades that ketamine has been used as an anesthetic, psychotomimetic, and abused drug, other potential targets have been reported, including dopamine, serotonin, opioid, and cholinergic receptors and HCN channels (37). However, ketamine has relatively low affinity for the majority of these targets, and in most cases, subanesthetic doses of ketamine are not expected to reach the brain levels necessary for effective interactions (37, 89, 90).
In addition to the pharmacological effects of ketamine itself, (R)- and (S)-ketamine stereoisomers are rapidly and stereoselectively metabolized in the liver by CYP450 enzymes to a number of metabolites, including the N-demethylated norketamines [(R)- and (S)-norketamine], dehydronorketamines [(R)- and (S)-dehydronorketamine], and also the hydroxynorketamines (HNKs) (37). The HNKs are rapidly formed by hydroxylation of the norketamine cyclohexyl ring. The 6-HNKs [primarily (2S,6S;2R,6R)-HNK] are the major HNK ketamine metabolites found in humans and rodents (Figure 1) (68, 91). Since the discovery of the rapid antidepressant actions of ketamine, most studies assessing ketamine’s antidepressant effects have been designed, conducted, and analyzed using a commonly accepted view that ketamine and, possibly, norketamine are the active agents with clinical effects that are due to inhibition of the NMDAR, and that other ketamine metabolites are inactive (Figure 1) (59, 92, 93). This focus originates from anesthesia-focused experiments, where an early seminal study found that ketamine and norketamine exert anesthetic actions, but that the 6-HNKs do not, which parallels the potencies of ketamine, norketamine, and the 6-HNKs to inhibit the NMDAR (93, 94).
It was more recently demonstrated that the metabolism of ketamine to (2S,6S;2R,6R)-HNK contributes to the antidepressant effects of ketamine in mice when the drug is administered peripherally (68). The (2R,6R)-HNK metabolite [formed exclusively from (R)-ketamine] exerts rapid and sustained ketamine-like behavioral and cellular actions in vivo (68, 95, 96). It was also reported that the (2S,6S)-HNK metabolite has antidepressant actions but possesses ketamine-like side effects (68). (2R,6R)-HNK, like ketamine, also produces an increase in electroencephalography (EEG)-measured gamma oscillations, which is indicative of high-frequency neural activity. Furthermore, when a nonmetabolizable deuterated analog of ketamine, which limits its metabolism to (2S,6S;2R,6R)-HNK, was compared to ketamine, less potent and sustained antidepressant-relevant actions were observed (68). (2R,6R)-HNK does not inhibit the NMDAR at antidepressant-relevant concentrations, as evidenced by the lack of displacement of [3H]-MK-801 binding in vitro by (2R,6R)-HNK, and it does not functionally inhibit NMDARs in cell culture or hippocampal slices at antidepressant-relevant concentrations (68, 97–100). (2R,6R)-HNK lacks the sensory dissociative effects and abuse liability side effects that limit the broader therapeutic use of ketamine (68). These data suggest that, in mice, ketamine is not acting primarily through NMDAR inhibition to exert its antidepressant actions; they also suggest the possibility of identifying rapid-acting antidepressants that are devoid of the most worrisome ketamine side effects based on the (2R,6R)-HNK metabolite. Additional studies have identified biological effects of (2R,6R)-HNK relevant to depression (98, 101–105) as well as effectiveness of (2R,6R)-HNK in ameliorating chronic pain symptoms in mice (106).
As discussed in the preceding section, there is some clinical evidence identifying other drugs as potential rapid-acting antidepressants in humans, including GLYX-13 and scopolamine. GLYX-13 was derived from a monoclonal antibody that enhanced long-term potentiation of hippocampal synapses, and it appears to act as a functional partial agonist of the NMDAR (49). Preclinical data suggest GLYX-13 has rapid antidepressant actions similar to those of ketamine (107–110). The antidepressant actions of GLYX-13 appear to require NMDAR activation, as administration of the NMDAR competitive antagonist 3-(2-carboxypiperazin-4-yl) propyl-1-phosphonic acid reversed the antidepressant actions of GLYX-13 (111). Furthermore, GLYX-13 administered at antidepressant-relevant doses prevented the NMDAR inhibition–mediated cognitive side effects of ketamine (112).
Scopolamine is a nonselective muscarinic receptor antagonist. Preclinical experiments utilizing mice in which each of the five muscarinic receptors (M1–M5) had been individually genetically deleted demonstrated that the M1 and M5 subtypes are required for the antidepressant actions of scopolamine (113). Antagonists of M1 and M2 receptors have also produced an antidepressant and antianhedonic effect in animal models, similar to the actions of scopolamine (113, 114). Scopolamine administered to rodents also increases mTORC1 signaling, synaptogenesis, and antidepressant behavioral responses that are mTORC1 activation–dependent (115) and that have been functionally linked to disinhibition of somatostatin interneurons in the medial prefrontal cortex, a result that converges with the disinhibition hypothesis of ketamine action (116) (Figure 1).
NEUROPHYSIOLOGICAL MECHANISMS UNDERLYING RAPID-ACTING ANTIDEPRESSANT ACTION
Ketamine’s beneficial actions—lasting up to a week or longer in patients—after a single administration far outlast its time in the body [it has a human terminal half-life of 155 minutes (37)]. This suggests that some rapid change is induced while ketamine is present, and this change leads to a long-lasting alteration in synaptic structure, function, and/or gene expression that persists long after ketamine is cleared. This situation is reminiscent of many forms of activity-dependent plasticity, such as long-term potentiation (LTP), in which a brief induction period (e.g., a high-frequency stimulus train) leads to long-lasting changes in synaptic strength. These parallels have led to suggestions that rapid-acting antidepressants rapidly engage endogenous processes that promote synaptic growth and strengthening (5, 117). Support for such a mechanism is evidenced by preclinical models of stress-induced depression symptomatology. In these models, excitatory synapses are weakened in multiple key relays within the corticomesolimbic structures that form the backbone of the brain’s reward and mood circuitry (Figure 2a), such as the nucleus accumbens (118), prefrontal cortex (119), and hippocampus (120); these perturbations are attenuated by rapid-acting antidepressants. Endogenous synapse-strengthening mechanisms are thus poised to restore what might be a core defect in brain circuitry underlying the symptoms of depression (Figure 3).
Figure 3.
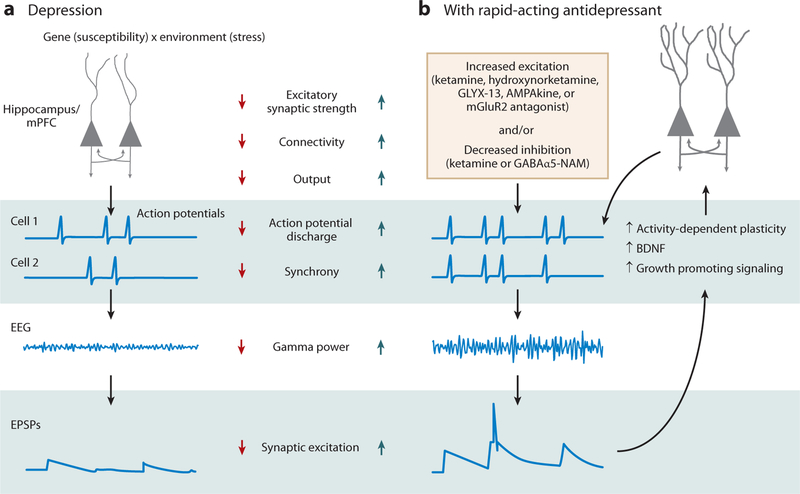
Neurophysiological mechanisms of rapid-acting antidepressant action. (a) Depression susceptibility is modulated by genetic risk and exposure to environmental factors, such as chronic stress. Such factors are proposed to interact to cause dendritic atrophy and decreased synaptic function in key circuits within the corticomesolimbic structures illustrated in Figure 2, thereby contributing to the symptoms of depression. These defects have the net effect of reducing neuronal output, weakening connectivity within and between circuit nodes, and decreasing synchronous action potential discharge, which can be detected as decreases in coherent oscillations with EEG. Decreased synchronous coherent activity will act to reduce synaptic strength. (b) Rapid-acting antidepressants are proposed to counteract these changes either by acutely increasing overall excitation, as do hydroxynorketamines, mGluR2 antagonists, AMPAkines, or GLYX-13, or by acutely decreasing synaptic inhibition, as do ketamine, GABAα5-NAMs or scopolamine. This shift in the balance of excitation and inhibition favors synchronous discharge, apparent as increased gamma power oscillations in the EEG, and strong synaptic excitation. Strong synaptic excitation, in turn, triggers the activation of long-term potentiation and a range of cellular growth–associated signaling cascades, including synthesis and release of BDNF, activation of mTORC1, and dephosphorylation of eEF2K, all of which act synergistically to restore dendritic strength, promote synaptogenesis, and strengthen excitatory connections, thereby restoring the function of the reward circuitry and relieving the symptoms of depression. Abbreviations: BDNF, brain-derived neurotrophic factor; eEF2K, eukaryotic elongation factor-2 kinase; EEG, electroencephalography; EPSPs, excitatory postsynaptic potentials; GABA, γ-aminobutyric acid; GABAα5-NAMs, GABA α5 receptor negative allosteric modulators; mTORC1, mechanistic target of rapamycin complex 1.
