

Abstract
The Journal of Biological Chemistry (JBC) has been a major vehicle for disseminating and recording the discovery and characterization of proteolytic enzymes. The pace of discovery in the protease field accelerated during the 1971–2010 period that Dr. Herb Tabor served as the JBC's editor-in-chief. When he began his tenure, the fine structure and kinetics of only a few proteases were known; now thousands of proteases have been characterized, and over 600 genes for proteases have been identified in the human genome. In this review, besides reflecting on Dr. Tabor's invaluable contributions to the JBC and the American Society for Biochemistry and Molecular Biology (ASBMB), I endeavor to provide an overview of the extensive history of protease research, highlighting a few discoveries and roles of proteases in vivo. In addition, metalloproteinases, particularly meprins of the astacin family, will be discussed with regard to structural characteristics, regulation, mechanisms of action, and roles in health and disease. Proteases and protein degradation play crucial roles in living systems, and I briefly address future directions in this highly diverse and thriving research area.
Keywords: proteinase, metalloprotease, protein degradation, protein complex, protein domain, astacins, meprins, metalloproteinase
Historical aspects of proteases and their role in protein degradation
In the very first issue of the Journal of Biological Chemistry (JBC)2 in 1905, P. A. Levene published studies on “The Cleavage Products of Proteoses” (1). The Journal continually published state-of-the-art work on proteases over the years, but the pace of discovery in the field accelerated during the 39 years that Herb Tabor served as Editor of the JBC. When Herb began his tenure as Chief Editor of the JBC (1971), we knew the fine structure and a substantial amount about the kinetics of only a few proteases. Some examples of the major classes of proteolytic enzymes (aspartic, serine, cysteine, metallo) that were well studied before 1970 are as follows.
Pepsin, an aspartic protease of the stomach, was one of the first enzymes to be discovered, characterized, and named (in 1825), and it was crystallized in 1930 (2). Studies of pepsin's action can be found in the JBC as far back as in 1907 (3), and mechanistic studies were well on the way in the 1970s.
The serine proteases, trypsin and chymotrypsin from pancreatic secretions, were also discovered in the 1800s and crystallized in the 1930s (4). Studies of the action of trypsin appeared in the JBC in 1907 (5), whereas those for chymotrypsin appeared in the 1930s (6).
Papain, the cysteine protease from papaya, was also discovered in the 1800s, and pure forms were reported in the JBC as early as 1954 (7).
Thermolysin, an extracellular metalloprotease from thermophilic bacteria, was the first metalloendoproteinase to be crystallized and to have its structure solved (8).
Carboxypeptidase A, isolated in 1937 (9), was kinetically characterized in 1970 (10).
Carboxypeptidase B was isolated in 1960 (11), and bacterial collagenase, now known as part of the matrixin family, matrix metalloproteinase 1 (MMP-1), was isolated in 1957 (12).
There are many excellent reviews available for individually characterized proteases and for clans and families of proteases, as well as for general insights into functional aspects of proteases (e.g. see Ref. 13). A comprehensive database, MEROPS, of the more than 1000 individual proteases is available to all and contains a wealth of information on the characterization and evolutionary relationships of the proteases and the current literature (https://www.ebi.ac.uk/merops/)3 (98). A degradome database of human proteases (14) and the Handbook of Proteolytic Enzymes (15) are also valuable resources.
There was ample new information coming forth in the 1960s and early 1970s on protease structure and function about small (20–35-kDa), secreted proteases (as those cited above), but little to nil was known about cell-associated proteases, cellular functions of proteases, or protein turnover. In an era when there were great advances and interest in the mechanisms of protein synthesis (the 1950s and 1960s), there was a comparative dearth of information and effort devoted to studies of protein degradation. That said, it had been known since the pioneering studies of Schoenheimer (1942) (16) that there was continuous turnover (synthesis and breakdown) of cellular proteins in eukaryotic cells. The extent of that turnover (intracellular protein degradative process) and its importance to the vitality of the cell, however, was unappreciated. Cell death was recognized to involve proteases, as were wasting diseases (e.g. type 1 diabetes), and lysosomes (17) were thought to handle these “downhill” processes through autophagy. Studies with individual proteins indicated great differences in turnover of specific proteins (18, 19), and the concept of short- and long-lived proteins grew with studies of many individual cellular proteins. There was expanding interest in intracellular protein degradation in the 1970s, and one of the first conferences in the United States that heralded that interest was organized by Bob Schimke (an Associate Editor of the JBC) and Nobuhiku Katunuma (a prominent biochemist in Japan) in 1973, the Conference on Protein Turnover in Palo Alto, California (20).
Intracellular protein degradation was clearly of international interest and activity, leading to several conferences in Europe in the 1970s. For example, Alan Barrett organized a meeting at Strangeways Research Laboratory in Cambridge, England, in 1970 on tissue proteinases; in 1973, a group of scientists at the Martin Luther University in Halle, German Democratic Republic (GDR), organized a symposium on intracellular protein catabolism in Reinhardsbrunn, GDR; Vito Turk organized a meeting in 1975 in Lubljana, Yugoslavia (now Slovenia); and Professors Horst Hanson and Peter Bohley organized additional conferences on intracellular proteolytic enzymes and protein turnover in vivo in 1977 and 1981. The 1970s were times in which GDR scientists could not leave their country for meetings, so scientists in Western countries went to the GDR, placing science above politics. This interest resulted in the formation of committees to increase communication among scientists who work on proteases and protein turnover. First there was ECOP, the European Committee on Proteolysis, in 1981, followed by ACOP (the American Committee on Proteolysis, which organized the 5th International Symposium on Intracellular Protein Catabolism) and then JCOP, the Japanese Committee on Proteolysis, and finally ICOP, the International Committee on Proteolysis. These were forerunners of the current International Proteolysis Society formed in 1999.
Before the 1970s, there were several myths, or misconceptions, regarding proteolytic enzymes and protein turnover.
There were many who thought the only function of proteases was to totally degrade proteins at certain stages of life (particularly end-stages) or that their only function was to be secreted in order to degrade extracellular proteins, thereby releasing amino acids so that other proteins could be synthesized.
It was thought that there were very few proteases in cells and that they could handle a great variety of degradative functions, similar to the trypsins and chymotrypsins along with some exopeptidases that could degrade almost anything in the intestinal tract.
There were bacteriologists who argued that protein degradation did not occur in growing procaryotes because there was no need to degrade proteins; it was thought that defective, damaged, or useless proteins could be diluted out as cells divided rapidly.
The known proteases were small (20–35 kDa), compact, uncomplicated (no carbohydrate, lipids, or cofactors) proteins, and it was assumed that this was generally true of all proteases.
Lysosomes were thought to be the primary or only site for degrading proteins in cells, as well as those taken up by endocytosis, and that this occurred through the merging of lysosomes and other cell components to form autophagic vacuoles.
But now we know that there are a large number of proteases in and secreted from cells. Proteinases are the largest enzyme gene family in vertebrates.
There are 641 protease genes in the human and 677 in the mouse (i.e. ∼3% of the human and mouse genome).
Proteolysis occurs in virtually all stages of a cell's life, in all cell compartments, and in many stages of a protein's existence: from processing of preproproteins coincident or soon after protein synthesis to total destruction of the protein.
There are a great variety of protease structures, from small to large (20 kDa to 6 MDa), highly complex structures, some containing multiple domains with many posttranslational moieties, such as carbohydrates and lipids.
