

Abstract
Amyloid precursor protein (APP) is processed along the amyloidogenic pathway by the β-secretase, BACE1, generating β-amyloid (Aβ), or along the nonamyloidogenic pathway by α-secretase, precluding Aβ production. The plasma membrane is considered the major site for α-secretase–mediated APP cleavage, but other cellular locations have not been rigorously investigated. Here, we report that APP is processed by endogenous α-secretase at the trans-Golgi network (TGN) of both transfected HeLa cells and mouse primary neurons. We have previously shown the adaptor protein complex, AP-4, and small G protein ADP-ribosylation factor–like GTPase 5b (Arl5b) are required for efficient post-Golgi transport of APP to endosomes. We found here that AP-4 or Arl5b depletion results in Golgi accumulation of APP and increased secretion of the soluble α-secretase cleavage product sAPPα. Moreover, inhibition of γ-secretase following APP accumulation in the TGN increases the levels of the membrane-bound C-terminal fragments of APP from both α-secretase cleavage (α-CTF, named C83 according to its band size) and BACE1 cleavage (β-CTF/C99). The level of C83 was ∼4 times higher than that of C99, indicating that α-secretase processing is the major pathway and that BACE1 processing is the minor pathway in the TGN. AP-4 silencing in mouse primary neurons also resulted in the accumulation of endogenous APP in the TGN and enhanced α-secretase processing. These findings identify the TGN as a major site for α-secretase processing in HeLa cells and primary neurons and indicate that both APP processing pathways can occur within the TGN compartment along the secretory pathway.
Keywords: β-secretase 1 (BACE1), amyloid precursor protein (APP), membrane trafficking, enzyme processing, protein trafficking (Golgi), neurodegenerative disease, secretion, adaptor protein, secretase, α-secretase, trans-Golgi network
Introduction
Amyloid precursor protein (APP)3 is a membrane protein that undergoes proteolytic processing to produce a variety of soluble secreted products, including the cytotoxic β-amyloid (Aβ) peptide that promotes the pathology of Alzheimer's disease. There are two processing pathways of APP, initiated by α- and β-secretases, resulting in the production of distinct products. Amyloidogenic processing of APP is initiated by the β-secretase, BACE1, to release the luminal or ectodomain of APP (sAPPβ) and is followed by γ-secretase cleavage to release soluble Aβ. There is substantial evidence that sAPPβ and Aβ can be generated in both the endosomal system (1–5) and the secretory pathway (6–8), in particular the TGN (9–11). In the alternative nonamyloidogenic processing pathway, APP is initially cleaved by an α-secretase, which then precludes the formation of the Aβ peptide and amyloid (12). The nonamyloidogenic processing of APP is generally considered to occur predominantly on the plasma membrane due to abundance of α-secretase at this location (13), although the possibility of α-secretase cleavage of APP along the secretory pathway has not been excluded (14). Given that only minor levels of APP appear to reach the PM (11), it is likely that intracellular sites of α-secretase cleavage are important. Defining the sites of action of α-secretase is relevant for understanding the regulation of APP processing by the two pathways and for the potential to divert APP processing along the nonamyloidogenic protective pathway and reduce the level of Aβ produced.
Cleavage of APP by α-secretase occurs at the so-called α-site, a site located within the Aβ domain, in order to generate a membrane-bound C-terminal fragment (α-CTF/C83) and a large secretory sAPPα luminal or ectodomain (12, 15). The subsequent cleavage of C83 by γ-secretase generates a truncated Aβ peptide (p3) and APP intracellular domain (16). A disintegrin and metalloprotease 10 (ADAM10) is considered the major α-secretase that regulates the constitutive cleavage of APP (15, 17). Newly synthesized ADAM10 is transported along the secretory pathway to the PM (18). Like other metalloproteinases, ADAM10 is synthesized as an inactive preprotein and then is activated following cleavage by a convertase within the secretory pathway, most likely in the late Golgi and/or during transport from the Golgi to the cell surface (19, 20).
It is not known whether α-secretase cleavage of APP can occur at the TGN or is restricted to post-Golgi compartments. The conclusion that the cell surface is the major site for APP processing by α-secretase is based on early studies, which have limitations. Parvathy et al. (21) showed no detectable sAPPα when transport along the secretory pathway was blocked at 20 °C, a nonpermissive temperature for the secretory pathway, or in the presence of a biotinylated zinc metalloproteinase inhibitor (AMG2380), considered to inhibit only α-secretase at the cell surface (22). However, there are potential complications associated with these studies. First, it remains unclear whether α-secretase activity had been compromised at 20 °C, and second, as biotin has been shown to diffuse freely across the PM and into intracellular compartments (23, 24), it is likely that biotinylated AMG2380 may also diffuse across the PM and inhibit intracellular α-secretase activity. In another study, enhanced secretion of APP species was detected when the endocytosis of APP was inhibited by mutating a cytosolic sorting signal (YENPTY) of APP (13). However, the sorting motif (YENPTY) of APP is also required for the regulation of intracellular trafficking of APP (25–32) in addition to endocytosis (33, 34), and mutation of this APP motif could alter the anterograde trafficking pathway of newly synthesized APP. Finally, recent studies have demonstrated that newly synthesized APP is diverted at the TGN and trafficked directly to early endosomes, with only low levels detected at the cell surface (11). Given these issues, the potential importance of intracellular sites of α-secretase cleavage of APP remains an open question and warrants further investigation.
Previously, we demonstrated that BACE1 cleavage of APP can occur in the TGN (11), and here we have assessed whether α-secretase processing of APP can also take place in the late Golgi. The TGN adaptor complex, AP-4, is required for exit of APP from the Golgi, and AP-4 silencing resulted in APP accumulating at the Golgi in cultured cells (6, 11). Our previous studies also showed that the small G protein, Arl5b, is required for recruitment of AP-4 to the Golgi (11). There is a block in post-Golgi cargo transport of APP upon silencing of either AP-4 or Arl5b, which is selective as BACE1 is not affected by either AP-4 or Arl5b depletion (11). Here, we have shown that retarding the exit of APP from the Golgi resulted in enhanced processing of APP by α-secretase in both transfected HeLa cells and primary mouse cortical neurons demonstrating that both amyloidogenic and nonamyloidogenic APP-processing pathways can occur in the late Golgi.
Results
Detection of secreted sAPPα ectodomain in APP stable HeLa cells
To assess the intracellular processing of APP by α-secretase, we used a HeLa cell line stably expressing human APP695 (HeLa–APP695WT) (11). The APP695 isoform was selected as this is the isoform predominantly expressed in the brain (35). The HeLa–APP695WT cell line expresses APP at only modest levels, and moreover, the expression level of APP does not saturate the trafficking machinery along the secretory pathway. To detect APP and secreted sAPPα in conditioned medium from HeLa–APP695WT, we used two commercial monoclonal antibodies to APP, namely W0-2 and Y188 (Fig. 1A). The monoclonal mouse anti-Aβ (W0-2), raised against residues 4–10 of the Aβ domain, recognizes full-length APP and the processed products of APP, including sAPPα, Aβ peptide, and β-CTF/C99 (Fig. 1A and Table 1). However, the monoclonal rabbit anti-APP (Y188), raised against residues 682–687 (YENPTY motif) of APP, recognizes full-length APP and the C-terminal fragments of APP, β-CTF/C99, and α-CTF/C83 (Fig. 1A and Table 1). When HeLa–APP695WT cell extracts were probed with antibody W0-2, two bands of ∼89 and 98 kDa were detected either in the presence or absence of the γ-secretase inhibitor DAPT (Fig. 1B). The upper band detected in the cell extract likely corresponds in size to the mature glycosylated full-length APP (98 kDa) and the lower band (89 kDa) to either the immature (i.e. biosynthetic precursor) and/or processed APP (6). As two bands of similar size were also detected in the cell extracts with Y188, they probably represent the mature and immature (precursor) full-length APP. A single band of ∼89 kDa was detected in the overnight conditioned medium with W0-2. The single band in the conditioned medium could represent either sAPPα or immature APP as the molecular sizes of sAPPα, and the immature full-length APPs are similar (Table 1 and Fig. 1B); in contrast, secreted sAPPβ would be not detected by antibody W0-2 as it lacks the epitope derived from the Aβ domain. No bands were detected in the conditioned medium with the Y188 antibody (Fig. 1B), suggesting that the ∼89-kDa band detected by W0-2 in the conditioned medium represents sAPPα. However, antibody W0-2 would also detect the secreted product of a minor and alternative BACE1 cleavage site of APP (β′-site) at residue 10 of the Aβ domain to release sAPPβ′. To distinguish between these products, we inhibited α-secretase activity in HeLa–APP695WT cells using the metalloprotease inhibitor, TAPI-1 (Fig. 1, C and D). There was a 78.0 ± 7.5% reduction in the level of the 89-kDa APP product in the 6-h culture medium of TAPI-1–treated cells, confirming that the majority of the APP ectodomain is sAPPα. As analyses in the subsequent experiments have analyzed the effect of intracellular processing in the presence of the γ-secretase inhibitor, DAPT, we initially assessed whether DAPT affects the levels of secreted sAPPα. There were no obvious changes in the levels of sAPPα in the presence of DAPT (Fig. 1B), suggesting that the treatment of cells with DAPT is unlikely to affect α-secretase activity.
Figure 1.

