

Abstract
Myosins are molecular motors that use a conserved ATPase cycle to generate force. We investigated two mutations in the converter domain of myosin V (R712G and F750L) to examine how altering specific structural transitions in the motor ATPase cycle can impair myosin mechanochemistry. The corresponding mutations in the human β-cardiac myosin gene are associated with hypertrophic and dilated cardiomyopathy, respectively. Despite similar steady-state actin-activated ATPase and unloaded in vitro motility–sliding velocities, both R712G and F750L were less able to overcome frictional loads measured in the loaded motility assay. Transient kinetic analysis and stopped-flow FRET demonstrated that the R712G mutation slowed the maximum ATP hydrolysis and recovery-stroke rate constants, whereas the F750L mutation enhanced these steps. In both mutants, the fast and slow power-stroke as well as actin-activated phosphate release rate constants were not significantly different from WT. Time-resolved FRET experiments revealed that R712G and F750L populate the pre- and post-power–stroke states with similar FRET distance and distance distribution profiles. The R712G mutant increased the mole fraction in the post-power–stroke conformation in the strong actin-binding states, whereas the F750L decreased this population in the actomyosin ADP state. We conclude that mutations in key allosteric pathways can shift the equilibrium and/or alter the activation energy associated with key structural transitions without altering the overall conformation of the pre- and post-power–stroke states. Thus, therapies designed to alter the transition between structural states may be able to rescue the impaired motor function induced by disease mutations.
Keywords: myosin, actin, muscle, fluorescence resonance energy transfer (FRET), cardiomyopathy, ATPase, mechanochemistry, motor protein, structural kinetics
Introduction
Myosins are molecular motors that generate force by utilizing a highly conserved ATPase cycle (1). Myosin motors perform diverse cellular functions, including cytokinesis, cell adhesion, organelle transport, cell polarization, and muscle contraction (2, 3). The structure of the myosin molecule is broadly divided into head, neck, and tail regions. The head contains the catalytic domain, which is conserved across the myosin superfamily and is responsible for ATP hydrolysis and actin binding. Depending on the myosin isoform, the neck contains a variable number of IQ motifs that serve as binding sites for light chains (e.g. calmodulin) and is also referred to as the lever arm domain. Force is generated as a result of the lever arm swinging at key points in the ATPase cycle, and it is the converter domain that communicates structural changes between the lever arm and the catalytic domain. Recently, the converter domain has been highlighted as hot spot for point mutations in cardiac myosin associated with inherited cardiomyopathies, indicating the importance of this region in mediating mechanochemical coupling in all myosin motors (4).
Hypertrophic cardiomyopathy (HCM)4 is an extremely prevalent inherited heart disease, affecting ∼1 in 200 individuals (5). HCM is also the most common cause of sudden cardiac death in young people (6). Clinically, HCM is characterized by left-ventricular hypertrophy, myocyte disarray, fibrosis, and diastolic dysfunction (7–9). Furthermore, the characteristic left-ventricular hypertrophy is associated with increased contractility. Dilated cardiomyopathy (DCM) affects ∼1 in 2500 individuals and is the most common cause of heart failure in young adults (10). In contrast to HCM, DCM is characterized by left-ventricular dilation, myocyte death, fibrosis, and systolic dysfunction (8, 11). However, it still remains unclear how these disease mutations impact key structural changes in the myosin ATPase cycle and how specific structure–function defects lead to disease phenotypes.
The atomic structure of the myosin motor domain from both muscle and nonmuscle myosin and various species from Dictyostelium to humans has revealed a conserved structural fold (12). The myosin motor consists of four structural domains, each connected by flexible linkers, an N-terminal 25-kDa domain, an upper and lower 50-kDa domain, and a 20-kDa C-terminal domain (Fig. 1A) (13). Allosteric coupling between these domains allows for coordination of ATP binding and hydrolysis in the active site with structural changes in the actin-binding region associated with actin binding/dissociation. In addition, the priming of the lever arm for force generation (recovery stroke) and the force-generating structural changes in the lever arm (power stroke) are coordinated by amplifying small structural changes in the active site with large conformational changes in the converter/lever arm (14) (Fig. 1, A and B). Specific allosteric pathways in the myosin motor have been proposed based on high resolution structural studies of myosin in different nucleotide states (15) and mutational structure–function studies (16). However, many of these hypothesized pathways have not been examined with techniques that can directly examine conformational changes in real-time and examine the impact of specific point mutations on myosin mechanochemistry. Myosin V is an exemplary model for examining key structural changes and defining important allosteric pathways because there is high-resolution structural information on many of the structural states in the myosin ATPase pathway (17). Additionally, many biochemical and biophysical studies have revealed important details about the mechanochemical cycle (18–20). Thus, introducing mutations in crucial amino acid residues and examining how structural changes are altered in real time will allow for a complete understanding of how point mutations can disrupt key allosteric pathways and specifically impair motor function.
Figure 1.

Structural model of converter mutations in myosin V. A, diagram of the domain structure of myosin and the movement of the lever arm in the pre- and post-power–stroke states (L50, lower 50-kDa domain; U50, upper 50-kDa domain; CNV, converter domain; NT, N-terminal domain). B, an alignment of pre- and post-power–stroke state crystal structures showing the conformational change of the converter/lever arm. Navy, pre-power–stroke state structure (PDB code 1BR4); orange, post-power–stroke state structure (PDB code 1OE9); beige, light chain; green, converter domain.
Interestingly, mutations in the converter domain of MYH7 (β-cardiac myosin (M2β)) have been linked to opposing phenotypes. Specifically, R723G and F764L are both located in the converter domain (Fig. 1B) of human cardiac myosin; R723G causes HCM, whereas F764L causes DCM (21, 22). Because the converter domain is proposed to communicate structural changes between the lever arm and motor domain during the catalytic cycle, we proposed that mutations in this region will differentially alter the movement of the lever arm. In addition, communication between the converter domain and the active site is thought to be a mechanism for mediating the sensitivity of the myosin ATPase cycle to external load. Using an extremely high-resolution FRET system developed by our laboratory, we have examined the dynamics of the lever arm during the ATPase cycle in WT myosin V, as well as for the converter domain mutants R712G and F750L that are analogous to the HCM/DCM mutations in M2β. Although the mutations will not likely have the same impact on M2β as they do in myosin V, this study highlights a powerful approach of directly examining the impact of mutations on key structural changes to allow for determining how structural impairments disrupt motor function. Specifically, this study presents direct evidence that mutations in the converter domain can differentially alter lever arm rotation, which ultimately leads to the impaired ability to adapt to external load. Our work also highlights important allosteric pathways in myosin that allow for communication between the active site and force-generating lever arm.
Results
Expression and purification of myosin V constructs
We examined the motor function, biochemical, and structural properties of myosin V 1IQ containing the N-terminal tetracysteine motif (MV) expressed in the baculovirus system as described (23–26). To examine the impact of the mutations on the steady-state kinetic, transient kinetic, and motile properties, we co-expressed MV with WT calmodulin. For the studies in which we examined the lever arm swing during the recovery and power stroke by FRET, we exchanged QSY-labeled calmodulin (T110C) during the purification procedure and labeled the N-terminal tetracysteine site with FlAsH (MV NTF:QSY-CaM) (25). A noncalmodulin-exchanged MV was labeled with FlAsH and served as a donor-only control. Each of the protein preparations had similar yields/purity and fluorescence labeling efficiencies as previously reported (25).
Impact of the mutations on steady-state motor properties
We first examined the impact of the R712G and F750L mutations on myosin motor properties. The actin-activated ATPase assay was examined in four protein preparations and revealed that the mutations only had a minor impact on the steady-state ATPase kinetics. The ATPase activity was plotted as function of actin concentration and fit to a Michaelis–Menten equation (Fig. S1 and Table 1). The actin concentration at which ATPase is one-half maximal (KATPase) was unchanged in both mutants compared with WT. The maximum rate of ATPase (kcat) was slightly increased (10%) in the F750L mutant but unchanged in the R712G mutant.
Table 1.
Summary of steady-state and transient kinetic results
*, p < 0.05; **, p < 0.01.
| WT | R712G | F750L | |
|---|---|---|---|
| Steady-state ATPase values ± S.D. (n = 4)a | |||
| v0 (s−1) | 0.09 ± 0.11 | 0.04 ± 0.03 | 0.07 ± 0.05 |
| kcat (s−1) | 9.4 ± 0.3 | 8.9 ± 0.7 | *10.3 ± 0.5 |
| KATPase (μm) | 2.4 ± 0.4 | 2.6 ± 0.7 | 1.8 ± 0.5 |
| Rate/equilibrium constants ± S.E. | |||
| ATP binding/hydrolysis (myosin)b | |||
| K1Tk+2T (μm·s−1) | 1.5 ± 0.1 | 1.4 ± 0.1 | 1.5 ± 0.1 |
| k+H + k−H (maximum rate, s−1) | 397 ± 46 | 230 ± 23 | 484 ± 25 |
| ATP binding (actomyosin)c | |||
| 1/K′1Tk′+2T (μm·s−1) | 1.6 ± 0.2 | 1.7 ± 0.2 | 1.6 ± 0.2 |
| k′+2T (s−1) | 660 ± 50 | 699 ± 46 | 631 ± 34 |
| Recovery stroke (n = 3–4)d | |||
| k+H + k−H (maximum rate, s−1) | 290 ± 8 | *219 ± 14 | **596 ± 44 |
| Actin-activated phosphate release (n = 3)e | |||
| k+Pi (maximum rate) | 206 ± 35 | 270 ± 69 | 181 ± 56 |
| Power stroke (n = 3)d | |||
| Kactin × kPWF (actin concentration dependence, μm·s−1) | 7.9 ± 0.5 | 8.1 ± 0.6 | 7.3 ± 0.6 |
| k+PWF (maximum rate, s−1) | ≥276 | ≥288 | ≥314 |
| k+PWS (maximum rate, s−1) | 78 ± 33 | 109 ± 75 | 38 ± 12 |
| Actomyosin ADP release | |||
| k′+D (s−1)f | 26.9 ± 0.4 | 25.3 ± 0.3 | 33.0 ± 0.6 |
a Steady-state ATPase measurements.
b Intrinsic tryptophan fluorescence.
c Pyrene actin.
d FRET.
e MDCC-PBP fluorescence.
f mantADP fluorescence.