Lysosomal proteases are not the only intracellular proteases and, under many circumstances, are not the major proteases responsible for intracellular protein degradation.
Evolutionary clans and families of proteases have been identified, and the classification of individual proteases is highly developed.
Proteases regulate fate, localization, and activity of many proteins.
Proteases are key factors in the health and viability of cells, involved in multiple processes, such as replication, transcription, cell proliferation, differentiation, extracellular matrix remodeling, and processing of hormones and biologically active peptides.
Proteases are highly regulated (e.g. transcriptionally, post-translationally, activated, inhibited, and compartmentalized).
Proteases are involved in many diseases (e.g. cancer, Alzheimer's, arthritis, blood clotting disorders, allergies, and infections, to name a few).
Protease inhibitors are useful medically (e.g. angiotensin-converting enzyme inhibitors for blood pressure, HIV inhibitors, proteasome inhibitors for myeloma, dipeptidyl peptidase IV inhibitors for type II diabetes).
Proteases are useful industrially (e.g. clarifying beer and wines, preparation of leather, tenderizing, and debraiding).
Herb Tabor's leadership and protease advances during his oversight of the JBC
The JBC has been a major vehicle for elucidating the structures and functions of proteases and especially the fundamental aspects of these enzymes. Herb was responsible for keeping the Journal focused on fundamental/basic science, not the “hot science” of the day. His emphasis was on high-quality science that stood the test of time and had the potential of long-range importance and impact.
Herb also has had a strong commitment to and influence on the ASBMB. I know this through my role as an Associate Editor of the JBC from 1999 to 2012 and as a president of the ASBMB (2004–2006). Herb participated in many activities of ASBMB, including business and financial meetings, publication committee meetings, centennial planning meetings, and Associate Editor and editorial board member activities. When it was time for the centennial celebration, he felt strongly that both the Society and the JBC should be celebrated together, even though the Journal started in 1905, one year before the Society was established (1906). He gave strong support to the Associate Editors and staff. He was always thinking ahead about issues, best ways to communicate, and new emerging areas. Herb always listened to various viewpoints, considered alternatives, and had an uncanny way of getting people to “agree” with his view. He has always been forward-looking and especially encouraged the online version of the Journal; the JBC was the first of the life science journals to appear online (in 1995).
There has been great excitement about proteases and their functions in the last half-century. A few examples of discoveries that created that excitement will be mentioned here.
The discovery of proteasomes and the ATP-ubiquitin proteolytic pathway certainly changed our view of the world of protein degradation. The role of ubiquitin and the proteasome in intracellular protein breakdown began to unfold in the 1970s (e.g. see Refs. 21–23) and expanded rapidly in the 1980s (e.g. see Refs. 24–28).
Signal peptidases that cleave signal peptides from secretory and membrane-associated proteins as they are translocated across membranes and into the endoplasmic reticulum were discovered in the 1970s and 1980s (e.g. see Refs. 29–31).
Caspases, proteases involved in programmed cell death (apoptosis), were discovered in Caenorhabditis elegans in the 1980s, and the complexity of the caspase family in humans and the role of these enzymes in apoptosis and cytokine processing was revealed in the 1990s (e.g. see Refs. 32 and 33).
The HIV-1 protease, the retroviral aspartic protease that is essential for the maturation of the AIDS virus, was discovered in the 1980s (see Ref. 34). This protease is a prime target for drug therapy, and inhibitors of the protease, along with other drugs, have greatly prolonged the lives of people infected with the virus. The development of inhibitors of the HIV-1 protease was accelerated by the large body of information available about aspartic proteases in many organisms, which allowed development of specific viral protease inhibitors. This is an example of the importance of basic science for therapeutic advances.
The great variety of cysteine proteases (e.g. cathepsins and calpains) and their diverse functions have come to light in recent decades (see, for example, Ref. 35). They participate in a variety of processes, including autophagy, the lysosomal degradation of cellular constituents. The discovery of the molecular players in the autophagic process has enhanced our understanding of this process in health and disease (36, 37).
Advances in metalloproteases, highlighting meprins
Metalloproteases have emerged as a fascinating group of enzymes. They are present in all kingdoms of living organisms and have expanded widely during evolution. In 1980, 11 metalloproteinases were identified (38). Now we know that the mouse and human genomes encode ∼200 metalloproteinases, the largest group in the proteolytic enzyme realm (39). Most of these enzymes are secreted from cells or plasma membrane–bound, and they act pericellularly and extracellularly. They are involved in tissue differentiation and remodeling during embryogenesis and in processing biologically active peptides and cytokines in adult tissues. Angiotensin-converting enzyme inhibitors to control blood pressure are among the most widely used inhibitors for humans. Metalloproteinases are also involved in many diseases, such as cancer and inflammatory diseases. They and their inhibitors (e.g. TIMPs (tissue inhibitors of metalloproteinases)) are of great medical interest and have provided optimism and disappointment in clinical trials. The use of synthetic inhibitors of metalloproteinases to inhibit cancer cell mobility provides great promise but has not yet reached its potential.
The metzincin superfamily contains most of the known metalloendoproteinases (zinc-containing enzymes that cleave peptide bonds internally on protein substrates) (39). The superfamily is composed of six evolutionarily related families: a disintegrin and metalloproteinases (ADAMs), MMPs, pappalysins (pregnancy-associated plasma proteins), serralysins (bacterial enzymes), leishmanolysins (protozoan proteinases), and astacins (Fig. 1). Each of these families has multiple individual enzymes and fascinating stories of discovery and functions. Interestingly, there are relatively low amino acid sequence similarities between the protease domains of different families. However, all of the catalytic domains have strikingly similar three-dimensional structures as well as a conserved zinc binding domain (HEXXHXX(G/N)XX(H/D)) at the active site and a conserved methionine-containing turn (Met-turn) (40). This review will focus on the astacin family (41, 42), and particularly meprins of this family.
Figure 1.

Metzinicins, astacins, and meprins. The metzincin superfamily includes the astacins, the ADAMs, the MMPs, the serralysins, and the papalysins and leishmanolysins (last two families not shown). Over 180 individual astacins have been identified in animals and bacteria, and several examples are shown, including the crayfish astacin, fly and human tolloids, and the meprins from hydra (HMP2), zebrafish, human, and mouse/rat. The ribbon diagram shows that the protease domain of meprins consists of five-stranded β-sheets, three α-helices, and a coil structure in the lower subdomain. The zinc (gray sphere) is pentacoordinated by three histidines of the motif HEXXHXXGXXH, a water molecule, and a tyrosine positioned by the Met-turn. Adapted from Ref. 39. This research was originally published in Molecular Aspects of Medicine. Sterchi, E. E., Stöcker, W., and Bond, J. S. Meprins, membrane-bound and secreted astacin metalloproteinases. Molecular Aspects of Medicine. 2008; 29, 309–328. © Elsevier Ltd.
