

Abstract
Herbert “Herb” Tabor, who celebrated his 100th birthday this past year, served the Journal of Biological Chemistry as a member of the Editorial Board beginning in 1961, as an Associate Editor, and as Editor-in-Chief for 40 years, from 1971 until 2010. Among the many discoveries in biological chemistry during this period was the identification of RNA modification by C6 deamination of adenosine (A) to produce inosine (I) in double-stranded (ds) RNA. This posttranscriptional RNA modification by adenosine deamination, known as A-to-I RNA editing, diversifies the transcriptome and modulates the innate immune interferon response. A-to-I editing is catalyzed by a family of enzymes, adenosine deaminases acting on dsRNA (ADARs). The roles of A-to-I editing are varied and include effects on mRNA translation, pre-mRNA splicing, and micro-RNA silencing. Suppression of dsRNA-triggered induction and action of interferon, the cornerstone of innate immunity, has emerged as a key function of ADAR1 editing of self (cellular) and nonself (viral) dsRNAs. A-to-I modification of RNA is essential for the normal regulation of cellular processes. Dysregulation of A-to-I editing by ADAR1 can have profound consequences, ranging from effects on cell growth and development to autoimmune disorders.
Keywords: innate immunity, RNA editing, interferon, double-stranded RNA (dsRNA), RIG-I–like receptor (RLR), 2'-5'-oligoadenylate synthetase, ADAR, adenosine deaminase acting on RNA, protein kinase PKR, RNA deamination
Introduction
Herbert “Herb” Tabor, M.D., served The Journal of Biological Chemistry as a member of the Editorial Board, as an Associate Editor, and then as Editor-in-Chief for 40 years until 2010 when he became Co-Editor (1). Among the many paradigm-shifting discoveries in biological chemistry during this period was the identification of RNA modification by C6 deamination of adenosine (A) to produce inosine (I) in double-stranded RNA (dsRNA)2 (2, 3). This process is known now as A-to-I editing (4–8). The focus of this JBC Review is on one of the mammalian enzymes that catalyzes A-to-I editing, the adenosine deaminase acting on RNA1 (ADAR1) (9). ADAR1 plays a major role in immunity, most notably as a suppressor of the innate immune interferon (IFN) responses triggered by cellular sensors of dsRNA. This article is dedicated to Herb Tabor on the occasion of his 100th birthday. Herb is a truly remarkable individual. He is synonymous with JBC. For me, beginning as a JBC author first when a graduate student and then continuing years later as a member of the JBC Editorial Board and subsequently as an Associate Editor, it has been a special privilege to work together with Herb and learn so much from him. Herb Tabor is a scholar, a leader, and a gentleman. Happy Birthday, Herb!
Deamination of adenosine in dsRNA structures by ADARs
The C6 deamination of adenosine to produce inosine in dsRNA (Fig. 1, upper) was discovered in Xenopus during antisense RNA studies. It was found that stable dsRNA hybrid structures were not formed and protected against digestion with ssRNA-specific RNases, and the structures displayed altered mobility under native gel electrophoresis conditions (10, 11). It was then shown that these changes in dsRNA behavior resulted from covalent deamination of adenosine to inosine that occurred in both Xenopus (2) and mammalian cells (3). Deamination of adenosine in dsRNA can destabilize the RNA structure as a resultant I-U mismatch pair is less stable than an A:U base pair (2, 3, 12). ADAR enzymatic activity was purified and characterized from bovine nuclear extracts (13) and from cultured HeLa cells (14). The findings, described in JBC, revealed that the nuclear dsRNA adenosine deaminase purified from cells not treated with interferon was a mixture of size forms, ranging from ∼80 to ∼100 kDa. Molecular cDNA and genomic cloning then established that there are three gene members of the mammalian ADAR family, designated ADAR1 (ADAR), ADAR2 (ADARB1), and ADAR3 (ADARB2) (4–8).
Figure 1.
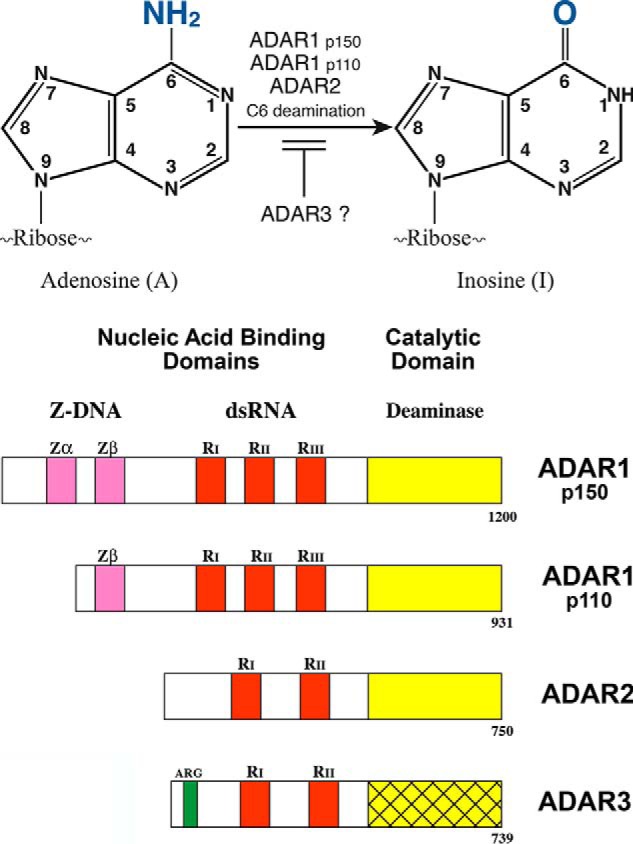
RNA editing by ADARs. Upper panel, C6 deamination of adenosine (A) in duplex RNA to produce inosine (I) catalyzed by ADARs. ADAR1, both the IFN-inducible p150 and the constitutively expressed p110, and ADAR2 possess deaminase activity. ADAR3 lacks deaminase activity and is implicated as a negative regulator of editing by ADAR1 and ADAR2. Lower panel, domain organization of ADAR proteins. The nucleic acid–binding domains include repeated dsRNA-binding domains (red, RI, RII, and RIII), either two (ADAR2 and ADAR3) or three (ADAR1 p110 and p150) copies. The N-terminal region of ADAR1 p150 possesses two copies of a Z-DNA–binding domain (pink, Zα and Zβ), and ADAR3 has an arginine-rich ssRNA-binding domain (green, ARG). The deaminase catalytic domain (yellow) is C terminus; ADAR3 (cross-hatched yellow) is not yet demonstrated to possess enzymatic activity. (Adapted from Ref. 5.)
ADAR1 proteins and their expression
The sequence for the human ADAR1 cDNA predicts an ORF of 1226 amino acids (15–17). There are two size isoforms of ADAR1, referred to as p110 and p150 (16). Antibodies prepared against recombinant ADAR1 recognize two proteins present in extracts from human cell lines: one ∼110 kDa (p110) that is constitutively expressed, and the other ∼150 kDa (p150) that is inducible by IFN (16). p110 is nuclear, whereas p150 is both cytoplasmic and nuclear (16–19). The gene for ADAR1 maps to a single locus, chromosome 1 q21 for human ADAR1 (20, 21) and chromosome 3 F2 for mouse Adar1 (22). Genomic and cDNA sequence analyses are consistent with a single ADAR1 gene, which in the human is about ∼40 kbp and includes 17 exons (23). Expression of the human (24–26) and mouse (27) ADAR1 genes is driven by multiple promoters, one of which is IFN-inducible and the others are constitutively active. The IFN-inducible p150 protein initiates from AUG1 present in exon 1A of the IFN-inducible human transcript, and the constitutive p110 protein initiates from the in-frame AUG296 present in exon 2 as the constitutive alternative exon 1B lacks an AUG (23). Alternative forms of exons 6 and 7 also occur (27–29). Expression of the mouse Adar1 gene and its exon organization involves strategies of alternative promoter usage and alternative splicing conceptually similar to that of the human ADAR1 gene (23, 27–31). Exon 7a is found in constitutively-expressed transcripts that specify p110, predicted to be 931 amino acids (human) or 903 amino acids (mouse). The smaller exon 7b is present in IFN-inducible human ADAR1 transcripts that specify p150, predicted to be 1200 amino acids (human) or 1152 amino acids (mouse) (23).
ADAR1 is both ubiquitously expressed (27, 32, 33) and inducible by IFN (16, 24, 27, 32–35). Canonical JAK–STAT signaling responsible for transcriptional activation of gene expression by IFNα/β involves binding of IFN to its cognate cell-surface receptor that is found on most types of cells. Activation of JAK1 and TYK2 kinases then mediates the phosphorylation of STAT1 and STAT2 transcription factors that associate with the IRF9 factor, translocate to the nucleus, and bind at the interferon-stimulated response DNA element to drive the inducible gene expression (36, 37). The IFN-inducible ADAR1 promoter possesses a consensus ISRE element (23), both the human (24) and mouse (29) genes. Induction of the p150-encoding transcripts by IFN depends upon both STAT2 and IRF9, but the requirement for STAT1 varies between cell lines; detectable induction occurs in the absence of STAT1 in mouse but not human cells (29, 35).