We examined the in vitro motility assay under unloaded conditions in six different protein preparations. The average velocity of 2–3 videos and more than 30 filaments from each video was determined, and the overall average and S.D. of all of the preparations were found to be similar in WT, R712G, and F750L (395 ± 19, 401 ± 17, and 399 ± 11 nm/s, respectively).
In two protein preparations, we examined loaded in vitro motility by determining the sliding velocity in the presence of increasing amounts of a tethering load (Fig. 2). The tethering load was produced by an MV mutant (G440A) that can bind actin with a high affinity even in the presence of saturating ATP (27). The advantage of using a mutant myosin as a tether is that its size and therefore interaction with the actin filament on the motility surface is identical to that of the active myosin. We found that the velocity and number of stuck filaments for both R712G and F750L were more sensitive to the presence of tethering loads. The average velocity, percentage of stuck filaments, and average velocity of moving filaments were plotted as a function of the fraction of G440A MV present ([G440A]/[total MV]) (Fig. 2, A–C). In the presence of 20% or more G440A MV, the average sliding velocity was reduced ∼2-fold more than WT in both mutants (WT = 226 ± 20, R712G = 98 ± 37, and F750L = 143 ± 12 nm/s) (Fig. 2A). A similar trend was observed by examining the percentage of stuck filaments (Fig. 2B). We also examined the velocity of only the moving filaments by eliminating the stuck filaments from the analysis and found that there was little difference in the velocity as a function of load in all three constructs (Fig. 2C).
Figure 2.
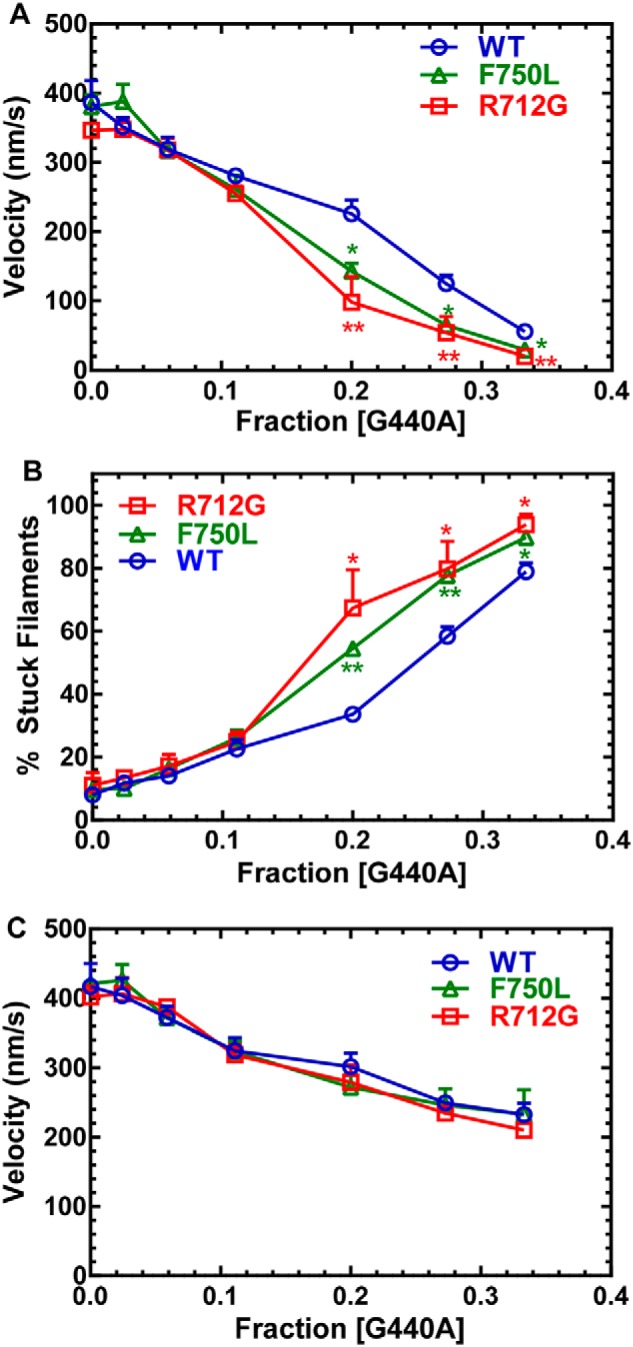
In vitro motility–sliding velocity. The sliding velocity was similar to WT for both mutants in the absence of load. The presence of G440A MV (nonhydrolyzing mutant) was used as a tethering load in the loaded in vitro motility assay. A, the average velocity of all of the filaments (moving and nonmoving) was determined and plotted as a function of the fraction of G440A MV present relative to the total MV added to the assay. B, the number of stuck filaments was determined and plotted as a function of the fraction of G440A. C, the velocity of only the moving filaments was plotted as a function of G440A fraction. Significant differences of the mutants compared with the WT are indicated by asterisks (*, p < 0.05; **, p < 0.005; n = 4).
Transient kinetic analysis
We performed transient kinetic analysis of key steps in the MV ATPase cycle using established methods (27–30). The conserved actomyosin ATPase pathway (Scheme 1) was used to model the transient kinetic studies with the actin-bound equilibrium constants represented by a prime and the dominant pathway outlined within the box. The transient and steady-state kinetic results are summarized in Table 1 and Table S1.
Scheme 1.

Conserved actomyosin ATPase pathway.
The rate of ATP binding and the maximum rate of ATP hydrolysis in the absence of actin were monitored by examining the increase in tryptophan fluorescence (1 μm MV) at varying ATP concentrations (Fig. 3). The fluorescence transients were fit to a single-exponential function, and the rate constants were hyperbolically dependent on ATP concentration (Fig. 3A). At concentrations less than 50 μm ATP, the rate constants were linearly dependent on ATP concentration and allowed us to determine the second-order binding constant for ATP binding, which was unchanged in both mutants compared with WT. However, the maximum rates of the tryptophan fluorescence transients were 20% faster in the F750L mutant and 40% slower in the R712G mutant (Fig. 3B), indicating a change in the apparent rate constant for ATP hydrolysis.
Figure 3.
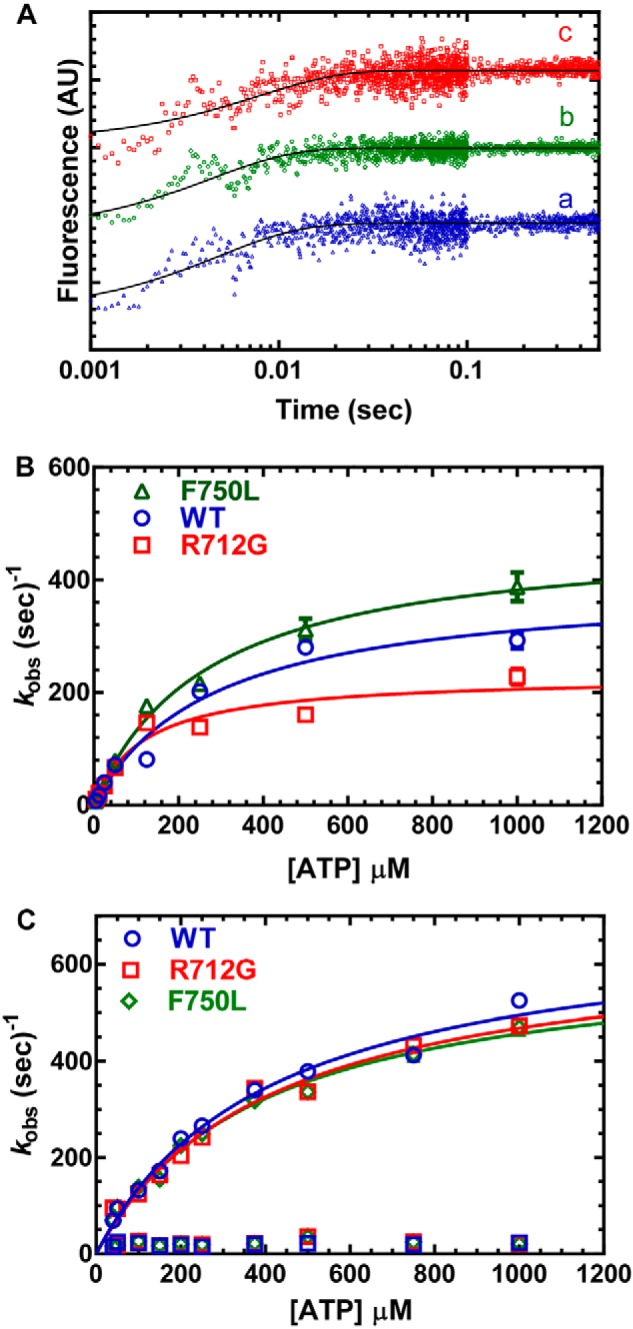
ATP binding and hydrolysis. A, MV (1 μm) was mixed with varying concentrations of ATP, and the tryptophan fluorescence increase was monitored as a function of time and fit to a single-exponential function. B, the observed rate constants were hyperbolically dependent on ATP concentration. C, the rate of ATP-induced dissociation from pyrene actin was examined by mixing MV:pyrene actin (0.25 μm) with varying concentrations of ATP. The fluorescence transients were fit to a two-exponential function with the fast phase being hyperbolically dependent on ATP concentration. The slow phase was a small component (5–10%) of the fluorescence signal and similar at each actin concentration (10–20 s−1). Fluorescence is reported in arbitrary units (AU).