Detection of sAPPα in conditioned medium and CTFs in cell extracts. A, schematic of APP695 and epitope sequences of commercial antibodies. Monoclonal mouse anti-Aβ (clone W0-2; red) was raised against residues 4–10 of the Aβ domain. W0-2 recognizes full-length APP, sAPPα, Aβ peptide, and β-CTF/C99 fragments of APP. Monoclonal rabbit anti-APP (clone Y188; green) was raised against residues 682-687 (YENPTY motif) of APP695. Y188 recognizes full-length APP, β-CTF/C99, and α-CTF/C83 fragments of APP. B, HeLa cells stably expressing APP695WT (HeLa–APP695WT) were treated with either DMSO (carrier control) (−) or 250 nm γ-secretase inhibitor, DAPT (+), for 16 h. Conditioned medium (CM) and cell extracts (CE) were obtained. CM (10 μl) and CE (20 μg) were analyzed by immunoblotting with W0-2 or Y188 antibodies, using a chemiluminescence detection. The bands that correspond to sAPPα is marked (*). C, HeLa–APP695WT cell monolayers were treated either DMSO (carrier) or with 50 μm TAPI-1 (α-secretase inhibitor) for 6 h. Conditioned medium was collected, and equivalent volumes of samples, based on protein content of extracts of cell monolayers, were analyzed by immunoblotting with W0-2 antibodies. Cell extracts were analyzed by immunoblotting with mouse anti-α-tubulin antibodies. D, bar graph representing the percentage reduction of sAPPα levels in C. Data are represented as the mean ± S.D. of three independent experiments and analyzed by paired, two-tailed Student's t test. ***, p < 0.001. E, HeLa–APP695WT cell monolayers were treated either DMSO (carrier) or 250 nm DAPT in the presence or absence of 2 μm C3 (BACE1 inhibitor) and/or 5 μm TAPI-1 for 16–20 h. CM was analyzed by immunoblotting with W0-2 antibodies. CE were immunoblotted with either mouse anti-α-tubulin antibodies or rabbit anti-APP (Y188) antibodies. F, bar graph representing the percentage reduction of α-CTF/C83 levels in E. Data are represented as the mean ± S.D. of three independent experiments and analyzed by paired, two-tailed Student's t test. ***, p < 0.001.
Table 1.
Detection of APP and APP cleavage products by immunoblotting
| Products | Size | Mouse α-APP (clone W0-2) | Rabbit α-APP (clone Y188) |
|---|---|---|---|
| kDa | |||
| Mature glycosylated APP695 | ∼98 | ✓ | ✓ |
| Immature glycosylated APP695 | ∼77 | ✓ | ✓ |
| sAPPα (residues 1–612 of APP695) | ∼89 | ✓ | Χ |
| sAPPβ (residues 1–596 of APP695) | ∼87 | Χ | Χ |
| β-CTF/C99 | ∼11 | ✓ | ✓ |
| α-CTF/C83 | ∼9 | Χ | ✓ |
To assess the intracellular levels of the membrane-bound product of α-secretase, namely α-CTF/C83, cell extracts were probed with Y188 antibody; Y188 will detect both α-CTF/C83 and the membrane-bound product of APP cleavage by BACE1, β-CTF/C99 (Table 1 and Fig. 1E). An ∼9-kDa band, the size of C83, was detected in cell extracts without the addition of inhibitors, whereas the addition of the γ-secretase inhibitor, DAPT, resulted in an increase in the level of the 9-kDa component as well as an 11-kDa component, the size of C99, at a considerably lower level. To confirm the identity of the 9- and 11-kDa components as α-CTF/C83 and β-CTF/C99, respectively, cells were treated with DAPT and the BACE1 inhibitor C3, or with DAPT, C3, and TAPI-1. In the presence of C3, there was a reduction in the 11-kDa component but not the 9-kDa component, whereas both components were dramatically reduced in the presence of both C3 and TAPI-1. Quantitation, by normalizing for tubulin levels in the cell extracts, revealed that TAPI-1 reduced the level of the 9-kDa component by ∼80% (Fig. 1F), a reduction similar to that of the secreted APPα product (Fig. 1D). These data confirm that the larger 11-kDa component of the cell extracts is β-CTF/C99 and the smaller 9-kDa component is α-CTF/C83.
Production of sAPPα in the secretory pathway
We have previously demonstrated that depletion of either AP-4 or Arl5b accumulated APP in the TGN of HeLa–APP695WT cells and resulted in the increased processing of APP by BACE1 (11). To determine whether APP could also be processed by endogenous α-secretase in the TGN, we depleted the ϵ subunit of AP-4 in HeLa–APP695WT cells using two independent AP-4ϵ siRNAs (Fig. 2). As expected, there was a dramatic increase in the level of intracellular APP that colocalized with the TGN marker, GCC88 (Fig. 2A). Quantitation of total APP pixels that overlapped with GCC88 showed an increase in colocalization of APP with GCC88 from 8.55 ± 2.27% in control cells to 24.10 ± 3.32 and 29.63 ± 2.93% in AP-4ϵ siRNA-1 and siRNA-2 treated cells, respectively (Fig. 2B). Two independent AP-4ϵ siRNAs demonstrate that the accumulation of APP in the TGN was specifically associated with AP-4 depletion. Given the higher level of APP in the TGN, AP-4ϵ siRNA-2 was chosen for the subsequent experiments. AP-4ϵ siRNA-2 showed >90% reduction in levels of AP-4ϵ, whereas AP-1, another adaptor of the TGN, was not affected in RNAi-treated cells (Fig. 3A). Analyses of the overnight conditioned medium for sAPPα by blotting with antibody W0-2 revealed a 1.37-fold increase in the level of secreted sAPPα in medium from cells treated with AP-4ϵ siRNA compared with the control siRNA-treated cells (Fig. 3, A and B). Arl5b, which is required for the TGN recruitment of AP-4 (11), was also silenced using conditions previously established to reduce Arl5b mRNA levels by 90% (36). Antibodies are not available to Arl5b, and we established in these experiments that Arl5b was effectively silenced in GFP–Arl5b stable HeLa cells (Fig. 3A). Treatment of HeLa–APP695WT cells with Arl5b siRNA also resulted in an increase in the level of secreted sAPPsα by 1.65-fold when compared with control conditions (Fig. 3, A and B). These findings demonstrate that the accumulation of APP in the TGN enhances APP processing by α-secretase and secretion of sAPPα.
Figure 2.
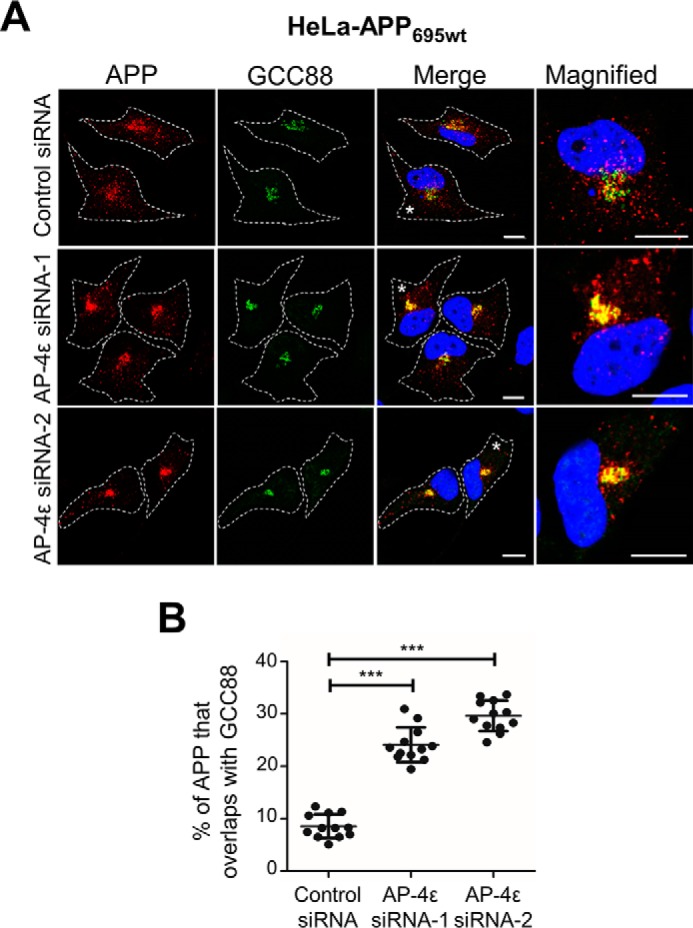
AP-4ϵ depletion results in APP accumulation in the TGN. A, HeLa–APP695WT cells were transfected with either control siRNA, AP-4ϵ siRNA-1, or AP-4ϵ siRNA-2, as indicated, for 72 h. Monolayers were fixed and stained with mouse anti-human APP antibodies (clone NAB228) (red), rabbit anti-GCC88 antibodies (green) as a TGN marker, and DAPI (blue). Higher magnification of the merged images of the cells marked * are also shown. Bars represent 10 μm. B, percentage of APP at the TGN was calculated as a percentage of total APP pixels that overlapped with GCC88 using the OBCOL plugin on ImageJ. Data are represented as the mean ± S.D. of three independent experiments (n = 12) and analyzed by unpaired, two-tailed Student's t test. ***, p < 0.001.
Figure 3.

Generation of sAPPα along the secretory pathway. A, HeLa–APP695WT cells or HeLa cells stably expressing constitutive active Arl5b(Q70L)-GFP were transfected with either control siRNA, AP-4ϵ siRNA, or Arl5b siRNA, as indicated, for 72 h and treated with either DMSO (carrier) or 250 nm DAPT in the final 16 h of siRNA transfection. Conditioned medium (10 μl) and cell extracts (20 μg) were subjected to SDS-PAGE and proteins transferred to a PVDF membrane. Conditional medium samples were probed with W0-2 antibodies and cell extract samples probed with either mouse anti-AP-4ϵ, mouse anti-AP1γ, mouse anti-GFP, or mouse anti-α-tubulin antibodies as indicated. B, bar graph representing fold change of sAPPα levels in AP-4ϵ siRNA or Arl5b siRNA-treated cells compared with control siRNA. Levels of sAPPα were normalized to total protein levels in cell extracts. Data are represented as the mean ± S.D. of three independent experiments and analyzed by paired, two-tailed Student's t test. ***, p < 0.001.
Nonamyloidogenic and amyloidogenic processing of APP in the TGN
Next, we compared the nonamyloidogenic and amyloidogenic processing of APP in the TGN following the accumulation of APP in this compartment. Upon depletion of AP-4ϵ in HeLa–APP695WT cells, there was a 1.40–1.50-fold increase in the level of full-length APP compared with control conditions when immunoblots were probed with either antibody Y188 or W0-2 (Fig. 4, A and B). qPCR analyses revealed that APP mRNA was not increased in AP-4ϵ siRNA–treated cells, rather APP mRNA was reduced by 30% (Fig. 4C); hence, the accumulation of APP in AP-4ϵ siRNA–treated cells was not due to an increase in APP expression, rather it is likely that the increase is due to a reduced rate of turnover. However, there was no obvious alteration in the level of full-length APP in the presence of the γ-secretase inhibitor (DAPT) or the BACE1 inhibitor, C3, in AP-4ϵ siRNA and control siRNA-treated samples (Fig. 4A).
Figure 4.