The domain structures of the mammalian ADARs are summarized in the Fig. 1 (lower panel) schematic. Both p110 and p150 ADAR1 are active dsRNA adenosine deaminases (16, 28, 38). The C-terminal region of ADAR1 specifies the catalytic domain; three copies of the dsRNA-binding domain are present in the central region of p150 and p110 (15–17, 39). p150 is N-terminally extended compared with p110; the additional p150 sequence includes the Zα Z-DNA–binding domain (5, 8). Substitution mutations of the His (H) and Glu (E) amino acid residues in the conserved CHAE sequence of the ADAR1 catalytic core abolishes A-to-I deaminase activity (39–41). Active ADAR1 is a dimer (42–44). The RNA-binding domains present in ADAR1 p110 and p150 (15–17, 39) are homologous to the repeated dsRNA-binding domain discovered earlier in the dsRNA-dependent protein kinase PKR (45–48). Substitution mutations of a conserved lysine residue within the core of each of the RNA-binding domains (39) as well as deletion mutations (40) revealed that the RIII copy linearly adjacent to the deaminase catalytic domain is the most important for enzymatic activity, and the central RII copy is the least important (39). The repeated Z-DNA–binding domain present in p150, Zα and Zβ, was identified as a domain homologous to the N-terminal region of the poxvirus E3L interferon antagonist protein (16, 49) and was shown to bind Z-DNA (50). Zα can also bind Z-structured dsRNA (51). The physiological significance of the Z-domains, and the nucleic acid bound by them within cells, is not fully understood. Functionally distinct dsRNA-binding domains are associated with splice variants of ADAR1 (28). The dsRNA-binding domains contribute to the A-to-I editing selectivity of the catalytic domains of ADAR1 and ADAR2 (38, 52–55). This is illustrated by the recombinant chimeric PKR–ADAR1 protein, where the dsRNA-binding domains from PKR replace those of ADAR1; significant deaminase editing activity is retained with a synthetic dsRNA substrate but not with natural (GluRB or 5HT-2cR) substrates (52). Human ADAR1 and ADAR2 have a 5′ nearest neighbor preference of U > A > C > G and a 3′ nearest neighbor preference of G > C ∼ A > U and G > C >U ∼ A, respectively (54). For ADAR2, the preferences appear to derive from differential base flipping of the targeted adenosine out of the double helix substrate rather than from direct recognition of neighboring bases (56).
ADAR2 and ADAR3 proteins
In addition to ADAR1, there are two more mammalian ADARs, ADAR2 and ADAR3 (4–8). The single mammalian gene for ADAR2 encodes multiple size isoforms of ADAR2 protein by alternative promoter usage and alternative splicing, with major isoforms predicted to be 701 and 750 amino acids for the human protein (23). ADAR2 is an active A-to-I deaminase, localizes to the nucleus, possesses two copies of the dsRNA-binding domain, and a C-terminal catalytic domain. As discussed in the following section, although ADAR1 is responsible for most of the A-to-I editing observed in mammalian cells, ADAR2 is generally responsible for the highly selective editing observed in the comparatively small number of identified exonic coding sites (8, 33). ADAR3 differs from ADAR1 and ADAR2 in an important manner: ADAR3 has not yet been demonstrated to possess enzymic activity (5–8). Expression of ADAR3 is limited to regions of the brain (57), unlike the ubiquitous expression seen for ADAR1 (4–8).
Substrate RNAs and roles of A-to-I editing
Transcripts encoding the glutamate and serotonin 2C receptors were among the first substrates identified, and the highly specific A-to-I editing of them is exquisitely well characterized (38, 58–63). The editing of GluRB and 5HT-2cR transcripts occurs within exonic sequences at the pre-mRNA level prior to splicing, thereby leading to amino acid substitutions in the expressed receptor proteins that alter their function. Editing specificity is dictated by unique cis-acting inverted repeat sequences predicted to form imperfect duplex structures in the substrate RNAs. Based on results of complementation studies, the glutamine (Q) to arginine (R) site of GluRB is the main target of ADAR2 as the knockin of GluRB encoding arginine at the Q/R site largely rescues the mouse Adar2 knockout phenotype (62). ADAR3 binding to the GluRB pre-RNA inhibits ADAR2 editing at the Q/R site, with elevated inhibition of Q/R editing in glioblastoma cells (63). In addition to specific editing events that recode mRNA genetic information, thereby leading to amino acid substitutions during translation (GluRB, 5HT-2cR), the editing of an amber UAG termination codon to generate a tryptophan UIG codon occurs in hepatitis D virus, thereby permitting synthesis of large delta antigen (5, 64).
RNA-seq strategies with high coverage and high accuracy have identified many A-to-I editing sites in RNA isolated from cultured cells and animal tissues (8, 33). Although a few additional nonrepetitive exon–coding sites were found, the vast majority of the ∼2 million-plus human A-to-I editing events occur in repetitive noncoding sequences (mostly Alu sequences in the human) (8, 33, 65–71). The extent of editing seen at a given site is typically partial, with editing at most sites less than 20% (7, 68, 73). However, the values vary widely, from near 100% (for the GluRB Q/R site) to less than 1% (7, 68). ADAR1 appears predominantly responsible for A-to-I editing of noncoding sites and ADAR2 for editing of coding sites (33). Furthermore, the knockin of the catalytically inactive E861A mutant of Adar1 gives normal mice when rescued by the concurrent knockout of Mda5, indicating that protein recoding by ADAR1 catalyzed A-to-I editing is not essential for normal mouse development and homeostasis (72). When 557 loci containing 11,103 editing sites were analyzed in untreated and IFN-treated WT and mutant mouse MEF cells, it was found that the vast majority of A-to-I editing events were dependent upon ADAR1 and not ADAR2; furthermore, this editing was enhanced by IFN treatment in a manner dependent upon the p150 isoform of ADAR1 (73).
Biochemical mechanisms
A-to-I editing of RNA transcripts affects multiple processes of mammalian cells and their viruses (Fig. 2). Among the mechanisms by which ADARs act, in addition to effects exerted on mRNA translation, are those on pre-mRNA splicing and microRNA processing and targeting. For RNA viruses, editing of viral RNA sequences also can potentially lead to genome mutation. For ADAR1, A-to-I editing events observed in mouse and human RNA transcripts largely occur in noncoding repetitive sequences that form duplex structures, and the major role of this editing is the suppression of innate immune interferon responses.
Figure 2.

Biochemical mechanisms by which A-to-I editing of RNA transcripts possessing double-stranded structure may affect gene expression and product function. Because I base-pairs as G instead of A, A-to-I RNA editing has the capacity to alter processes, including mRNA translation by altering codons and hence coding potential, pre-mRNA splicing by changing splice site recognition sequences, and RNA silencing by altering microRNA production or targeting. A-to-I editing may also lead to RNA mutations of viral genomes and transcripts by changing template and hence product sequences during RNA-dependent RNA replication. Finally, A-to-I editing may lead to I-U mismatches in place of A:U bp, thereby destabilizing dsRNA structures and hence affecting the activity of dsRNA sensing proteins of the interferon response, including the MDA5 RIG-I–like receptor, protein kinase PKR, and 2′-5′-oligoadenylate synthetase OAS–RNase L. (Adapted from Ref. 146.)
mRNA translation
Because inosine generated by deamination of adenosine base-pairs as if the I were a G instead of an A, A-to-I RNA editing has the capacity to alter decoding during mRNA translation, thereby leading to amino acid substitution in the protein product. As discussed above, among the earliest and best-characterized examples of editing that affects translation and gives rise to protein products with altered function are the editing of the cellular GluRB and 5HT2cR transcripts encoding neurotransmitter receptors and hepatitis D virus RNA encoding delta antigen. In these cases, the editing is highly selective, changing a codon and thereby recoding genetic information within the pre-mRNA, leading to amino acid substitutions or elimination of a termination codon. Only a very few exon recoding sites have been identified among the many A-to-I sites in the human transcriptome (8, 33, 70, 71).
Pre-mRNA splicing
Most introns are of the U2-type and are flanked by GT-AG splice site dinucleotides. Because I is recognized as G by the spliceosome machinery, alternative 5′-AT or AA-3′ sites can be converted into canonical sites by editing. A-to-I editing leading to alternative splicing during the processing of pre-mRNA was first demonstrated for ADAR2 transcripts. This ADAR2 autoediting of its own pre-mRNA creates a 3′ AG splice site (AA changed to AI (AG)) for alternative splicing (74). Human nuclear prelamin A recognition factor transcript includes an Alu-exon that depends upon A-to-I editing for exonization in a tissue-dependent manner, again by creation of a functional 3′ AG splice site (75). Tumor-associated intronic editing of the HNRPLL splicing factor transcript by ADAR1 p110 and ADAR2 generates a novel variant containing an additional exon 12A (76). A comprehensive survey of noncanonical splice sights using deep transcriptome profiling identified seven U2/U12-like noncanonical sites that are converted to canonical sites by A-to-I editing (77). Three of the noncanonical sites are AT-AG, and four are GT-AA, and all are involved in alternative splicing (77).
MicroRNA silencing
The discovery that A-to-I editing may affect RNA silencing by microRNAs (miRs) provides an additional manner by which ADARs impact gene expression. Editing effects are observed at the level of processing of miR precursor RNA to produce a functional miR (illustrated by miR-142 and miR-151) or by altering the targeting of the miR (illustrated by miR-376 and miR-378). Pri-miRNA precursors are processed by Drosha and Dicer endonucleases together with dsRNA-binding proteins to produce mature miRs. Estimates are that ∼20% of the pri-miRs are subject to editing by the ADARs (78). Editing of the pri-miR-142 impairs processing by the Drosha DGCR8 complex, reducing the amount of mature miR-142 produced (79). Editing of the pri-miR-151 impairs cleavage by Dicer-TAR RNA-binding protein complex (80). Editing within the seed sequence of miR-376 by ADAR2 alters targeting and subsequent silencing (81), whereas ADAR1 creates an miR-378 recognition site in the 3′-UTR of the human aryl hydrocarbon receptor transcript (82). Furthermore, comprehensive analyses of miRNA-seq datasets of human cancer tissues identified multiple editing sites in miRs and their 3′-UTR targets (83, 84). ADARs are potent dsRNA-binding proteins and in some instances impair knockdown efficiency of siRNAs independent of their catalytic activity (85, 86).