Antidepressant Induction Mechanisms
As discussed above, ketamine is well characterized as an NMDAR antagonist, and over a decade of research has dogmatically pursued the answer to how this action initiates the drug’s beneficial antidepressant actions. Hippocampal and cortical circuits are composed of both excitatory glutamatergic pyramidal neurons and inhibitory GABAergic interneurons, which together create a delicate balance of excitation and inhibition. All of these cells are excited, in part, by NMDAR activation, suggesting that ketamine should broadly decrease overall activity of both cell types. Paradoxically, however, experimental observations have consistently revealed that ketamine promotes (net) excitation, detected as an increase in extracellular levels of glutamate in rodent dialysis studies (69) or as an increase in high-frequency gamma oscillations in EEG recordings in both humans and rodents (43, 100, 121). As mentioned above, one of the most widely proposed theories of ketamine action postulates that NMDARs play a more important role in the excitation of inhibitory interneurons than they do in pyramidal cells, so that blocking them with ketamine exerts a net disinhibitory action (72, 122, 123). This differential action could arise if NMDAR-mediated excitation is disproportionately more important in driving the firing of interneurons than in driving the firing of pyramidal cells, for example, if interneurons were more depolarized than pyramidal cells, resulting in a lower degree of Mg2+ block of the NMDAR ion channel localized to interneurons. Given that ketamine acts as an open channel blocker, this would make the inactivation of rapidly firing interneurons more sensitive to an NMDAR antagonist such as ketamine acting by blocking the channel pore. More specifically, parvalbumin-expressing, fast-spiking interneurons control the excitability of pyramidal cells and the synchrony of their discharge (124). Partially impaired excitation of these interneurons by ketamine is predicted to increase overall activity and gamma oscillations, as has been observed, and there is preclinical evidence revealing that inhibition of NMDARs by ketamine reduces interneuron firing, resulting in increased pyramidal neuron firing in rats (70, 125). However, there is also evidence that genetic deletion of NMDARs from parvalbumin interneurons does not induce anhedonia or impair behavioral responses to ketamine (126), perhaps because chronic genetic deletion induces compensatory mechanisms that acute NMDAR inhibition by ketamine does not.
We have recently suggested that ketamine’s initial actions are mediated not by its ability to block NMDARs, but rather by a more direct potentiation of AMPA receptor (AMPAR)-mediated synaptic transmission by its HNK metabolites (68). Application of (2R,6R)-HNK to hippocampal brain slices does not significantly inhibit NMDAR function, but it does robustly increase the strength of AMPAR-mediated synaptic excitation within an hour of application; this increase persists beyond drug washout (68). Similar to administration of ketamine, (2R,6R)-HNK administration acutely increases gamma power measured via skull surface electrodes in vivo, independent of locomotor activity changes and without altering alpha, beta, delta or theta oscillations (68). The mechanisms underlying this potentiation and activity remain to be determined but might entail a change in presynaptic glutamate release mechanisms, an increased postsynaptic sensitivity to glutamate secondary to some change in another pre- or postsynaptic ion channel, or a combination of these effects. Regardless of the initial direct target of ketamine or its (2R,6R)-HNK metabolite, the majority of hypotheses regarding ketamine’s acute downstream effects converge on AMPAR-mediated synaptic excitation (Figure 1). Probably the best evidence for this theory derives from preclinical studies, where preadministration of the AMPAR antagonist NBQX prevents the antidepressant actions of ketamine, as well as those of other classes of putative rapid-acting antidepressants (68, 71, 74, 81, 84, 115, 127–130). One explanation for these results is that the concentrations of NBQX, at the doses given, are not sufficiently high to block the AMPAR-mediated excitation necessary for normal synaptic transmission but are sufficient to disrupt the balance between excitation and inhibition underlying the gamma oscillations produced by these compounds. Supporting this interpretation, we have reported that preadministration of NBQX prevented (2R,6R)-HNK-induced increases in gamma power, potentially providing a human translational biomarker of the central nervous system response to (2R,6R)-HNK (68).
BDNF signaling mediated via tropomyosin receptor kinase B (TrkB) is another prime candidate for the initiation of antidepressant-induced plasticity responses (72, 131, 132). Both traditional antidepressants (e.g., SSRIs) and many rapid-acting antidepressants are reported to increase expression of BDNF mRNA or protein (e.g., 68, 74, 108, 110, 133–136). Deletion of forebrain BDNF or TrkB and prevention of TrkB activation hinder the actions of rapid-acting antidepressant compounds in preclinical models (74, 110, 131, 132, 135). Furthermore, intrahippocampal administration of BDNF exerts antidepressant-like effects (e.g., 137). There is considerable evidence that expression of BDNF is upregulated in response to high-frequency correlated activity (138), such as that underlying gamma rhythms, making it well suited to be a candidate for contributing to the mechanism initiating these effects.
Antidepressant Expression Mechanisms
A primary mechanism underlying expression of canonical LTP is activity-dependent increase in postsynaptic AMPAR expression. There is evidence that, in addition to being required for induction, AMPARs are required for the expression of rapid antidepressant actions. Long-term antidepressant effects are associated with upregulation of synaptic levels of AMPARs, similar to what occurs during LTP induced in brain slices. Upregulation of AMPAR synaptic expression has been measured in immunoblots from animals within 24 hours after treatment with ketamine and other putative rapid-acting antidepressant drugs (68, 71, 107, 129, 139). Consistent with an increase in synaptic AMPARs being involved in sustained antidepressant actions, administration of NBQX immediately prior to antidepressant behavioral testing—at a time point distant from drug administration—has been shown to prevent expression of the antidepressant actions of both ketamine and (2R,6R)-HNK (68, 127). Taken together with evidence of decreased excitation and AMPAR downregulation in reward circuits in stress-based depression models (e.g., 120), AMPAR upregulation represents a logical synaptic restorative mechanism. Furthermore, AMPAR upregulation and membrane insertion are likely to be triggered by synchronous, high-frequency activity, such as the gamma discharge induced by ketamine and other rapid-acting antidepressants, making it a plausible expression mechanism.