We also examined the rate of ATP-induced dissociation of actomyosin using pyrene-labeled actin (0.25 μm MV and 0.25 μm pyrene actin) (Fig. 3C). We found that the rate of pyrene fluorescence increase as a function of ATP concentration was best fit to a two-exponential function. The slow phase was ∼10% of the fluorescence signal and independent of ATP concentration (kobs ∼20 s−1). The fast phase of the fluorescence transient was hyperbolically dependent on ATP concentration and allowed us to determine the equilibrium constant for the initial rapid-equilibrium interaction between ATP and actomyosin (K1T) and the maximum rate of transition into the weak binding states (k+2T). We found that there was little difference in K1T and k+2T for the three constructs.
The rate constants for actin-activated phosphate release were measured by mixing MV (0.5 μm) with substoichiometric ATP (0.45 μm) and then mixing with varying actin concentrations in the presence of phosphate-binding protein (5 μm; MDCC-PBP) (Fig. 4, A and B). The fluorescence transients were best fit by a single-exponential function (phosphate burst) followed by a linear or slow exponential phase (Fig. 4A). The phosphate burst rate constant was hyperbolically dependent on actin concentration for WT and both mutants (Fig. 4B). The actin concentration required to reach one-half maximal phosphate release (13 ± 8, 11 ± 9, and 10 ± 4 μm, respectively) and the maximum rate of phosphate release (270 ± 69, 181 ± 56, and 206 ± 35 s−1, respectively) in R712G and F750L were not significantly different from WT.
Figure 4.

Actin-activated product release. Sequential mix single-turnover experiments were performed by mixing MV with ATP, aging the reaction to form the M.ADP.Pi state (0.45 μm), and then mixing with different concentrations of actin in the presence of MDCC-PBP (5 μm). A, the fluorescence transients were fit to a single-exponential function (phosphate burst) followed by a linear or slow exponential rise. B, the phosphate release rate constants were plotted as a function of actin concentration and fit to a hyperbolic function to estimate the maximum rate of phosphate release. C, acto-MV (0.25 μm) in presence mantADP (5 μm) was mixed with saturating ATP (1 mm) at 25 °C to determine the ADP release rate constant. D, the mantADP release rate constant was examined as a function of temperature, and the associated Eyring plot (natural log of the ADP release rate constant as a function of inverse temperature) was fit to a linear regression. Fluorescence is reported in arbitrary units (AU).
The rate constants for ADP release from acto-MV were determined by mixing a complex of MV, mantADP, and actin (0.25 μm, 5 μm, and 0.5 μm, respectively) with excess ATP (1 mm) (Fig. 4, C and D). The mant fluorescence decrease for most transients followed a single-exponential function, with some transients displaying a slow fluorescence decrease that was less than 10% of the signal (Fig. 4C). There was an increase (23%) in the mantADP release rate constant in the F750L mutant, whereas the R712G was similar to WT. We also measured the mantADP release rate constant as a function of temperature and observed that the F750L mutant maintained a faster mantADP release rate constant at all temperatures, whereas the Eyring plots yielded a similar energy of activation for all three constructs (ΔH ∼80–82 kJ·mol−1) (Fig. 4D).
Structural kinetics of lever arm swing
In our previous work, we established a method of measuring the movement of the lever arm during the recovery and power-stroke steps using FRET between FlAsH labeled at the N terminus and the nonfluorescent acceptor QSY-labeled at T110C on calmodulin of the first IQ motif (25). In the current work, we performed similar measurements in the R712G and F750L mutant and compared these results with repeated measurements of WT MV.
The rate constants for the movement of the lever arm during the recovery stroke were monitored by mixing MV NTF:QSY-CaM (0.25 μm) with varying concentrations of ATP (data represent 3–4 protein preparations) (Fig. 5). The fluorescence transients were best fit by a one- or two-exponential function (Fig. 5A). The fast phase of the fluorescence transient was hyperbolically dependent on ATP concentration (Fig. 5B), whereas the slow phase (5% or less of the fluorescence signal) was also hyperbolically dependent on ATP concentration (Fig. 5C). We found that compared with WT, the R712G mutant reduced the maximum rate constant of the fast recovery stroke (25%), whereas the F750L had a 2-fold increase in the maximum rate constant of the recovery stroke. The slow phase of the recovery stroke was unchanged in the two mutants compared with WT.
Figure 5.
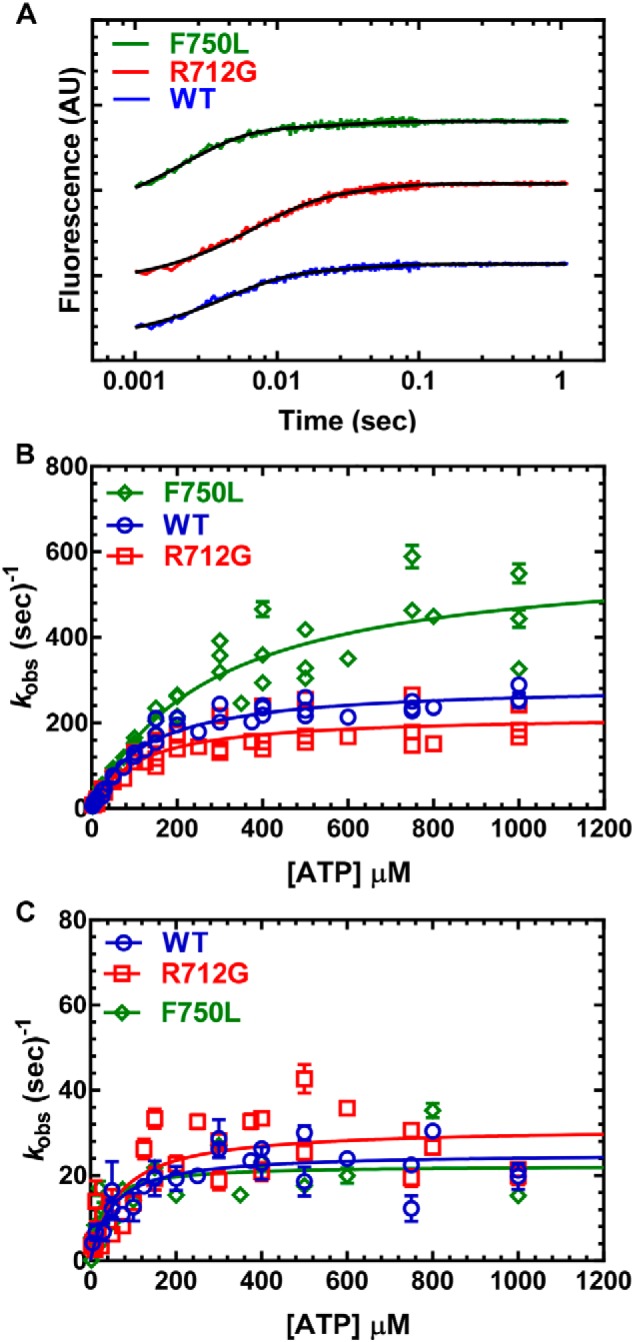
Recovery stroke. 0.25 μm MV FlAsH.QSY-CaM was mixed with increasing concentrations of ATP, and the decrease in the FRET signal was monitored by following the increase in FlAsH fluorescence. A, the fluorescence transients were best fit by a two-exponential function at all ATP concentrations. B, the fast phase (recovery stroke) was the majority of the fluorescence signal (≥90%) and was hyperbolically dependent on ATP concentration. C, the slow phase was also hyperbolically dependent on ATP concentration. Fluorescence is reported in arbitrary units (AU).
The lever arm swing during the power stroke was measured with a sequential mix stopped-flow experiment (Fig. 6) (similar conditions to the phosphate release experiments in Fig. 4, A and B). We first mixed MV NTF:QSY-CaM (0.25 μm) with substoichiometric ATP (0.2 μm) and allowed the reaction to age (10 s) and then mixed with varying concentrations of actin. The resulting fluorescence transients were best fit by a three-exponential function (Fig. 6A). The fluorescence decrease contained a fast phase (k+PWF, fast power stroke) and slow phase (k+PWS slow power stroke) that were both dependent on actin concentration. The relative amplitudes of the fast/slow phase were similar in the WT and two mutants (fast phase 85–90% of the fluorescence transients at 40 μm actin). We also observed a very slow fluorescence increase in many of the fluorescence transients that was similar at all actin concentrations (kobs ∼0.1–0.3 s−1). The actin dependence of the fast power stroke was similar in both mutants and WT (Fig. 6B). Also, the maximum rate of the slow power stroke in both mutants was found to be not significantly different from WT MV (Fig. 6C).
Figure 6.