Viral RNA mutation
Viruses that possess RNA genomes typically encode a viral RNA-dependent RNA polymerase that transcribes and replicates the genome. If complementary sequences are generated, dsRNA structures may arise that would provide targets for editing. This, then, might generate viral RNA mutations. Editing within a template RNA strand by ADAR to substitute an “I” for an “A” would lead to a complementary change in the product RNA strand following replication by the viral polymerase. ADAR A-to-I editing then would generate either A-to-G or U-to-C transitions dependent upon the strand sequenced. Studies of a number of viruses reveal that ADARs are both antiviral and proviral, dependent upon the virus–host combination, a subject that has been reviewed (5). Viral A-to-I substitution editing has a long history, beginning with measles virus. Viral RNAs from brain autopsies of subacute sclerosing pan-encephalitis patients were found to possess extensive A-to-G (U-to-C) transitions characteristic of ADAR editing (87). For measles virus deficient in C protein expression, defective interfering dsRNAs are generated frequently and early, activate PKR, and impair virus growth (88, 89). These dsRNA structures are destabilized by ADAR-mediated hypermutations, and ADAR1 suppresses measles virus-induced apoptosis and PKR activation (90, 91). Increased A-to-G and U-to-C mutations characteristic of ADAR editing are described for lymphocytic choriomeningitis virus (LCMV), with bias to the glycoprotein region of the S segment RNA under conditions of increased p150 expression during infection (92). Possibly editing of a virion surface component, such as the LCMV glycoprotein, might create changes in a neutralization epitope thereby facilitating escape from immune surveillance.
Suppression of innate immune interferon responses
dsRNA has a long history in the interferon field (93). dsRNA is an inducer of IFN production, and dsRNA is an activator of some IFN-induced proteins responsible for IFN's actions. Interferon was the first cytokine discovered, identified by Isaacs and Lindenmann (94) during studies on virus interference. They observed that viral infection (with influenza A virus) induced the production of a secreted cellular factor that possessed the ability to interfere with virus growth, both of the homologous inducing virus and also of heterologous (Sendai and Newcastle disease) viruses. The interferon system now is recognized as the cornerstone of innate antiviral immunity. Considerable detail has been learned about the signal transduction pathways by which viral infection through the production of dsRNA leads to the induction of IFN (95–98). Likewise, two cellular responses that play central roles in the antiviral and proapoptotic actions of IFN are triggered by dsRNA: the activation of protein kinase PKR, and the activation of 2′-5′-oligoadenylate synthetases OAS. PKR (47, 99, 100) and OAS (101, 102) sense dsRNA that leads to their enzymatic activation or, in some instances, antagonism of activation.
MDA5–MAVS
Among the sensors that detect the presence of viral (nonself) dsRNA and trigger the production of IFN are the family of cytosolic receptor proteins known as RIG-I–like helicase receptors. These include MDA5 and RIG-I (Fig. 3). Different characteristic features of viral dsRNAs are sensed by MDA5 and RIG-I (95, 97, 98). However, it is now apparent that in the absence of ADAR1, endogenous cellular (self) dsRNA also triggers MDA5 signaling via the MAVS adaptor to activate innate immune proinflammatory responses (32, 41, 103–105). The crystal structure of MDA5 bound to dsRNA provides insight into the structural basis of dsRNA recognition, filament formation, and signal activation via the MAVS adaptor. MDA5 recognizes the internal duplex structure, whereas RIG-I recognizes the terminus of dsRNA (106). Alu:Alu dsRNAs formed by inverted repeat Alu-containing transcripts are ligands of MDA5. A-to-I editing suppresses filament formation of WT MDA5, whereas unmodified Alu:Alu dsRNAs activate WT MDA5 under conditions of ADAR1 deficiency (107). Gain-of-function mutation of MDA5 allows mis-recognition of self, cytosolic inverted repeat Alu dsRNAs (107).
Figure 3.

Model summarizing the role of ADAR1 as a suppressor of dsRNA-triggered innate immune responses. Cytoplasmic RLR and endosomal membrane-associated TLR3 sense dsRNA to mediate the production of type I IFN through activation of interferon-regulatory (IRF) and NF-κB transcription factors. The RLR family of proteins includes the MDA5 sensor that detects cytoplasmic dsRNAs, both viral (nonself) and cellular (self), and signals via the mitochondrial adaptor MAVS (IPS-1 and VISA) to produce IFN. Among the IFN-induced proteins are the PKR protein kinase and OAS synthetases, also cytoplasmic dsRNA-binding proteins. PKR, when activated by dsRNA-dependent autophosphorylation, phosphorylates translation initiation factor eIF2α thereby leading to an inhibition of translation. OAS, when activated by dsRNA, produces 2′-5′-oligoadenylates, which then activate the 2–5A–dependent RNase L thereby leading to RNA degradation. The p150 isoform of ADAR1 is IFN-inducible and both cytoplasmic and nuclear, whereas ADAR1 p110 and ADAR2 are both nuclear proteins and constitutively expressed. Under conditions of ADAR1 p150 deficiency, cellular RNAs (self) with double-stranded structure accumulate to sufficiently high concentration, above the threshold, and trigger activation of cytoplasmic dsRNA sensors, including MDA5, PKR, and OAS. In the presence of ADAR1 p150, A-to-I editing leads to inactivation of cellular (self) dsRNAs and impairment of dsRNA-triggered innate immune responses, as the dsRNA concentration is below the threshold. Infection leads to increased levels of viral dsRNA (nonself) present in infected cells compared with the cellular dsRNA (self) present in uninfected cells, thereby triggering activation of dsRNA sensors MDA5, PKR, and OAS. (Adapted from Ref. 73.)
ADAR1 down-regulates the sensing of both cellular (self) and viral (nonself) dsRNAs. Measles virus is an example of an RNA virus that activates RIG-I–like receptor signaling via MAVS to induce IFNβ gene transcription (108). Although C mutant virus is a robust inducer of IFNβ in human cell lines expressing ADAR1, wildtype (WT) measles virus by contrast is a poor inducer (108). However, WT virus becomes an excellent IFN inducer, comparable with that of the C mutant, under conditions of ADAR1 deficiency (109). Optimal suppression of IFNβ induction in WT virus-infected cells by ADAR1 p150 requires deaminase catalytic activity but not Z-DNA–binding activity (110). PKR kinase enhances both measles virus induction of IFNβ and apoptosis, mediated by cytosolic sensor signaling through MAVS, with the amplification of IFNβ induction occurring through eIF2α-mediated translational control (111, 112).
Mouse models of ADAR1 deficiency provided novel insight into the functional significance of ADAR1-mediated suppression of dsRNA detection and hence suppression of triggering of type I interferon and autoinflammatory responses. Genetic disruption of Adar1 in mice achieved by deleting both p150 and p110 (30, 31), by knocking out only p150 expression (104) or by knocking in the expression of the catalytic-deficient Adar1 E861A mutant (41), results in embryonic lethality characterized by disintegration of the fetal liver. The conditional knockout of Adar1 (32) and the knockin of the E861A mutant lacking editing activity (41) both result in an interferon signature of gene expression and high levels of cell death. ADAR1 is essential for normal murine erythropoiesis (113). However, the embryonic lethality and interferon signature phenotypes of Adar1 mutant mice can be rescued by concurrent deletion of either the MDA5 RIG-I–like receptor (41) or the MAVS mitochondrial adaptor (103, 105) but not the RIG-I receptor (105). An Ifnar interferon receptor mutation only partially rescues the Adar1 mutant embryonic lethality, and while the Mavs mutation rescues embryo survival to live birth, the mutant mice die shortly after birth (103, 105). By contrast, the concurrent Mda5 mutation rescues the Adar1 E861A mutation to live birth and well beyond. Adar1(E861A)-Mda5 double mutant mice appear normal, including their life span (7, 72). This suggests that the absence of ADAR1-editing activity is tolerated in the absence of MDA5. One possibility, given the differences between the MDA5 and MAVS rescues, is that under conditions of ADAR1 enzyme deficiency, the MDA5 dsRNA sensor may possibly function by an additional mechanism that is independent of the well established signaling via mitochondrial MAVS. Another possibility is that the knockin of the E861A catalytic mutant protein, while lacking editing activity, still possesses dsRNA-binding activity, and it is this dsRNA-binding activity that in the absence of functional MDA5–MAVS signaling contributes to the rescue phenotype beyond birth. However, the fact that the concurrent deletion of either Mda5 or Mavs does rescue the Adar1 embryonic lethality phenotype strongly suggests that the accumulation of self dsRNA structures recognized by MDA5 and signaling through MAVS is responsible in large part for the Adar1 mutant phenotypes.
Independent roles of the ADAR1 p110 and p150 proteins have been identified, with regulation of the MDA5 sensor activity by p150, whereas both p150 and p110 affect multiorgan development in the mouse (41, 104, 105, 113). Evidence also has been provided that ADAR1 regulates dsRNA sensing in the setting of ischemic stress in the liver, where ADAR deficiency leads to increased RIG-I–dependent IFN production, inflammation, and organ damage following ischemic stress (114). By contrast to the embryonic lethality phenotype of Adar1 ablation (30–32, 41, 104), the Adar2 and Adar3 mouse knockouts do not display embryonic lethality. Furthermore, postnatal death of the Adar2 mutant mouse remarkably is rescued by knockin of the edited form of the GluR-B Q/R site (62), although ADAR2 is required for normal physiology more broadly (115). An extended phenotypic analysis of 320 parameters showed the mice were hypermetabolic, had a hearing deficit, and displayed increased serum IgE levels (115). RNA editing by ADAR2 is metabolically regulated, for example in pancreatic islets and beta cells (116). Adar3 knockout mice are born following predicted Mendelian ratios and do not display any atypical developmental characteristics (117). Mice with the Adar3 gene disrupted by deletion of exon 3 (which includes the two copies of the dsRNA-binding domain, Fig. 1, lower panel) do, however, show deficits in learning and memory (117).