AP-4ϵ depletion results in accumulation of APP in cells. A, HeLa–APP695WT cells were transfected with either control siRNA or AP-4ϵ siRNA for 72 h and treated with either DMSO (carrier), 250 nm DAPT, 2 μm C3 (BACE1 inhibitor), or 250 nm DAPT + 2 μm C3 in the last 16 h of siRNA transfection. Cell extracts (20 μg) were analyzed by immunoblotting with either rabbit anti-APP (Y188), mouse anti-APP (W0-2), AP-4ϵ, or α-tubulin antibodies, as indicated. B, bar graph representing fold change of full-length APP levels in AP-4ϵ siRNA-treated cells compared with control siRNA. Levels of APP were normalized to α-tubulin. Data are represented as the mean ± S.D. of three independent experiments and analyzed by paired, two-tailed Student's t test. **, p < 0.01. C, quantitative PCR (qRT-PCR) analysis of human APP. Total RNA was purified from control siRNA and AP-4ϵ siRNA-treated cells, converted to cDNA, and singleplex reactions performed, in quadruplicate, for APP and the internal reference gene, GAPDH. qRT-PCR was performed and analyzed using the comparative ΔΔCT method. Relative quantification of mRNA levels used control siRNA cells as normalizer and GAPDH as the internal reference gene. Data are represented as the mean ± S.E. of two independent experiments.
To assess the intracellular levels of α-CTF/C83 and β-CTF/C99, the membrane-bound products of α-secretase and BACE1, respectively, cells were treated with DAPT and cell extracts probed with Y188 and W0-2 antibodies. Y188 will detect both β-CTF/C99 and α-CTF/C83, and W0-2 will detect only β-CTF/C99 (Table 1 and Fig. 4A). In the absence of DAPT and C3 inhibitors, the 9-kDa α-CTF/C83 was detected with the Y188 antibody in both control siRNA- and AP-4ϵ siRNA-treated cells (Fig. 4A). A slightly higher level of the α-CTF/C83 component was detected in AP-4ϵ–depleted cells compared with control conditions (Figs. 4A and 5A). In the presence of DAPT (for 16 h), which protects CTFs from further processing, a 2.05-fold increase in the level of α-CTF/C83 was detected in AP-4ϵ–depleted cells compared with control conditions (Figs. 4A and 5B). These results demonstrate that the C83 α-secretase product is rapidly cleaved by γ-secretase.
Figure 5.

Nonamyloidogenic processing of APP at the Golgi. A–D, quantitation of α-CTF/C83 levels from Fig. 4A. Shown are bar graphs representing fold change of α-CTF/C83 levels in AP-4ϵ siRNA–treated cells compared with control siRNA, under various treatment conditions as indicated. Levels of α-CTF/C83 were normalized to α-tubulin. Data are represented as the mean ± S.D. of three independent experiments and analyzed by paired two-tailed Student's t test. ***, p < 0.001.
In addition, a component of ∼11 kDa was detected with both Y188 and W0-2 antibodies, consistent with the identity of β-CTF/C99. β-CTF/C99 was not detected in control siRNA-treated cells and only at very low levels in cells treated with AP-4ϵ siRNA in the absence of DAPT (Fig. 4A). In the presence of DAPT, both Y188 and W0-2 antibodies revealed a 2.60–2.80-fold increase in the level of intracellular β-CTF/C99 in AP-4ϵ–depleted cells compared with control conditions (Fig. 4A). Notably, when γ-secretase is inhibited by DAPT, α-CTF/C83 levels were considerably higher than β-CTF/C99, indicating that α-secretase processing of APP was the major processing pathway.
Cells were also treated with the BACE1 inhibitor C3 and/or DAPT. As expected, in the presence of C3, with or without DAPT, β-CTF/C99 was not detected by either Y188 or W0-2 antibody even in cells depleted of AP-4ϵ (Fig. 4A). This observation further verifies the identity of the 11-kDa component as β-CTF/C99, and the ∼9-kDa component detected by the Y188 antibody, and not by W0-2, as α-CTF/C83. In the presence of C3 and DAPT, a 2.44-fold increase in the level of α-CTF/C83 was detected in AP-4ϵ–depleted cells compared with control conditions (Figs. 4A and 5D), again demonstrating that APP is processed by α-secretase in the TGN. Depletion of Arl5b in HeLa–APP695WT cells also showed similar increases in β-CTF/C99 and α-CTF/C83 (Fig. S1), as for AP-4ϵ–depleted cells.
We also analyzed the level of sAPPα in AP-4ϵ–depleted cells treated with DAPT and C3. As expected from Fig. 3, increased levels (>2-fold) of 89-kDa sAPP were detected in the culture supernatant following AP-4 knockdown of HeLa–APP695WT cells (Fig. 6A). The absence of detectable α-tubulin in the conditioned media indicates the 89-kDa protein is a secreted product and is not derived from cell lysis (Fig. 6, A and B). Similar levels of 89-kDa sAPPα were detected in the culture supernatant regardless of C3 treatment (Fig. 6A), indicating that the inhibition of BACE1 does not enhance α-secretase processing. In contrast, treatment of AP-4 knockdown cells with TAPI-1 dramatically reduced the level of the 89-kDa secreted product by 75–80% (Fig. 6B), confirming the identity of the 89-kDa sAPP from AP-4ϵ–depleted cells as sAPPα.
Figure 6.

Effect of α-secretase and γ-secretase inhibitors on secretion of the luminal domain of APP. A and B, HeLa–APP695WT cells were transfected with either control siRNA or AP-4ϵ siRNA for 72 h. Cells were also treated with either DMSO carrier (−), 250 nm DAPT (γ-secretase inhibitor), 2 μm C3 (BACE1 inhibitor), or 2 μm C3 and 250 nm DAPT (A) or 50 μm TAPI-1 (α-secretase inhibitor) or 50 μm TAPI-1 and 250 nm DAPT (B) in the last 16–20 h of siRNA transfection. Conditioned medium was collected, and the equivalent volume of samples based on protein content of extracts of cell monolayers were analyzed by immunoblotting with W0-2 antibodies or mouse anti-α-tubulin antibodies. CE were analyzed by immunoblotting with mouse anti-α-tubulin antibodies (the CM collected for analysis in A was carried out in conjunction with the experiment as Fig. 4A; therefore, the immunoblot for α-tubulin in the CE in Fig. 6A is the same as Fig. 4A).
Enhanced nonamyloidogenic processing of endogenous APP by AP-4 knockdown in neurons
To determine whether α-secretase processing occurs in the TGN of neurons, we initially assessed whether silencing AP-4 in primary mouse neurons resulted in retention of APP in the TGN. To silence AP-4 in primary mouse cortical neurons, neurons were transduced with AP-4ϵ shRNA recombinant lentivirus, and transduced neurons were detected by GFP expression. GFP-positive neurons had a dramatically reduced staining of AP-4ϵ, as detected by confocal microscopy (Fig. 7A). In addition, staining for endogenous APP showed an increase in APP that colocalized with the TGN marker, p230/golgin-245 (Fig. 7B). Quantitative analyses, using Manders' coefficient, revealed a significant increase in APP colocalization with the TGN marker, p230/golgin245, in AP-4–depleted neurons (Fig. 7C). Hence, depletion of AP-4 in primary neurons results in an accumulation of endogenous APP in the TGN.
Figure 7.
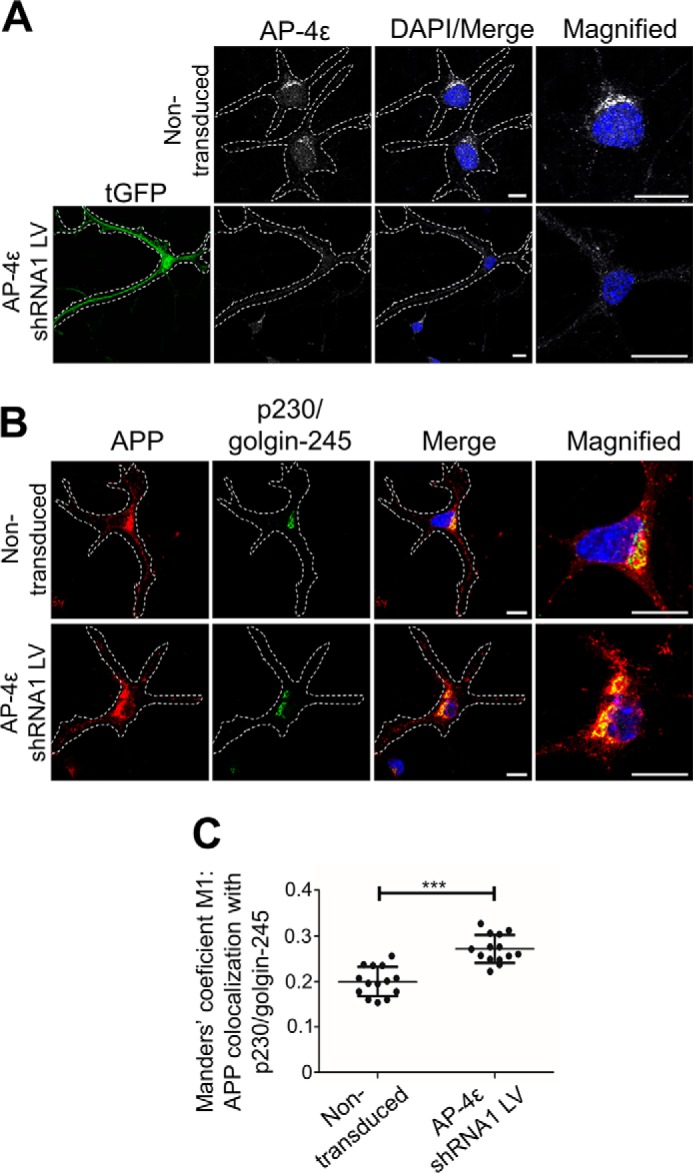
Depletion of AP-4ϵ in mouse primary cortical neurons results in accumulation of APP in the TGN and increased APP processing. A and B, transduction of E16 primary mouse cortical neurons at DIV 3 with AP-4ϵ–shRNA lentivirus for 96 h. Neurons were fixed at DIV 7 and stained with either mouse anti-AP-4ϵ (gray) (A) or rabbit anti-APP (Y188; red) (B) and the TGN marker, human anti-p230/golgin-245 (far-red converted to green), antibodies overnight at 4 °C. Nuclei was stained with DAPI (blue). Bar represents 10 μm. C, VolocityTM software was used to calculate Manders' coefficient M1 values of APP colocalization with the TGN marker, p230/golgin-245. Data are represented as the mean ± S.D. of three independent experiments (n = 14) and analyzed by unpaired, two-tailed Student's t test. ***, p < 0.001.