These processes may also impact neosynaptogenesis (Figure 3). A consistent brain imaging finding in human depression is atrophy of the hippocampus and medial prefrontal cortex (140). This atrophy is believed to reflect the stress-induced pruning and shrinkage of pyramidal cell dendrites, as described by McEwen and colleagues (141) in preclinical studies and observed in human postmortem tissue samples (140). Taken together, this evidence of decreases in synaptic function in rodent models suggests that synapse loss contributes to the pathology of depression. Promoting restoration of synaptic number and size would thus be a beneficial mechanism of antidepressant action. Indeed, there is extensive evidence that numerous growth-promoting signaling pathways are activated by ketamine and other rapid-acting antidepressants (72, 122, 123, 131). Such signaling pathways may be activated by high-frequency correlated activity and either directly or indirectly in response to BDNF–TrkB or signaling via alternative neurotrophic factors. There is strong evidence that growth-associated signaling cascades converging on the protein mTOR are important in the responses of several rapid-acting antidepressant compounds (71). These same pathways have been implicated in activity-dependent synaptic potentiation, making them attractive effector pathways mediating rapid antidepressant responses that are subsequently maintained. Overall, these sustained changes in synaptic strength likely underlie the continued expression of antidepressant effects long after the time point of drug exposure.
FUTURE MEDICATION DEVELOPMENT FOR THE RAPID TREATMENT OF DEPRESSION
There is persuasive evidence that existing and putative rapid-acting antidepressant compounds converge on a similar mechanism to restore pathological processes that underlie the symptoms of depression (Figures 1 and 3). Depression in preclinical models is accompanied by dysfunction of synapse structure and function in multiple loci within corticomesolimbic circuits regulating mood and reward behaviors, including the medial prefrontal cortex, hippocampus, and nucleus accumbens (Figure 2a), as well as circuits mediating aversion (Figure 2b). Such dysfunction will decrease communication between these structures and can reasonably be expected to produce dysphoria and anhedonia, which are cardinal symptoms of depression. While they are present in the body, compounds with known or probable rapid-acting antidepressant properties share an ability to produce—directly or indirectly—rapid, short-lived increases in excitability that favor high-frequency correlated action potential discharge in large populations of cells, apparent as increases in the power of gamma frequency activity measured by EEG. This relatively transient increase in synchronous discharge has the potential to activate many of the brain’s endogenous capabilities to strengthen synapse number, structure, and function, thereby producing a persistent correction of the defects underlying the symptoms of depression. It is hypothesized that antidepressant-mediated synaptic potentiation occurs selectively in circuits that are abnormally weakened in depression (5, 123) (Figure 3). Thus, new treatments may potentiate these connections while leaving synapses involved primarily in other brain functions unaltered. This knowledge, and the body of research into ketamine’s actions, helps us to mechanistically understand, and has motivated the development of, other drugs that exert rapid antidepressant actions in preclinical models. Candidate targets for novel antidepressant drugs include those that disinhibit inhibitory input to excitatory neurons, strengthen excitatory transmission by increasing the probability of glutamate release, or trigger postsynaptic activation of glutamate receptors (Figure 1).
One such target is the metabotropic glutamate receptor 2 (mGluR2) (142), which, along with mGluR3, is categorized as a group II receptor. mGluR2 receptors are highly expressed in the corticolimbic brain regions implicated in regulation of mood and reward (143). The mGluR2 receptor acts as an autoreceptor, localized primarily on glutamatergic presynaptic nerve terminals, where it regulates glutamate release via negative feedback. When the mGluR2 receptor is activated by glutamate, it acts via Gi/Go coupling to decrease adenylyl cyclase activity, resulting in a decrease in the probability of glutamate release (144). Conversely, antagonists at this receptor increase the probability of glutamate release (Figure 1). Although development of compounds with specificity for the mGluR2 receptor has proven elusive, a number of compounds have been developed that either activate or inhibit mGluR2/3 receptors (144). mGluR2/3 antagonists have been shown to exert antidepressant effects in many preclinical assays (142). Studies indicate that mGluR2/3 antagonists exert rapid effects following chronic stress or corticosterone paradigms (145–148), similar to ketamine and unlike SSRIs, which require weeks to exert effects in these paradigms. Furthermore, mGluR2/3 antagonists may increase high-frequency gamma oscillations in rodents, similar to ketamine (149). A phase II, randomized controlled trial evaluating the antidepressant efficacy of adjunctive RO4995819 (decoglurant, an mGluR2/3 negative allosteric modulator) in patients with treatment-resistant MDD has been completed (150). This agent did not show antidepressant efficacy, and its development for treatment-resistant MDD by Roche (under the name RG1578) was therefore ended (150). However, there may still be reasons to pursue other mGluR2/3 antagonists. For example, RO4995819 is a negative allosteric modulator, rather than a competitive antagonist. Furthermore, no marker of target engagement by RO4995819 was included in this study to confirm appropriate dosing.
As mentioned above, extensive data indicate that the antidepressant actions of ketamine, as well as those of ketamine’s (2R,6R)-HNK metabolite, are blocked by inhibition of the activity of AMPARs by coadministration of the competitive AMPAR antagonist NBQX. The antidepressant actions of mGluR2/3 antagonists are similarly blocked by NBQX, indicating that AMPAR activation is important for the antidepressant actions of this class of compounds (127, 151). However, the antidepressant actions of classical, monoamine-acting antidepressants appear to be insensitive to acute administration of NBQX (84, 152). These and other, earlier data suggest the possibility that AMPAR activation may be targeted directly to exert antidepressant actions, independent of pre-synaptic glutamate release mechanisms (153). AMPAR positive allosteric modulators, also known as AMPAkines, offer a particularly promising approach. The rationale for developing AMPAR modulators is that direct stimulation of AMPARs can provoke seizures or otherwise be neurotoxic (154). To overcome this potential issue, drug development in this field has focused on the use of AMPAkines, which potentiate currents mediated by AMPARs. Preclinical studies have found that these agents exhibit antidepressant actions (136, 155–157). Additionally, selective activation of NMDARs should similarly enhance synaptic potentiation and, as established by GLYX-13, act as an antidepressant (77, 158).
Another strategy for promoting excitation is to weaken inhibition mediated by GABA receptors. Several compounds have been developed that are selective negative allosteric modulators of GABA receptors containing α5 subunits. Such selective targeting limits side effects related to broadly targeting GABA receptors, including anxiety and seizures (159). In rodents, the expression of α5 subunits is particularly high in pyramidal cells in the hippocampus and deep layers of the neocortex, particularly the prefrontal cortex, and low throughout the rest of the brain. Human positron emission tomography experiments have similarly identified enhanced expression of α5 subunits in limbic structures (160). This unique distribution is ideal for targeting key nodes of the reward circuitry (Figure 2) and, because the extent of expression is limited, also minimizes unwanted side effects (161). α5-containing GABA receptors contribute to both phasic synaptic inhibition and tonic extrasynaptic inhibition (162). This provides an explanation of why an α5 subunit–selective GABA negative allosteric modulator (GABA-NAM) increases pyramidal cell excitability and promotes coherent oscillatory activity, particularly in the gamma frequency band (20–60 Hz) (161). As discussed above, gamma frequency oscillations are predicted to facilitate the induction of synaptic plasticity in synapses suppressed by stress, which then yields antidepressant effects. Indeed, rapid antidepressant actions of α5 GABA-NAMs in several preclinical models of depression have been reported and persist for several days following a single administration, along with a strengthening of stress-weakened hippocampal synapses (139, 161, 163, 164). Several GABA-NAMs have been given to humans for other indications, and no unanticipated side effects have yet been uncovered, but they have yet to be tested in patients with depression (159).
CONCLUSIONS
Depression is among the most damaging of all human conditions, and patients currently have limited treatment options. Current treatments are deficient in terms of both the time scale over which these medications act and the fact that a significant number of ill individuals do not respond to the currently available pharmacopeia. The discovery of the rapid actions of ketamine in TRD has resulted in a reconceptualization of how depression could be treated in the future. Past treatment approaches (such as total sleep deprivation and ECT) and emerging findings with other medications, including scopolamine, (S)-ketamine, and GLYX-13, support the notion that depression can be treated rapidly, provided that the correct receptors, synapses, and/or circuits are targeted. While there have been limited treatment advances over the past decades, there is currently reason to be optimistic that the future treatment of depression will be accomplished within hours, rather than the current standard of weeks or months.