The astacin family was recognized as a consequence of extensive cloning and sequencing that occurred in the 1980s and 1990s (see Fig. 1). The original members of the family, identified by sequence similarities, were as follows: the crayfish digestive enzyme astacin, bone morphogenetic protein-1 (BMP-1) from human bone, meprins from mouse kidney and human intestine, and UVS.2, a partial sequence from Xenopus laevis embryos (41). The name “astacin family” was chosen because the crayfish Astacus astacus enzyme was the first to be sequenced and characterized (43, 44). Astacins are present in animals and bacteria; none have yet been found in plants and fungi. Hundreds of astacins have been identified as genome sequencing expands to many species (45). In the human and mouse genomes, six astacin family genes have been identified, which includes two meprin genes, three BMP-1/tolloid-like genes, and one ovastacin gene. However, in Drosophila melanogaster, there are 16, and in C. elegans there are 40 astacins. The functions of most of the astacin genes in D. melanogaster and C. elegans have not been determined, but in parasitic nematodes, astacin enzymes are involved in moving through extracellular matrixes and in Hydra in head regeneration (46).
Of the characterized astacins, the crayfish astacin is the smallest, containing a 200-amino acid residue catalytic domain. From cDNA sequencing, it is known that there is a prepro sequence that is cleaved off during protein synthesis. Pre or signal sequences are found in all of the astacin family members examined thus far, presumably to direct the protein into the endoplasmic reticulum and the secretory pathway. Pro sequences keep the enzymes inactive as a regulatory mechanism. Whereas the active crayfish protein contains only the ∼20-kDa catalytic domain, most of the astacin family members contain one or more noncatalytic domains, C-terminal to the protease domain. Many contain one or more copies of an epidermal growth factor (EGF)-like domain, and a CUB (complement subcomponents Clr/Cls, embryonic sea urchin protein UEGF, BMP-1) domain (42). These are important for protein–protein or protein–substrate interactions. The noncatalytic domains are responsible for the variety of sizes of family members, which range from 200 (crayfish astacin) to 900 amino acids (mouse BMP-1). In addition, many of the more complex astacins are highly glycosylated proteins, further increasing their molecular mass and complexity.
Meprins, members of the astacin family, are unique oligomeric metalloproteases, containing homo- and hetero-oligomers of two evolutionarily related subunits, α and β (see Fig. 2). They exemplify the complexity of the metzincin superfamily members.
Figure 2.

Domain and oligomeric structure of meprins α and β. Domains are as follows: S (signal sequence), Pro (prosequence), protease, catalytic domain, MAM (meprin), A5 protein, protein-tyrosine phosphatase μ, TRAF homology, I (inserted), EGF (epidermal growth factor-like), TM (transmembrane-spanning), and C (cytoplasmic). During maturation, the meprin α subunit is cleaved in the I domain, separating the subunit from the membrane. As a result, three isoforms of meprin exist: membrane-bound meprin B (a homodimer of β subunits), membrane-bound meprin A (heterotetramers of α and β subunits, found in ratios of α2β2 and α1β3), and secreted meprin A (homomeric multimers of α subunit dimers). The secreted forms of meprin α dimers tend to self-associate and form large multimers (1–6 MDa) extracellularly.
Meprins were discovered in 1980 as a consequence of a search for proteolytic enzymes in diabetic mice (47, 48). In the process of searching for changes in proteolytic activity and protein turnover in streptozotocin-induced diabetes in BALB/c mice, proteolytic activities were measured in liver, kidney, and muscle tissues using a variety of substrates. No fundamental changes were found in degradation rates in the liver or in the proteolytic activities measured in diabetic mouse tissues compared with controls. However, it was noted that the kidneys of these mice had a relatively high activity using azocasein as substrate at basic pH values (pH 9). The enzyme was then purified from BALB/c mouse kidney and found to be a glycosylated membrane-bound metalloprotease with a subunit molecular mass of 85–90 kDa (48). The subunits formed disulfide-bridged dimers, and the dimers formed tetramers of 320 kDa. The human equivalent of mouse meprin, an intestinal enzyme called PABA-peptide hydrolase (named after the substrate hydrolyzed) was reported in 1982 (49), but the similarities of the mouse and human enzymes were not recognized until both were cloned and sequenced (41).
In 1983, BALB/c mice were unavailable for a period, and members of my laboratory collected kidneys from two other inbred mouse strains, C3H/He and CBA mice. In contrast to BALB/c mice, these mice had very low kidney azocaseinase activity. This led to a publication (50) describing the “deficiency” in many “C stock” mice. The publication was noticed by Chella David, a mouse immunogeneticist at the Mayo Clinic, who informed us that many C stock mice were bred for transplantation studies and had differences in the major histocompatibility genes (H-2 genes). These observations led to collaborative studies and the discovery that a gene (the Mep-1a gene) on mouse chromosome 17 near the H-2 complex was responsible for the level of expression of meprin activity in mouse kidney (51, 52). It is now known that the Mep-1a gene codes for the meprin α subunit (53). Further studies with the “deficient” or low-meprin mouse strains revealed that they expressed a latent form of meprin (54), containing meprin β subunits. The catalytic domain of meprin β is 58% identical to that of meprin α, and is encoded on mouse chromosome 18 (55), an example of divergent evolution from a single gene (53). The studies of meprin α and β in different strains of mice have led us to understand that there are several different combinations of meprin α and β that exist in mouse kidney to form the quaternary structure of the meprins, and the isoforms of meprin A and B (56), EC 3.4.24.63 and EC 3.4.24.18, respectively. The reason for the lack of expression of the α subunit in adults of some inbred mouse strains is unknown, but it is known that this is developmentally regulated because all mouse strains express both meprin α and β in embryonic kidney and until puberty (57).
The work on meprins in the 1980s was with proteins isolated from kidney brush-border membranes of adult mice; those from mice with high azocaseinase activity contain both α and β subunits, whereas those with the low meprin activity (“deficient” strains) contain only β subunits. The mouse α subunit is fully active at the plasma membrane, whereas the β subunit is predominantly latent but can be activated by trypsin-like enzymes. Studies of the activation of meprin α indicate that removal of the prosequence allows for formation of hydrogen bonds involving the two N-terminal residues that are critical for enzyme structure (58). With the advances of molecular biology in the 1980s and 1990s, especially cloning, sequencing, and site-directed mutagenesis, progress on structure and function proceeded at a rapid pace.
Meprin isoforms are structurally quite complex, with multidomain, multimeric structures, as shown diagrammatically in Fig. 2. The oligomers are composed of meprin α and/or β disulfide-linked dimers that may self-associate to form higher-molecular weight isoforms. Homomeric meprin A contains only α subunits, heteromeric meprin A contains both α and β subunits, and homomeric meprin B contains only β subunits. Both subunits are glycosylated, and the asparagine-linked sugars are important for disulfide bond formation, oligomerization, stability, secretion, and enzymatic activity (59, 60). As for the domain structure, both subunits contain a signal sequence, prosequence, protease (catalytic) domain, MAM domain, TRAF domain, EGF-like domain, transmembrane domain, and small C-terminal tail (6–26 amino acids). The noncatalytic domains are important for transport, structure, and activity of the proteases (Fig. 3) (61, 62). One notable difference between the α and β domain structures is that meprin α contains an I (inserted) domain between the EGF and TRAF domains that is missing in the β subunit. There is a proteolytic cleavage within the I domain during maturation in the secretory pathway that results in the release of this subunit from the membrane (63–65). Removing the I domain from the meprin α subunit by site-directed mutagenesis showed that the I domain is necessary and sufficient for proteolysis and release of the subunit from the membrane (Fig. 3). Therefore, meprin isomers containing only meprin α are secreted into the extracellular space. The disulfide-linked meprin α dimers tend to associate noncovalently into high-molecular weight complexes of 1–6 MDa, among the largest proteolytic complexes secreted in living systems (66) (Fig. 4). This self-association concentrates the monomer meprin A in the extracellular environment and may be important for stability and action, particularly at sites of infection and in the harsh environment of the intestine. Membrane-associated forms of meprins all contain meprin β subunits and may consist of meprin β disulfide–linked dimers, meprin α/β dimers that form tetramers, or meprin α/β dimers that associate noncovalently with meprin α dimers. Cross-linking studies with the meprin B dimer have revealed a compact structure with inter- and intradomain contacts within the protein, including TRAF–TRAF interactions (67). An X-ray structure of human meprin B is available showing that the active site is close to the membrane, which has implications for the shedding activity of this isoform (68).