To determine whether AP-4 increased the level of either α-secretase or BACE1 processing, we first assessed the level of 11-kDa β-CTF/C99 and 9-kDa α-CTF/C83 in neurons with and without DAPT. As for HeLa–APP695WT cells, 11 kDa (marked *) and 9 kDa (marked **), components were detected in the extracts of primary mouse neurons with the Y188 antibody, both of which increased in the presence of the γ-secretase inhibitor DAPT (Fig. 8A), demonstrating that the C-terminal fragments of APP are rapidly processed by γ-secretase in primary neurons as for HeLa cells. In the presence of the C3 inhibitor, there was a reduction in the 11-kDa component, whereas both components were dramatically reduced in the presence of both C3 and TAPI-1. These data confirm that the larger 11-kDa component of neuron cell extracts recognized by the Y188 antibody is β-CTF/C99, and the smaller 9-kDa component is α-CTF/C83.
Figure 8.

Depletion of AP-4ϵ in mouse primary cortical neurons results in increased APP processing. A, E16 primary mouse cortical neurons were treated either DMSO (carrier) or 2 μm DAPT in the presence or absence of 2 μm C3 (BACE1 inhibitor) and/or 20 μm TAPI-1 for 16–20 h. Cell extracts were immunoblotted with either mouse anti-α-tubulin antibodies or rabbit anti-APP (Y188) antibodies to C99*/C83**. B, E16 primary mouse cortical neurons were transduced at DIV 3 with either AP-4ϵ shRNA1 or AP-4ϵ shRNA2 lentivirus (LV) for 96 h and treated with DMSO (carrier control) (−) or 2 μm DAPT in the last 16–20 h of transduction. Neurons were lysed in RIPA buffer and cell extracts (10 μg) subjected to SDS-PAGE, as described under “Experimental procedures.” Proteins transferred onto PVDF membrane were probed with mouse antibodies to either AP-4ϵ or α-tubulin. Proteins transferred onto nitrocellulose membrane were probed with rabbit anti-APP (Y188) antibodies to C99*/C83**. C–E, bar graph representing fold change of α-CTF/C83 (C), β-CTF/C99 (D), and full-length APP (E) levels in AP-4ϵ shRNA1 or AP-4ϵ shRNA2 lentiviral transduction compared with nontransduced controls. Levels of α-CTF/C83, β-CTF/C99, and full-length APP were normalized to α-tubulin. Data are represented as the mean ± S.D. of three independent experiments and analyzed by paired, two-tailed Student's t test. *, p < 0.05; **, p < 0.01.
Next, we determined whether depletion of AP-4 increased the level of either α-secretase or BACE1 processing. We used two different shRNA targets to AP-4 and assessed the reduction of AP-4 in the population of recombinant lentivirus–transduced neurons by immunoblotting. AP-4ϵ was depleted by 37–41% in AP-4ϵ shRNA1 and AP-4ϵ shRNA2 lentivirus–transduced neuron population compared with nontransduced neurons (Fig. 8B). The transduction efficiency of the transduced neuronal populations, based on GFP expression, was 30–40%, indicating that the depletion of AP-4 in the transduced cells was >90%. In the presence of DAPT, there was an increase in the level of both β-CTF/C99 and α-CTF/C83 in AP-4ϵ–shRNA lentivirus–transduced neurons compared with nontransduced neurons (Fig. 8, B–D). α-CTF/C83 was increased by 1.93- and 2.17-fold, and β-CTF/C99 was increased by 1.65- and 1.61-fold in AP-4ϵ shRNA1 and AP-4ϵ shRNA2 lentivirus–transduced neuron populations, respectively, compared with nontransduced neurons (Fig. 8, B–D). In addition, we also determined the increase in total full-length APP in AP-4–depleted neurons (Fig. 8, B, and E). There is a trend for an increase in the total level of APP (122.3 ± 10.0% for shRNA1 and 132.7 ± 17.6% for shRNA2), although not statistically significant from the control sample based on a paired t test (p = 0.061 and p = 0.094, respectively). Nonetheless, given that the transduction efficiency was 30–40% in these neuronal populations, the data are consistent with the findings in HeLa–APP695WT cells depleted of AP-4. Taken together, AP-4 depletion retains APP in the TGN and results in an increased level of both β-CTF/C99 and α-CTF/C83, demonstrating that AP-4 depletion enhances intracellular APP processing by both BACE1 and α-secretase, respectively.
Discussion
Under physiological conditions, the nonamyloidogenic and amyloidogenic processing pathways of APP are considered to represent the major and minor pathway in cells, respectively (6, 37). The cleavage and shedding of the luminal, or ecto, domain of APP by the metalloprotease α-secretase is a key processing event, as it is considered to minimize the generation of the pathogenic Aβ peptides associated with Alzheimer's disease (38, 39). In addition, soluble sAPPα is known to have neurotrophic (40) and neuroprotective (41–43) properties. APP cleavage by membrane-bound α-secretase can occur at the plasma membrane (13, 44); however, other cellular location(s) of α-secretase cleavage remain poorly defined. An important unresolved question is whether α-secretase can cleave APP along the anterograde transport pathway. Our findings demonstrate that α-secretase cleavage of APP occurs in the TGN; moreover, the TGN may represent a major site for nonamyloidogenic processing. This conclusion is based on the following observations presented in the paper. 1) Depletion of AP-4 or Arl5b retarded the exit of APP from the TGN, resulting in APP accumulation in the TGN, and dramatically enhanced the cleavage of APP by endogenous α-secretase, as well as BACE1, in stable HeLa cells expressing APP695WT and primary neurons. 2) Depletion of AP-4 or Arl5b resulted in an increase in the secretion of soluble luminal domain of APPα in stable HeLa cells. 3) Depletion of AP-4 also resulted in the accumulation of endogenous APP in the TGN of primary mouse cortical neurons and enhanced α-secretase processing of APP in neurons. 4) Overall, a 1.5–3.0-fold increase in APP at the TGN resulted in an increase in the α-secretase APP product, α-CTF/C83, in the cell by >200% in both stable HeLa cells and primary neurons, indicating that the TGN represents a major site for α-secretase processing.
The use of the two anti-APP antibodies, Y188 and W0-2, together with inhibitors of the three secretases allowed the identification of sAPPα in the conditioned medium, and α-CTF/C83 and β-CTF/C99 in cell lysates, based on immunoblotting analyses. sAPPα in the conditioned medium is recognized by the W0-2 antibody, whereas sAPPβ is not detected by antibody W0-2 as it lacks the epitope. However, an alternative minor BACE1 cleavage site of APP, the β′-site at residue 10 within the Aβ domain (45), would result in the release of the product sAPPβ′ and would be recognized by the W0-2 antibody. We demonstrated that most if not all the secreted 98-kDa ectodomain of APP in the culture medium represents sAPPα, as the BACE1 inhibitor C3 did not affect the level of sAPP; moreover, the α-secretase inhibitor, TAPI, reduced the levels of sAPP by 78–85%. Using the γ-secretase inhibitor (DAPT) and/or BACE1 inhibitor (C3), we have also verified the identity of the C-terminal fragments of secretase cleavage of APP as α-CTF/C83 and β-CTF/C99 by immunoblotting analyses with the two antibodies, Y188 and W0-2. The C83 product was unaffected by treatment with the BACE1 inhibitor, whereas the C99 product was sensitive.
Treatment with TAPI inhibited the majority of the α-secretase cleavage of APP. There are three members of the ADAM family, namely ADAM9 (46), ADAM10 (20), and ADAM17 (TACE) (47), that have been reported to exhibit α-secretase activity. Moreover, ADAM10, but not ADAM9 and ADAM17, has been identified as the physiologically essential α-secretase that regulates the constitutive cleavage of APP in primary mouse neurons (15, 17).
In contrast to a number of earlier studies, which have proposed that APP is processed by α-secretase mainly at the cell surface (13), we have shown that APP can also be processed by α-secretase in the TGN. Depletion of AP-4 and Arl5b allows the selective block of cargo transport from the TGN to endosomes. We and others have shown that post-Golgi transport of APP in HeLa cells depends on AP-4 and Arl5b (6, 11). The block in post-Golgi cargo transport of APP is selective as BACE1 is not affected by either AP-4 or Arl5b depletion (11). Upon depletion of AP-4 in HeLa cells, there was increased processing of APP by α-secretase as demonstrated by an increased level of secreted sAPPα and, in the presence of DAPT, a 2-fold increase in intracellular α-CTF/C83, compared with control conditions. In neurons there was a similar increase, given the transduction efficiency of 30–40%. These findings demonstrate that the cleavage of APP by endogenous α-secretase is enhanced when the Golgi export of APP was impaired. Depletion of AP-4 also resulted in increased cleavage of APP by BACE1; however, the cleavage of APP by α-secretase in the TGN was four times higher than BACE1. These results indicate that the nonamyloidogenic processing of APP represents the major pathway and the amyloidogenic processing the minor pathway in this compartment. In addition, as only low levels of CTFs were detected in either untreated cells or in AP-4–depleted cells in the absence of DAPT, these findings indicate that γ-secretase rapidly processes α-CTF/C83 and β-CTF/C99. As the CTFs contain the cytoplasmic tail of APP with the motifs for binding AP-4, the post-Golgi transport of these C-terminal membrane fragments are also likely to be retarded in AP-4–depleted cells; hence, the rapid processing by γ-secretase is also probably occurring in the TGN. Rapid processing of C83 by γ-secretase has been reported previously (15) and is consistent with the known intracellular location of γ-secretases (7, 48, 49). Given that the increase in APP levels in the Golgi parallels the increase in α-secretase processing detected in the entire cell, and that the ratio of α-secretase processing and BACE1 processing remains unchanged following AP-4/Arl5b depletion, our findings indicate that the Golgi is likely to represent a major site under physiological conditions.
A key finding in our study is that the TGN adaptor, AP-4, plays an important role in the post-Golgi export of APP in primary neurons. To date, the impact of AP-4 on post-Golgi trafficking of APP has only been assessed in nonpolarized HeLa cells (6, 11). Our finding that AP-4 is also relevant to post-Golgi trafficking of APP in primary neurons is an important verification of the previous work in non-neuronal cells and has broader impact to the relevance of AP-4 mutations in neurological diseases and disorders in mice and humans (50–52).