A-to-I editing by ADAR1 may lead to I-U mismatches in place of A:U bp, thereby destabilizing dsRNA structures and hence suppressing the activity of dsRNA-sensing proteins of the interferon response, including not only the MDA5 RIG-I–like receptor considered above, but also the protein kinase PKR and the 2′-5′-oligoadenylate synthetase OAS–RNase L (41, 109, 110, 118, 119). In addition to destabilizing effects of deamination mediated by ADAR catalytic activity, ADARs are also potent dsRNA-binding proteins (16, 23). The potential effect of sequestration of dsRNA or perturbation of intermolecular interactions involving dsRNA in the absence of editing may also occur (120, 121).
PKR
The PKR protein is a dsRNA sensor (Fig. 3). Binding of dsRNA by PKR leads to dimerization and activation by autophosphorylation. Activated PKR then catalyzes the phosphorylation of serine 51 of the α-subunit of protein synthesis initiation factor eIF2, which leads to an inhibition of translation (47, 93, 100, 122–125). PKR is both antiviral and pro-apoptotic. ADAR1 deficiency leads to increased activation of PKR and reduced virus growth (90, 119). The formation of stress granules, cytoplasmic aggregates of stalled translation initiation complexes, is a hallmark of viral infection. Stress granule formation is PKR-dependent for several viruses, including measles virus and hepatitis C virus (110, 126). ADAR1 suppresses both the activation of PKR and the formation of stress granules in cells infected with either WT or V mutant measles virus but not the C mutant (90, 109, 110). C mutant measles virus produces large amounts of viral dsRNA and grows poorly compared with either WT or V mutant virus. The C mutant is an efficient activator of PKR even in the presence of ADAR1, leading to an inhibition of viral protein synthesis and reduced C mutant virus growth (88, 89). In the absence of infection, activation of PKR by cellular (self) RNAs is suppressed by ADAR1 (73). In uninfected mouse Adar1 null MEFs (lacking p110 and p150) and Adar1 p150 null MEFs, but not Adar2 null or WT MEFs, PKR becomes activated following IFN treatment as measured by increased phosphorylation of eIF2α and formation of stress granules (73). Deep sequencing of mouse exonic loci containing A-to-I editing sites using RNA from mutant and WT cells reveals that the majority of editing in MEFs is by ADAR1, with hyper-edited sites found in predicted duplex structures of cellular (self) RNAs (33, 73). Likewise, conceptually similar conclusions are reached with human 293 cell lines generated using CRISPR-Cas technology lacking both p110 and p150, or only lacking p150: ADAR1 blocks translational shutdown by preventing hyperactivation of the PKR kinase triggered by endogenous (self) RNA (127). Both dsRNA binding and catalytic activity of ADAR1 p150, but not Z-DNA–binding activity, appear necessary to maximally prevent activation of PKR (110, 127).
OAS–RNase L
In humans there are three IFN-inducible and catalytically active OAS enzymes, OAS1, -2, and -3. OAS is a dsRNA sensor (Fig. 3). Upon binding dsRNA, OAS is activated and synthesizes 2–5A from ATP. The 2–5A oligomers then are bound by RNase L, which mediates dimerization and activation of RNase L (93, 102). Activation of RNase L is mainly dependent upon OAS3 during infection with a range of human viruses (128). Activated RNase L is an endonuclease and cleaves both viral and cellular RNAs. Like PKR, RNase L is both antiviral and proapoptotic (102). The cell lethal phenotype of ADAR1 deletion in the human A549 cell line is rescued by knockout of either RNase L or MAVS or by expression of a viral 2′-5′-phosphodiesterase antagonist that degrades 2–5A and prevents activation of RNase L (118). RNase L, at least in A549 cells, is a major determinant of the cell death phenotype triggered by ADAR1 deficiency following accumulation of endogenous (self) dsRNA and activation of OAS (118). RNase L and PKR share homology in their pseudokinase (RNase L) and kinase (PKR) domains, and both are inhibited by sunitinib, an ATP competitive inhibitor used to suppress angiogenesis and tumor growth (129). Activation of RNase L by 2–5A also leads to the production of small cleavage products from endogenous self-RNA that amplify IFNβ production by the RIG-I–like receptor–MAVS pathway (130). The effect of sunitinib on inflammation triggered by ADAR1 deficiency is unknown.
Human disease and ADAR
Altered A-to-I editing is linked to a variety of human diseases and is an area of increased investigation (8, 33, 131–137). Among the disorders associated with changes in ADAR-editing activity is Aicardi-Goutieres syndrome, a childhood autoimmune disorder characterized by an elevated type I interferon signature and caused in part by mutations in ADAR1 that reduce activity (138, 139), similar to the elevated interferon signature seen in mice lacking functional Adar1 (32, 41). Dysregulation of A-to-I editing also is observed in cancers, often with A-to-I RNA editing levels elevated in tumor tissue compared with normal tissue (131–137). For example, depending upon the cell type, cancer progression has been linked to an up-regulation of ADAR1 in liver, lung, and esophageal cancers and myeloma (137, 140) or down-regulation of ADAR2 in glioblastoma (63, 137), although exceptions occur where ADAR1 is down-regulated, for example in metastatic melanoma (137, 142). Most studies to date have used sequencing strategies to identify changes attributed to A-to-I editing at the transcriptome level. Recent findings extend RNA-seq studies and demonstrate that editing contributes to proteomic diversity in a breast cancer model through coding sequence changes, including of the COPA transcript where increased editing correlates with poorer survival time (141).
Summary
A model by which ADAR1 p150 may suppress innate immune interferon responses triggered by cellular (self) RNA, but yet permits activation of the responses by viral (nonself) pathogen RNA, is summarized in Fig. 3. The findings described herein are consistent with the notion that A-to-I editing activity by ADAR1 in uninfected IFN-treated cells is capable of reducing the effective steady-state concentration of endogenous cellular (self) dsRNA structures to levels below the threshold concentration ordinarily required to trigger activation of the cellular dsRNA sensors (MDA5, PKR, and OAS). By contrast, in the absence of ADAR1 p150, the functional concentration of endogenous cytoplasmic self dsRNA increases to a level above the threshold necessary to trigger activation of MDA5, PKR, and OAS. Likewise, pathogen infection produces substantially elevated levels of dsRNA, well above the threshold both in the presence and absence of ADAR1. This then triggers activation of the MDA5, PKR, and OAS sensors, with the efficiency dependent upon the robustness of pathogen dsRNA production, as illustrated by the differences observed between WT (low dsRNA production) and C mutant (high dsRNA production) measles virus. Thus, ADAR1 regulates sensing of cellular (self) dsRNA structures, minimizing autotriggering of innate immune responses under conditions of low concentrations of dsRNA, yet permitting sensor activation by high concentrations of viral (nonself) dsRNAs produced in infected cells (73, 90, 127).
Challenges and opportunities in the A-to-I editing field
Although considerable progress has been made toward understanding the regulation of mammalian ADAR genes, the activities of their encoded ADAR proteins, and the functional roles that the A-to-I editing events play in biologic processes, much remains to be learned.
The major and possibly sole essential role of ADAR1 is the suppression of dsRNA-triggered innate immune IFN responses through editing of cellular dsRNA structures, with a few million editing sites identified in human transcripts mostly occurring in noncoding repetitive sequences (7, 8, 33, 143). For MDA5, PKR, and OAS, whether combinations of cellular transcripts are unique or overlapping and to what extent the editing must occur to cross the threshold necessary to suppress dsRNA sensing by MDA5, PKR, and OAS is largely unknown. Likewise, it is unclear whether the suppression of individual dsRNA sensors by ADAR1 results solely from a destabilization of duplex regions of RNA structure by generating base pair mismatches. The converse relates to which cellular (self) dsRNA transcripts in the unedited form have sufficient duplex character and abundance to activate a given dsRNA sensor. This is not yet delineated under conditions of ADAR1 deficiency for MDA5, PKR, or OAS. In the case of ADAR1, another need is to more fully define the functional roles of the p110 constitutively expressed nuclear isoform compared with the IFN-inducible p150 isoform that is the only known cytoplasmic ADAR. The molecular basis of the RNA substrate selectivity of the catalytically active ADAR1 and ADAR2 proteins and the roles played by the repeated RNA-binding domain copies compared with the catalytic domain in conferring substrate selectivity are not fully resolved. The role of the Z-DNA binding domains, Zα and Zβ, is not clear in p150. Comparatively little is known regarding the functional role of the ADAR3 protein, an ADAR not yet shown to possess catalytic activity that seems to act as a negative regulator of the enzymatically active ADARs. ADAR1 deficiency is linked to human diseases, exemplified by Aicardi-Goutieres syndrome. Changes in ADAR activity, typically an increased ADAR1 activity, are seen in some cancers. Assessing the effect of therapeutic modulation of ADAR activity as an approach to regulate innate immunity and inflammatory responses is largely unexplored. Finally, opportunity exists to utilize ADAR as a tool. Engineered nucleotide substitution of an “I” (= G) for an “A” by targeted adenosine deamination catalyzed by an ADAR catalytic domain using a site-directed guide strategy has potential for creating RNA mutations (144, 145).
Acknowledgments
I thank Dr. Cyril George for reviewing the manuscript, and the many investigators in the A-to-I editing field for their contributions that made the review possible.
This work was supported in part by National Institutes of Health Research Grants AI-12520 and AI-20611 from the NIAID. This JBC Review is part of a collection honoring Herbert Tabor on the occasion of his 100th birthday. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- dsRNA
- double-stranded RNA
- ADAR
- adenosine deaminase acting on RNA
- IFN
- interferon
- MAVS
- mitochondrial antiviral-signaling protein
- MDA5
- melanoma differentiation associated gene 5 protein (IFIH1)
- OAS
- oligoadenylate synthetase
- PKR
- RNA-dependent protein kinase
- RIG-I
- retinoic acid-inducible gene-1
- RLR
- RIG-I–like receptor
- 2-5A
- 2′-5′-oligoadenylate
- LCMV
- lymphocytic choriomeningitis virus
- miR
- microRNA
- pri
- primary
- MEF
- mouse embryo fibroblast
- ssRNA
- single-stranded RNA.