An improved understanding of the synaptic and circuit mechanisms underlying ketamine’s efficacy in preclinical models has provided insight into other approaches that may be useful as novel efficacious treatments. Candidate targets for novel treatments include those that disinhibit excitatory neurons; increase the probability of glutamate release selectively in limbic synapses; or result in postsynaptic potentiation affecting biological processes similar to those understood, via preclinical studies, to be necessary and sufficient for LTP (Figures 1 and 3), counteracting depression-induced changes (Figure 2). Ketamine’s anesthetic effects, as well as worrisome side effects—including dissociation and abuse potential—appear to be due to its interactions with the relatively widespread NMDAR. Clinical studies indicate that other NMDAR antagonists do not replicate the rapid, robust, and sustained clinical profile of ketamine (Table 1), suggesting that NMDAR inhibition is not the mechanism, or not the complete mechanism, by which ketamine acts as an antidepressant. We have recently shown that ketamine’s HNK metabolite, (2R,6R)-HNK, exerts antidepressant actions in preclinical models that are independent of NMDAR inhibition (68). These findings raise hope that (2R,6R)-HNK (or other ketamine metabolites or prodrugs) and other novel compounds having NMDAR-independent actions, such as mGluR2 receptor antagonists and AMPAkines, may act as rapid antidepressants without ketamine’s NMDAR inhibition–dependent side effects. The favorable preclinical antidepressant and side effect profiles of GABA-NAMs are encouraging for their further development as rapid-acting antidepressant medications. Overall, improved pharmacological treatments for depression have never seemed so near.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH) grant R01MH107615 and a Harrington Discovery Institute Scholar-Innovator grant to T.D.G., an NIH Bench-to-Bedside award to T.D.G. and C.A.Z., and NIH grant R01MH086828 and a Kahlert Foundation grant to S.M.T. C.A.Z. is supported by the National Institute of Mental Health Intramural Research Program.
Footnotes
DISCLOSURE STATEMENT
T.D.G. received research funding from Janssen, Allergan, and Roche Pharmaceuticals during the three years preceding the writing of this review. The authors declare competing financial interests: T.D.G. and C.A.Z. are listed as coinventors on a patent application for the use of (2R,6R)-hydroxynorketamine and (2S,6S)-hydroxynorketamine in the treatment of depression, anxiety, anhedonia, suicidal ideation and post-traumatic stress disorders. C.A.Z. is listed as a coinventor on a patent for the use of (2R,6R)-hydroxynorketamine, (S)-dehydronorketamine, and other stereoisomeric dehydro- and hydroxylated metabolites of ketamine in the treatment of depression and neuropathic pain. S.M.T. is listed as a coinventor on a patent application for the use of negative allosteric modulators of GABAA receptors containing alpha 5 subunits as fast-acting antidepressants. C.A.Z. has assigned his patent rights to the US government but will share a percentage of any royalties that may be received by the government. T.D.G. and S.M.T. have assigned their patent rights to the University of Maryland, Baltimore but will share a percentage of any royalties that may be received by the University of Maryland, Baltimore.
LITERATURE CITED
- 1.Kessler RC, Berglund P, Demler O, Jin R, Koretz D, et al. 2003. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 289:3095–105 [DOI] [PubMed] [Google Scholar]
- 2.Duman RS, Heninger GR, Nestler EJ. 1997. A molecular and cellular theory of depression. Arch. Gen. Psychiatry 54:597–606 [DOI] [PubMed] [Google Scholar]
- 3.Gould TD, Manji HK. 2002. Signaling networks in the pathophysiology and treatment of mood disorders. J. Psychosom. Res 53:687–97 [DOI] [PubMed] [Google Scholar]
- 4.Hyman SE, Nestler EJ. 1996. Initiation and adaptation: a paradigm for understanding psychotropic drug action. Am. J. Psychiatry 153:151–62 [DOI] [PubMed] [Google Scholar]
- 5.Thompson SM, Kallarackal AJ, Kvarta MD, Van Dyke AM, LeGates TA, Cai X. 2015. An excitatory synapse hypothesis of depression. Trends Neurosci 38:279–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, et al. 2006. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR∗D report. Am. J. Psychiatry 163:1905–17 [DOI] [PubMed] [Google Scholar]
- 7.UK ECT Rev. Group. 2003. Efficacy and safety of electroconvulsive therapy in depressive disorders: a systematic review and meta-analysis. Lancet 361:799–808 [DOI] [PubMed] [Google Scholar]
- 8.Segman RH, Shapira B, Gorfine M, Lerer B. 1995. Onset and time course of antidepressant action: psychopharmacological implications of a controlled trial of electroconvulsive therapy. Psychopharmacology 119:440–48 [DOI] [PubMed] [Google Scholar]
- 9.Wu JC, Bunney WE. 1990. The biological basis of an antidepressant response to sleep deprivation and relapse: review and hypothesis. Am. J. Psychiatry 147:14–21 [DOI] [PubMed] [Google Scholar]
- 10.Kellner CH, Greenberg RM, Murrough JW, Bryson EO, Briggs MC, Pasculli RM. 2012. ECT in treatment-resistant depression. Am. J. Psychiatry 169:1238–44 [DOI] [PubMed] [Google Scholar]
- 11.Reich DL, Silvay G. 1989. Ketamine: an update on the first twenty-five years of clinical experience. Can. J. Anaesth 36:186–97 [DOI] [PubMed] [Google Scholar]
- 12.Green SM, Rothrock SG, Lynch EL, Ho M, Harris T, et al. 1998. Intramuscular ketamine for pediatric sedation in the emergency department: safety profile in 1,022 cases. Ann. Emerg. Med 31:688–97 [DOI] [PubMed] [Google Scholar]
- 13.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, et al. 2000. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 47:351–54 [DOI] [PubMed] [Google Scholar]
- 14.Iadarola ND, Niciu MJ, Richards EM, Vande Voort JL, Ballard ED, et al. 2015. Ketamine and other N-methyl-D-aspartate receptor antagonists in the treatment of depression: a perspective review. Ther. Adv. Chronic Dis 6:97–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newport DJ, Carpenter LL, McDonald WM, Potash JB, Tohen M, et al. 2015. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am. J. Psychiatry 172:950–66 [DOI] [PubMed] [Google Scholar]
- 16.Kishimoto T, Chawla JM, Hagi K, Zarate CA, Kane JM, et al. 2016. Single-dose infusion ketamine and non-ketamine N-methyl-D-aspartate receptor antagonists for unipolar and bipolar depression: a meta-analysis of efficacy, safety and time trajectories. Psychol. Med 46:1459–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zarate CA Jr., Singh JB, Carlson PJ, Brutsche NE, Ameli R, et al. 2006. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 63:856–64 [DOI] [PubMed] [Google Scholar]
- 18.Singh JB, Fedgchin M, Daly EJ, De Boer P, Cooper K, et al. 2016. A double-blind, randomized, placebo-controlled, dose-frequency study of intravenous ketamine in patients with treatment-resistant depression. Am. J. Psychiatry 173:816–26 [DOI] [PubMed] [Google Scholar]
- 19.Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, et al. 2013. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am. J. Psychiatry 170:1134–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diazgranados N, Ibrahim L, Brutsche NE, Newberg A, Kronstein P, et al. 2010. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch. Gen. Psychiatry 67:793–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zarate CA, Brutsche N, Ibrahim L, Franco-Chaves J, Diazgranados N, et al. 2012. Replication of ketamine’s antidepressant efficacy in bipolar depression: a randomized controlled add-on trial. Biol. Psychiatry 71:939–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newport DJ, Carpenter LL, McDonald WM, Potash JB, Tohen M, et al. 2015. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am. J. Psychiatry 172:950–66 [DOI] [PubMed] [Google Scholar]