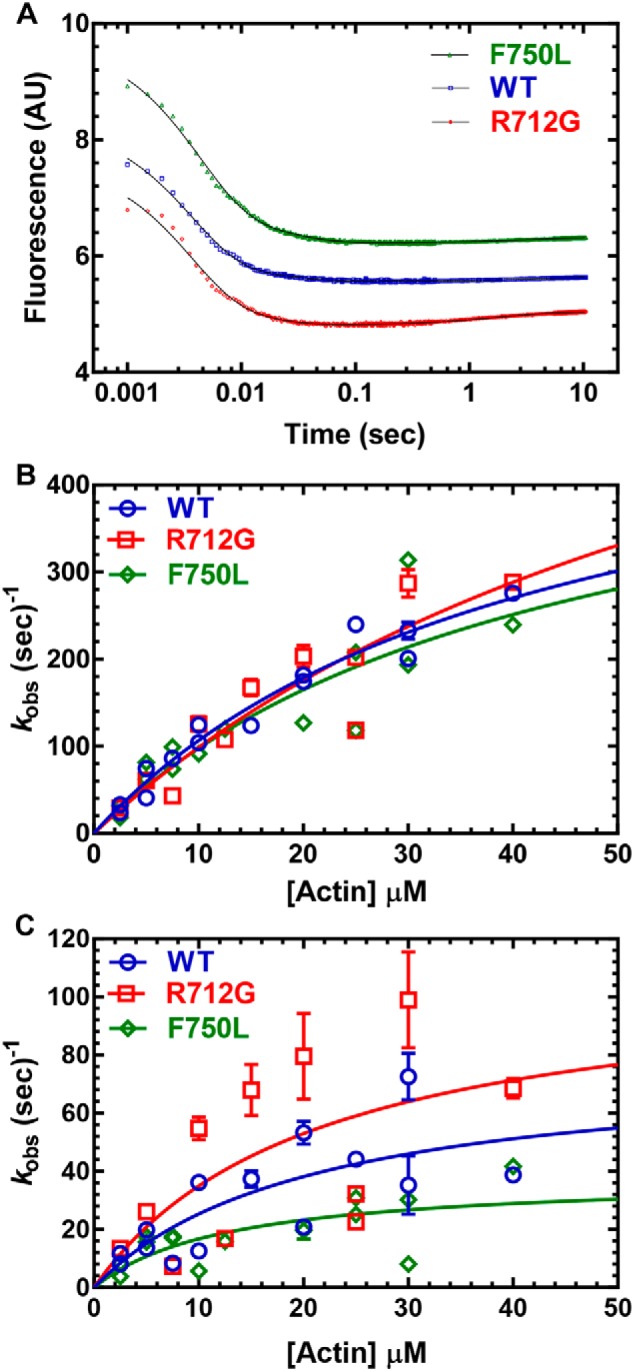
Power stroke. Sequential mix single-turnover experiments were performed in which 0.25 μm MV FlAsH.QSY-CaM was mixed with ATP (0.2 μm), aged for 10 s, and then mixed with different concentrations of actin. A, the increase in FRET was monitored by the decrease in FlAsH fluorescence and was best fit by a three-exponential function. B, the fast phase of the fluorescence transients (fast power stroke) was plotted as a function of actin concentration and fit to a hyperbolic or linear function. C, the slow phase (slow power stroke) was hyperbolically dependent on actin concentration, whereas the very slow fluorescence increase was similar at all actin concentrations. Fluorescence is reported in arbitrary units (AU).
Structural dynamics of the lever arm
To examine the impact of the converter mutations on the distribution of structural states of the lever arm in each nucleotide state, we performed transient time-resolved FRET ((TR)2FRET) (Fig. 7, Fig. S2, and Tables 2 and 3, and Table S2). In addition, (TR)2FRET allows interpretation of the fast- and slow-phase amplitudes observed in the total fluorescence studies. This approach utilizes nanosecond-resolved time-resolved fluorescence of the donor probe (TR-FRET) to follow changes in the structural state of the donor + acceptor–labeled protein acquiring individual time-resolved fluorescence waveforms every 0.1 ms following rapid mixing by stopped flow. A detailed description of the acquired data and instrumentation is given in previous work (31, 32). We analyzed the resulting data as described in those studies, fitting a structure-based model to each data set.
Figure 7.

Transient time-resolved FRET: time-resolved fluorescence waveform acquisition of structural kinetics. A, (TR)2FRET of the recovery stroke. The fluorescence of 160 nm MV FlAsH.QSY-CaM changes upon rapid stopped-flow mixing with varying concentrations of ATP (0.01 mm shown). Both the peak fluorescence intensity and the waveform shape change over the reaction time. The three-dimensional plot depicts the nanosecond-resolved waveforms (xy-plane) evolving with reaction time (z axis). Each waveform was fit to a two-Gaussian distance distribution. The three-dimensional plot depicts each distance distribution (xy-plane) evolving with reaction time (z axis). B, the summary of rate constants from fitting the recovery-stroke (TR)2FRET data to a double-exponential function. C, (TR)2FRET of the power stroke. 160 nm MV and 10 μm ATP were mixed with 5.0 mm ADP and varying actin concentrations. D, summary of rate constants from fitting the power-stroke (TR)2FRET data to a single-exponential function.
Table 2.
(TR)2FRET parameters determined by simultaneous fitting of a two-Gaussian distance distribution model to power-stroke and recovery-stroke data
Error estimates are ±67% confidence interval determined by error plane analysis.

Table 3.
Mole fractions of MV lever arm orientation in the post-power—stroke (M*) state determined by simultaneous fitting of a two-Gaussian distance distribution model to power-stroke and recovery-stroke data
Error estimates are ±67% confidence interval determined by error plane analysis.

Our results revealed that both mutants and WT MV could be fit to a model in which there are two structural states of the lever arm, a pre-power–stroke state (M**) and a post-power–stroke state (M*). Neither mutation impaired the distance or distance distribution profile of the pre- and post-power–stroke states as a function of nucleotides (Table 3 and Fig. S3). Interestingly, the R712G mutation caused a shift in the mole fraction of structural states, which demonstrated an increase in the population of the post-power–stroke conformation in the strong actin-binding nucleotide states (AM.ADP and AM) (Table 3 and Fig. S4). In contrast, the F750L mutation decreased the mole fraction of the post-power–stroke conformation in the ADP states (AM.ADP and M.ADP). In the M.ADP.Pi state, there was no difference in pre- and post-power–stroke mole fractions when comparing R712G with WT, whereas the F750L mutant had a slight increase in the post-power–stroke conformation in this state, suggesting the mutations do not dramatically alter the equilibrium constant for hydrolysis (KH).
We examined the structural kinetics of the recovery stroke with (TR)2FRET (Fig. 7, A and B) and found results similar to those of our other stopped-flow FRET studies (Fig. 5). We observed fluorescence transients (fluorescence intensity and lifetime) that were best fit by a two-exponential function. The maximum rate of the fast phase of the recovery stroke was enhanced 2-fold in F750L (891 ± 141 s−1), whereas it was reduced 35% in R712G (266 ± 6 s−1) compared with WT (408 ± 10 s−1) (Fig. 7B). The structural kinetics of the power stroke were examined with a single-mix experiment, in which MV and ATP were mixed with actin and ADP to induce a single turnover, as described previously (31). We observed a single-exponential function at each actin concentration, and the rate constants were linearly dependent on actin concentration. The rate constants of the power stroke were likely an average of the fast and slow phases we observed in our sequential mix stopped-flow experiments. The slopes of the linear dependence of the power-stroke rate constant on actin concentration were slightly higher for the F750L mutant (4.3 ± 0.4 μm−1·s−1) and slightly lower for the R712G mutant (2.3 ± 0.1 μm−1·s−1) compared with WT (3.3 ± 0.2 μm−1·s−1). Overall, the (TR)2FRET results demonstrate that the mutants dramatically changed the recovery stroke but did not induce major changes in the power stroke, which is similar to what we observed in our stopped-flow FRET studies.
Discussion
The structural mechanism of actomyosin-based force generation has been studied with many different structural and biophysical methods. One crucially important question that remains is how disease-associated mutations alter conserved allosteric pathways and how therapies can be designed to correct these impairments. Myosin V is one of the best-studied myosin motors and thus is thought to be an excellent model to investigate conserved allosteric pathways. In the current study, we investigated two mutations in the converter domain of myosin V and found that they both impair motor-based force generation. We demonstrate how these mutations disrupt the structural kinetics of lever arm rotation during the recovery and power strokes as well as how they alter other steps in the myosin ATPase cycle. We conclude that disruption in the allosteric pathway that coordinates structural changes in the lever arm with changes in the nucleotide-binding pocket can impair the ability of myosin to adapt to external loads. The mutations do not change the intrinsic structure of the pre- and post-power–stroke states based on (TR)2FRET measurements, but they do alter the rate and equilibrium constants associated with the transition between these two structural states. Our results have implications for understanding how conserved communication pathways in myosin can be altered by disease-associated mutations.
Impact on mechanosensitivity
Myosin motors and other motor proteins have the unique ability to alter their catalytic cycle in the presence of external loads (33). This feature was demonstrated in early studies of muscle contraction, which found a decrease in heat production (myosin ATPase activity) when muscle contracts against a higher load (Fenn effect) (34). Thus, myosins working within a contractile unit can fine-tune their mechanical properties by altering their load-dependent kinetics (e.g. slowing the detachment rate at higher loads to increase the time myosin is attached to actin). The structural mechanism of myosin mechanosensitivity has been investigated in recent studies using X-ray crystallography and cryo-EM, which demonstrated different conformations of the lever arm in nucleotide states that are thought to play a crucial role in mediating force production (35–38). The two converter mutants we investigated in the current study demonstrated little difference in unloaded sliding velocity but displayed increased sensitivity to external loads examined with load-dependent mixed motor motility assays (Fig. 2). It is expected that the tethering load would cause the myosin to slow the ADP release rate constant and thus slow the detachment and sliding velocity. Interestingly, the converter mutants were not able to overcome the tethering loads as well as WT MV, whereas the sliding velocity of moving filaments was indistinguishable from WT. Thus, when the converter mutants experienced higher tethering loads, they displayed the largest and most robust differences in mechanosensitivity compared with WT MV. The ADP release rate constant typically correlates well with unloaded in vitro motility, whereas in the case of the F750L mutant, the increased ADP release rate constant (k′+D) did not lead to a corresponding increase in unloaded sliding velocity. However, the changes in the slow power-stroke (k′PWS) and/or ADP isomerization (k′+D′) steps may contribute to the overall detachment rate. It is possible that the mutants reduce ensemble force (FE) which is the product of intrinsic force (f), duty ratio (r), and the number of myosin motors (N) working in the contractile unit (FE = f × r × N) (7). Kinetic simulations did not reveal a major change in duty ratio in either mutant compared with WT (Table S1 and Fig. S7). However, the duty ratio simulations did not take into account the impact of load on the individual rate constants. It is conceivable that the mutants impair intrinsic force, which was observed in several converter mutants in human M2β associated with HCM (39). The number of force-generating myosin motors in a contractile unit could be changed by altering the hydrolysis equilibrium constant (KH), but because we observed only minor changes in the mole fraction values of the pre- and post-power–stroke state conformation in the M.ADP.Pi state (Table 3), we argue that this is unlikely. A final possibility is that the R712G and F750L mutants have impaired strain-dependent detachment, which has been shown to be crucial for myosin motors to adapt to different loads (33, 40). Interestingly, both the R712G and F750L mutant demonstrate alterations in the actomyosin.ADP states, which are known to be critical for strain-dependent ADP release. Compared with WT, the R712G mutant displays an increase in the post-power–stroke conformation, whereas the F750L mutant displays a decrease in the post-power–stroke conformation in the AM.ADP state. In addition, F750L demonstrates an altered ADP-release rate constant (k′+D).