References
- 1. Guengerich E. P. (2019) Cytochrome P450 research and The Journal of Biological Chemistry. J. Biol. Chem. 294, 1671–1680 10.1074/jbc.TM118.004144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bass B. L., and Weintraub H. (1988) An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55, 1089–1098 10.1016/0092-8674(88)90253-X [DOI] [PubMed] [Google Scholar]
- 3. Wagner R. W., Smith J. E., Cooperman B. S., and Nishikura K. (1989) A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs. Proc. Natl. Acad. Sci. U.S.A. 86, 2647–2651 10.1073/pnas.86.8.2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bass B. L. (2002) RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 71, 817–846 10.1146/annurev.biochem.71.110601.135501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Samuel C. E. (2011) Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 411, 180–193 10.1016/j.virol.2010.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nishikura K. (2016) A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 17, 83–96 10.1038/nrm.2015.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walkley C. R., and Li J. B. (2017) Rewriting the transcriptome: adenosine-to-inosine RNA editing by ADARs. Genome Biol. 18, 205 10.1186/s13059-017-1347-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eisenberg E., and Levanon E. Y. (2018) A-to-I RNA editing-immune protector and transcriptome diversifier. Nat. Rev. Genet. 19, 473–490 10.1038/s41576-018-0006-1 [DOI] [PubMed] [Google Scholar]
- 9. Bass B. L., Nishikura K., Keller W., Seeburg P. H., Emeson R. B., O'Connell M. A., Samuel C. E., and Herbert A. (1997) A standardized nomenclature for adenosine deaminases that act on RNA. RNA 9, 947–949 [PMC free article] [PubMed] [Google Scholar]
- 10. Bass B. L., and Weintraub H. (1987) A developmentally regulated activity that unwinds RNA duplexes. Cell 48, 607–613 10.1016/0092-8674(87)90239-X [DOI] [PubMed] [Google Scholar]
- 11. Rebagliati M. R., and Melton D. A. (1987) Antisense RNA injections in fertilized frog eggs reveal an RNA duplex unwinding activity. Cell 48, 599–605 10.1016/0092-8674(87)90238-8 [DOI] [PubMed] [Google Scholar]
- 12. Strobel S. A., Cech T. R., Usman N., and Beigelman L. (1994) The 2,6-diaminopurine riboside.5-methylisocytidine wobble base pair: an isoenergetic substitution for the study of G.U pairs in RNA. Biochemistry 33, 13824–13835 10.1021/bi00250a037 [DOI] [PubMed] [Google Scholar]
- 13. Kim U., Garner T. L., Sanford T., Speicher D., Murray J. M., and Nishikura K. (1994) Purification and characterization of double-stranded RNA adenosine deaminase from bovine nuclear extracts. J. Biol. Chem. 269, 13480–13489 [PubMed] [Google Scholar]
- 14. O'Connell M. A., Gerber A., and Keller W. (1997) Purification of human double-stranded RNA-specific editase 1 (hRED1) involved in editing of brain glutamate receptor B pre-mRNA. J. Biol. Chem. 272, 473–478 10.1074/jbc.272.1.473 [DOI] [PubMed] [Google Scholar]
- 15. Kim U., Wang Y., Sanford T., Zeng Y., and Nishikura K. (1994) Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc. Natl. Acad. Sci. U.S.A. 91, 11457–11461 10.1073/pnas.91.24.11457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Patterson J. B., and Samuel C. E. (1995) Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol. Cell. Biol. 15, 5376–5388 10.1128/MCB.15.10.5376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. O'Connell M. A., Krause S., Higuchi M., Hsuan J. J., Totty N. F., Jenny A., and Keller W. (1995) Cloning of cDNAs encoding mammalian double-stranded RNA-specific adenosine deaminase. Mol. Cell. Biol. 15, 1389–1397 10.1128/MCB.15.3.1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Poulsen H., Nilsson J., Damgaard C. K., Egebjerg J., and Kjems J. (2001) Crm1 mediates the export of ADAR1 through a nuclear export signal within the Z-DNA–binding domain. Mol. Cell. Biol. 21, 7862–7871 10.1128/MCB.21.22.7862-7871.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Strehblow A., Hallegger M., and Jantsch M. F. (2002) Nucleocytoplasmic distribution of human RNA-editing enzyme ADAR1 is modulated by double-stranded RNA-binding domains, a leucine-rich export signal, and a putative dimerization domain. Mol. Biol. Cell 13, 3822–3835 10.1091/mbc.e02-03-0161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weier H. U., George C. X., Greulich K. M., and Samuel C. E. (1995) The interferon-inducible, double-stranded RNA-specific adenosine deaminase gene (DSRAD) maps to human chromosome 1q21.1–21.2. Genomics 30, 372–375 10.1006/geno.1995.0034 [DOI] [PubMed] [Google Scholar]
- 21. Wang Y., Zeng Y., Murray J. M., and Nishikura K. (1995) Genomic organization and chromosomal location of the human dsRNA adenosine deaminase gene: the enzyme for glutamate-activated ion channel RNA editing. J. Mol. Biol. 254, 184–195 10.1006/jmbi.1995.0610 [DOI] [PubMed] [Google Scholar]
- 22. Weier H. U., George C. X., Lersch R. A., Breitweser S., Cheng J. F., and Samuel C. E. (2000) Assignment of the RNA-specific adenosine deaminase gene (Adar) to mouse chromosome 3f2 by in situ hybridization. Cytogenet. Cell Genet. 89, 214–215 10.1159/000015615 [DOI] [PubMed] [Google Scholar]
- 23. George C. X., Gan Z., Liu Y., and Samuel C. E. (2011) Adenosine deaminases acting on RNA (ADARs), RNA editing and interferon action. J. Interferon Cytokine Res. 31, 99–117 10.1089/jir.2010.0097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. George C. X., and Samuel C. E. (1999a) Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc. Natl. Acad. Sci. U.S.A. 96, 4621–4626 10.1073/pnas.96.8.4621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. George C. X., and Samuel C. E. (1999b) Characterization of the 5′-flanking region of the human RNA-specific adenosine deaminase ADAR1 gene and identification of an interferon-inducible ADAR1 promoter. Gene 229, 203–213 10.1016/S0378-1119(99)00017-7 [DOI] [PubMed] [Google Scholar]
- 26. Kawakubo K., and Samuel C. E. (2000) Human RNA-specific adenosine deaminase (ADAR1) gene specifies transcripts that initiate from a constitutively active alternative promoter. Gene 258, 165–172 10.1016/S0378-1119(00)00368-1 [DOI] [PubMed] [Google Scholar]
- 27. George C. X., Wagner M. V., and Samuel C. E. (2005) Expression of interferon-inducible RNA adenosine deaminase ADAR1 during pathogen infection and mouse embryo development involves tissue-selective promoter utilization and alternative splicing. J. Biol. Chem. 280, 15020–15028 10.1074/jbc.M500476200 [DOI] [PubMed] [Google Scholar]
- 28. Liu Y., George C. X., Patterson J. B., and Samuel C. E. (1997) Functionally distinct double-stranded RNA-binding domains associated with alternative splice site variants of the interferon-inducible double-stranded RNA-specific adenosine deaminase. J. Biol. Chem. 272, 4419–4428 10.1074/jbc.272.7.4419 [DOI] [PubMed] [Google Scholar]
- 29. George C. X., Das S., and Samuel C. E. (2008) Organization of the mouse RNA-specific adenosine deaminase Adar1 gene 5′-region and demonstration of STAT1-independent, STAT2-dependent transcriptional activation by interferon. Virology 380, 338–343 10.1016/j.virol.2008.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hartner J. C., Schmittwolf C., Kispert A., Müller A. M., Higuchi M., and Seeburg P. H. (2004) Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J. Biol. Chem. 279, 4894–4902 10.1074/jbc.M311347200 [DOI] [PubMed] [Google Scholar]
- 31. Wang Q., Miyakoda M., Yang W., Khillan J., Stachura D. L., Weiss M. J., and Nishikura K. (2004) Stress-induced apoptosis associated with null mutation of Adar1 RNA editing deaminase gene. J. Biol. Chem. 279, 4952–4961 10.1074/jbc.M310162200 [DOI] [PubMed] [Google Scholar]
- 32. Hartner J. C., Walkley C. R., Lu J., and Orkin S. H. (2009) ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol. 10, 109–115 10.1038/ni.1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tan M. H., Li Q., Shanmugam R., Piskol R., Kohler J., Young A. N., Liu K. I., Zhang R., Ramaswami G., Ariyoshi K., Gupte A., Keegan L. P., George C. X., Ramu A., Huang N., et al. (2017) Dynamic landscape and regulation of RNA editing in mammals. Nature 550, 249–254 10.1038/nature24041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Patterson J. B., Thomis D. C., Hans S. L., and Samuel C. E. (1995) Mechanism of interferon action: double-stranded RNA-specific adenosine-deaminase from human cells is inducible by α-interferon and γ-interferon. Virology 210, 508–511 10.1006/viro.1995.1370 [DOI] [PubMed] [Google Scholar]
- 35. George C. X., and Samuel C. E. (2015) STAT2-dependent induction of RNA adenosine deaminase ADAR1 by type I interferon differs between mouse and human cells in the requirement for STAT1. Virology 485, 363–370 10.1016/j.virol.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schindler C., Levy D. E., and Decker T. (2007) JAK–STAT signaling: from interferons to cytokines. J. Biol. Chem. 282, 20059–20063 10.1074/jbc.R700016200 [DOI] [PubMed] [Google Scholar]
- 37. Stark G. R., and Darnell J. E. (2012) The JAK–STAT pathway at twenty. Immunity 36, 503–514 10.1016/j.immuni.2012.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu Y., and Samuel C. E. (1999) Editing of glutamate receptor subunit B pre-mRNA by splice-site variants of interferon-inducible double-stranded RNA-specific adenosine deaminase ADAR1. J. Biol. Chem. 274, 5070–5077 10.1074/jbc.274.8.5070 [DOI] [PubMed] [Google Scholar]