Significantly, primary cortical neurons showed enhanced levels of α-secretase processing after AP-4 depletion, with results very similar to the HeLa–APP695WT stable cell line. The finding that α-secretase processing of endogenous APP in mouse neurons also occurs in the TGN, with a similar ratio of α-secretase processing and BACE1 processing as for HeLa cells, supports the relevance of our findings using HeLa–APP695WT–transfected cells and also indicates shared characteristics of the TGN between primary neurons and cultured HeLa cells.
A previous study also indicated that α-secretase can cleave APP in the TGN by using an APP construct with the cytoplasmic domain of furin replacing the APP tail (53). This APP/furin construct is concentrated in the Golgi under steady-state conditions (53) and resulted in enhanced α-secretase processing compared with WT APP. However, a complication of this earlier study, which was not addressed, is that the sorting motifs in the cytoplasmic domain of furin directs recycling between the plasma membrane and the TGN via the late endosomes (54); therefore, the APP/furin hybrid construct will be recycling between the Golgi and the plasma membrane with a different intracellular itinerary compared with WT APP. Hence, it was not clear in this earlier study (53) whether the increased α-secretase processing occurred in the Golgi or elsewhere in the intracellular recycling pathway. Our approach overcomes the limitation of the previous study by modulating the trafficking machinery and by characterizing the processing of endogenous APP in primary cells.
The majority of APP is processed by the α-secretase pathway (6, 37). It is of interest that the balance of α-secretase and BACE1 cleavage of APP is similar in untreated and AP-4–depleted cells, indicating that the ratio of α-secretase and BACE1 processing remains unchanged. This finding suggests that there is a tight regulation in APP processing along the two pathways, even within the one compartment. Of relevance to this observation is that the TGN is known to be organized into distinct membrane domains (55, 56). Perhaps the different secretases and APP are segregated into distinct domains within the TGN, which in turn would dictate the level of physical proximity between the enzymes and substrate and thereby regulate the cleavage events. A consequence of this scenario is that any perturbation of the partitioning of the individual membrane proteins, or of the subdomain organization of the TGN, would then influence the relative levels of processing by the α- and β-secretases.
A potential caveat to the interpretation of the data presented in this study is that depletion of the TGN-localized AP-4 may have disrupted the distribution of α-secretase, ADAM10, thereby enhancing the cleavage of APP. However, we consider this unlikely as AP-4 is an adaptor that selectively transports cargo to the early endosomes and is not required for transport of cargo directly to the cell surface (6, 57, 58). Of relevance is that α-secretases are most likely transported directly from the Golgi to the cell surface (39).
In conclusion, our findings demonstrate that α-secretase can process APP in the TGN, and the accumulation of APP in this Golgi compartment enhances processing of APP along both the amyloidogenic and nonamyloidogenic pathways.
Experimental procedures
Plasmids and antibodies
pGIPZ-shRNA lentiviral constructs against mouse AP-4ϵ (V2LMM_89028 and V2LMM_89029) were purchased from Dharmacon, GE Healthcare. pCMV-VSV-G (Addgene plasmid no. 8454) and psPAX2 (Addgene plasmid no. 12260) were kind gifts from Bob Weinberg (59) and Didier Trono, respectively. Mouse monoclonal antibody to AP-1γ (clone 88, no. 610385) and AP-4ϵ (clone 32, no. 612019) was purchased from BD Biosciences (New South Wales, Australia). Mouse monoclonal antibody to human APP (clone NAB228) was purchased from ThermoFisher Scientific. Mouse monoclonal antibody to α-tubulin (clone DM1A, no. T9026) was bought from Sigma (Merck). Mouse monoclonal antibody to Aβ(4–10) (W0-2, no. MABN10) was from Merck Millipore. Rabbit monoclonal antibody to APP (clone Y188, no. ab32136) was obtained from Abcam (Cambridge, UK). Rabbit polyclonal anti-GCC88 antibodies (60) and human autoantibodies to p230/golgin-245 (61) have been described.
Secondary antibodies used for immunofluorescence were goat anti-mouse IgG–Alexa Fluor 488 nm, goat anti-mouse IgG–Alexa Fluor 568 nm, goat anti-mouse IgG–Alexa Fluor 647 nm, goat anti-rabbit IgG–Alexa Fluor 488 nm, goat anti-rabbit IgG–Alexa Fluor 568 nm, goat anti-rabbit IgG–Alexa Fluor 647 nm, goat anti-rat IgG–Alexa Fluor 568 nm (for rat anti-LAMP1 antibodies), and goat anti-human IgG–Alexa Fluor 647 nm (for human anti-p230/golgin-245 antibodies) were obtained from ThermoFisher Scientific. Horseradish peroxidase (HRP)-conjugated rabbit anti-mouse Ig and HRP-conjugated swine anti-rabbit Ig were bought from DAKO Corp. (Carpentaria, CA).
RNAi
The silencing of Arl5b and AP-4 ϵ-adaptin was conducted using short-interfering RNA (siRNA) duplexes manufactured by Sigma-Proligo (Australia). Cell monolayers were transfected with siRNA using DharmaFECT1 (ThermoFisher Scientific) according to the manufacturer's protocol and incubated for 72 h in a humidified 10% CO2 incubator at 37 °C. Arl5b siRNA (36), AP-4ϵ siRNA-1, and AP-4ϵ siRNA-2 duplexes (11) have been previously described.
Cell culture
Mycoplasma-free and authentic HeLa cells (62) were maintained as semi-confluent monolayers in Dulbecco's modified Eagle's medium (DMEM) (ThermoFisher Scientific, Australia) supplemented with 10% v/v fetal calf serum (FCS) (Gibco®, ThermoFisher Scientific), 2 mm l-glutamine, 100 units/μl penicillin, and 0.1% w/v streptomycin (complete DMEM (C-DMEM)). HEK293T packaging cells were maintained as semi-confluent monolayers in C-DMEM without penicillin and streptomycin. All cells were cultured in a humidified 10% CO2 incubator at 37 °C.
Primary mouse cortical neuronal cultures
All experiments carried out on animals were approved by the Animal Ethics Committee, University of Melbourne (approval number 1212502.1), in accordance with animal ethics guidelines. Pregnant mice (C57BL/6) at gestational days 15–16 were euthanized by CO2 asphyxiation, and the embryos collected by cesarean section. Primary cortical neurons were prepared from the collected embryos, as described previously (63), and resuspended in Neurobasal medium supplemented with 2.5% B-27, 0.25% GlutaMAX, and 100 units/μl penicillin and 0.1% streptomycin (complete NBM) (Life Technologies, Inc.). Cells were plated at a density of 0.6 × 105 and 5 × 105 cells/well in 12- and 6-well plates, respectively. After 24 h, the medium was replaced with fresh complete NBM. Cells were grown for 3 days (DIV 3) before transduction. All experiments conducted on animals were approved by the Animal Ethics Committee, University of Melbourne (Ethics ID: 1613960).
Generation of lentivirus
Lentivirus was generated as described previously (64). HEK293T packaging cells were seeded at a density of 2 × 106 cells (DMEM, 10% v/v FCS, 1% v/v GlutaMAXTM) in 10 cm by 10-cm tissue culture dishes. Prior to transfection, the existing medium was replaced with 6 ml of fresh medium (DMEM, 10% v/v FCS, 1% v/v GlutaMAXTM). For calcium phosphate transfection of each 10 × 10-cm dish, 7.5 μg of pCMV-VSV-G, 3 μg of psPAX2, 10 μg of pGIPZ-shRNA, 250 μl of UltraPureTM H2O (ThermoFisher Scientific), and 250 μl of 0.5 m CaCl2 were prepared in this order (DNA mix). The DNA mix was added dropwise to 500 μl of 2× HEPES-buffered saline, pH 7.0 (VWR, Avantor). The transfection mix was vortexed for 30 s and incubated for 10 min at room temperature. The transfection mix was added to cells and incubated overnight in a humidified 10% CO2 incubator at 37 °C. The medium was then replaced with 6 ml of fresh medium (DMEM, 10% v/v FCS, 1% v/v GlutaMAXTM). Cells were incubated for another 48 h in the 37 °C incubator to allow the production of viral particles. Virus-containing medium was harvested and centrifuged at 3000 × g for 30 min at 4 °C to pellet cell debris, and the viral supernatant was filtered using Steriflip-HV sterile centrifuge tube top filter unit (Merck). PEG-it Virus Precipitation Solution (Integrated Sciences, Australia) was added to the filtered viral medium and incubated overnight at 4 °C. Viral particles were pelleted by centrifugation at 1500 × g for 30 min at 4 °C, and the viral pellet was resuspended in DMEM/NBM supplemented with 10 mm HEPES (Gibco® ThermoFisher Scientific) at 1:100 of its original harvested volume. Concentrated virus was allocated into cryovials, snap-frozen on dry ice, and stored at −80 °C. All lentiviral waste was discarded in sodium hypochlorite.
Transduction of primary mouse cortical neuronal cultures
Primary mouse cortical neurons were seeded and cultured as monolayers in 12-well plates with or without coverslips for lentivirus transduction. Concentrated lentivirus was thawed at room temperature before use. Neurons were transduced at DIV 3 with 10–15 μl/well of AP-4ϵ shRNA or AP-1γ shRNA lentivirus diluted with fresh C-NBM to a final volume of 200 μl/well. Transduced neurons were incubated for 24 h (DIV 4) in a 37 °C incubator with 10% CO2, followed by replacement of lentiviral medium with conditioned C-NBM (half-fresh C-NBM and half-C-NBM recovered from neuronal culture) and incubated for a further 72 h (DIV 7).