Figure 3.
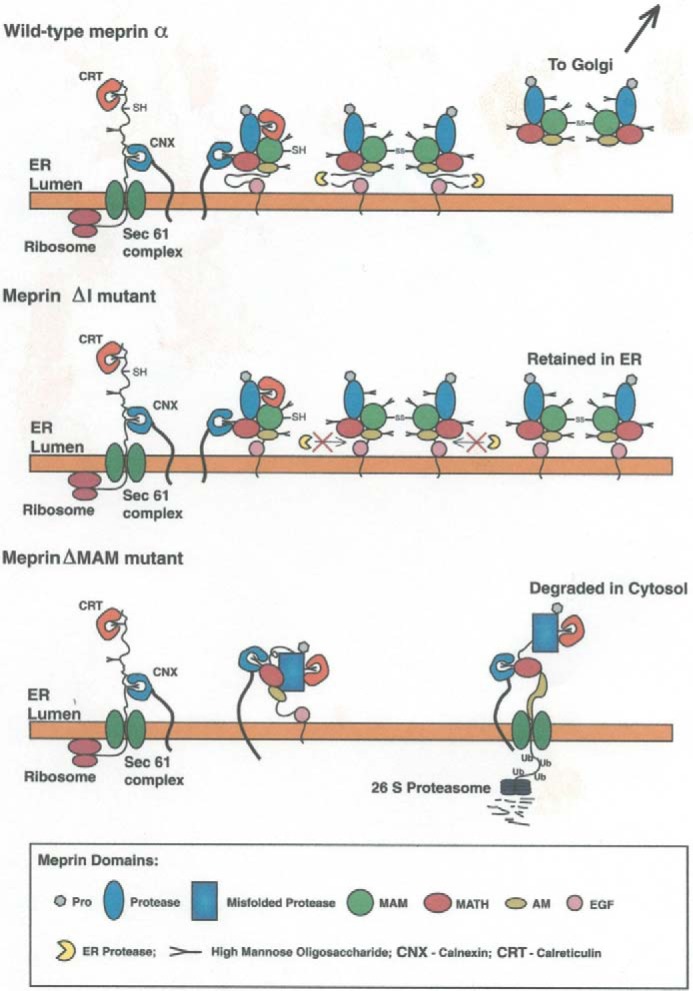
Intracellular trafficking of WT meprin α subunits and mutants. WT meprin α is secreted from cells after maturation in the endoplasmic reticulum (ER) and Golgi; if the I domain is deleted by site-directed mutagenesis (ΔI mutant), the subunit is retained in the endoplasmic reticulum/cis-Golgi; if the MAM domain is deleted (ΔMAM mutant) the subunit misfolds, and this triggers retrograde transport to the cytosol and degradation by the proteasome (64).
Figure 4.
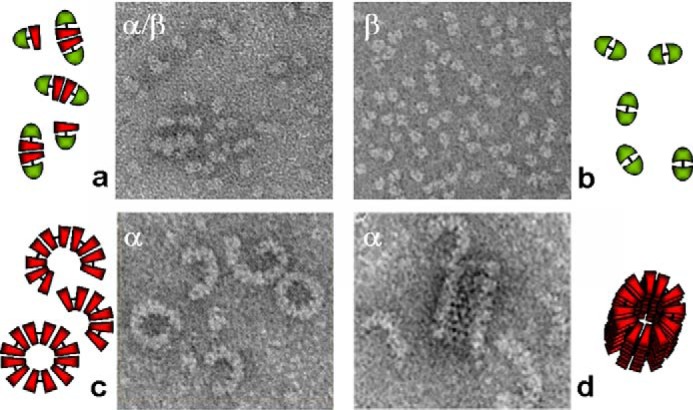
Oligomerization of meprin A and B. Shown are electron micrographs of rat meprin A and B expressed in human embryonic kidney 293 cells. Negatively stained samples of various isoforms are shown. Shown clockwise from left to right are the following: heteromeric meprin A containing tetramers of α and β subunits (a); homomeric meprin B (dimers of β subunits) (b); latent homomeric meprin A containing homodimers of meprin α subunits that associate noncovalently to form crescents, tubes, and spirals containing up to 100 subunits (d); and activated homomeric meprin A forming primarily rings and crescents containing about 10–12 subunits (c). Adapted from Refs. 39 and 66. This research was originally published in Molecular Aspects of Medicine. Sterchi, E. E., Stöcker, W., and Bond, J. S. Meprins, membrane-bound and secreted astacin metalloproteinases. Molecular Aspects of Medicine. 2008; 29, 309–328. © Elsevier Ltd. and the Journal of Biological Chemistry. Bertenshaw, G. P., Norcum, M. T., and Bond, J. S. Structure of homo- and hetero-oligomeric meprin metalloproteases: dimers, tetramers, and high molecular mass multimers. Journal of Biological Chemistry. 2003; 278, 2522–2532. © the American Society for Biochemistry and Molecular Biology.
The localization of meprins to kidney and intestinal brush-border membranes was originally deduced from cell fractionation studies and later through immunohistochemical studies (69, 70). Meprins are also expressed in leukocytes, and studies with these cells from mice with a deleted meprin β gene showed a diminished ability to move through extracellular matrix (71). Meprin expression in monocytes and natural killer cells affects homeostasis of these cells (72). Human meprin α and β subunits are expressed in different layers of the epidermis and are involved in cell proliferation and terminal differentiation of human skin cells (73). Through mRNA studies, meprin subunits have been detected in pancreas, testis, fetal liver, embryonic stem cells, and brain tissues. The highest expression levels are in kidney and intestine. For this reason, the functions of meprins have primarily been studied in these tissues. Additional insights into the function of the meprins have been gathered by creating knockout mice and challenging the mice (74, 75).
Meprins are capable of hydrolyzing a wide variety of substrates, from peptides to proteins. Mouse meprin B has a clear preference for acidic residues at cleavage sites; by contrast, homomeric meprin A prefers to cleave bonds containing small or hydrophobic amino acid residues (76, 77). These preferences lead to clear differences in the hydrolysis of peptide substrates. For example, meprin B, but not homomeric meprin A, cleaves gastrin and osteopontin; homomeric meprin A, but not meprin B, cleaves bradykinin and substance P. Meprin B also cleaves cell surface proteins, such as E-cadherin and ENaC (epithelial sodium channel) and thereby can affect cell–cell interactions and ion transport (78). Meprin A cleaves the tight junction protein occludin, which impairs epithelial barrier function and enhances monocyte migration (79). The meprin isoforms also have different preferences for cytokine activation and degradation, and the balance of the meprin isoforms could have important implications in cytokine profiles and the progression of inflammatory responses (80, 81, 82). High-throughput techniques, in a search for substrates of human meprins, have revealed many substrates and interesting links between meprins and ADAMs (83).