- 23.Diazgranados N, Ibrahim LA, Brutsche NE, Ameli R, Henter ID, et al. 2010. Rapid resolution of suicidal ideation after a single infusion of an N-methyl-D-aspartate antagonist in patients with treatment-resistant major depressive disorder. J. Clin. Psychiatry 71:1605–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price RB, Nock MK, Charney DS, Mathew SJ. 2009. Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol. Psychiatry 66:522–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price RB, Iosifescu DV, Murrough JW, Chang LC, Al Jurdi RK, et al. 2014. Effects of ketamine on explicit and implicit suicidal cognition: a randomized controlled trial in treatment-resistant depression. Depression Anxiety 31:335–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murrough JW, Soleimani L, DeWilde KE, Collins KA, Lapidus KA, et al. 2015. Ketamine for rapid reduction of suicidal ideation: a randomized controlled trial. Psychol. Med 45:3571–80 [DOI] [PubMed] [Google Scholar]
- 27.Wilkinson ST, Ballard ED, Bloch MH, Mathew SJ, Murrough JW, et al. 2017. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am. J. Psychiatry 175:150–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ballard ED, Ionescu DF, Vande Voort JL, Niciu MJ, Richards EM, et al. 2014. Improvement in suicidal ideation after ketamine infusion: relationship to reductions in depression and anxiety. J. Psychiatr. Res 58:161–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lally N, Nugent AC, Luckenbaugh DA, Ameli R, Roiser JP, Zarate CA Jr. 2014. Anti-anhedonic effect of ketamine and its neural correlates in treatment-resistant bipolar depression. Transl. Psychiatry 4:e469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lally N, Nugent AC, Luckenbaugh DA, Niciu MJ, Roiser JP, Zarate CA Jr. 2015. Neural correlates of change in major depressive disorder anhedonia following open-label ketamine. J. Psychopharmacol 29:596–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ballard ED, Wills K, Lally N, Richards EM, Luckenbaugh DA, et al. 2017. Anhedonia as a clinical correlate of suicidal thoughts in clinical ketamine trials. J. Affect. Disord 218:195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilkinson ST, Toprak M, Turner MS, Levine SP, Katz RB, Sanacora G. 2017. A survey of the clinical, off-label use of ketamine as a treatment for psychiatric disorders. Am. J. Psychiatry 174:695–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanacora G, Frye MA, McDonald W, Mathew SJ, Turner MS, et al. 2017. A consensus statement on the use of ketamine in the treatment of mood disorders. JAMA Psychiatry 74:399–405 [DOI] [PubMed] [Google Scholar]
- 34.Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, et al. 1994. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 51:199–214 [DOI] [PubMed] [Google Scholar]
- 35.Singh JB, Fedgchin M, Daly E, Xi L, Melman C, et al. 2015. Intravenous esketamine in adult treatment-resistant depression: a double-blind, double-randomization, placebo-controlled study. Biol. Psychiatry 80:424–31 [DOI] [PubMed] [Google Scholar]
- 36.Daly EJ, Singh JB, Fedgchin M, Cooper K, Lim P, et al. 2018. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry 75:139–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zanos P, Moaddel R, Morris PJ, Riggs LM, Highland JN, et al. 2018. Ketamine and ketamine metabolite pharmacology: insights into therapeutic mechanisms. Pharmacol. Rev 70:621–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zarate CA Jr., Singh JB, Quiroz JA, De Jesus G, Denicoff KK, et al. 2006. A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am. J. Psychiatry 163:153–55 [DOI] [PubMed] [Google Scholar]
- 39.Lee SY, Chen SL, Chang YH, Chen PS, Huang SY, et al. 2013. Add-on memantine to valproate treatment increased HDL-C in bipolar II disorder. J. Psychiatr. Res 47:1343–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith EG, Deligiannidis KM, Ulbricht CM, Landolin CS, Patel JK, Rothschild AJ. 2013. Antidepressant augmentation using the N-methyl-D-aspartate antagonist memantine: a randomized, double-blind, placebo-controlled trial. J. Clin. Psychiatry 74:966–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Omranifard V, Shirzadi E, Samandari S, Afshar H, Maracy MR. 2014. Memantine add on to citalopram in elderly patients with depression: a double-blind placebo-controlled study. J. Res. Med. Sci 19:525–30 [PMC free article] [PubMed] [Google Scholar]
- 42.Zarate CA Jr., Mathews D, Ibrahim L, Chaves JF, Marquardt C, et al. 2013. A randomized trial of a low-trapping nonselective N-methyl-D-aspartate channel blocker in major depression. Biol. Psychiatry 74:257–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanacora G, Smith MA, Pathak S, Su HL, Boeijinga PH, et al. 2014. Lanicemine: a low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Mol. Psychiatry 19:978–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanacora G, Johnson MR, Khan A, Atkinson SD, Riesenberg RR, et al. 2017. Adjunctive lanicemine (AZD6765) in patients with major depressive disorder and history of inadequate response to antidepressants: a randomized, placebo-controlled study. Neuropsychopharmacology 42:844–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. 2008. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J. Clin. Psychopharmacol 28:631–37 [DOI] [PubMed] [Google Scholar]
- 46.Ibrahim L, Diazgranados N, Jolkovsky L, Brutsche N, Luckenbaugh D, et al. 2012. A randomized, placebo-controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. J. Clin. Psychopharmacol 32:551–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cerecor. 2016. Cerecor reports top-line data from CERC-301 phase 2 study for major depressive disorder Press Release, Nov. 29. https://ir.cerecor.com/press-releases/detail/30/cerecor-reports-top-line-data-from-cerc-301-phase-2-study [Google Scholar]
- 48.Paterson B, Fraser H, Wang C, Marcus R. 2015. A randomized, double-blind, placebo-controlled, sequential parallel study of CERC-301 in the adjunctive treatment of subjects with severe depression and recent active suicidal ideation despite antidepressant treatment Poster presented at National Network of Depression Centers Annual Conference, Ann Arbor, MI, [Google Scholar]
- 49.Moskal JR, Burch R, Burgdorf JS, Kroes RA, Stanton PK, et al. 2014. GLYX-13, an NMDA receptor glycine site functional partial agonist enhances cognition and produces antidepressant effects without the psychotomimetic side effects of NMDA receptor antagonists. Expert Opin. Invest. Drugs 23:243–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Preskorn S, Macaluso M, Mehra DO, Zammit G, Moskal JR, et al. 2015. Randomized proof of concept trial of GLYX-13, an N-methyl-D-aspartate receptor glycine site partial agonist, in major depressive disorder nonresponsive to a previous antidepressant agent. J. Psychiatr. Pract 21:140–49 [DOI] [PubMed] [Google Scholar]
- 51.Murrough JW, Abdallah CG, Mathew SJ. 2017. Targeting glutamate signalling in depression: progress and prospects. Nat. Rev. Drug Discov 16:472–86 [DOI] [PubMed] [Google Scholar]
- 52.Gillin JC, Sutton L, Ruiz C, Darko D, Golshan S, et al. 1991. The effects of scopolamine on sleep and mood in depressed patients with a history of alcoholism and a normal comparison group. Biol. Psychiatry 30:157–69 [DOI] [PubMed] [Google Scholar]
- 53.Furey ML, Drevets WC. 2006. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch. Gen. Psychiatry 63:1121–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drevets WC, Furey ML. 2010. Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol. Psychiatry 67:432–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khajavi D, Farokhnia M, Modabbernia A, Ashrafi M, Abbasi SH, et al. 2012. Oral scopolamine augmentation in moderate to severe major depressive disorder: a randomized, double-blind, placebo-controlled study. J. Clin. Psychiatry 73:1428–33 [DOI] [PubMed] [Google Scholar]