Quantitative real-time PCR
Total RNA was purified using Qiagen RNeasy mini kit (Qiagen, Germany) from ∼2 × 106 cells treated with either control siRNA or AP-4ϵ siRNA-1 for 72 h. RNA preparations were treated with RNase-free DNase set (Qiagen) during the purification process as per manufacturer's protocol. RNA samples were stored at −80 °C. The RNA integrity number (RIN) of the RNA samples was determined (65). Briefly, RNA samples were diluted to 250 ng/μl and prepared in RNA ScreenTape sample buffer (Agilent Technologies). The RNA sample mix was loaded on the RNA analysis ScreenTape (Agilent Technologies) and analyzed by the 4200 TapeStation instrument (Agilent Technologies) located at the Department of Anatomy and Neuroscience, University of Melbourne. Total RNA (1 μg) was then converted to cDNA using a SuperScript III First-Strand Synthesis kit (Life Technologies, Inc., and ThermoFisher Scientific) according to manufacturer's protocol. To set up singleplex reactions (20 μl/reaction) in quadruplicate for APP (Hs00169098_m1) and internal reference gene GAPDH (Hs02758991_g1), the cDNA sample (1:20 dilution) was prepared with TaqMan Fast Advanced Master mix (Applied Biosystems and ThermoFisher Scientific) and human-specific TaqMan gene expression assays (Applied Biosystems and ThermoFisher Scientific). qRT-PCR was conducted using QuantStudio 6 qPCR instrument (ThermoFisher Scientific) and analyzed using the comparative ΔΔCT method. Control siRNA cells were used as normalizer, and GAPDH was used as the internal reference gene for the relative quantification of mRNA levels.
γ-Secretase inhibitor (DAPT) treatment
HeLa–APP695WT cells and primary mouse cortical neurons (DIV 6) were seeded and cultured as monolayers. Cells were treated with either 0.01% v/v DMSO (carrier control), 250 nm γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT) (Sigma, Merck) for HeLa–APP695WT cells or 2 μm DAPT for primary mouse cortical neurons. Cells were incubated for 16 h (HeLa cells) or 20 h (neurons) in a 37 °C incubator with 10% CO2.
BACE1 inhibitor (C3) treatment
HeLa–APP695WT cells were seeded and cultured as monolayers in 6-well plates. Cells were treated with either 0.01% v/v of DMSO (carrier control) or 2 μm β-secretase/BACE1 inhibitor C3 (Calbiochem, Merck) and incubated for 16 h in a 37 °C incubator with 10% CO2.
α-Secretase inhibitor (TAPI) treatment
HeLa–APP695WT cells were seeded and cultured as monolayers in 6-well plates. Cells were treated with either 0.01% v/v of DMSO (carrier control) or 5–50 μm metalloprotease inhibitor TAPI-1 (N-(R)-[2-(hydroxyaminocarbonyl)methyl]-4-methylpentanoyl-l-naphthylalanyl-l-alanine, 2-aminoethyl amide) (Calbiochem, Merck) and incubated for either 6 or 20 h in a 37 °C incubator with 10% CO2.
Indirect immunofluorescence
Monolayers of cultured mammalian cells and primary mouse cortical neurons were fixed in 4% paraformaldehyde (PFA) (Wako Pure Chemical Industries, Japan) for 15 min at room temperature, quenched in 50 mm NH4Cl/PBS for 10 min at room temperature, permeabilized with 0.1% v/v Triton X-100/PBS for 4 min at room temperature, and blocked in blocking solution (5% v/v FCS and 0.02% v/v sodium azide, in PBS) for 30 min to reduce nonspecific binding. For AP-4ϵ staining, when required cells were fixed and permeabilized with chilled 100% methanol at −20 °C for 5 min, washed four times in PBS, and blocked in blocking solution for 30 min at room temperature. Cultured cells were stained with primary antibodies diluted in blocking solution for 1 h at room temperature and washed six times with PBS. Neurons were stained overnight at 4 °C with primary antibodies and washed six times with PBS. Cultured cells and neurons were then stained with fluorophore-conjugated secondary antibodies diluted in blocking solution for 30 min at room temperature and washed six times with PBS. Coverslips were washed with distilled water and mounted in Mowiol (10% w/v Hopval 5-88, 25% w/v glycerol, 0.1 m Tris in milli-Q water) on microscope glass slide. Images were acquired sequentially for multicolor imaging on a confocal laser-scanning microscope (Leica TCS SP8 confocal imaging system) using a ×63 1.4 NA HCX PL APO CS oil immersion objective and a Leica HyD photodetector. GFP and Alexa Fluor 488 were excited using the 488-nm line source of an argon laser. Alexa Fluor 568 and Alexa Fluor 647 were excited with the 543- and 633-nm helium–neon (HeNe) laser, respectively. DAPI was excited with a 405-nm UV laser.
Cell extracts
HeLa cells and primary mouse cortical neurons were extracted in radioimmunoprecipitation (RIPA) lysis buffer (50 mm Tris-HCl, pH 7.3, 150 mm NaCl, 0.1 mm EDTA, 1% w/v sodium deoxycholate, 1% v/v Triton X-100, 0.2% w/v NaF, and 100 μm Na3VO4) supplemented with 1× cOmpleteTM Mini Protease Inhibitor Mixture (Roche Applied Science, Sigma, and Merck). Cell lysates were incubated on ice for 10 min followed by centrifugation at 16,000 × g at 40 °C for 10 min. The supernatant was collected and protein concentration determined by the Bradford protein assay (Bio-Rad) using bovine serum albumin (BSA) protein standards (Pierce BSA standard, ThermoFisher Scientific).
β-CTF/C99 and α-CTF/C83 blots
To detect levels of β-CTF/C99 and α-CTF/C83 in HeLa–APP695WT cells, 15 μg of cell extracts were prepared in 2× reducing sample buffer containing 10% β-mercaptoethanol and boiled at 100 °C for 7 min. Samples were resolved on 12% NuPAGETM BisTris SDS-polyacrylamide gel (Invitrogen and ThermoFisher Scientific) at 125 V for ∼2.5 h. Transfer of proteins onto 0.2-μm nitrocellulose membrane (Bio-Rad) was conducted at 400 mA for 1 h on ice. The membrane was incubated in warm PBS for 5 min and blocked with 10% w/v skim milk/PBS Tween 20 for 1 h at room temperature to reduce nonspecific binding. To probe for β-CTF/C99, the membrane was incubated overnight at 4 °C with W0-2 antibodies diluted 1:6000 in PBS Tween 20. To probe for β-CTF/C99 and α-CTF/C83, the membrane was incubated overnight at 4 °C with Y188 antibodies diluted in PBS Tween 20 (1:6000). The membrane was washed three times with PBS Tween 20 at 10 min/wash and incubated with 1:500 diluted secondary antibodies conjugated with HRP for 1 h and washed as above. An ECL Western blotting chemiluminescence detection system (GE Healthcare) was used to detect bound antibodies, and the protein band signals were imaged using the Gel-ProTM Analyzer version 4.5 software (Media Cybernetics, Bethesda, MD). The Gel-ProTM Analyzer software was used to measure the densitometry of the protein bands.
To detect levels of β-CTF/C99 and α-CTF/C83 in primary mouse cortical neurons, 10 μg of cell extracts were prepared as above. Samples were resolved on 12% NuPAGETM BisTris SDS-polyacrylamide gel and proteins transferred onto 0.2-μm nitrocellulose membrane as described above. The membrane was incubated overnight at 4 °C with Y188 antibodies diluted in PBS Tween 20 (1:6000) and washed three times with PBS Tween 20 at 10 min/wash. The membrane was then incubated with 1:500 diluted secondary antibodies conjugated with horseradish peroxidase (HRP) for 1 h and washed as above. Bound antibodies were detected and imaged as described.
sAPPα blots
To measure levels of secreted sAPPα, conditioned medium from HeLa–APP695WT cells was collected and mixed with 2× reducing sample buffer containing 10% β-mercaptoethanol. Samples were boiled at 100 °C for 7 min, and 15 μl of samples were resolved on 4–12% NuPAGETM BisTris SDS-polyacrylamide gel (Invitrogen and ThermoFisher Scientific) at 125 V for ∼2 h. Proteins were transferred onto a 0.2-μm nitrocellulose membrane (Bio-Rad) at 400 mA for 1 h on ice. The membrane was then incubated on warm PBS for 5 min and blocked with 10% w/v skim milk/PBS Tween 20 for 1 h at room temperature. The membrane was incubated overnight at 4 °C with W0-2 antibodies diluted in PBS Tween 20 (1:6000) and washed three times with PBS Tween 20 at 10 min/wash. The membrane was then incubated with 1:500 diluted secondary antibodies conjugated with HRP for 1 h and washed as above. Bound antibodies were detected and imaged as described.
Quantitation of colocalization
Quantitation of the colocalization between APP695WT and endogenous organelle markers in HeLa cells was conducted using an organelle-based colocalization (OBCOL) plugin (66) on FIJI/ImageJ (National Institutes of Health public domain software). Images were thresholded to remove background fluorescence signals, and organelles were segmented into individual structures. The percentage of colocalization of cargo with various organelles was calculated by dividing the total cargo pixels that overlapped with each organelle marker with total cargo pixels within each cell. VolocityTM imaging software (PerkinElmer Life Sciences, UK) was used to calculate Manders' coefficient M1 values (67) to determine the percentage colocalization of the endogenous APP with endogenous organelle markers in primary mouse cortical neurons. All analysis was carried out on the indicated number of cells over three independent experiments.
The generation of graphs and statistical analyses were conducted using Prism (GraphPad Software). Data for fold change of α-CTF/C83 levels and sAPPα levels were plotted as bar graphs and analyzed by paired, two-tailed, Student's t test. Data from quantitation of colocalization were plotted as dot-plots and analyzed by unpaired, two-tailed, Student's t test. A p value of <0.05 (*) was considered significant; a p value of <0.01 (**) was considered highly significant; and a p value of <0.001 (***) was considered very highly significant.
Author contributions
J. Z. A. T. and P. A. G. formal analysis; J. Z. A. T. investigation; J. Z. A. T. methodology; J. Z. A. T. writing-review and editing; P. A. G. conceptualization; P. A. G. funding acquisition; P. A. G. writing-original draft.
Supplementary Material
Acknowledgments
We gratefully acknowledge Dr. Wei Hong Toh and Fiona Houghton for expert technical advice.
This work was supported in part by the National Health and Medical Research Council of Australia. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Fig. S1.