There is good evidence that meprins are involved in several disease processes, and these are areas that will be explored in the future and have therapeutic possibilities. For example, meprins are present at sites of inflammation, where they affect migration of leukocytes, degrade tight junction proteins, and activate/degrade cytokines. Studies of meprin α knockout mice have shown that decreased expression of this subunit is associated with increased intestinal inflammation in an experimental model of intestinal bowel disease. Furthermore, the human MEP1A gene is a susceptibility gene for inflammatory bowel disease, particularly ulcerative colitis (84). Meprins also influence the course of urinary tract infections in mice (85). Other studies with mice have implicated meprins in the pathogenesis of kidney diseases (75, 85–89), and polymorphisms in the human MEP1B gene are associated with diabetic nephropathy in Pima Indians (90). Meprins are expressed in various cancer cells (e.g. colon and breast) and are thought to play a role in tumor cell invasion and migration (91–94). Meprins have also been found to cleave amyloid precursor protein (APP) in vivo, implying a role in neurodegenerative diseases, such as Alzheimer's (95). Recent studies of the interaction of meprins with mucins in the intestine imply a role in protecting the host epithelium from bacteria and affecting the microbiome (96).
One of the challenges of the future is to understand the function of proteases in living organisms and to be able to activate and inhibit them selectively in specific tissues. Proteases exist in the context of networks of other molecules and other proteases, in cellular compartments, at cellular membranes, and in the extracellular milieu, and these environments are no doubt critical in determining function. System-wide approaches, such as degradomics that uses a combination of genetics, cell biology, and proteomics to identify substrates and active proteases, will be necessary to understand and regulate proteolytic systems (97). Herb Tabor has built the foundation for the JBC to move past the identification and characterization of individual enzymes and into complex multicomponent systems to shed more light on the role of proteases in the fabric of life.
This JBC Review is part of a collection honoring Herbert Tabor on the occasion of his 100th birthday. The author declares that she has no conflicts of interest with the contents of this article.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- JBC
- Journal of Biological Chemistry
- MMP
- matrix metalloproteinase
- ADAM
- a disintegrin and metalloproteinase
- ASBMB
- American Society for Biochemistry and Molecular Biology
- Met-turn
- methionine-containing turn
- BMP
- bone morphogenetic protein
- EGF
- epidermal growth factor.
References
- 1. Levene P. A. (1905) The cleavage products of proteoses. J. Biol. Chem. 1, 45–58 [Google Scholar]
- 2. Northrup J. H. (1930) Crystalline pepsin. I. Isolation and tests of purity. J. Gen. Physiol. 13, 739–766 10.1085/jgp.13.6.739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Robertson T. B. (1907) Note on the synthesis of a protein through the action of pepsin. J. Biol. Chem. 3, 95–99 [Google Scholar]
- 4. Northrop J. H., and Kunitz M. (1931) Isolation of protein crystals processing tryptic activity. Science 73, 262–263 10.1126/science.73.1888.262 [DOI] [PubMed] [Google Scholar]
- 5. Robertson T. B. (1907) Studies in the chemistry of the ion-proteid compounds: IV. On some chemical properties of casein and their possible relation to the chemical behavior of other protein bodies, with especial reference to hydrolysis of casein by trypsin. J. Biol. Chem. 2, 317–383 [Google Scholar]
- 6. Bergman M., and Fruton J. S. (1937) On proteolytic enzymes: XIII. Synthetic substrates for chymotrypsin. J. Biol. Chem. 118, 405–415 [Google Scholar]
- 7. Kimmel J. R., and Smith E. L. (1954) Crystalline papain: I. Preparation, specificity, and activation. J. Biol. Chem. 207, 515–531 [PubMed] [Google Scholar]
- 8. Matthews B. W., Jansonius J. N., Colman P. M., Schoenborn B. P., and Dupourque D. (1972) Three dimensional structure of thermolysin. Nature 238, 37–41 [DOI] [PubMed] [Google Scholar]
- 9. Anson M. L. (1937) The preparation of crystalline carboxypeptidase. J. Gen. Physiol. 20, 663–669 10.1085/jgp.20.5.663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Auld D. S., and Vallee B. L. (1970) Kinetics of carboxypeptidase A. II. Inhibitors of the hydrolysis of oligopeptides. Biochemistry 9, 602–609 10.1021/bi00805a022 [DOI] [PubMed] [Google Scholar]
- 11. Folk J. E., Piez K. A., Carroll W. R., and Gladner J. A. (1960) Carboxy-peptidase B: IV. Purification and characterization of the porcine enzyme. J. Biol. Chem. 235, 2272–2277 [PubMed] [Google Scholar]
- 12. Gallop P. M., Seifter S., and Meilman E. (1957) Studies on collagen: I. The partial purification, assay, and mode of activation of bacterial collagenase. J. Biol. Chem. 227, 891–906 [PubMed] [Google Scholar]
- 13. López-Otín C., and Bond J. S. (2008) Proteases: multifunctional enzymes in life and disease. J. Biol. Chem. 283, 30433–30437 10.1074/jbc.R800035200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. López-Otín C., and Matrisian L. M. (2007) Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 7, 800–808 10.1038/nrc2228 [DOI] [PubMed] [Google Scholar]
- 15. Rawlings N. D., and Salvesen G. (eds) (2013) Handbook of Proteolytic Enzymes, 3rd Ed., Academic Press, Inc., New York [Google Scholar]
- 16. Schoenheimer R. (1942) The Dynamic State of Body Constituents, Harvard University Press, Cambridge, MA [Google Scholar]
- 17. Coffey J. W., and De Duve C. (1968) Digestive activity of lysosomes: I. The digestion of protein by extracts of rat liver lysosomes. J. Biol. Chem. 243, 3255–3263 [PubMed] [Google Scholar]
- 18. Schimke R. T., Sweeney E. W., and Berlin C. M. (1965) Studies of the stability in vivo and in vitro of rat liver tryptophan pyrrolase. J. Biol. Chem. 240, 4609–4620 [PubMed] [Google Scholar]
- 19. Berlin C. M., and Schimke R. T. (1965) Influence of turnover rates on the responses of enzymes to cortisone. Mol. Pharmacol. 1, 149–156 [PubMed] [Google Scholar]
- 20. Schimke R. T., and Katumuma N. (eds) (1975) Intracellular Protein Turnover, Academic Press, New York [Google Scholar]
- 21. Ciechanover A., Hod Y., and Hershko A. (1978) A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem. Biophys. Res. Commun. 81, 1100–1105 10.1016/0006-291X(78)91249-4 [DOI] [PubMed] [Google Scholar]
- 22. Rose I. A., Warms J. V., and Hershko A. (1979) A high molecular weight protease in liver cytosol. J. Biol. Chem. 254, 8135–8138 [PubMed] [Google Scholar]