- 56.Park L, Furey ML, Nugent AC, Farmer C, Ellis J, et al. 2018. Neurophysiological changes associated with antidepressant response to ketamine not observed in a negative trial of scopolamine in major depressive disorder. Int. J. Neuropsychopharmacol [DOI] [PMC free article] [PubMed]
- 57.Domino EF, Chodoff P, Corssen G. 1965. Pharmacologic effects of Ci-581, a new dissociative anesthetic, in man. Clin. Pharmacol. Ther 6:279–91 [DOI] [PubMed] [Google Scholar]
- 58.Anis NA, Berry SC, Burton NR, Lodge D. 1983. The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methyl-aspartate. Br. J. Pharmacol 79:565–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Domino EF. 2010. Taming the ketamine tiger. Anesthesiology 113:678–84 [DOI] [PubMed] [Google Scholar]
- 60.Hansen KB, Yi F, Perszyk RE, Menniti FS, Traynelis SF. 2017. NMDA receptors in the central nervous system. In Methods in Molecular Biology, Vol. 1677, ed. N Burnashev P Szepetowski, pp. 1–80. New York: Humana Press; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kotermanski SE, Johnson JW. 2009. Mg2+ imparts NMDA receptor subtype selectivity to the Alzheimer’s drug memantine. J. Neurosci 29:2774–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dravid SM, Erreger K, Yuan H, Nicholson K, Le P, et al. 2007. Subunit-specific mechanisms and proton sensitivity of NMDA receptor channel block. J. Physiol 581:107–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yamakura T, Mori H, Masaki H, Shimoji K, Mishina M. 1993. Different sensitivities of NMDA receptor channel subtypes to non-competitive antagonists. Neuroreport 4:687–90 [DOI] [PubMed] [Google Scholar]
- 64.Irifune M, Shimizu T, Nomoto M, Fukuda T. 1992. Ketamine-induced anesthesia involves the N-methyl-D-aspartate receptor-channel complex in mice. Brain Res 596(1–2):1–9 [DOI] [PubMed] [Google Scholar]
- 65.Petrenko AB, Yamakura T, Sakimura K, Baba H. 2014. Defining the role of NMDA receptors in anesthesia: Are we there yet? Eur. J. Pharmacol 723:29–37 [DOI] [PubMed] [Google Scholar]
- 66.Chen X, Shu S, Bayliss DA. 2009. HCN1 channel subunits are a molecular substrate for hypnotic actions of ketamine. J. Neurosci 29:600–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rocha BA, Ward AS, Egilmez Y, Lytle DA, Emmett-Oglesby MW. 1996. Tolerance to the discriminative stimulus and reinforcing effects of ketamine. Behav. Pharmacol 7:160–68 [PubMed] [Google Scholar]
- 68.Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, et al. 2016. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533:481–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moghaddam B, Adams B, Verma A, Daly D. 1997. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci 17:2921–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Homayoun H, Moghaddam B. 2007. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J. Neurosci 27:11496–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, et al. 2010. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329:959–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Duman RS. 2014. Pathophysiology of depression and innovative treatments: remodeling glutamatergic synaptic connections. Dialogues Clin. Neurosci 16:11–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Monteggia LM, Gideons E, Kavalali ET. 2013. The role of eukaryotic elongation factor 2 kinase in rapid antidepressant action of ketamine. Biol. Psychiatry 73:1199–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, et al. 2011. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475:91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Miller OH, Yang L, Wang CC, Hargroder EA, Zhang Y, et al. 2014. GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. eLife 3:e03581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miller OH, Moran JT, Hall BJ. 2016. Two cellular hypotheses explaining the initiation of ketamine’s antidepressant actions: direct inhibition and disinhibition. Neuropharmacology 100:17–26 [DOI] [PubMed] [Google Scholar]
- 77.Zanos P, Gould TD. 2018. Mechanisms of ketamine action as an antidepressant. Mol. Psychiatry 23:801–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang Y, Cui Y, Sang K, Dong Y, Ni Z, et al. 2018. Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 554:317–22 [DOI] [PubMed] [Google Scholar]
- 79.Yang Y, Wang H, Hu J, Hu H. 2018. Lateral habenula in the pathophysiology of depression. Curr. Opin. Neurobiol 48:90–96 [DOI] [PubMed] [Google Scholar]
- 80.Zhang JC, Li SX, Hashimoto K. 2014. R (−)-ketamine shows greater potency and longer lasting antidepressant effects than S (+)-ketamine. Pharmacol. Biochem. Behav 116:137–41 [DOI] [PubMed] [Google Scholar]
- 81.Yang C, Shirayama Y, Zhang JC, Ren Q, Yao W, et al. 2015. R-ketamine: a rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl. Psychiatry 5:e632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fukumoto K, Toki H, Iijima M, Hashihayata T, Yamaguchi JI, et al. 2017. Antidepressant potential of (R)-ketamine in rodent models: comparison with (S)-ketamine. J. Pharmacol. Exp. Ther 361:9–16 [DOI] [PubMed] [Google Scholar]
- 83.Zanos P, Gould TD. 2018. Intracellular signaling pathways involved in (S)- and (R)-ketamine antidepressant actions. Biol. Psychiatry 83:2–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maeng S, Zarate CA Jr., Du J, Schloesser RJ, McCammon J, et al. 2008. Cellular mechanisms underlying the antidepressant effects of ketamine: role of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol. Psychiatry 63:349–52 [DOI] [PubMed] [Google Scholar]
- 85.Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, et al. 2011. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol. Psychiatry 69:754–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jimenez-Sanchez L, Campa L, Auberson YP, Adell A. 2014. The role of GluN2A and GluN2B subunits on the effects of NMDA receptor antagonists in modeling schizophrenia and treating refractory depression. Neuropsychopharmacology 39:2673–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kiselycznyk C, Jury NJ, Halladay LR, Nakazawa K, Mishina M, et al. 2015. NMDA receptor subunits and associated signaling molecules mediating antidepressant-related effects of NMDA-GluN2B antagonism. Behav. Brain Res 287:89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zanos P, Piantadosi SC, Wu HQ, Pribut HJ, Dell MJ, et al. 2015. The prodrug 4-chlorokynurenine causes ketamine-like antidepressant effects, but not side effects, by NMDA/glycineB-site inhibition. Pharmacol. Exp. Ther 355:76–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shaffer CL, Osgood SM, Smith DL, Liu J, Trapa PE. 2014. Enhancing ketamine translational pharmacology via receptor occupancy normalization. Neuropharmacology 86:174–80 [DOI] [PubMed] [Google Scholar]
- 90.Can A, Zanos P, Moaddel R, Kang HJ, Dossou KS, et al. 2016. Effects of ketamine and ketamine metabolites on evoked striatal dopamine release, dopamine receptors, and monoamine transporters. J. Pharmacol. Exp. Ther 359:159–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zarate CA Jr., Brutsche N, Laje G, Luckenbaugh DA, Venkata SL, et al. 2012. Relationship of ketamine’s plasma metabolites with response, diagnosis, and side effects in major depression. Biol. Psychiatry 72:331–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hirota K, Lambert DG. 2011. Ketamine: new uses for an old drug? Br. J. Anaesth 107:123–26 [DOI] [PubMed] [Google Scholar]
- 93.Singh NS, Zarate CA Jr., Moaddel R, Bernier M, Wainer IW. 2014. What is hydroxynorketamine and what can it bring to neurotherapeutics? Expert Rev. Neurother 14:1239–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Leung LY, Baillie TA. 1986. Comparative pharmacology in the rat of ketamine and its two principal metabolites, norketamine and (Z)-6-hydroxynorketamine. J. Med. Chem 29:2396–99 [DOI] [PubMed] [Google Scholar]