- APP
- amyloid precursor protein
- Aβ
- amyloid β
- BACE1
- β-site APP-cleaving enzyme 1
- sAPP
- soluble large extracellular N-terminal domain of APP truncated at the primary cleavage site
- CTF
- C-terminal fragment
- TGN
- trans-Golgi network
- ADAM10
- a disintegrin and metalloprotease 10
- DIV
- days in vitro
- PFA
- paraformaldehyde
- NBM
- neurobasal medium
- DAPT
- N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester
- TAPI-1
- (N-(R)-[2-(hydroxyaminocarbonyl)methyl]-4-methylpentanoyl-l-naphthylalanyl-l-alanine, 2-aminoethyl amide)
- CE
- cell extract
- CM
- conditioned medium
- PVDF
- polyvinylidene difluoride
- qRT
- quantitative RT
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- DAPI
- 4′,6-diamidino-2-phenylindole
- DMEM
- Dulbecco's modified Eagle's medium
- C-DMEM
- complete DMEM
- FCS
- fetal calf serum
- HRP
- horseradish peroxidase
- qPCR
- quantitative PCR
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PM
- plasma membrane.
References
- 1. Thinakaran G., and Koo E. H. (2008) Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619 10.1074/jbc.R800019200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kinoshita A., Fukumoto H., Shah T., Whelan C. M., Irizarry M. C., and Hyman B. T. (2003) Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J. Cell Sci. 116, 3339–3346 10.1242/jcs.00643 [DOI] [PubMed] [Google Scholar]
- 3. Koo E. H., Squazzo S. L., Selkoe D. J., and Koo C. H. (1996) Trafficking of cell-surface amyloid β-protein precursor. I. Secretion, endocytosis and recycling as detected by labeled monoclonal antibody. J. Cell Sci. 109, 991–998 [DOI] [PubMed] [Google Scholar]
- 4. Rajendran L., Honsho M., Zahn T. R., Keller P., Geiger K. D., Verkade P., and Simons K. (2006) Alzheimer's disease β-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 103, 11172–11177 10.1073/pnas.0603838103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Small S. A., and Gandy S. (2006) Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron 52, 15–31 10.1016/j.neuron.2006.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burgos P. V., Mardones G. A., Rojas A. L., daSilva L. L., Prabhu Y., Hurley J. H., and Bonifacino J. S. (2010) Sorting of the Alzheimer's disease amyloid precursor protein mediated by the AP-4 complex. Dev. Cell 18, 425–436 10.1016/j.devcel.2010.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Siman R., and Velji J. (2003) Localization of presenilin-nicastrin complexes and γ-secretase activity to the trans-Golgi network. J. Neurochem. 84, 1143–1153 10.1046/j.1471-4159.2003.01616.x [DOI] [PubMed] [Google Scholar]
- 8. Xu H., Sweeney D., Wang R., Thinakaran G., Lo A. C., Sisodia S. S., Greengard P., and Gandy S. (1997) Generation of Alzheimer β-amyloid protein in the trans-Golgi network in the apparent absence of vesicle formation. Proc. Natl. Acad. Sci. U.S.A. 94, 3748–3752 10.1073/pnas.94.8.3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Choy R. W., Cheng Z., and Schekman R. (2012) Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in the trans-Golgi network. Proc. Natl. Acad. Sci. U.S.A. 109, E2077–2082 10.1073/pnas.1208635109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huse J. T., Liu K., Pijak D. S., Carlin D., Lee V. M., and Doms R. W. (2002) β-Secretase processing in the trans-Golgi network preferentially generates truncated amyloid species that accumulate in Alzheimer's disease brain. J. Biol. Chem. 277, 16278–16284 10.1074/jbc.M111141200 [DOI] [PubMed] [Google Scholar]
- 11. Toh W. H., Tan J. Z., Zulkefli K. L., Houghton F. J., and Gleeson P. A. (20176) Amyloid precursor protein traffics from the Golgi directly to early endosomes in an Arl5b- and AP4-dependent pathway. Traffic 18, 159–175 10.1111/tra.12465 [DOI] [PubMed] [Google Scholar]
- 12. Esch F. S., Keim P. S., Beattie E. C., Blacher R. W., Culwell A. R., Oltersdorf T., McClure D., and Ward P. J. (1990) Cleavage of amyloid β peptide during constitutive processing of its precursor. Science 248, 1122–1124 10.1126/science.2111583 [DOI] [PubMed] [Google Scholar]
- 13. Sisodia S. S. (1992) β-Amyloid precursor protein cleavage by a membrane-bound protease. Proc. Natl. Acad. Sci. U.S.A. 89, 6075–6079 10.1073/pnas.89.13.6075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tomita S., Kirino Y., and Suzuki T. (1998) Cleavage of Alzheimer's amyloid precursor protein (APP) by secretases occurs after O-glycosylation of APP in the protein secretory pathway. Identification of intracellular compartments in which APP cleavage occurs without using toxic agents that interfere with protein metabolism. J. Biol. Chem. 273, 6277–6284 10.1074/jbc.273.11.6277 [DOI] [PubMed] [Google Scholar]
- 15. Kuhn P. H., Wang H., Dislich B., Colombo A., Zeitschel U., Ellwart J. W., Kremmer E., Rossner S., and Lichtenthaler S. F. (2010) ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 29, 3020–3032 10.1038/emboj.2010.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haass C., Hung A. Y., Schlossmacher M. G., Teplow D. B., and Selkoe D. J. (1993) β-Amyloid peptide and a 3-kDa fragment are derived by distinct cellular mechanisms. J. Biol. Chem. 268, 3021–3024 [PubMed] [Google Scholar]
- 17. Jorissen E., Prox J., Bernreuther C., Weber S., Schwanbeck R., Serneels L., Snellinx A., Craessaerts K., Thathiah A., Tesseur I., Bartsch U., Weskamp G., Blobel C. P., Glatzel M., De Strooper B., and Saftig P. (2010) The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J. Neurosci. 30, 4833–4844 10.1523/JNEUROSCI.5221-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vincent B. (2016) Regulation of the α-secretase ADAM10 at transcriptional, translational and post-translational levels. Brain Res. Bull 126, 154–169 10.1016/j.brainresbull.2016.03.020 [DOI] [PubMed] [Google Scholar]
- 19. Gutwein P., Mechtersheimer S., Riedle S., Stoeck A., Gast D., Joumaa S., Zentgraf H., Fogel M., and Altevogt D. P. (2003) ADAM10-mediated cleavage of L1 adhesion molecule at the cell surface and in released membrane vesicles. FASEB J. 17, 292–294 10.1096/fj.02-0430fje [DOI] [PubMed] [Google Scholar]
- 20. Lammich S., Kojro E., Postina R., Gilbert S., Pfeiffer R., Jasionowski M., Haass C., and Fahrenholz F. (1999) Constitutive and regulated α-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. U.S.A. 96, 3922–3927 10.1073/pnas.96.7.3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guo X., Mattera R., Ren X., Chen Y., Retamal C., González A., and Bonifacino J. S. (2013) The adaptor protein-1 mu1B subunit expands the repertoire of basolateral sorting signal recognition in epithelial cells. Dev. Cell 27, 353–366 10.1016/j.devcel.2013.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parvathy S., Hussain I., Karran E. H., Turner A. J., and Hooper N. M. (1999) Cleavage of Alzheimer's amyloid precursor protein by α-secretase occurs at the surface of neuronal cells. Biochemistry 38, 9728–9734 10.1021/bi9906827 [DOI] [PubMed] [Google Scholar]
- 23. Boncompain G., Divoux S., Gareil N., de Forges H., Lescure A., Latreche L., Mercanti V., Jollivet F., Raposo G., and Perez F. (2012) Synchronization of secretory protein traffic in populations of cells. Nat. Methods 9, 493–498 10.1038/nmeth.1928 [DOI] [PubMed] [Google Scholar]
- 24. Boncompain G., and Perez F. (2012) Synchronizing protein transport in the secretory pathway. Curr. Protoc. Cell Biol. Chapter 15, Unit 15.19 10.1002/0471143030.cb1519s57 [DOI] [PubMed] [Google Scholar]
- 25. Borg J. P., Ooi J., Levy E., and Margolis B. (1996) The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol. Cell. Biol. 16, 6229–6241 10.1128/MCB.16.11.6229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McLoughlin D. M., Irving N. G., Brownlees J., Brion J. P., Leroy K., and Miller C. C. (1999) Mint2/X11-like colocalizes with the Alzheimer's disease amyloid precursor protein and is associated with neuritic plaques in Alzheimer's disease. Eur. J. Neurosci. 11, 1988–1994 10.1046/j.1460-9568.1999.00610.x [DOI] [PubMed] [Google Scholar]
- 27. Tanahashi H., and Tabira T. (1999) X11L2, a new member of the X11 protein family, interacts with Alzheimer's β-amyloid precursor protein. Biochem. Biophys. Res. Commun. 255, 663–667 10.1006/bbrc.1999.0265 [DOI] [PubMed] [Google Scholar]
- 28. Rogelj B., Mitchell J. C., Miller C. C., and McLoughlin D. M. (2006) The X11/Mint family of adaptor proteins. Brain Res. Rev. 52, 305–315 10.1016/j.brainresrev.2006.04.005 [DOI] [PubMed] [Google Scholar]
- 29. Shrivastava-Ranjan P., Faundez V., Fang G., Rees H., Lah J. J., Levey A. I., and Kahn R. A. (2008) Mint3/X11γ is an ADP-ribosylation factor-dependent adaptor that regulates the traffic of the Alzheimer's precursor protein from the trans-Golgi network. Mol. Biol. Cell 19, 51–64 10.1091/mbc.e07-05-0465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chaufty J., Sullivan S. E., and Ho A. (2012) Intracellular amyloid precursor protein sorting and amyloid-β secretion are regulated by Src-mediated phosphorylation of Mint2. J. Neurosci. 32, 9613–9625 10.1523/JNEUROSCI.0602-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Caster A. H., and Kahn R. A. (2013) Recruitment of the Mint3 adaptor is necessary for export of the amyloid precursor protein (APP) from the Golgi complex. J. Biol. Chem. 288, 28567–28580 10.1074/jbc.M113.481101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dunning C. J., Black H. L., Andrews K. L., Davenport E. C., Conboy M., Chawla S., Dowle A. A., Ashford D., Thomas J. R., and Evans G. J. (2016) Multisite tyrosine phosphorylation of the N terminus of Mint1/X11α by Src kinase regulates the trafficking of amyloid precursor protein. J. Neurochem. 137, 518–527 10.1111/jnc.13571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lai A., Sisodia S. S., and Trowbridge I. S. (1995) Characterization of sorting signals in the β-amyloid precursor protein cytoplasmic domain. J. Biol. Chem. 270, 3565–3573 10.1074/jbc.270.8.3565 [DOI] [PubMed] [Google Scholar]