It was previously determined that mutations in the converter domain can impair the stiffness of a myosin head and thus alter its force-generating properties without altering cross-bridge kinetics (41–43). Studies of the R723G mutation in human M2β, which corresponds to R712G in MV, found an increase in the rigidity of the strongly bound myosin heads, which led to a 1.3-fold increase in force per myosin head (43). Interestingly, the increased stiffness did not alter Vmax or the rate of force redevelopment. Another study found that the F764L mutation in mouse α cardiac myosin resulted in decreased ATPase and in vitro motility but no change in ensemble force (44, 45). Thus, the current work highlights that mutations can have different effects in different myosin isoforms, and it is important to evaluate their impact on both unloaded and loaded myosin mechanochemistry.
Impact of converter mutations on the recovery stroke
Overall, our results demonstrate that the mutations do not alter the conformation of the pre- and post-power–stroke states, as the distance and distance distributions of these two states are identical (Table 2). However, the mutations do alter the mole fractions of the pre- and post-power–stroke structural states (Table 3). The shift in the mole fractions indicates that the mutation alters the free energy difference between the pre- and post-power–stroke states, whereas the change in the recovery-stroke rate constant indicates an impact on the activation energy associated with the structural transition. The recovery stroke is an important determinant of the number of myosin motors in an ensemble that are primed to generate force (N). In addition, the rate constant of the recovery stroke can alter the duty ratio by altering the period of time myosin remains in the weakly bound states. We find that the F750L mutation increases the recovery-stroke rate constant 2-fold, and this correlates well with the increase in the maximum rate of ATP hydrolysis measured by tryptophan fluorescence. Thus, the mutation lowers the activation energy associated with the transition from the post- to pre-power–stroke states. The region where Phe-750 is located has been identified as a site of hydrophobic interactions between the converter domain, SH1-SH2 helix, and relay helix/loop (13, 46). As depicted in Fig. 8A, this pathway allows for communication between the lever arm and active site because the relay helix extends into the nucleotide-binding pocket and is connected to the switch II region, which participates in ATP binding/hydrolysis. The formation of the pre-power–stroke state is driven by a change in the relay helix from a straight to a bent conformation, which then causes a repositioning of the converter/lever arm domain into the pre-power–stroke conformation (47). A salt bridge that forms between the converter and relay loop is thought to be important for communication (Fig. 8A, blue). In addition, hydrophobic interactions between the relay helix region and the converter domain play a particularly important role, and Phe-750 is a highly conserved converter domain residue (Fig. 8C) that has been proposed in structural and molecular modeling studies to be crucial (47). The leucine that replaces the phenylalanine at this position likely reduces the hydrophobic surface because of the loss of the bulky aromatic side chain that interacts with other hydrophobic residues (e.g. Tyr-477, Phe-698, Pro-699, Ile-749, and Phe-751). However, the substitution of leucine at this position appears to improve the efficiency of this structural transition, possibly because a smaller hydrophobic side chain allows a more rapid reorganization of the hydrophobic cluster. A recent study that mutated a nearby residue R778K in nonmuscle myosin IIC (Arg-762 in MV) found a 50% increase in the maximum rate of ATP hydrolysis, indicating that this region is important for tuning the recovery stroke, which is coupled to formation of the hydrolysis-competent state (48). Other studies in insect flight muscle found that mutations that disrupt the converter–relay interactions have a major impact on myosin and muscle function (49, 50). Thus, modulating the charge–charge and hydrophobic interactions in the relay/converter interface can alter the recovery-stroke kinetics.
Figure 8.

Summary of allosteric interactions associated with the converter/relay helix/lever arm region. A, a diagram of the key structural elements that allow communication between the nucleotide-binding region and the lever arm (switch II, relay helix/loop, SH1-SH2 helix, converter domain; see “Discussion” for allosteric pathway description). B, graph representing the correlation between the ATP hydrolysis rate constant and potential interactions between the converter domain (Arg-712 site) and the light chain bound to the first IQ motif. C, an alignment of several myosin heavy chains demonstrating conservation of the Arg-712 (red box) and Phe-750 (green box) residues. D, an alignment of several myosin light chains demonstrating negatively charged residues in close proximity to the Arg-712 site. The red underlined residues were found to be in close proximity to the Arg-712 site in several crystal structures (PDB codes 1OE9, 1W7J, 4ZLK and 5N69). The following abbreviations were used for the myosin heavy chains: chicken MYO5A (cMVa), mouse MYO5A (mMVa), human MYO5A (hMVc), human MYH9 (hNMMIIa), human MYH10 (hNMMIIb), human MYH14 (hNMMIIc), chicken MYSS (cSkelMII), rabbit MYSS (rSkel), human MYH7 (hCardMIIb), porcine MYH7 (pCardiac). The following abbreviations were used for the myosin light chains: human MYL6B (hLC1sa), mouse CALM3 (mCaM), bovine ventricular MYL3 (bVELC), human ventricular MYL3 (hVELC), bovine non-muscle MYL6 (bLC17b), chicken skeletal MLRS (cSkelELC). The data in B were taken from the following references: cMVa:LC1sa (30), cMVa:CaM (51), mMVa:CaM (64), rSkel:ELC (65), pCardiac:vELC (66), hMVc:CaM (52), hNMMIIc:NoLC (67), hNMMIIb:17b (68), hNMMIIa:17b (69).
The R712G mutation causes a reduction in the recovery-stroke rate constant and shifts the mole fraction of structural states to favor the post-power–stroke conformation in the strong actin-binding states (M, AM.D, and M.D). The Arg-712 residue is not well-conserved in myosins (Fig. 8C) and is located at the region of the converter that is in close proximity to the light chain bound to the first IQ motif. Furthermore, we found that in many myosin structures, the region of the converter near Arg-712 is often in close proximity to charged residues on the essential light chain (Fig. 8, C and D). Thus, Arg-712 may form charge–charge interactions with light-chain residues (e.g. Asp-118, Glu-119, and Glu-120 in CaM), which could stabilize the pre-power–stroke state (see Fig. S8). Studies in myosin V demonstrated that altering the light chain associated with the first IQ motif can impact the ATP hydrolysis rate and equilibrium constant (51). Interestingly, myosin Vc has a naturally occurring glycine at the 712 site, and it was found to have a reduced maximum rate of ATP hydrolysis compared with myosin Va (52). We evaluated the correlation between the maximum rate of ATP hydrolysis (k+H + k−H) and the number of potential complementary charges between the converter domain and essential light chain in different myosin isoforms (Fig. 8). We found that myosins with a positively charged amino acid at the position corresponding to Arg-712, which allows for a potential interaction with at least one other negatively charged residue on the ELC, tend to have a faster ATP hydrolysis rate constant, whereas those that have no charge or a negative charge at the Arg-712 position have a slower ATP hydrolysis rate constant (Fig. 8B). In the highly mechanosensitive Myo1b, an insert in the converter domain called the WPH motif (near the corresponding Arg-712 residue) was found to directly interact with calmodulin (36). Thus, our results suggest that interactions between the converter and essential light chain are important for fine tuning the recovery-stroke rate constant and may play a role in mechanosensitivity.
Impact of converter mutations on the power stroke
Compared with the recovery stroke, the converter mutations had less of an impact on the power stroke. A possible explanation is that the allosteric coupling pathway (active site–relay helix–converter–lever) is crucial for the recovery stroke, whereas other unknown allosteric pathways are important for the power stroke. In our previous work, we found that the there is a fast structural change in the lever arm that occurs upon myosin binding to actin in the M.ADP.Pi state (k+PWF), which is closely followed by phosphate release (k+Pi) and then a slow power stroke (k+PWS) that occurs before ADP release (25). Although there has been considerable controversy, a recent review article describes a unifying theme for how actin activates the power stroke based on much of the current data available (15). The authors suggest that actin binding triggers a rapid structural change in the active site that opens up the phosphate release tunnel, allowing phosphate to enter the tunnel and the fast power stroke to occur. The observed rate of phosphate release measured in solution is slower because dissociation of phosphate from the exit tunnel and binding to the phosphate-binding protein are being monitored. There is a second power stroke that occurs during the transition between actomyosin. ADP states, which was identified in structural studies of smooth muscle myosin, myosin V, and myosin IB (35, 38, 53). Kinetic simulations demonstrate that the power stroke and phosphate release transients are best fit by a fast power stroke (k+PWF) followed by rapid phosphate release (k+Pi) (Figs. S5 and S6). However, we cannot rule out the possibility that phosphate release and the fast power stroke occur simultaneously, because phosphate may remain in the exit tunnel for a period of time before being released into solution and detected by the phosphate-binding protein. We find that the converter mutants do not disrupt the fast power stroke associated with the phosphate release step, and no significant differences were observed in the slow power stroke that occurs during the transition between actomyosin.ADP states (Table 1). The actin dependence of phosphate release is also similar in the mutants and WT (see Table S1 and Fig. 6C). Overall, the observed transient kinetic parameters do not alter the steady-state ATPase activity, as the KATPase and kcat are not dramatically altered from WT. Interestingly, altering the slow power-stroke rate constant may alter the period of time myosin spends in the force-generating actomyosin.ADP states, which is thought to be an important determinant of the myosin duty ratio. However, a reduction in the slow power-stroke rate constant could be accompanied by a faster ADP release rate constant (k′+D), which overall results in little change in duty ratio.