- 23. DeMartino G. N., and Goldberg A. L. (1979) Identificaton and partial purification of an ATP-stimulated alkaline protease in rat liver. J. Biol. Chem. 254, 3712–3715 [PubMed] [Google Scholar]
- 24. Wilk S., and Orlowski M. (1980) Cation-sensitive endopeptidase: isolation and specificity of the bovine pituitary enzyme. J. Neurochem. 35, 1172–1182 10.1111/j.1471-4159.1980.tb07873.x [DOI] [PubMed] [Google Scholar]
- 25. Hershko A., Ciechanover A., Heller H., Haas A. L., and Rose I. A. (1980) Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc. Natl. Acad. Sci. U.S.A. 77, 1783–1786 10.1073/pnas.77.4.1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ciechanover A., Finley D., and Varshavsky A. (1984) Ubiquitin dependence of selective protein degradation demonstrated in the mammalian cell cycle mutant ts85. Cell 37, 57–66 10.1016/0092-8674(84)90300-3 [DOI] [PubMed] [Google Scholar]
- 27. Hough R., Pratt G., and Rechsteiner M. (1987) Purification of two high molecular weight proteases from rabbit reticulocyte lysate. J. Biol. Chem. 262, 8303–8313 [PubMed] [Google Scholar]
- 28. Waxman L., Fagan J. M., and Goldberg A. L. (1987) Demonstration of two distinct high molecular weight proteases in rabbit reticulocytes, one of which degrades ubiquitin conjugates. J. Biol. Chem. 262, 2451–2457 [PubMed] [Google Scholar]
- 29. Milstein C., Brownlee G. G., Harrison T. M., and Mathews M. B. (1972) A possible precursor of immunoglobulin light chains. Nat. New Biol. 239, 117–120 10.1038/newbio239117a0 [DOI] [PubMed] [Google Scholar]
- 30. Wolfe P. B., Silver P., and Wickner W. (1982) The isolation of homogeneous leader peptidase from a strain of Escherichia coli which overproduces the enzyme. J. Biol. Chem. 257, 7898–7902 [PubMed] [Google Scholar]
- 31. Evans E. A., Gilmore R., and Blobel G. (1986) Purification of microsomal signal peptidase as a complex. Proc. Natl. Acad. Sci. U.S.A. 83, 581–585 10.1073/pnas.83.3.581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ellis H. M., and Horvitz H. R. (1986) Genetic control of programmed cell death in the nematode C. elegans. Cell 44, 817–829 10.1016/0092-8674(86)90004-8 [DOI] [PubMed] [Google Scholar]
- 33. Orth K., O'Rourke K., Salvesen G. S., and Dixit V. M. (1996) Molecular ordering of apoptotic mammalian CED-3/ICE-like proteases. J. Biol. Chem. 271, 20977–20980 10.1074/jbc.271.35.20977 [DOI] [PubMed] [Google Scholar]
- 34. De Clercq E. (2007) The design of drugs for HIV and HCV. Nat. Rev. Drug Discov. 6, 1001–1018 10.1038/nrd2424 [DOI] [PubMed] [Google Scholar]
- 35. Turk V., Stoka V., Vasiljeva O., Renko M., Sun T., Turk B., and Turk D. (2012) Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim. Biophys. Acta 1824, 68–88 10.1016/j.bbapap.2011.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsukada M., and Ohsumi Y. (1993) Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 333, 169–174 10.1016/0014-5793(93)80398-E [DOI] [PubMed] [Google Scholar]
- 37. DeMartino G. N. (2018) Introduction to the Thematic Minireview series: autophagy. J. Biol. Chem. 293, 5384–5385 10.1074/jbc.TM118.002429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barrett A. J., and McDonald J. K. (1980) in Mammalian Proteases: A Glossary and Bibliography, Vol. 1, pp. 359–378, Academic Press, Inc., New York [Google Scholar]
- 39. Sterchi E. E., Stöcker W., and Bond J. S. (2008) Meprins, membrane-bound and secreted astacin metalloproteinases. Mol. Aspects Med. 29, 309–328 10.1016/j.mam.2008.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bode W., Gomis-Rüth F. X., and Stöcker W. (1993) Astacins, serralysins, snake venom and matrix metalloproteinases exhibit identical zinc-binding environments (HEXXHXXGXXH and Met-turn) and topologies and should be grouped into a common family, the “metzincins”. FEBS Lett. 331, 134–140 10.1016/0014-5793(93)80312-I [DOI] [PubMed] [Google Scholar]
- 41. Dumermuth E., Sterchi E. E., Jiang W. P., Wolz R. L., Bond J. S., Flannery A. V., and Beynon R. J. (1991) The astacin family of metalloendopeptidases. J. Biol. Chem. 266, 21381–21385 [PubMed] [Google Scholar]
- 42. Bond J. S., and Beynon R. J. (1995) The astacin family of metalloendopeptidases. Protein Sci. 4, 1247–1261 10.1002/pro.5560040701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Titani K., Torff H. J., Hormel S., Kumar S., Walsh K. A., Rödl J., Neurath H., and Zwilling R. (1987) Amino acid sequence of a unique protease from the crayfish Astacus. Biochemistry 26, 222–226 10.1021/bi00375a029 [DOI] [PubMed] [Google Scholar]
- 44. Bode W., Gomis-Rüth F. X., Huber R., Zwilling R., and Stöcker W. (1992) Structure of astacin and implications for activation of astacins and zinc-ligation of collagenases. Nature 358, 164–167 10.1038/358164a0 [DOI] [PubMed] [Google Scholar]
- 45. Möhrlen F., Maniura M., Plickert G., Frohme M., and Frank U. (2006) Evolution of astacin-like metalloproteases in animals and their function in development. Evol. Dev. 8, 223–231 10.1111/j.1525-142X.2006.00092.x [DOI] [PubMed] [Google Scholar]
- 46. Yan L., Leontovich A., Fei K., and Sarras M. P. Jr. (2000) Hydra metalloproteinase 1: a secreted astacin metalloproteinase whose apical axis expression is differentially regulated during head regeneration. Dev. Biol. 219, 115–128 10.1006/dbio.1999.9568 [DOI] [PubMed] [Google Scholar]
- 47. Bond J. S. (1980) Failure to demonstrate increased protein turnover and intracellular proteinase activity in livers of mice with streptozotocin-induced diabetes. Diabetes 29, 648–654 10.2337/diab.29.8.648 [DOI] [PubMed] [Google Scholar]
- 48. Beynon R. J., Shannon J. D., and Bond J. S. (1981) Purification and characterization of a metalloendoproteinase from mouse kidney. Biochem. J. 199, 591–598 10.1042/bj1990591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sterchi E. E., Green J. R., and Lentze M. J. (1982) Non-pancreatic hydrolysis of N-benzoyl-l-tyrosyl-p-aminobenzoic acid (PABA-peptide) in the human small intestine. Clin. Sci. 62, 557–560 10.1042/cs0620557 [DOI] [PubMed] [Google Scholar]
- 50. Beynon R. J., and Bond J. S. (1983) Deficiency of a kidney metalloproteinase activity in inbred mouse strains. Science 219, 1351–1353 10.1126/science.6338590 [DOI] [PubMed] [Google Scholar]
- 51. Bond J. S., Beynon R. J., Reckelhoff J. F., and David C. S. (1984) Mep-1 gene controlling a kidney metalloendopeptidase is linked to the major histocompatibility complex in mice. Proc. Natl. Acad. Sci. U.S.A. 81, 5542–5545 10.1073/pnas.81.17.5542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reckelhoff J. F., Bond J. S., Beynon R. J., Savarirayan S., and David C. S. (1985) Proximity of the Mep-1 gene to H-2D on chromosome 17 in mice. Immunogenetics 22, 617–623 10.1007/BF00430310 [DOI] [PubMed] [Google Scholar]