- 39. Liu Y., and Samuel C. E. (1996) Mechanism of interferon action: functionally distinct RNA-binding and catalytic domains in the interferon-inducible, double-stranded RNA-specific adenosine deaminase. J. Virol. 70, 1961–1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lai F., Drakas R., and Nishikura K. (1995) Mutagenic analysis of double-stranded-RNA adenosine-deaminase, a candidate enzyme for RNA editing of glutamate-gated ion-channel transcripts. J. Biol. Chem. 270, 17098–17105 10.1074/jbc.270.29.17098 [DOI] [PubMed] [Google Scholar]
- 41. Liddicoat B. J., Piskol R., Chalk A. M., Ramaswami G., Higuchi M., Hartner J. C., Li J. B., Seeburg P. H., and Walkley C. R. (2015) RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120 10.1126/science.aac7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cho D. S., Yang W., Lee J. T., Shiekhattar R., Murray J. M., and Nishikura K. (2003) Requirement of dimerization for RNA editing activity of adenosine deaminases acting on RNA. J. Biol. Chem. 278, 17093–17102 10.1074/jbc.M213127200 [DOI] [PubMed] [Google Scholar]
- 43. Chilibeck K. A., Wu T., Liang C., Schellenberg M. J., Gesner E. M., Lynch J. M., and MacMillan A. M. (2006) FRET analysis of in vivo dimerization by RNA-editing enzymes. J. Biol. Chem. 281, 16530–16535 10.1074/jbc.M511831200 [DOI] [PubMed] [Google Scholar]
- 44. Valente L., and Nishikura K. (2007) RNA binding-independent dimerization of adenosine deaminases acting on RNA and dominant negative effects of nonfunctional subunits on dimer functions. J. Biol. Chem. 282, 16054–16061 10.1074/jbc.M611392200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McCormack S. J., Thomis D. C., and Samuel C. E. (1992) Mechanism of interferon action: identification of a RNA-binding domain within the N-terminal region of the human RNA-dependent P1/eIF-2α protein kinase. Virology 188, 47–56 10.1016/0042-6822(92)90733-6 [DOI] [PubMed] [Google Scholar]
- 46. McMillan N. A., Carpick B. W., Hollis B., Toone W. M., Zamanian-Daryoush M., and Williams B. R. (1995) Mutational analysis of the double-stranded RNA (dsRNA) binding domain of the dsRNA-activated protein kinase, PKR. J. Biol. Chem. 270, 2601–2606 10.1074/jbc.270.6.2601 [DOI] [PubMed] [Google Scholar]
- 47. Samuel C. E. (1993) The eIF-2α protein kinases, regulators of translation in eukaryotes from yeasts to humans. J. Biol. Chem. 268, 7603–7606 [PubMed] [Google Scholar]
- 48. Fierro-Monti I., and Mathews M. B. (2000) Proteins binding to duplexed RNA: one motif, multiple functions. Trends Biochem. Sci. 25, 241–246 10.1016/S0968-0004(00)01580-2 [DOI] [PubMed] [Google Scholar]
- 49. Herbert A., Alfken J., Kim Y. G., Mian I. S., Nishikura K., and Rich A. (1997) A Z-DNA–binding domain present in the human editing enzyme, double-stranded RNA adenosine deaminase. Proc. Natl. Acad. Sci. U.S.A. 94, 8421–8426 10.1073/pnas.94.16.8421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schwartz T., Rould M. A., Lowenhaupt K., Herbert A., and Rich A. (1999) Crystal structure of the Z domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science 284, 1841–1845 10.1126/science.284.5421.1841 [DOI] [PubMed] [Google Scholar]
- 51. Placido D., Brown B. A. 2nd., Lowenhaupt K., Rich A., and Athanasiadis A. (2007) A left-handed RNA double helix bound by the Zα domain of the RNA-editing enzyme ADAR1. Structure 15, 395–404 10.1016/j.str.2007.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu Y., Lei M., and Samuel C. E. (2000) Chimeric double-stranded RNA-specific adenosine deaminase ADAR1 proteins reveal functional selectivity of double-stranded RNA-binding domains from ADAR1 and protein kinase PKR. Proc. Natl. Acad. Sci. U.S.A. 97, 12541–12546 10.1073/pnas.97.23.12541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stephens O. M., Haudenschild B. L., and Beal P. A. (2004) The binding selectivity of ADAR2's dsRBMs contributes to RNA-editing selectivity. Chem. Biol. 11, 1239–1250 10.1016/j.chembiol.2004.06.009 [DOI] [PubMed] [Google Scholar]
- 54. Eggington J. M., Greene T., and Bass B. L. (2011) Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun. 2, 319 10.1038/ncomms1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang Y., Park S., and Beal P. A. (2018) Selective recognition of RNA substrates by ADAR deaminase domains. Biochemistry 57, 1640–1651 10.1021/acs.biochem.7b01100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kuttan A., and Bass B. L. (2012) Mechanistic insights into editing-site specificity of ADARs. Proc. Natl. Acad. Sci. U.S.A. 109, E3295–E3304 10.1073/pnas.1212548109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen C. X., Cho D. S., Wang Q., Lai F., Carter K. C., and Nishikura K. (2000) A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA-binding domains. RNA 6, 755–767 10.1017/S1355838200000170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sommer B., Köhler M., Sprengel R., and Seeburg P. H. (1991) RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67, 11–19 10.1016/0092-8674(91)90568-J [DOI] [PubMed] [Google Scholar]
- 59. Burns C. M., Chu H., Rueter S. M., Hutchinson L. K., Canton H., Sanders-Bush E., and Emeson R. B. (1997) Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature 387, 303–308 10.1038/387303a0 [DOI] [PubMed] [Google Scholar]
- 60. Liu Y., Emeson R. B., and Samuel C. E. (1999) Serotonin-2c receptor pre-mRNA editing in rat brain and in vitro by splice site variants of the interferon-inducible double-stranded RNA-specific adenosine deaminase ADAR1. J. Biol. Chem. 274, 18351–18358 10.1074/jbc.274.26.18351 [DOI] [PubMed] [Google Scholar]
- 61. Hood J. L., and Emeson R. B. (2012) Editing of neurotransmitter receptor and ion channel RNAs in the nervous system. Curr. Top. Microbiol. Immunol. 353, 61–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Higuchi M., Maas S., Single F. N., Hartner J., Rozov A., Burnashev N., Feldmeyer D., Sprengel R., and Seeburg P. H. (2000) Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406, 78–81 10.1038/35017558 [DOI] [PubMed] [Google Scholar]
- 63. Oakes E., Anderson A., Cohen-Gadol A., and Hundley H. A. (2017) Adenosine deaminase that acts on RNA 3 (ADAR3) binding to glutamate receptor subunit B pre-mRNA inhibits RNA editing in glioblastoma. J. Biol. Chem. 292, 4326–4335 10.1074/jbc.M117.779868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Casey J. L. (2006) RNA editing in hepatitis delta virus. Curr. Top. Microbiol. Immunol. 307, 67–89 [DOI] [PubMed] [Google Scholar]
- 65. Bazak L., Haviv A., Barak M., Jacob-Hirsch J., Deng P., Zhang R., Isaacs F. J., Rechavi G., Li J. B., Eisenberg E., and Levanon E. Y. (2014) A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 24, 365–376 10.1101/gr.164749.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Neeman Y., Levanon E. Y., Jantsch M. F., and Eisenberg E. (2006) RNA editing level in the mouse is determined by the genomic repeat repertoire. RNA 12, 1802–1809 10.1261/rna.165106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ramaswami G., Lin W., Piskol R., Tan M. H., Davis C., and Li J. B. (2012) Accurate identification of human Alu and non-Alu RNA editing sites. Nat. Methods 9, 579–581 10.1038/nmeth.1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li J. B., Levanon E. Y., Yoon J. K., Aach J., Xie B., Leproust E., Zhang K., Gao Y., and Church G. M. (2009) Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 324, 1210–1213 10.1126/science.1170995 [DOI] [PubMed] [Google Scholar]
- 69. Pinto Y., Cohen H. Y., and Levanon E. Y. (2014) Mammalian conserved ADAR targets comprise only a small fragment of the human editosome. Genome Biol. 15, R5 10.1186/gb-2014-15-1-r5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ramaswami G., and Li J. B. (2014) RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 42, D109–D113 10.1093/nar/gkt996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ramaswami G., and Li J. B. (2016) Identification of human RNA editing sites: a historical perspective. Methods 107, 42–47 10.1016/j.ymeth.2016.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Heraud-Farlow J. E., Chalk A. M., Linder S. E., Li Q., Taylor S., White J. M., Pang L., Liddicoat B. J., Gupte A., Li J. B., and Walkley C. R. (2017) Protein recoding by ADAR1-mediated RNA editing is not essential for normal development and homeostasis. Genome Biol. 18, 166 10.1186/s13059-017-1301-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. George C. X., Ramaswami G., Li J. B., and Samuel C. E. (2016) Editing of cellular self-RNAs by adenosine deaminse ADAR1 suppresses innate immune stress responses. J. Biol. Chem. 291, 6158–6168 10.1074/jbc.M115.709014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dawson T. R., Sansam C. L., and Emeson R. B. (2004) Structure and sequence determinants required for the RNA editing of ADAR2 substrates. J. Biol. Chem. 279, 4941–4951 10.1074/jbc.M310068200 [DOI] [PubMed] [Google Scholar]