Conclusions
The current study demonstrates direct evidence for an allosteric pathway in myosin associated with positioning the lever arm in the pre- and post-power–stroke states. We find that the converter mutations disrupt ensemble force, likely by altering intrinsic force or strain-dependent detachment. The impact of the converter mutations on cardiac myosin are likely to be different from what we found in myosin V. However, our work highlights that mutations in key allosteric pathways can change the kinetics of the recovery and power stroke as well as the equilibrium between the pre- and post-power–stroke states. In addition, our studies demonstrate that point mutations can alter the rate and equilibrium constants that control the population of structural states without altering the overall conformation of the lever arm. Thus, therapies designed to shift the equilibrium between major structural states may be effective at rescuing the structural impact of the disease mutations. Our work provides a basis for future studies designed to examine the impact of disease mutations on key structural changes as a method of determining the mechanism of motor impairment and the potential for therapeutic strategies to correct specific impairments.
Experimental procedures
Reagents
Reagents used for all experiments were commercially available and of the highest purity. ATP and ADP stocks were freshly prepared from powder, and their concentrations were measured by absorbance at 259 nm (ϵ259 = 15,400 m−1·cm−1). The nonfluorescent acceptor QSY-9 and pyrene iodoacetamide were purchased from Invitrogen, Inc. FlAsH dye was a generous gift from Roger Tsien and Stephen Adams at the University of California (San Diego, CA). The fluorescently labeled phosphate-binding protein (MDCC-PBP) (54) was a generous gift from Howard White (Eastern Virginia Medical School). The mantADP was purchased from Jena Biosciences.
Protein construction, expression, and purification
A chicken MV construct containing a single IQ motif (residues 1–792) was used for this study. This construct was further modified to add a tetracysteine motif (CCPGCC) to the extreme N terminus to label with FlAsH and to add a C-terminal FLAG tag (DYKDDDDK) for purification as described previously (23, 55). The two converter domain mutations, R712G and F750L, were separately introduced into this construct. All constructs were coexpressed with chicken calmodulin in the baculovirus system. MV R712G, MV F750L, and WT MV were labeled with FlAsH as described previously. A single cysteine (T110C) was introduced into calmodulin (CaM) for site-specific labeling with the nonfluorescent acceptor QSY-9. During purification, the MV constructs were stripped of native calmodulin, and QSY-9–labeled CaM was exchanged onto the lever arm at the single IQ motif (25). Actin was purified from rabbit skeletal muscle by the acetone powder method (56) and labeled with pyrene actin when necessary (57). All experiments were performed in KMg50 TCEP buffer (10 mm imidazole-HCl, 50 mm KCl, 1 mm EGTA, 1 mm MgCl2, 1 mm TCEP) at pH 7.0 and 25 °C, except where noted.
In vitro motility
In vitro motility measurements were carried out as described previously (24–26). Briefly, MV WT, R712G, and F750L were attached to a nitrocellulose-coated coverslip using anti-cMYC antibody (Invitrogen). The coverslip surface was blocked with 1 mg·ml−1 BSA. Motility was initiated by adding activation buffer that consisted of 0.35% methylcellulose, 1 mg·ml−1 BSA, 2 mm ATP, 45 units·ml−1 pyruvate kinase, 0.5 mm phosphoenolpyruvate and 10 μm calmodulin. An oxygen-scavenging system consisting of 5 mg·ml−1 glucose, 0.2 mg·ml−1 glucose oxidase, and 0.02 mg·ml−1 catalase was used to prevent photobleaching. Time-lapse images of the motility of rhodamine-phalloidin–labeled F-actin filaments were recorded in 2-s intervals for 4 min using a Nikon TE2000 microscope. The loaded in vitro motility assay was performed as described above except that different amounts of the nonhydrolyzing strong actin-binding mutant (G440A) (27) (58) were mixed with MV (the amount of MV remained constant at 0.4 μm, whereas G440A MV was varied between 0 and 0.2 μm) and added to the flow cell. The sliding velocities were determined using the program FAST (59), which allowed determination of the total number of filaments moving and the velocity of each moving filament. The velocities of the moving filaments were binned and fit to a Gaussian function to determine the average velocity and S.E. at each condition. An unpaired student t test was performed to determine significant differences between WT and the two mutants.
Steady-state ATPase measurements
We performed actin-activated ATPase measurements of MV constructs (0.1 μm) as a function of actin concentration (0, 2.5, 5, 10, 20, and 30 μm) using the NADH-coupled assay (23–26, 60). Experiments were performed in an Applied Photophysics (Surrey, UK) stopped flow at 25 °C for a 200-s period. A standard curve of known ADP concentrations was used to determine the relationship between the NADH absorbance and ADP concentration. The ATPase rate was plotted as a function of actin concentration and fit to the Michaelis–Menten equation to determine the maximum ATPase rate (kcat) and actin concentration at which the ATPase activity is one-half maximal (KATPase). Data from several protein preparations were used to determine the average ATPase rate ± S.D. at each actin concentration. An unpaired Student's t test was performed to determine significant differences between WT and the two mutants.
Transient kinetic measurements
Measurements of the individual steps in the myosin ATPase cycle were examined using an Applied Photophysics stopped flow with a 1.2-ms dead time and a 9.3-nm band pass. The mantADP fluorescence was examined by exciting intrinsic protein fluorescence at 290 nm and examining the mant emission with a 395-nm-long pass filter. Pyrene-actin fluorescence was examined by exciting at 365 nm and measuring the emission with a 395-nm-long pass filter. To examine tryptophan fluorescence, an excitation of 290 nm and a 320-nm-long pass emission filter were used (performed in a modified KMg50 TCEP buffer to reduce background fluorescence: 10 mm imidazole replaced with 10 mm MOPS, pH 7.0). To examine MDCC-PBP, an excitation of 380 nm and a 395- or 425-nm long pass emission filter were used. We examined the FRET signal with the FlAsH-QSY donor/acceptor pair by exciting FlAsH at 488 nm and examining the emission with a 515-nm long-pass filter (because QSY is nonfluorescent). An unpaired Student's t test was performed to determine significant differences between WT and the two mutants (n ≥ 3 separate protein preparations).
Transient time-resolved FRET
(TR)2FRET (0.2-ms–resolved transient biochemical experiments with 125-ps–resolved fluorescence decay detection) was measured using a transient time-resolved fluorescence spectrophotometer (32, 61, 62). This instrument utilizes a Biologic USA SFM/20 single-mix stopped-flow accessory coupled to a transient time-resolved fluorescence spectrophotometer. The dead time for the instrument is 1.8 ms, calibrated using the 8-hydroxyquinoline + Mg2+ control reaction (63).
In the recovery-stroke experiments (post-mix concentrations), 50–80 nm MV.FlAsH-CaM.QSY was mixed with 0–1.0 mm ATP. In the power-stroke experiments, 50–80 nm MV.FlAsH-CaM.QSY and 5.0 μm ATP were mixed with 0–40 μm actin and 5.0 mm ADP. Biochemical mixes were prepared with 600 μl per syringe and shot at 40 μl per syringe for acquisition. 10–12 shots were averaged for each biochemical mix, with replicates of n = 3–6.
Time-resolved FRET transients for WT, F750L, and R712G donor-only labeled samples in n = 12 were globally fit to obtain amplitudes and lifetimes for the donor-only fluorescence (Table S2). These parameters were used to fit a distance-dependent FRET model. One-, two-, and three-distance models were tested, with the best fit obtained using a two-Gaussian distance distribution (Table 2), corresponding to pre- and post-power–stroke structural states, extensively discussed in our earlier work (31). Each biochemical mix, containing 500+ waveforms acquired after excitation of the sample from the pulsed laser, was then globally fit to this two-Gaussian structural model, allowing the mole fractions of the two states to vary. Reported error estimates reflect the 67% confidence interval for each parameter determined by error plane analysis with all parameters allowed to vary, as described in previous publications (31).
Author contributions
L. K. G., J. A. R., W. T., S. D. W., W. C. U., D. V. T., and C. M. Y. data curation; L. K. G., J. A. R., W. T., S. D. W., D. V. T., J. M. M., and C. M. Y. formal analysis; L. K. G., J. A. R., W. T., S. D. W., J. M. M., D. D. T., and C. M. Y. writing-review and editing; J. A. R., S. D. W., and C. M. Y. writing-original draft; W. C. U. resources; W. C. U. methodology; J. M. M., D. D. T., and C. M. Y. supervision; D. D. T. and C. M. Y. funding acquisition; C. M. Y. conceptualization.
Supplementary Material
Acknowledgment
We recognize the outstanding technical efforts of Rohini Desetty.