- 53. Jiang W., Gorbea C. M., Flannery A. V., Beynon R. J., Grant G. A., and Bond J. S. (1992) The α subunit of meprin A: molecular cloning and sequencing, differential expression in inbred mouse strains, and evidence for divergent evolution of the α and β subunits. J. Biol. Chem. 267, 9185–9193 [PubMed] [Google Scholar]
- 54. Butler P. E., and Bond J. S. (1988) A latent proteinase in mouse kidney membranes. Characterization and relationship to meprin. J. Biol. Chem. 263, 13419–13426 [PubMed] [Google Scholar]
- 55. Gorbea C. M., Marchand P., Jiang W., Copeland N. G., Gilbert D. J., Jenkins N. A., and Bond J. S. (1993) Cloning, expression, and chromosomal localization of the mouse meprin β subunit. J. Biol. Chem. 268, 21035–21043 [PubMed] [Google Scholar]
- 56. Kounnas M. Z., Wolz R. L., Gorbea C. M., and Bond J. S. (1991) Meprin-A and -B: cell surface endopeptidases of the mouse kidney. J. Biol. Chem. 266, 17350–17357 [PubMed] [Google Scholar]
- 57. Kumar J. M., and Bond J. S. (2001) Developmental expression of meprin metalloprotease subunits in ICR and C3H/He mouse kidney and intestine in the embryo, postnatally and after weaning. Biochim. Biophys. Acta 1518, 106–114 10.1016/S0167-4781(01)00188-9 [DOI] [PubMed] [Google Scholar]
- 58. Johnson G. D., and Bond J. S. (1997) Activation mechanism of meprins, members of the astacin metalloendopeptidase family. J. Biol. Chem. 272, 28126–28132 10.1074/jbc.272.44.28126 [DOI] [PubMed] [Google Scholar]
- 59. Kadowaki T., Tsukuba T., Bertenshaw G. P., and Bond J. S. (2000) N-Linked oligosaccharides on the meprin A metalloprotease are important for secretion and enzymatic activity but not for apical targeting. J. Biol. Chem. 275, 25577–25584 10.1074/jbc.M003521200 [DOI] [PubMed] [Google Scholar]
- 60. Ishmael S. S., Ishmael F. T., Jones A. D., and Bond J. S. (2006) Protease domain glycans affect oligomerization, disulfide bond formation, and stability of the meprin A metalloprotease homooligomer. J. Biol. Chem. 281, 37404–37415 10.1074/jbc.M602769200 [DOI] [PubMed] [Google Scholar]
- 61. Tsukuba T., and Bond J. S. (1998) Role of the COOH-terminal domains of meprin A in folding, secretion, and activity of the metalloendopeptidase. J. Biol. Chem. 273, 35260–35267 10.1074/jbc.273.52.35260 [DOI] [PubMed] [Google Scholar]
- 62. Hengst J. A., and Bond J. S. (2004) Transport of meprin subunits through the secretory pathway: role of the transmembrane and cytoplasmic domains and oligomerization. J. Biol. Chem. 279, 34856–34864 10.1074/jbc.M405774200 [DOI] [PubMed] [Google Scholar]
- 63. Marchand P., Tang J., Johnson G. D., and Bond J. S. (1995) COOH-terminal proteolytic processing of secreted and membrane forms of the α subunit of the metalloprotease meprin A: requirement of the I domain for processing in the endoplasmic reticulum. J. Biol. Chem. 270, 5449–5456 10.1074/jbc.270.10.5449 [DOI] [PubMed] [Google Scholar]
- 64. Tsukuba T., Kadowaki T., Hengst J. A., and Bond J. S. (2002) Chaperone interactions of the metalloproteinase meprin A in the secretory or proteasomal-degradative pathway. Arch. Biochem. Biophys. 397, 191–198 10.1006/abbi.2001.2672 [DOI] [PubMed] [Google Scholar]
- 65. Tang J., and Bond J. S. (1998) Maturation of secreted meprin α during biosynthesis: role of the furin site, and identification of the COOH-terminal amino acids of the mouse kidney metalloprotease subunit. Arch. Biochem. Biophys. 349, 192–200 10.1006/abbi.1997.0453 [DOI] [PubMed] [Google Scholar]
- 66. Bertenshaw G. P., Norcum M. T., and Bond J. S. (2003) Structure of homo- and hetero-oligomeric meprin metalloproteases: dimers, tetramers, and high molecular mass multimers. J. Biol. Chem. 278, 2522–2532 10.1074/jbc.M208808200 [DOI] [PubMed] [Google Scholar]
- 67. Ishmael F. T., Shier V. K., Ishmael S. S., and Bond J. S. (2005) Intersubunit and domain interactions of the meprin B metalloproteinase: disulfide bonds and protein-protein interactions in the MAM and TRAF domains. J. Biol. Chem. 280, 13895–13901 10.1074/jbc.M414218200 [DOI] [PubMed] [Google Scholar]
- 68. Arolas J. L., Broder C., Jefferson T., Guevara T., Sterchi E. E., Bode W., Stöcker W., Becker-Pauly C., and Gomis-Rüth F. X. (2012) Structural basis for the sheddase function of human meprin β metalloproteinase at the plasma membrane. Proc. Natl. Acad. Sci. U.S.A. 109, 16131–16136 10.1073/pnas.1211076109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Craig S. S., Reckelhoff J. F., and Bond J. S. (1987) Distribution of meprin, in kidneys from mice with high- and low-meprin activity. Am. J. Physiol. 253, C535–C540 10.1152/ajpcell.1987.253.4.C535 [DOI] [PubMed] [Google Scholar]
- 70. Lottaz D., Hahn D., Müller S., Müller C., and Sterchi E. E. (1999) Secretion of human meprin from intestinal epithelial cells depends on differential expression of the α and β subunits. Eur. J. Biochem. 259, 496–504 10.1046/j.1432-1327.1999.00071.x [DOI] [PubMed] [Google Scholar]
- 71. Crisman J. M., Zhang B., Norman L. P., and Bond J. S. (2004) Deletion of the mouse meprin β metalloproteinase gene diminishes the ability of leukocytes to disseminate through extracellular matrix. J. Immunol. 172, 4510–4519 10.4049/jimmunol.172.7.4510 [DOI] [PubMed] [Google Scholar]
- 72. Sun Q., Jin H.-J., and Bond J. S. (2009) Disruption of the meprin α and β genes in mice alters homeostasis of monocytes and natural killer cells. Exp. Hematol. 37, 346–356 10.1016/j.exphem.2008.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Becker-Pauly C., Höwel M., Walker T., Vlad A., Aufenvenne K., Oji V., Lottaz D., Sterchi E. E., Debela M., Magdolen V., Traupe H., and Stöcker W. (2007) The α and β subunits of the metalloprotease meprin are expressed in separate layers of human epidermis, revealing different functions in keratinocyte proliferation and differentiation. J. Invest. Dermatol. 127, 1115–1125 10.1038/sj.jid.5700675 [DOI] [PubMed] [Google Scholar]
- 74. Norman L. P., Jiang W., Han X., Saunders T. L., and Bond J. S. (2003) Targeted disruption of the meprin β gene in mice leads to under representation of knock-out mice and changes in renal gene expression profiles. Mol. Cell. Biol. 23, 1221–1230 10.1128/MCB.23.4.1221-1230.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bylander J., Li Q., Ramesh G., Zhang B., Reeves W. B., and Bond J. S. (2008) Targeted disruption of the meprin metalloproteinase beta gene protects against renal ischemia/reperfusion injury in mice. Am. J. Physiol. Renal Physiol. 294, F480–F490 10.1152/ajprenal.00214.2007 [DOI] [PubMed] [Google Scholar]