- 95.Pham TH, Defaix C, Xu X, Deng SX, Fabresse N, et al. 2017. Common neurotransmission recruited in (R,S)-ketamine and (2R,6R)-hydroxynorketamine-induced sustained antidepressant-like effects. Biol. Psychiatry 84:e3–6 [DOI] [PubMed] [Google Scholar]
- 96.Chou D, Peng HY, Lin TB, Lai CY, Hsieh MC, et al. 2018. (2R,6R)-hydroxynorketamine rescues chronic stress-induced depression-like behavior through its actions in the midbrain periaqueductal gray. Neuropharmacology 139:1–12 [DOI] [PubMed] [Google Scholar]
- 97.Morris PJ, Moaddel R, Zanos P, Moore CE, Gould T, et al. 2017. Synthesis and N-methyl-D-aspartate (NMDA) receptor activity of ketamine metabolites. Org. Lett 19:4572–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Moaddel R, Abdrakhmanova G, Kozak J, Jozwiak K, Toll L, et al. 2013. Sub-anesthetic concentrations of (R,S)-ketamine metabolites inhibit acetylcholine-evoked currents in α7 nicotinic acetylcholine receptors. Eur. J. Pharmacol 698:228–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Suzuki K, Nosyreva E, Hunt KW, Kavalali ET, Monteggia LM. 2017. Effects of a ketamine metabolite on synaptic NMDAR function. Nature 546:E1–3 [DOI] [PubMed] [Google Scholar]
- 100.Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, et al. 2017. Reply to: Effects of a ketamine metabolite on synaptic NMDAR function. Nature 546:E4–5 [DOI] [PubMed] [Google Scholar]
- 101.Paul RK, Singh NS, Khadeer M, Moaddel R, Sanghvi M, et al. 2014. (R,S)-Ketamine metabolites (R,S)-norketamine and (2S,6S)-hydroxynorketamine increase the mammalian target of rapamycin function. Anesthesiology 121:149–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Singh NS, Rutkowska E, Plazinska A, Khadeer M, Moaddel R, et al. 2016. Ketamine metabolites enantioselectively decrease intracellular D-serine concentrations in PC-12 cells. PLOS ONE 11:e0149499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wray NH, Schappi JM, Singh H, Senese NB, Rasenick MM. 2018. NMDAR-independent, cAMP-dependent antidepressant actions of ketamine. Mol. Psychiatry [DOI] [PMC free article] [PubMed]
- 104.Yao N, Skiteva O, Zhang X, Svenningsson P, Chergui K. 2018. Ketamine and its metabolite (2R,6R)-hydroxynorketamine induce lasting alterations in glutamatergic synaptic plasticity in the mesolimbic circuit. Mol. Psychiatry [DOI] [PubMed]
- 105.Cavalleri L, Merlo Pich E, Millan MJ, Chiamulera C, Kunath T, et al. 2018. Ketamine enhances structural plasticity in mouse mesencephalic and human iPSC-derived dopaminergic neurons via AMPAR-driven BDNF and mTOR signaling. Mol. Psychiatry 23:812–23 [DOI] [PubMed] [Google Scholar]
- 106.Kroin J, Das V, Moric M, Buvanendran A. 2018. Efficacy of the ketamine metabolite (2R,6R)-hydroxynorketamine in mice models of pain. Reg. Anesth. Pain Manag [DOI] [PubMed]
- 107.Liu RJ, Duman C, Kato T, Hare B, Lopresto D, et al. 2017. GLYX-13 produces rapid antidepressant responses with key synaptic and behavioral effects distinct from ketamine. Neuropsychopharmacology 42:1231–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lepack AE, Bang E, Lee B, Dwyer JM, Duman RS. 2016. Fast-acting antidepressants rapidly stimulate ERK signaling and BDNF release in primary neuronal cultures. Neuropharmacology 111:242–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Burgdorf J, Zhang XL, Nicholson KL, Balster RL, Leander JD, et al. 2013. GLYX-13, a NMDA receptor glycine-site functional partial agonist, induces antidepressant-like effects without ketamine-like side effects. Neuropsychopharmacology 38:729–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kato T, Fogaca MV, Deyama S, Li XY, Fukumoto K, Duman RS. 2018. BDNF release and signaling are required for the antidepressant actions of GLYX-13. Mol. Psychiatry [DOI] [PMC free article] [PubMed]
- 111.Burgdorf J, Zhang XL, Weiss C, Gross A, Boikess SR, et al. 2015. The long-lasting antidepressant effects of rapastinel (GLYX-13) are associated with a metaplasticity process in the medial prefrontal cortex and hippocampus. Neuroscience 308:202–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rajagopal L, Burgdorf JS, Moskal JR, Meltzer HY. 2016. GLYX-13 (rapastinel) ameliorates subchronic phencyclidine- and ketamine-induced declarative memory deficits in mice. Behav. Brain Res 299:105–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Witkin JM, Overshiner C, Li X, Catlow JT, Wishart GN, et al. 2014. M1 and M2 muscarinic receptor subtypes regulate antidepressant-like effects of the rapidly acting antidepressant scopolamine. J. Pharmacol. Exp. Ther 351:448–56 [DOI] [PubMed] [Google Scholar]
- 114.Navarria A, Wohleb ES, Voleti B, Ota KT, Dutheil S, et al. 2015. Rapid antidepressant actions of scopolamine: role of medial prefrontal cortex and M1-subtype muscarinic acetylcholine receptors. Neurobiol. Dis 82:254–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Voleti B, Navarria A, Liu RJ, Banasr M, Li N, et al. 2013. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol. Psychiatry 74:742–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wohleb ES, Wu M, Gerhard DM, Taylor SR, Picciotto MR, et al. 2016. GABA interneurons mediate the rapid antidepressant-like effects of scopolamine. J. Clin. Investig 126:2482–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pittenger C, Duman RS. 2008. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 33:88–109 [DOI] [PubMed] [Google Scholar]
- 118.Lim BK, Huang KW, Grueter BA, Rothwell PE, Malenka RC. 2012. Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature 487:183–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yuen EY, Wei J, Liu W, Zhong P, Li X, Yan Z. 2012. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron 73:962–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kallarackal AJ, Kvarta MD, Cammarata E, Jaberi L, Cai X, et al. 2013. Chronic stress induces a selective decrease in AMPA receptor-mediated synaptic excitation at hippocampal temporoammonic-CA1 synapses. J. Neurosci 33:15669–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hong LE, Summerfelt A, Buchanan RW, O’Donnell P, Thaker GK, et al. 2010. Gamma and delta neural oscillations and association with clinical symptoms under subanesthetic ketamine. Neuropsychopharmacology 35:632–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zanos P, Thompson SM, Duman RS, Zarate CA Jr., Gould TD. 2018. Convergent mechanisms underlying rapid antidepressant action. CNS Drugs 32:197–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hare BD, Ghosal S, Duman RS. 2018. Rapid acting antidepressants in chronic stress models: molecular and cellular mechanisms. Chronic Stress [DOI] [PMC free article] [PubMed]
- 124.Sohal VS, Zhang F, Yizhar O, Deisseroth K. 2009. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459:698–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Widman AJ, McMahon LL. 2018. Disinhibition of CA1 pyramidal cells by low-dose ketamine and other antagonists with rapid antidepressant efficacy. PNAS 115:E3007–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pozzi L, Pollak Dorocic I, Wang X, Carlen M, Meletis K. 2014. Mice lacking NMDA receptors in parvalbumin neurons display normal depression-related behavior and response to antidepressant action of NMDAR antagonists. PLOS ONE 9:e83879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Koike H, Chaki S. 2014. Requirement of AMPA receptor stimulation for the sustained antidepressant activity of ketamine and LY341495 during the forced swim test in rats. Behav. Brain Res 271:111–15 [DOI] [PubMed] [Google Scholar]
- 128.Koike H, Iijima M, Chaki S. 2011. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav. Brain Res 224:107–11 [DOI] [PubMed] [Google Scholar]
- 129.Yu H, Li M, Zhou D, Lv D, Liao Q, et al. 2018. Vesicular glutamate transporter 1 (VGLUT1)-mediated glutamate release and membrane GluA1 activation is involved in the rapid antidepressant-like effects of scopolamine in mice. Neuropharmacology 131:209–22 [DOI] [PubMed] [Google Scholar]