This work was supported by National Institutes of Health Grant HL127699 (to C. M. Y.) and Grants R01AR32961 and R37AG26160 (to D. D. T.) and American Heart Association Grant 14SDG20480032 (to J. M. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2 and Figs. S1–S8.
- HCM
- hypertrophic cardiomyopathy
- M2β
- human β-cardiac myosin
- MV
- chicken myosin Va
- DCM
- dilated cardiomyopathy
- FlAsH
- fluorescein bis-arsenical hairpin binding dye
- QSY
- QSYTM 9 C5-maleimide
- CaM
- calmodulin
- mantADP
- 2′-deoxy-ADP labeled with N-methylanthraniloy at the 3′-ribose position
- mant
- N-methylanthraniloy
- PBP-MDCC
- phosphate-binding protein labeled with 7-diethylamino-3-((((2-maleimidyl)ethyl)amino)carbonyl)coumarin
- (TR)2FRET
- transient time-resolved FRET
- ELC
- essential light chain
- PDB
- Protein Data Bank.
References
- 1. De La Cruz E. M., and Ostap E. M. (2004) Relating biochemistry and function in the myosin superfamily. Curr. Opin. Cell Biol. 16, 61–67 10.1016/j.ceb.2003.11.011 [DOI] [PubMed] [Google Scholar]
- 2. Syamaladevi D. P., Spudich J. A., and Sowdhamini R. (2012) Structural and functional insights on the Myosin superfamily. Bioinform. Biol. Insights 6, 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Heissler S. M., and Sellers J. R. (2014) Myosin light chains: Teaching old dogs new tricks. Bioarchitecture 4, 169–188 10.1080/19490992.2015.1054092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Homburger J. R., Green E. M., Caleshu C., Sunitha M. S., Taylor R. E., Ruppel K. M., Metpally R. P., Colan S. D., Michels M., Day S. M., Olivotto I., Bustamante C. D., Dewey F. E., Ho C. Y., Spudich J. A., and Ashley E. A. (2016) Multidimensional structure-function relationships in human β-cardiac myosin from population-scale genetic variation. Proc. Natl. Acad. Sci. U.S.A. 113, 6701–6706 10.1073/pnas.1606950113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Semsarian C., Ingles J., Maron M. S., and Maron B. J. (2015) New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 65, 1249–1254 10.1016/j.jacc.2015.01.019 [DOI] [PubMed] [Google Scholar]
- 6. Maron B. J., Haas T. S., Ahluwalia A., Murphy C. J., and Garberich R. F. (2016) Demographics and epidemiology of sudden deaths in young competitive athletes: from the United States National Registry. Am. J. Med. 129, 1170–1177 10.1016/j.amjmed.2016.02.031 [DOI] [PubMed] [Google Scholar]
- 7. Spudich J. A. (2014) Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys. J. 106, 1236–1249 10.1016/j.bpj.2014.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Watkins H., Ashrafian H., and Redwood C. (2011) Inherited cardiomyopathies. N. Engl. J. Med. 364, 1643–1656 10.1056/NEJMra0902923 [DOI] [PubMed] [Google Scholar]
- 9. Maron B. J., and Maron M. S. (2013) Hypertrophic cardiomyopathy. Lancet 381, 242–255 10.1016/S0140-6736(12)60397-3 [DOI] [PubMed] [Google Scholar]
- 10. Park H. Y. (2017) Hereditary dilated cardiomyopathy: recent advances in genetic diagnostics. Korean Circ. J. 47, 291–298 10.4070/kcj.2016.0017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tayal U., Prasad S., and Cook S. A. (2017) Genetics and genomics of dilated cardiomyopathy and systolic heart failure. Genome Med. 9, 20 10.1186/s13073-017-0410-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sweeney H. L., and Houdusse A. (2010) Structural and functional insights into the myosin motor mechanism. Annu. Rev. Biophys. 39, 539–557 10.1146/annurev.biophys.050708.133751 [DOI] [PubMed] [Google Scholar]
- 13. Houdusse A., Szent-Gyorgyi A. G., and Cohen C. (2000) Three conformational states of scallop myosin S1. Proc. Natl. Acad. Sci. U.S.A. 97, 11238–11243 10.1073/pnas.200376897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Preller M., and Manstein D. J. (2013) Myosin structure, allostery, and mechano-chemistry. Structure 21, 1911–1922 10.1016/j.str.2013.09.015 [DOI] [PubMed] [Google Scholar]
- 15. Houdusse A., and Sweeney H. L. (2016) How myosin generates force on actin filaments. Trends Biochem. Sci. 41, 989–997 10.1016/j.tibs.2016.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Manstein D. J. (2004) Molecular engineering of myosin. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 1907–1912 10.1098/rstb.2004.1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sweeney H. L., and Houdusse A. (2004) The motor mechanism of myosin V: insights for muscle contraction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 1829–1841 10.1098/rstb.2004.1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mehta A. D., Rock R. S., Rief M., Spudich J. A., Mooseker M. S., and Cheney R. E. (1999) Myosin-V is a processive actin-based motor. Nature 400, 590–593 10.1038/23072 [DOI] [PubMed] [Google Scholar]
- 19. Rief M., Rock R. S., Mehta A. D., Mooseker M. S., Cheney R. E., and Spudich J. A. (2000) Myosin-V stepping kinetics: a molecular model for processivity. Proc. Natl. Acad. Sci. U.S.A. 97, 9482–9486 10.1073/pnas.97.17.9482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trybus K. M. (2008) Myosin V from head to tail. Cell Mol. Life Sci. 65, 1378–1389 10.1007/s00018-008-7507-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Enjuto M., Francino A., Navarro-López F., Viles D., Paré J. C., and Ballesta A. M. (2000) Malignant hypertrophic cardiomyopathy caused by the Arg723Gly mutation in beta-myosin heavy chain gene. J. Mol. Cell Cardiol. 32, 2307–2313 10.1006/jmcc.2000.1260 [DOI] [PubMed] [Google Scholar]
- 22. Kamisago M., Sharma S. D., DePalma S. R., Solomon S., Sharma P., McDonough B., Smoot L., Mullen M. P., Woolf P. K., Wigle E. D., Seidman J. G., and Seidman C. E. (2000) Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 343, 1688–1696 10.1056/NEJM200012073432304 [DOI] [PubMed] [Google Scholar]
- 23. Jacobs D. J., Trivedi D., David C., and Yengo C. M. (2011) Kinetics and thermodynamics of the rate-limiting conformational change in the actomyosin V mechanochemical cycle. J. Mol. Biol. 407, 716–730 10.1016/j.jmb.2011.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Swenson A. M., Trivedi D. V., Rauscher A. A., Wang Y., Takagi Y., Palmer B. M., Málnási-Csizmadia A., Debold E. P., and Yengo C. M. (2014) Magnesium modulates actin binding and ADP release in myosin motors. J. Biol. Chem. 289, 23977–23991 10.1074/jbc.M114.562231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Trivedi D. V., Muretta J. M., Swenson A. M., Davis J. P., Thomas D. D., and Yengo C. M. (2015) Direct measurements of the coordination of lever arm swing and the catalytic cycle in myosin V. Proc. Natl. Acad. Sci. U.S.A. 112, 14593–14598 10.1073/pnas.1517566112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Trivedi D. V., Muretta J. M., Swenson A. M., Thomas D. D., and Yengo C. M. (2013) Magnesium impacts myosin V motor activity by altering key conformational changes in the mechanochemical cycle. Biochemistry 52, 4710–4722 10.1021/bi4004364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yengo C. M., De la Cruz E. M., Safer D., Ostap E. M., and Sweeney H. L. (2002) Kinetic characterization of the weak binding states of myosin V. Biochemistry 41, 8508–8517 10.1021/bi015969u [DOI] [PubMed] [Google Scholar]
- 28. Yengo C. M., and Sweeney H. L. (2004) Functional role of loop 2 in myosin V. Biochemistry 43, 2605–2612 10.1021/bi035510v [DOI] [PubMed] [Google Scholar]
- 29. De La Cruz E. M., and Ostap E. M. (2009) Kinetic and equilibrium analysis of the myosin ATPase. Methods Enzymol. 455, 157–192 10.1016/S0076-6879(08)04206-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. De La Cruz E. M., Wells A. L., Rosenfeld S. S., Ostap E. M., and Sweeney H. L. (1999) The kinetic mechanism of myosin V. Proc. Natl. Acad. Sci. U.S.A. 96, 13726–13731 10.1073/pnas.96.24.13726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Muretta J. M., Rohde J. A., Johnsrud D. O., Cornea S., and Thomas D. D. (2015) Direct real-time detection of the structural and biochemical events in the myosin power stroke. Proc. Natl. Acad. Sci. U.S.A. 112, 14272–14277 10.1073/pnas.1514859112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nesmelov Y. E., Agafonov R. V., Negrashov I. V., Blakely S. E., Titus M. A., and Thomas D. D. (2011) Structural kinetics of myosin by transient time-resolved FRET. Proc. Natl. Acad. Sci. U.S.A. 108, 1891–1896 10.1073/pnas.1012320108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Greenberğ M. J., Arpag G., Tüzel E., and Ostap E. M. (2016) A perspective on the role of myosins as mechanosensors. Biophys. J. 110, 2568–2576 10.1016/j.bpj.2016.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fenn W. O. (1924) The relation between the work performed and the energy liberated in muscular contraction. J. Physiol. 