- 34. Perez R. G., Soriano S., Hayes J. D., Ostaszewski B., Xia W., Selkoe D. J., Chen X., Stokin G. B., and Koo E. H. (1999) Mutagenesis identifies new signals for β-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Aβ42. J. Biol. Chem. 274, 18851–18856 10.1074/jbc.274.27.18851 [DOI] [PubMed] [Google Scholar]
- 35. Kang J., and Müller-Hill B. (1990) Differential splicing of Alzheimer's disease amyloid A4 precursor RNA in rat tissues: preA4(695) mRNA is predominantly produced in rat and human brain. Biochem. Biophys. Res. Commun. 166, 1192–1200 10.1016/0006-291X(90)90992-V [DOI] [PubMed] [Google Scholar]
- 36. Houghton F. J., Bellingham S. A., Hill A. F., Bourges D., Ang D. K., Gemetzis T., Gasnereau I., and Gleeson P. A. (2012) Arl5b is a Golgi-localised small G protein involved in the regulation of retrograde transport. Exp. Cell Res. 318, 464–477 10.1016/j.yexcr.2011.12.023 [DOI] [PubMed] [Google Scholar]
- 37. Penke B., Bogar F., and Fulop L. (2017) β-Amyloid and the pathomechanisms of Alzheimer's disease: a comprehensive view. Molecules 10, E1692 10.3390/molecules22101692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Postina R., Schroeder A., Dewachter I., Bohl J., Schmitt U., Kojro E., Prinzen C., Endres K., Hiemke C., Blessing M., Flamez P., Dequenne A., Godaux E., van Leuven F., and Fahrenholz F. (2004) A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Invest. 113, 1456–1464 10.1172/JCI20864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lichtenthaler S. F. (2011) α-Secretase in Alzheimer's disease: molecular identity, regulation and therapeutic potential. J. Neurochem. 116, 10–21 10.1111/j.1471-4159.2010.07081.x [DOI] [PubMed] [Google Scholar]
- 40. Ninomiya H., Roch J. M., Jin L. W., and Saitoh T. (1994) Secreted form of amyloid β/A4 protein precursor (APP) binds to two distinct APP binding sites on rat B103 neuron-like cells through two different domains, but only one site is involved in neuritotropic activity. J. Neurochem. 63, 495–500 [DOI] [PubMed] [Google Scholar]
- 41. Smith-Swintosky V. L., Pettigrew L. C., Craddock S. D., Culwell A. R., Rydel R. E., and Mattson M. P. (1994) Secreted forms of β-amyloid precursor protein protect against ischemic brain injury. J. Neurochem. 63, 781–784 [DOI] [PubMed] [Google Scholar]
- 42. Hefter D., Kaiser M., Weyer S. W., Papageorgiou I. E., Both M., Kann O., Müller U. C., and Draguhn A. (2016) Amyloid precursor protein protects neuronal network function after hypoxia via control of voltage-gated calcium channels. J. Neurosci. 36, 8356–8371 10.1523/JNEUROSCI.4130-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Corrigan F., Thornton E., Roisman L. C., Leonard A. V., Vink R., Blumbergs P. C., van den Heuvel C., and Cappai R. (2014) The neuroprotective activity of the amyloid precursor protein against traumatic brain injury is mediated via the heparin binding site in residues 96–110. J. Neurochem. 128, 196–204 10.1111/jnc.12391 [DOI] [PubMed] [Google Scholar]
- 44. Bauereiss A., Welzel O., Jung J., Grosse-Holz S., Lelental N., Lewczuk P., Wenzel E. M., Kornhuber J., and Groemer T. W. (2015) Surface trafficking of APP and BACE in live cells. Traffic 16, 655–675 10.1111/tra.12270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kimura A., Hata S., and Suzuki T. (2016) Alternative selection of β-site APP-cleaving enzyme 1 (BACE1) cleavage sites in amyloid β-protein precursor (APP) harboring protective and pathogenic mutations within the Aβ sequence. J. Biol. Chem. 291, 24041–24053 10.1074/jbc.M116.744722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Koike H., Tomioka S., Sorimachi H., Saido T. C., Maruyama K., Okuyama A., Fujisawa-Sehara A., Ohno S., Suzuki K., and Ishiura S. (1999) Membrane-anchored metalloprotease MDC9 has an α-secretase activity responsible for processing the amyloid precursor protein. Biochem. J. 343, 371–375 [PMC free article] [PubMed] [Google Scholar]
- 47. Slack B. E., Ma L. K., and Seah C. C. (2001) Constitutive shedding of the amyloid precursor protein ectodomain is up-regulated by tumour necrosis factor-α converting enzyme. Biochem. J. 357, 787–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Baulac S., LaVoie M. J., Kimberly W. T., Strahle J., Wolfe M. S., Selkoe D. J., and Xia W. (2003) Functional γ-secretase complex assembly in Golgi/trans-Golgi network: interactions among presenilin, nicastrin, Aph1, Pen-2, and γ-secretase substrates. Neurobiol. Dis. 14, 194–204 10.1016/S0969-9961(03)00123-2 [DOI] [PubMed] [Google Scholar]
- 49. Vetrivel K. S., Cheng H., Lin W., Sakurai T., Li T., Nukina N., Wong P. C., Xu H., and Thinakaran G. (2004) Association of γ-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 279, 44945–44954 10.1074/jbc.M407986200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. De Pace R., Skirzewski M., Damme M., Mattera R., Mercurio J., Foster A. M., Cuitino L., Jarnik M., Hoffmann V., Morris H. D., Han T. U., Mancini G. M. S., Buonanno A., and Bonifacino J. S. (2018) Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP-4 deficiency syndrome. PLoS Genet. 14, e1007363 10.1371/journal.pgen.1007363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Matsuda S., Miura E., Matsuda K., Kakegawa W., Kohda K., Watanabe M., and Yuzaki M. (2008) Accumulation of AMPA receptors in autophagosomes in neuronal axons lacking adaptor protein AP-4. Neuron 57, 730–745 10.1016/j.neuron.2008.02.012 [DOI] [PubMed] [Google Scholar]
- 52. Verkerk A. J. M. H., Schot R., Dumee B., Schellekens K., Swagemakers S., Bertoli-Avella A. M., Lequin M. H., Dudink J., Govaert P., van Zwol A. L., Hirst J., Wessels M. W., Catsman-Berrevoets C., Verheijen F. W., de Graaff E., et al. (2009) Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am. J. Hum. Genet. 85, 40–52 10.1016/j.ajhg.2009.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Skovronsky D. M., Moore D. B., Milla M. E., Doms R. W., and Lee V. M. (2000) Protein kinase C-dependent α-secretase competes with β-secretase for cleavage of amyloid-β precursor protein in the trans-Golgi network. J. Biol. Chem. 275, 2568–2575 10.1074/jbc.275.4.2568 [DOI] [PubMed] [Google Scholar]
- 54. Chia P. Z., Gasnereau I., Lieu Z. Z., and Gleeson P. A. (2011) Rab9-dependent retrograde transport and endosomal sorting of the endopeptidase furin. J. Cell Sci. 124, 2401–2413 10.1242/jcs.083782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Goud B., and Gleeson P. A. (2010) TGN golgins, Rabs and cytoskeleton: regulating the Golgi trafficking highways. Trends Cell Biol. 20, 329–336 10.1016/j.tcb.2010.02.006 [DOI] [PubMed] [Google Scholar]
- 56. Guo Y., Sirkis D. W., and Schekman R. (2014) Protein sorting at the trans-Golgi network. Annu. Rev. Cell Dev. Biol. 30, 169–206 10.1146/annurev-cellbio-100913-013012 [DOI] [PubMed] [Google Scholar]
- 57. Heuer D., Rejman Lipinski A., Machuy N., Karlas A., Wehrens A., Siedler F., Brinkmann V., and Meyer T. F. (2009) Chlamydia causes fragmentation of the Golgi compartment to ensure reproduction. Nature 457, 731–735 10.1038/nature07578 [DOI] [PubMed] [Google Scholar]
- 58. Hirst J., Bright N. A., Rous B., and Robinson M. S. (1999) Characterization of a fourth adaptor-related protein complex. Mol. Biol. Cell 10, 2787–2802 10.1091/mbc.10.8.2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stewart S. A., Dykxhoorn D. M., Palliser D., Mizuno H., Yu E. Y., An D. S., Sabatini D. M., Chen I. S., Hahn W. C., Sharp P. A., Weinberg R. A., and Novina C. D. (2003) Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501 10.1261/rna.2192803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Luke M. R., Kjer-Nielsen L., Brown D. L., Stow J. L., and Gleeson P. A. (2003) GRIP domain-mediated targeting of two new coiled-coil proteins, GCC88 and GCC185, to subcompartments of the trans-Golgi network. J. Biol. Chem. 278, 4216–4226 10.1074/jbc.M210387200 [DOI] [PubMed] [Google Scholar]
- 61. Kooy J., Toh B. H., Pettitt J. M., Erlich R., and Gleeson P. A. (1992) Human autoantibodies as reagents to conserved Golgi components. Characterization of a peripheral, 230-kDa compartment-specific Golgi protein. J. Biol. Chem. 267, 20255–20263 [PubMed] [Google Scholar]
- 62. Scherer W. F., Syverton J. T., and Gey G. O. (1953) Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J. Exp. Med. 97, 695–710 10.1084/jem.97.5.695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Toh W. H., Chia P. Z. C., Hossain M. I., and Gleeson P. A. (2018) GGA1 regulates signal-dependent sorting of BACE1 to recycling endosomes, which moderates Aβ production. Mol. Biol. Cell 29, 191–208 10.1091/mbc.E17-05-0270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dull T., Zufferey R., Kelly M., Mandel R. J., Nguyen M., Trono D., and Naldini L. (1998) A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72, 8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Brisco M. J., and Morley A. A. (2012) Quantification of RNA integrity and its use for measurement of transcript number. Nucleic Acids Res. 40, e144 10.1093/nar/gks588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Woodcroft B. J., Hammond L., Stow J. L., and Hamilton N. A. (2009) Automated organelle-based colocalization in whole-cell imaging. Cytometry A. 75, 941–950 [DOI] [PubMed] [Google Scholar]
- 67. Manders E. M. M., Verbeek F. J., and Aten J. A. (1993) Measurement of co-localization of objects in dual-colour confocal images. J. Microscopy 169, 375–382 10.1111/j.1365-2818.1993.tb03313.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.