- 75. Lev-Maor G., Sorek R., Levanon E. Y., Paz N., Eisenberg E., and Ast G. (2007) RNA-editing-mediated exon evolution. Genome Biol. 8, R29 10.1186/gb-2007-8-2-r29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chen Y. T., Chang I. Y., Liu H., Ma C. P., Kuo Y. P., Shih C. T., Shih Y. H., Kang L., and Tan B. C. (2018) Tumor-associated intronic editing of HNRPLL generates a novel splicing variant linked to cell proliferation. J. Biol. Chem. 293, 10158–10171 10.1074/jbc.RA117.001197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Parada G. E., Munita R., Cerda C. A., and Gysling K. (2014) A comprehensive survey of non-canonical splice sites in the human transcriptome. Nucleic Acids Res. 42, 10564–10578 10.1093/nar/gku744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kawahara Y., Megraw M., Kreider E., Iizasa H., Valente L., Hatzigeorgiou A. G., and Nishikura K. (2008) Frequency and fate of microRNA editing in human brain. Nucleic Acids Res. 36, 5270–5280 10.1093/nar/gkn479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yang W., Chendrimada T. P., Wang Q., Higuchi M., Seeburg P. H., Shiekhattar R., and Nishikura K. (2006) Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 13, 13–21 10.1038/nsmb1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kawahara Y., Zinshteyn B., Chendrimada T. P., Shiekhattar R., and Nishikura K. (2007) RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 8, 763–769 10.1038/sj.embor.7401011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kawahara Y., Zinshteyn B., Sethupathy P., Iizasa H., Hatzigeorgiou A. G., and Nishikura K. (2007) Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 315, 1137–1140 10.1126/science.1138050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nakano M., Fukami T., Gotoh S., Takamiya M., Aoki Y., and Nakajima M. (2016) RNA editing modulates human hepatic aryl hydrocarbon receptor expression by creating microRNA recognition sequence. J. Biol. Chem. 291, 894–903 10.1074/jbc.M115.699363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pinto Y., Buchumenski I., Levanon E. Y., and Eisenberg E. (2018) Human cancer tissues exhibit reduced A-to-I editing of miRNAs coupled with elevated editing of their targets. Nucleic Acids Res. 46, 71–82 10.1093/nar/gkx1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang Y., Xu X., Yu S., Jeong K. J., Zhou Z., Han L., Tsang Y. H., Li J., Chen H., Mangala L. S., Yuan Y., Eterovic A. K., Lu Y., Sood A. K., Scott K. L., Mills G. B., and Liang H. (2017) Systematic characterization of A-to-I RNA editing hotspots in microRNAs across human cancers. Genome Res. 27, 1112–1125 10.1101/gr.219741.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yang W., Wang Q., Howell K. L., Lee J. T., Cho D. S., Murray J. M., and Nishikura K. (2005) ADAR1 RNA deaminase limits short interfering RNA efficacy in mammalian cells. J. Biol. Chem. 280, 3946–3953 10.1074/jbc.M407876200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Heale B. S., Keegan L. P., McGurk L., Michlewski G., Brindle J., Stanton C. M., Caceres J. F., and O'Connell M. A. (2009) Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J. 28, 3145–3156 10.1038/emboj.2009.244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cattaneo R., and Billeter M. A. (1992) Mutations and A/I hypermutations in measles virus persistent infections. Curr. Top. Microbiol. Immunol. 176, 63–74 [DOI] [PubMed] [Google Scholar]
- 88. Toth A. M., Devaux P., Cattaneo R., and Samuel C. E. (2009) Protein kinase PKR mediates the apoptosis induction and growth restriction phenotypes of C protein-deficient measles virus. J. Virol. 83, 961–968 10.1128/JVI.01669-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Pfaller C. K., Radeke M. J., Cattaneo R., and Samuel C. E. (2014) Measles virus C protein impairs production of defective copyback double-stranded viral RNA and activation of protein kinase R. J. Virol. 88, 456–468 10.1128/JVI.02572-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Toth A. M., Li Z., Cattaneo R., and Samuel C. E. (2009) RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. J. Biol. Chem. 284, 29350–29356 10.1074/jbc.M109.045146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Pfaller C. K., Mastorakos G. M., Matchett W. E., Ma X., Samuel C. E., and Cattaneo R. (2015) Measles virus defective-interfering RNAs are generated frequently and early in the absence of C protein and can be destabilized by adenosine deaminase acting on RNA 1-like hypermutations. J. Virol. 89, 7735–7747 10.1128/JVI.01017-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zahn R. C., Schelp I., Utermöhlen O., and von Laer D. (2007) A-to-G hypermutation in the genome of lymphocytic choriomeningitis virus. J. Virol. 81, 457–464 10.1128/JVI.00067-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Samuel C. E. (2001) Antiviral actions of interferons. Clin. Microbiol. Rev. 14, 778–809 10.1128/CMR.14.4.778-809.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Isaacs A., and Lindenmann J. (1957) Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 147, 258–267 [PubMed] [Google Scholar]
- 95. Yoneyama M., and Fujita T. (2007) Function of RIG-I–like receptors in antiviral innate immunity. J. Biol. Chem. 282, 15315–15318 10.1074/jbc.R700007200 [DOI] [PubMed] [Google Scholar]
- 96. Kawai T., and Akira S. (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- 97. Wu B., and Hur S. (2015) How RIG-I like receptors activate MAVS. Curr. Opin. Virol. 12, 91–98 10.1016/j.coviro.2015.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lässig C., and Hopfner K. P. (2017) Discrimination of cytosolic self and non-self RNA by RIG-I–like receptors. J. Biol. Chem. 292, 9000–9009 10.1074/jbc.R117.788398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Samuel C. E. (1979) Mechanism of interferon action: phosphorylation of protein synthesis initiation factor eIF-2 in interferon-treated human cells by a ribosome-associated kinase possessing site specificity similar to hemin-regulated rabbit reticulocyte kinase. Proc. Natl. Acad. Sci. U.S.A. 76, 600–604 10.1073/pnas.76.2.600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sadler A. J., and Williams B. R. (2007) Structure and function of the protein kinase R. Curr. Top. Microbiol. Immunol. 316, 253–292 [DOI] [PubMed] [Google Scholar]
- 101. Hovanessian A. G., Brown R. E., and Kerr I. M. (1977) Synthesis of low molecular weight inhibitor of protein synthesis with enzyme from interferon-treated cells. Nature 268, 537–540 10.1038/268537a0 [DOI] [PubMed] [Google Scholar]
- 102. Silverman R. H. (2007) Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 81, 12720–12729 10.1128/JVI.01471-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Mannion N. M., Greenwood S. M., Young R., Cox S., Brindle J., Read D., Nellåker C., Vesely C., Ponting C. P., McLaughlin P. J., Jantsch M. F., Dorin J., Adams I. R., Scadden A. D., Ohman M., et al. (2014) The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 9, 1482–1494 10.1016/j.celrep.2014.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ward S. V., George C. X., Welch M. J., Liou L. Y., Hahm B., Lewicki H., de la Torre J. C., Samuel C. E., and Oldstone M. B. (2011) RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc. Natl. Acad. Sci. U.S.A. 108, 331–336 10.1073/pnas.1017241108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Pestal K., Funk C. C., Snyder J. M., Price N. D., Treuting P. M., and Stetson D. B. (2015) Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43, 933–944 10.1016/j.immuni.2015.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Wu B., Peisley A., Richards C., Yao H., Zeng X., Lin C., Chu F., Walz T., and Hur S. (2013) Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 152, 276–289 10.1016/j.cell.2012.11.048 [DOI] [PubMed] [Google Scholar]
- 107. Ahmad S., Mu X., Yang F., Greenwald E., Park J. W., Jacob E., Zhang C.-Z., and Hur S. (2018) Breaching self-tolerance to Alu duplex RNA underlies MDA5-mediated inflammation. Cell 172, 797–810.e13 10.1016/j.cell.2017.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. McAllister C. S., Toth A. M., Zhang P., Devaux P., Cattaneo R., and Samuel C. E. (2010) Mechanisms of protein kinase PKR-mediated amplification of β interferon induction by C protein-deficient measles virus. J. Virol. 84, 380–386 10.1128/JVI.02630-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Li Z., Okonski K. M., and Samuel C. E. (2012) Adenosine deaminase acting on RNA 1 (ADAR1) suppresses the induction of interferon by measles virus. J. Virol. 86, 3787–3794 10.1128/JVI.06307-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Okonski K. M., and Samuel C. E. (2013) Stress granule formation induced by measles virus is protein kinase PKR-dependent and impaired by RNA adenosine deaminase ADAR1. J. Virol. 87, 756–766 10.1128/JVI.02270-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. McAllister C. S., and Samuel C. E. (2009) The RNA-activated protein kinase enhances the induction of interferon-β and apoptosis mediated by cytoplasmic RNA sensors. J. Biol. Chem. 284, 1644–1651 10.1074/jbc.M807888200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. McAllister C. S., Taghavi N., and Samuel C. E. (2012) Protein kinase PKR amplification of interferon β induction occurs through initiation factor eIF-2α-mediated translational control. J. Biol. Chem. 287, 36384–36392 10.1074/jbc.M112.390039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Liddicoat B. J., Hartner J. C., Piskol R., Ramaswami G., Chalk A. M., Kingsley P. D., Sankaran V. G., Wall M., Purton L. E., Seeburg P. H., Palis J., Orkin S. H., Lu J., Li J. B., and Walkley C. R. (2016) Adenosine-to-inosine RNA editing by ADAR1 is essential for normal murine erythropoiesis. Exp. Hematol. 44, 947–963 10.1016/j.exphem.2016.06.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wang H., Wang G., Zhang L., Zhang J., Zhang J., Wang Q., and Billiar T. R. (2016) ADAR1 suppresses the activation of cytosolic RNA-sensing signaling pathways to protect the liver from ischemia/reperfusion injury. Sci. Rep. 6, 20248 10.1038/srep20248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Horsch M., Seeburg P. H., Adler T., Aguilar-Pimentel J. A., Becker L., Calzada-Wack J., Garrett L., Götz A., Hans W., Higuchi M., Hölter S. M., Naton B., Prehn C., Puk O., Rácz I., et al. (2011) Requirement of the RNA-editing enzyme ADAR2 for normal physiology in mice. J. Biol. Chem. 286, 18614–18622 10.1074/jbc.M110.200881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Gan Z., Zhao L., Yang L., Huang P., Zhao F., Li W., and Liu Y. (2006) RNA editing by ADAR2 is metabolically regulated in pancreatic islets and β-cells. J. Biol. Chem. 281, 33386–33394 10.1074/jbc.M604484200 [DOI] [PubMed] [Google Scholar]