58, 373–395 10.1113/jphysiol.1924.sp002141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mentes A., Huehn A., Liu X., Zwolak A., Dominguez R., Shuman H., Ostap E. M., and Sindelar C. V. (2018) High-resolution cryo-EM structures of actin-bound myosin states reveal the mechanism of myosin force sensing. Proc. Natl. Acad. Sci. U.S.A. 115, 1292–1297 10.1073/pnas.1718316115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shuman H., Greenberg M. J., Zwolak A., Lin T., Sindelar C. V., Dominguez R., and Ostap E. M. (2014) A vertebrate myosin-I structure reveals unique insights into myosin mechanochemical tuning. Proc. Natl. Acad. Sci. U.S.A. 111, 2116–2121 10.1073/pnas.1321022111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Greenberg M. J., Lin T., Shuman H., and Ostap E. M. (2015) Mechanochemical tuning of myosin-I by the N-terminal region. Proc. Natl. Acad. Sci. U.S.A. 112, E3337–E3344 10.1073/pnas.1506633112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wulf S. F., Ropars V., Fujita-Becker S., Oster M., Hofhaus G., Trabuco L. G., Pylypenko O., Sweeney H. L., Houdusse A. M., and Schröder R. R. (2016) Force-producing ADP state of myosin bound to actin. Proc. Natl. Acad. Sci. U.S.A. 113, E1844–E1852 10.1073/pnas.1516598113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kawana M., Sarkar S. S., Sutton S., Ruppel K. M., and Spudich J. A. (2017) Biophysical properties of human β-cardiac myosin with converter mutations that cause hypertrophic cardiomyopathy. Sci. Adv. 3, e1601959 10.1126/sciadv.1601959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nyitrai M., and Geeves M. A. (2004) Adenosine diphosphate and strain sensitivity in myosin motors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 1867–1877 10.1098/rstb.2004.1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brenner B., Seebohm B., Tripathi S., Montag J., and Kraft T. (2014) Familial hypertrophic cardiomyopathy: functional variance among individual cardiomyocytes as a trigger of FHC-phenotype development. Front. Physiol. 5, 392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Köhler J., Winkler G., Schulte I., Scholz T., McKenna W., Brenner B., and Kraft T. (2002) Mutation of the myosin converter domain alters cross-bridge elasticity. Proc. Natl. Acad. Sci. U.S.A. 99, 3557–3562 10.1073/pnas.062415899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Seebohm B., Matinmehr F., Köhler J., Francino A., Navarro-Lopéz F., Perrot A., Ozcelik C., McKenna W. J., Brenner B., and Kraft T. (2009) Cardiomyopathy mutations reveal variable region of myosin converter as major element of cross-bridge compliance. Biophys. J. 97, 806–824 10.1016/j.bpj.2009.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Debold E. P., Schmitt J. P., Patlak J. B., Beck S. E., Moore J. R., Seidman J. G., Seidman C., and Warshaw D. M. (2007) Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse α-cardiac myosin in the laser trap assay. Am. J. Physiol. Heart Circ. Physiol. 293, H284–H291 10.1152/ajpheart.00128.2007 [DOI] [PubMed] [Google Scholar]
- 45. Schmitt J. P., Debold E. P., Ahmad F., Armstrong A., Frederico A., Conner D. A., Mende U., Lohse M. J., Warshaw D., Seidman C. E., and Seidman J. G. (2006) Cardiac myosin missense mutations cause dilated cardiomyopathy in mouse models and depress molecular motor function. Proc. Natl. Acad. Sci. U.S.A. 103, 14525–14530 10.1073/pnas.0606383103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Llinas P., Isabet T., Song L., Ropars V., Zong B., Benisty H., Sirigu S., Morris C., Kikuti C., Safer D., Sweeney H. L., and Houdusse A. (2015) How actin initiates the motor activity of Myosin. Dev. Cell 33, 401–412 10.1016/j.devcel.2015.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fischer S., Windshügel B., Horak D., Holmes K. C., and Smith J. C. (2005) Structural mechanism of the recovery stroke in the myosin molecular motor. Proc. Natl. Acad. Sci. U.S.A. 102, 6873–6878 10.1073/pnas.0408784102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chinthalapudi K., Heissler S. M., Preller M., Sellers J. R., and Manstein D. J. (2017) Mechanistic insights into the active site and allosteric communication pathways in human nonmuscle myosin-2C. Elife 6, e32742 10.7554/eLife.32742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bloemink M. J., Melkani G. C., Bernstein S. I., and Geeves M. A. (2016) The relay/converter interface influences hydrolysis of ATP by skeletal muscle myosin II. J. Biol. Chem. 291, 1763–1773 10.1074/jbc.M115.688002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kronert W. A., Melkani G. C., Melkani A., and Bernstein S. I. (2014) Mapping interactions between myosin relay and converter domains that power muscle function. J. Biol. Chem. 289, 12779–12790 10.1074/jbc.M114.550673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. De La Cruz E. M., Wells A. L., Sweeney H. L., and Ostap E. M. (2000) Actin and light chain isoform dependence of myosin V kinetics. Biochemistry 39, 14196–14202 10.1021/bi001701b [DOI] [PubMed] [Google Scholar]
- 52. Takagi Y., Yang Y., Fujiwara I., Jacobs D., Cheney R. E., Sellers J. R., and Kovács M. (2008) Human myosin Vc is a low duty ratio, nonprocessive molecular motor. J. Biol. Chem. 283, 8527–8537 10.1074/jbc.M709150200 [DOI] [PubMed] [Google Scholar]
- 53. Whittaker M., Wilson-Kubalek E. M., Smith J. E., Faust L., Milligan R. A., and Sweeney H. L. (1995) A 35-A movement of smooth muscle myosin on ADP release. Nature 378, 748–751 10.1038/378748a0 [DOI] [PubMed] [Google Scholar]
- 54. White H. D., Belknap B., and Webb M. R. (1997) Kinetics of nucleoside triphosphate cleavage and phosphate release steps by associated rabbit skeletal actomyosin, measured using a novel fluorescent probe for phosphate. Biochemistry 36, 11828–11836 10.1021/bi970540h [DOI] [PubMed] [Google Scholar]
- 55. Trivedi D. V., David C., Jacobs D. J., and Yengo C. M. (2012) Switch II mutants reveal coupling between the nucleotide- and actin-binding regions in myosin V. Biophys. J. 102, 2545–2555 10.1016/j.bpj.2012.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pardee J. D., and Spudich J. A. (1982) Purification of muscle actin. Methods Enzymol. 85, 164–181 10.1016/0076-6879(82)85020-9 [DOI] [PubMed] [Google Scholar]
- 57. Criddle A. H., Geeves M. A., and Jeffries T. (1985) The use of actin labelled with N-(1-pyrenyl)iodoacetamide to study the interaction of actin with myosin subfragments and troponin/tropomyosin. Biochem. J. 232, 343–349 10.1042/bj2320343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sasaki N., and Sutoh K. (1998) Structure-mutation analysis of the ATPase site of Dictyostelium discoideum myosin II. Adv. Biophys. 35, 1–24 10.1016/S0065-227X(98)80002-6 [DOI] [PubMed] [Google Scholar]
- 59. Aksel T., Choe Yu E., Sutton S., Ruppel K. M., and Spudich J. A. (2015) Ensemble force changes that result from human cardiac myosin mutations and a small-molecule effector. Cell Rep. 11, 910–920 10.1016/j.celrep.2015.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. De La Cruz E. M., Sweeney H. L., and Ostap E. M. (2000) ADP inhibition of myosin V ATPase activity. Biophys. J. 79, 1524–1529 10.1016/S0006-3495(00)76403-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Muretta J. M., Petersen K. J., and Thomas D. D. (2013) Direct real-time detection of the actin-activated power stroke within the myosin catalytic domain. Proc. Natl. Acad. Sci. U.S.A. 110, 7211–7216 10.1073/pnas.1222257110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Muretta J. M., Kyrychenko A., Ladokhin A. S., Kast D. J., Gillispie G. D., and Thomas D. D. (2010) High-performance time-resolved fluorescence by direct waveform recording. Rev. Sci. Instrum. 81, 103101 10.1063/1.3480647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brissette P., Ballou D. P., and Massey V. (1989) Determination of the dead time of a stopped-flow fluorometer. Anal. Biochem. 181, 234–238 10.1016/0003-2697(89)90235-2 [DOI] [PubMed] [Google Scholar]
- 64. Trybus K. M., Krementsova E., and Freyzon Y. (1999) Kinetic characterization of a monomeric unconventional myosin V construct. J. Biol. Chem. 274, 27448–27456 10.1074/jbc.274.39.27448 [DOI] [PubMed] [Google Scholar]
- 65. Marston S. B., and Taylor E. W. (1980) Comparison of the myosin and actomyosin ATPase mechanisms of the four types of vertebrate muscles. J. Mol. Biol. 139, 573–600 10.1016/0022-2836(80)90050-9 [DOI] [PubMed] [Google Scholar]
- 66. Liu Y., White H. D., Belknap B., Winkelmann D. A., and Forgacs E. (2015) Omecamtiv Mecarbil modulates the kinetic and motile properties of porcine β-cardiac myosin. Biochemistry 54, 1963–1975 10.1021/bi5015166 [DOI] [PubMed] [Google Scholar]
- 67. Heissler S. M., and Manstein D. J. (2011) Comparative kinetic and functional characterization of the motor domains of human nonmuscle myosin-2C isoforms. J. Biol. Chem. 286, 21191–21202 10.1074/jbc.M110.212290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang F., Kovacs M., Hu A., Limouze J., Harvey E. V., and Sellers J. R. (2003) Kinetic mechanism of non-muscle myosin IIB: functional adaptations for tension generation and maintenance. J. Biol. Chem. 278, 27439–27448 10.1074/jbc.M302510200 [DOI] [PubMed] [Google Scholar]
- 69. Kovács M., Wang F., Hu A., Zhang Y., and Sellers J. R. (2003) Functional divergence of human cytoplasmic myosin II: kinetic characterization of the non-muscle IIA isoform. J. Biol. Chem. 278, 38132–38140 10.1074/jbc.M305453200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.