- 130.Dwyer JM, Lepack AE, Duman RS. 2012. mTOR activation is required for the antidepressant effects of mGluR2/3 blockade. Int. J. Neuropsychopharmacol 15:429–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Autry AE, Monteggia LM. 2012. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol. Rev 64:238–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Castren E, Kojima M. 2017. Brain-derived neurotrophic factor in mood disorders and antidepressant treatments. Neurobiol. Dis 97:119–26 [DOI] [PubMed] [Google Scholar]
- 133.Nibuya M, Morinobu S, Duman RS. 1995. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci 15:7539–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ghosal S, Bang E, Yue W, Hare BD, Lepack AE, et al. 2018. Activity-dependent brain-derived neurotrophic factor release is required for the rapid antidepressant actions of scopolamine. Biol. Psychiatry 83:29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lepack AE, Fuchikami M, Dwyer JM, Banasr M, Duman RS. 2014. BDNF release is required for the behavioral actions of ketamine. Int. J. Neuropsychopharmacol 18: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhou W, Wang N, Yang C, Li XM, Zhou ZQ, Yang JJ. 2014. Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur. Psychiatry 29:419–23 [DOI] [PubMed] [Google Scholar]
- 137.Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS. 2002. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J. Neurosci 22:3251–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Park H, Poo MM. 2013. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci 14:7–23 [DOI] [PubMed] [Google Scholar]
- 139.Fischell J, Van Dyke AM, Kvarta MD, LeGates TA, Thompson SM. 2015. Rapid antidepressant action and restoration of excitatory synaptic strength after chronic stress by negative modulators of alpha5-containing GABAA receptors. Neuropsychopharmacology 40:2499–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Manji HK, Drevets WC, Charney DS. 2001. The cellular neurobiology of depression. Nat. Med 7:541–47 [DOI] [PubMed] [Google Scholar]
- 141.McEwen BS, Nasca C, Gray JD. 2016. Stress effects on neuronal structure: hippocampus, amygdala, and prefrontal cortex. Neuropsychopharmacology 41:3–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chaki S 2017. mGlu2/3 receptor antagonists as novel antidepressants. Trends Pharmacol. Sci 38:569–80 [DOI] [PubMed] [Google Scholar]
- 143.Gu G, Lorrain DS, Wei H, Cole RL, Zhang X, et al. 2008. Distribution of metabotropic glutamate 2 and 3 receptors in the rat forebrain: implication in emotional responses and central disinhibition. Brain Res 1197:47–62 [DOI] [PubMed] [Google Scholar]
- 144.Nicoletti F, Bockaert J, Collingridge GL, Conn PJ, Ferraguti F, et al. 2011. Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology 60:1017–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ago Y, Yano K, Araki R, Hiramatsu N, Kita Y, et al. 2013. Metabotropic glutamate 2/3 receptor antagonists improve behavioral and prefrontal dopaminergic alterations in the chronic corticosterone-induced depression model in mice. Neuropharmacology 65:29–38 [DOI] [PubMed] [Google Scholar]
- 146.Koike H, Iijima M, Chaki S. 2013. Effects of ketamine and LY341495 on the depressive-like behavior of repeated corticosterone-injected rats. Pharmacol. Biochem. Behav 107:20–23 [DOI] [PubMed] [Google Scholar]
- 147.Dong C, Zhang JC, Yao W, Ren Q, Ma M, et al. 2017. Rapid and sustained antidepressant action of the mGlu2/3 receptor antagonist MGS0039 in the social defeat stress model: comparison with ketamine. Int. J. Neuropsychopharmacol 20:228–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dwyer JM, Lepack AE, Duman RS. 2013. mGluR2/3 blockade produces rapid and long-lasting reversal of anhedonia caused by chronic stress exposure. J. Mol. Psychiatry 1:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ahnaou A, Ver Donck L, Drinkenburg WHIM. 2014. Blockade of the metabotropic glutamate (mGluR2) modulates arousal through vigilance states transitions: evidence from sleep–wake EEG in rodents. Behav. Brain Res 270:56–67 [DOI] [PubMed] [Google Scholar]
- 150.Henter ID, de Sousa RT, Zarate CA Jr. 2018. Glutamatergic modulators in depression. Harv. Rev. Psychiatry [DOI] [PMC free article] [PubMed]
- 151.Karasawa J, Shimazaki T, Kawashima N, Chaki S. 2005. AMPA receptor stimulation mediates the antidepressant-like effect of a group II metabotropic glutamate receptor antagonist. Brain Res 1042:92–98 [DOI] [PubMed] [Google Scholar]
- 152.Wolak M, Siwek A, Szewczyk B, Poleszak E, Pilc A, et al. 2013. Involvement of NMDA and AMPA receptors in the antidepressant-like activity of antidepressant drugs in the forced swim test. Pharmacol. Rep 65:991–97 [DOI] [PubMed] [Google Scholar]
- 153.Alt A, Nisenbaum ES, Bleakman D, Witkin JM. 2006. A role for AMPA receptors in mood disorders. Biochem. Pharmacol 71:1273–88 [DOI] [PubMed] [Google Scholar]
- 154.O’Neill MJ, Bleakman D, Zimmerman DM, Nisenbaum ES. 2004. AMPA receptor potentiators for the treatment of CNS disorders. Curr. Drug Targets CNS Neurol. Disord 3:181–94 [DOI] [PubMed] [Google Scholar]
- 155.O’Neill MJ, Witkin JM. 2007. AMPA receptor potentiators: application for depression and Parkinson’s disease. Curr. Drug Targets 8:603–20 [DOI] [PubMed] [Google Scholar]
- 156.Fukumoto K, Iijima M, Chaki S. 2014. Serotonin-1A receptor stimulation mediates effects of a metabotropic glutamate 2/3 receptor antagonist, 2S-2-amino-2-(1S,2S-2-carboxycycloprop-1-yl)-3-(xanth-9-yl)propanoic acid (LY341495), and an N-methyl-D-aspartate receptor antagonist, ketamine, in the novelty-suppressed feeding test. Psychopharmacology 231:2291–98 [DOI] [PubMed] [Google Scholar]
- 157.Li X, Tizzano JP, Griffey K, Clay M, Lindstrom T, Skolnick P. 2001. Antidepressant-like actions of an AMPA receptor potentiator (LY392098). Neuropharmacology 40:1028–33 [DOI] [PubMed] [Google Scholar]
- 158.Abdallah CG, Sanacora G, Duman RS, Krystal JH. 2018. The neurobiology of depression, ketamine and rapid-acting antidepressants: Is it glutamate inhibition or activation? Pharmacol. Ther 190:148–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Atack JR, Maubach KA, Wafford KA, O’Connor D, Rodrigues AD, et al. 2009. In vitro and in vivo properties of 3-tert-butyl-7-(5-methylisoxazol-3-yl)-2-(1-methyl-1H-1,2,4-triazol-5-ylmethoxy)-pyrazolo[1,5-d]-[1,2,4]triazine (MRK-016), a GABAA receptor α5 subtype-selective inverse agonist. Pharmacol. Exp. Ther 331:470–84 [DOI] [PubMed] [Google Scholar]
- 160.Lingford-Hughes A, Hume SP, Feeney A, Hirani E, Osman S, et al. 2002. Imaging the GABA-benzodiazepine receptor subtype containing the α5-subunit in vivo with [11C]Ro15 4513 positron emission tomography. J. Cereb. Blood Flow Metab 22:878–89 [DOI] [PubMed] [Google Scholar]
- 161.Zanos P, Nelson ME, Highland JN, Krimmel SR, Georgiou P, et al. 2017. A negative allosteric modulator for α5 subunit-containing GABA receptors exerts a rapid and persistent antidepressant-like action without the side effects of the NMDA receptor antagonist ketamine in mice. eNeuro 4: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Prenosil GA, Schneider Gasser EM, Rudolph U, Keist R, Fritschy JM, Vogt KE. 2006. Specific subtypes of GABAA receptors mediate phasic and tonic forms of inhibition in hippocampal pyramidal neurons. Neurophysiol 96:846–57 [DOI] [PubMed] [Google Scholar]
- 163.Carreno FR, Collins GT, Frazer A, Lodge DJ. 2017. Selective pharmacological augmentation of hippocampal activity produces a sustained antidepressant-like response without abuse-related or psychotomimetic effects. Int. J. Neuropsychopharmacol 20:504–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Xu NZ, Ernst M, Treven M, Cerne R, Wakulchik M, et al. 2018. Negative allosteric modulation of alpha 5-containing GABAA receptors engenders antidepressant-like effects and selectively prevents age-associated hyperactivity in tau-depositing mice. Psychopharmacology 235:1151–61 [DOI] [PubMed] [Google Scholar]