- 117. Mladenova D., Barry G., Konen L. M., Pineda S. S., Guennewig B., Avesson L., Zinn R., Schonrock N., Bitar M., Jonkhout N., Crumlish L., Kaczorowski D. C., Gong A., Pinese M., Franco G. R., Walkley C. R., Vissel B., and Mattick J. S. (2018) Adar3 is involved in learning and memory in mice. Front. Neurosci. 12, 243 10.3389/fnins.2018.00243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Li Y., Banerjee S., Goldstein S. A., Dong B., Gaughan C., Rath S., Donovan J., Korennykh A., Silverman R. H., and Weiss S. R. (2017) Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. Elife 6, e25687 10.7554/eLife.25687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Li Z., Wolff K. C., and Samuel C. E., (2010) RNA adenosine deaminase ADAR1 deficiency leads to increased activation of protein kinase PKR and reduced vesicular stomatitis virus growth following interferon treatment. Virology 396, 316–322 10.1016/j.virol.2009.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wang Y., and Samuel C. E., (2009) Adenosine deaminase ADAR1 increases gene expression at the translational level by decreasing protein kinase PKR-dependent eIF-2α phosphorylation. J. Mol. Biol. 393, 777–787 10.1016/j.jmb.2009.08.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Yang S., Deng P., Zhu Z., Zhu J., Wang G., Zhang L., Chen A. F., Wang T., Sarkar S. N., Billiar T. R., and Wang Q. (2014) Adenosine deaminase acting on RNA 1 limits RIG-I RNA detection and suppresses IFN production responding to viral and endogenous RNAs. J. Immunol. 193, 3436–3445 10.4049/jimmunol.1401136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. García M. A., Meurs E. F., and Esteban M. (2007) The dsRNA protein kinase PKR: virus and cell control. Biochimie 89, 799–811 10.1016/j.biochi.2007.03.001 [DOI] [PubMed] [Google Scholar]
- 123. Berry M. J., Knutson G. S., Lasky S. R., Munemitsu S. M., and Samuel C. E. (1985) Mechanism of interferon action. Purification and substrate specificities of the double-stranded RNA-dependent protein kinase from untreated and interferon-treated mouse fibroblasts. J. Biol. Chem. 260, 11240–11247 [PubMed] [Google Scholar]
- 124. Samuel C. E., Duncan R., Knutson G. S., and Hershey J. W. (1984) Mechanism of interferon action. Increased phosphorylation of protein synthesis initiation factor eIF-2α in interferon-treated, reovirus-infected mouse L929 fibroblasts in vitro and in vivo. J. Biol. Chem. 259, 13451–13457 [PubMed] [Google Scholar]
- 125. Scheuner D., Patel R., Wang F., Lee K., Kumar K., Wu J., Nilsson A., Karin M., and Kaufman R. J. (2006) Double-stranded RNA-dependent protein kinase phosphorylation of the α-subunit of eukaryotic translation initiation factor 2 mediates apoptosis. J. Biol. Chem. 281, 21458–21468 10.1074/jbc.M603784200 [DOI] [PubMed] [Google Scholar]
- 126. Ruggieri A., Dazert E., Metz P., Hofmann S., Bergeest J. P., Mazur J., Bankhead P., Hiet M. S., Kallis S., Alvisi G., Samuel C. E., Lohmann V., Kaderali L., Rohr K., Frese M., et al. (2012) Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe 12, 71–85 10.1016/j.chom.2012.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Chung H., Calis J. J. A., Wu X., Sun T., Yu Y., Sarbanes S. L., Dao Thi V. L., Shilvock A. R., Hoffmann H. H., Rosenberg B. R., and Rice C. M. (2018) Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell 172, 811–824.e14 10.1016/j.cell.2017.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Li Y., Banerjee S., Wang Y., Goldstein S. A., Dong B., Gaughan C., Silverman R. H., and Weiss S. R. (2016) Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc. Natl. Acad. Sci. U.S.A. 113, 2241–2246 10.1073/pnas.1519657113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Jha B. K., Polyakova I., Kessler P., Dong B., Dickerman B., Sen G. C., and Silverman R. H. (2011) Inhibition of RNase L and RNA-dependent protein kinase (PKR) by sunitinib impairs antiviral innate immunity. J. Biol. Chem. 286, 26319–26326 10.1074/jbc.M111.253443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Malathi K., Dong B., Gale M. Jr., and Silverman R. H. (2007) Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448, 816–819 10.1038/nature06042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Paz-Yaacov N., Bazak L., Buchumenski I., Porath H. T., Danan-Gotthold M., Knisbacher B. A., Eisenberg E., and Levanon E. Y. (2015) Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep. 13, 267–276 10.1016/j.celrep.2015.08.080 [DOI] [PubMed] [Google Scholar]
- 132. Fumagalli D., Gacquer D., Rothé F., Lefort A., Libert F., Brown D., Kheddoumi N., Shlien A., Konopka T., Salgado R., Larsimont D., Polyak K., Willard-Gallo K., Desmedt C., Piccart M., et al. (2015) Principles governing A-to-I RNA editing in the breast cancer transcriptome. Cell Rep. 13, 277–289 10.1016/j.celrep.2015.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Han L., Diao L., Yu S., Xu X., Li J., Zhang R., Yang Y., Werner H. M. J., Eterovic A. K., Yuan Y., Li J., Nair N., Minelli R., Tsang Y. H., Cheung L. W. T., et al. (2015) The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell 28, 515–528 10.1016/j.ccell.2015.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Wang Y., and Liang H. (2018) When microRNAs meet RNA editing in cancer: a nucleotide change can make a difference. Bioessays 40, 10.1002/bies.201700188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Wang C., Zou J., Ma X., Wang E., and Peng G. (2017) Mechanisms and implications of ADAR-mediated RNA editing in cancer. Cancer Lett. 411, 27–34 10.1016/j.canlet.2017.09.036 [DOI] [PubMed] [Google Scholar]
- 136. Jiang Q., Crews L. A., Holm F., and Jamieson C. H. M. (2017) RNA editing-dependent epitranscriptome diversity in cancer stem cells. Nat. Rev. Cancer 17, 381–392 10.1038/nrc.2017.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Fritzell K., Xu L. D., Lagergren J., and Öhman M. (2018) ADARs and editing: the role of A-to-I RNA modification in cancer progression. Semin. Cell Dev. Biol. 79, 123–130 10.1016/j.semcdb.2017.11.018 [DOI] [PubMed] [Google Scholar]
- 138. Rice G. I., Kasher P. R., Forte G. M., Mannion N. M., Greenwood S. M., Szynkiewicz M., Dickerson J. E., Bhaskar S. S., Zampini M., Briggs T. A., Jenkinson E. M., Bacino C. A., Battini R., Bertini E., Brogan P. A., et al. (2012) Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat. Genet. 44, 1243–1248 10.1038/ng.2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Gallo A., Vukic D., Michalík D., O'Connell M. A., and Keegan L. P. (2017) ADAR RNA editing in human disease; more to it than meets the I. Hum. Genet. 136, 1265–1278 10.1007/s00439-017-1837-0 [DOI] [PubMed] [Google Scholar]
- 140. Lazzari E., Mondala P. K., Santos N. D., Miller A. C., Pineda G., Jiang Q., Leu H., Ali S. A., Ganesan A. P., Wu C. N., Costello C., Minden M., Chiaramonte R., Stewart A. K., Crews L. A., and Jamieson C. H. M. (2017) Alu-dependent RNA editing of GL:I1 promotes malignant regeneration in multiple myeloma. Nat. Commun. 8, 1922 10.1038/s41467-017-01890-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Peng X., Xu X., Wang Y., Hawke D. H., Yu S., Han L., Zhou Z., Mojumdar K., Jeong K. J., Labrie M., Tsang Y. H., Zhang M., Lu Y., Hwu P., Scott K. L., Liang H., and Mills G. B. (2018) A-to-I RNA editing contributes to proteomic diversity in cancer. Cancer Cell 33, 817–828.e7 10.1016/j.ccell.2018.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Nemlich Y., Baruch E. N., Besser M. J., Shoshan E., Bar-Eli M., Anafi L., Barshack I., Schachter J., Ortenberg R., and Markel G. (2018) ADAR1-mediated regulation of melanoma invasion. Nat. Commun. 9, 2154 10.1038/s41467-018-04600-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Wang Q., Li X., Qi R., and Billiar T. (2017) RNA editing, ADAR1, and the innate immune response. Genes 8, E41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Montiel-González M. F., Vallecillo-Viejo I. C., and Rosenthal J. J. (2016) An efficient system for selectively altering genetic information within mRNAs. Nucleic Acids Res. 44, e157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Cox D. B. T., Gootenberg J. S., Abudayyeh O. O., Franklin B., Kellner M. J., Joung J., and Zhang F. (2017) RNA editing with CRISPR-Cas13. Science 358, 1019–1027 10.1126/science.aaq0180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Samuel C. E. (2003) RNA editing minireview series. J. Biol. Chem. 278, 1389–1390 10.1074/jbc.R200032200 [DOI] [PubMed] [Google Scholar]