- 76. Bertenshaw G. P., Turk B. E., Hubbard S. J., Matters G. L., Bylander J. E., Crisman J. M., Cantley L. C., and Bond J. S. (2001) Marked differences between metalloproteases meprin A and B in substrate and peptide bond specificity. J. Biol. Chem. 276, 13248–13255 10.1074/jbc.M011414200 [DOI] [PubMed] [Google Scholar]
- 77. Villa J. P., Bertenshaw G. P., and Bond J. S. (2003) Critical amino acids in the active site of meprin metalloproteinases for substrate and peptide bond specificity. J. Biol. Chem. 278, 42545–42550 10.1074/jbc.M303718200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Garcia-Caballero A., Ishmael S. S., Dang Y., Gillie D., Bond J. S., Milgram S. L., and Stutts M. J. (2011) Activation of the epithelial sodium channel by the metalloprotease meprin β subunit. Channels 5, 14–22 10.4161/chan.5.1.13759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bao J., Yura R. E., Matters G. L., Bradley S. G., Shi P., Tian F., and Bond J. S. (2013) Meprin A impairs epithelial barrier function, enhances monocyte migration, and cleaves the tight junction protein occludin. Am. J. Physiol. Renal Physiol. 305, F714–F726 10.1152/ajprenal.00179.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Banerjee S., and Bond J. S. (2008) Prointerleukin-18 is activated by meprin β in vitro and in vivo in intestinal inflammation. J. Biol. Chem. 283, 31371–31377 10.1074/jbc.M802814200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Banerjee S., Jin G., Bradley S. G., Matters G. L., Gailey R. D., Crisman J. M., and Bond J. S. (2011) Balance of meprin A and B in mice affects the progression of experimental inflammatory bowel disease. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G273–G282 10.1152/ajpgi.00504.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Keiffer T. R., and Bond J. S. (2014) Meprin metalloproteinases inactivate interleukin 6. J. Biol. Chem. 289, 7580–7588 10.1074/jbc.M113.546309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jefferson T., Auf dem Keller U., Bellac C., Metz V. V., Broder C., Hedrich J., Ohler A., Maier W., Magdolen V., Sterchi E., Bond J. S., Jayakumar A., Traupe H., Chalaris A., Rose-John S., et al. (2013) The substrate degradome of meprin metalloproteases reveals an unexpected proteolytic link between meprin β and ADAM10. Cell Mol. Life Sci. 70, 309–333 10.1007/s00018-012-1106-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Banerjee S., Oneda B., Yap L. M., Jewell D. P., Matters G. L., Fitzpatrick L. R., Seibold F., Sterchi E. E., Ahmad T., Lottaz D., and Bond J. S. (2009) MEP1A for meprin A metalloprotease subunit is a susceptibility gene for inflammatory bowel disease. Mucosal Immunol. 2, 220–231 10.1038/mi.2009.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yura R. E., Bradley S. G., Ramesh G., Reeves W. B., and Bond J. S. (2009) Meprin A metalloproteases enhance renal damage and bladder inflammation after LPS challenge. Am. J. Physiol. Renal Physiol. 296, F135–F144 10.1152/ajprenal.90524.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Trachtman H., Valderrama E., Dietrich J. M., and Bond J. S. (1995) The role of meprin A in the pathogenesis of acute renal failure. Biochem. Biophys. Res. Commun. 208, 498–505 10.1006/bbrc.1995.1366 [DOI] [PubMed] [Google Scholar]
- 87. Mathew R., Futterweit S., Valderrama E., Tarectecan A. A., Bylander J. E., Bond J. S., and Trachtman H. (2005) Meprin-α in chronic diabetic nephropathy: interaction with the rennin-angiotensin axis. Am. J. Physiol. Renal Physiol. 289, F911–F921 10.1152/ajprenal.00037.2005 [DOI] [PubMed] [Google Scholar]
- 88. Ongeri E. M., Anyanwu O., Reeves W. B., and Bond J. S. (2011) Villin and actin in the mouse kidney brush border membrane bind to and are degraded by meprins, an interaction that contributes to injury in ischemia reperfusion. Am. J. Physiol. Renal Physiol. 301, F871–F882 10.1152/ajprenal.00703.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Niyitegeka J.-M. V., Bastidas A. C., Newman R. H., Taylor S. S., and Ongeri E. M. (2015) Isoform-specific interactions between meprin metalloproteases and the catalytic subunit of protein kinase A: significance in acute and chronic kidney injury. Am. J. Physiol. Renal Physiol. 308, F56–F68 10.1152/ajprenal.00167.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Red Eagle A. R., Hanson R. L., Jiang W., Han X., Matters G. L., Imperatore G., Knowler W. C., and Bond J. S. (2005) Meprin β metalloprotease gene (MEP1B) polymorphisms associated with diabetic nephropathy in the Pima Indians. Hum. Genet. 118, 12–22 10.1007/s00439-005-0019-7 [DOI] [PubMed] [Google Scholar]
- 91. Dietrich J. M., Jiang W., and Bond J. S. (1996) A novel meprin β mRNA in mouse embryonal and human colon carcinoma cells. J. Biol. Chem. 271, 2271–2278 10.1074/jbc.271.4.2271 [DOI] [PubMed] [Google Scholar]
- 92. Lottaz D., Maurer C. A., Hahn D., Büchler M. W., and Sterchi E. E. (1999) Nonpolarized secretion of human meprin α in colorectal cancer generates an increased proteolytic potential in the stroma. Cancer Res. 59, 1127–1133 [PubMed] [Google Scholar]
- 93. Matters G. L., and Bond J. S. (1999) Expression and regulation of the meprin β gene in human cancer cells. Mol. Carcinog. 25, 169–178 10.1002/(SICI)1098-2744(199907)25:3<169::AID-MC3>3.0.CO;2-Y [DOI] [PubMed] [Google Scholar]
- 94. Matters G. L., Manni A., and Bond J. S. (2005) Inhibitors of polyamine biosynthesis selectively decrease the expression of the metalloprotease meprin α in hormone-independent human breast cancer cells. Clin. Exp. Metastasis 22, 331–339 10.1007/s10585-005-0660-5 [DOI] [PubMed] [Google Scholar]
- 95. Broder C., and Becker-Pauly C. (2013) The metalloproteases meprin α and meprin β: unique enzymes in inflammation, neurodegeneration, cancer and fibrosis. Biochem. J. 450, 253–264 10.1042/BJ20121751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Schütte A., Ermund A., Becker-Pauly C., Johansson M. E. V., Rodriguez-Pineiro A. M., Bäckhed F., Müller S., Lottaz D., Bond J. S., and Hansson G. C. (2014) Microbial-induced meprn β cleavage in MUC2 mucin and functional CFTR channel are required to release anchored small intestinal mucus. Proc. Natl. Acad. Sci. U.S.A. 111, 12396–12401 10.1073/pnas.1407597111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. López-Otín C., and Overall C. M. (2002) Protease degradomics, a new challenge for proteomics. Nat. Rev. Mol. Cell Biol. 3, 509–519 10.1038/nrm858 [DOI] [PubMed] [Google Scholar]
- 98. Rawlings N. D., Barrett A. J., Thomas P. D., Huang X., Bateman A., and Finn R. D. (2018) The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 46, D624–D632 10.1093/nar/gkx1134 [DOI] [PMC free article] [PubMed] [Google Scholar]