

Abstract
Prostaglandin endoperoxide H synthases-1 and -2, commonly called cyclooxygenases-1 and -2 (COX-1 and -2), catalyze the committed step in prostaglandin biosynthesis—the conversion of arachidonic acid to prostaglandin endoperoxide H2. Both COX isoforms are sequence homodimers that function as conformational heterodimers having allosteric (Eallo) and catalytic (Ecat) subunits. At least in the case of COX-2, the enzyme becomes folded into a stable Eallo/Ecat pair. Some COX inhibitors (i.e. nonsteroidal anti-inflammatory drugs and coxibs) and common fatty acids (FAs) modulate Ecat activity by binding Eallo. However, the interactions and outcomes often differ between isoforms. For example, naproxen directly and completely inhibits COX-1 by binding Ecat but indirectly and incompletely inhibits COX-2 by binding Eallo. Additionally, COX-1 is allosterically inhibited up to 50% by common FAs like palmitic acid, whereas COX-2 is allosterically activated 2-fold by palmitic acid. FA binding to Eallo also affects responses to COX inhibitors. Thus, COXs are physiologically and pharmacologically regulated by the FA tone of the milieu in which each operates—COX-1 in the endoplasmic reticulum and COX-2 in the Golgi apparatus. Cross-talk between Eallo and Ecat involves a loop in Eallo immediately downstream of Arg-120. Mutational studies suggest that allosteric modulation requires a direct interaction between the carboxyl group of allosteric effectors and Arg-120 of Eallo; however, structural studies show some allosterically active FAs positioned in COX-2 in a conformation lacking an interaction with Arg-120. Thus, many details about the biological consequences of COX allosterism and how ligand binding to Eallo modulates Ecat remain to be resolved.
Keywords: prostaglandin, arachidonic acid (AA) (ARA), eicosanoid, cyclooxygenase (COX), allosteric regulation, eicosapentaenoic acid, naproxen, NSAID, oleic acid, palmitic acid
Cyclooxygenase basics
The most recent detailed reviews of cyclooxygenase catalysis were published by Tsai and Kulmacz (1) and Smith et al. (2). Prostaglandin endoperoxide H synthases (PGHSs)3 efficiently convert the ω-6 polyunsaturated fatty acid (FA) arachidonic acid (5c,8c,11c,14c-eicosatetraenoic acid; AA), two O2 molecules, and two electrons to prostaglandin endoperoxide H2 (PGH2) in two steps: (a) a bis-oxygenase (“cyclooxygenase” (COX)) reaction to form prostaglandin endoperoxide G2 (PGG2), and (b) a peroxidase (POX) reaction that reduces PGG2 to PGH2 (Fig. 1). These reactions occur at separate but interconnected COX and POX active sites of the enzymes. AA utilized in the COX reaction is probably derived mainly through the actions of various phospholipase A2 forms (3, 4). The physiological source(s) of the electrons used by the POX activity of PGHSs to form PGG2 is unknown.
Figure 1.

COX and POX reactions catalyzed by PGHSs. There are two PGHS isoforms that are commonly known as cyclooxygenases-1 and -2 (COX-1 and COX-2). Downstream PG products are formed from PGH2 via different synthases. The COX activity of the two COX isoforms is inhibited differently by nsNSAIDs, such as ibuprofen, naproxen, and aspirin, and by COX-2–selective inhibitors called coxibs, such as celecoxib and rofecoxib, and a variety of Fas, including ω-3 fish oil FAs such as EPA and DHA. This figure and the legend are modified from ASBMB Today (http://www.asbmb.org/asbmbtoday/201508/LipidNews/).
The formation of PGH2 is typically shown as a COX reaction followed by a POX reaction (Fig. 1); however, the initiation of COX activity requires an initial oxidation of the heme group at the POX site by ambient H2O2, an alkyl hydroperoxide or nitric oxide (1). Oxidation of the heme leads to generation of a heme radical cation in the POX active site and a tyrosyl radical on Tyr-385 in the COX active site. This tyrosyl radical abstracts the ω8 allylic hydrogen from AA bound in the COX site in the first step in COX catalysis. The first O2 insertion then occurs at C-11. Hydrogen abstraction from the ω8 position of dihomo-γ-linolenic acid (8c,11c,14c-eicosatrienoic acid; DHLA), the precursor of the “1 series” PGs, occurs with an isotope effect, which suggested that hydrogen abstraction is the rate-limiting step in COX catalysis (5). Recent evidence indicates that the first irreversible step is later in the complex reaction sequence when the hydrogen from a reduced Tyr-385 is transferred to generate PGG2 and regenerate the Tyr-385 radical (6).
There are two PGHS isoforms: PGHS-1 and PGHS-2, also called COX-1 and COX-2. COX-1 has a narrow substrate specificity, preferentially oxygenating AA. COX-2 has a broader substrate specificity and can oxygenate several FAs more efficiently than does COX-1, including DHLA, eicosapentaenoic acid (EPA), and adrenic acid (22:4ω6) at 115, 45, and 57% of the rate of AA. One functional difference between COX-1 and COX-2 is the ability of COX-2 to efficiently oxygenate neutral derivatives of AA, such as 2-arachidonoylglycerol (2-AG) and anandamide (AEA). 2-AG and AEA have a wide tissue distribution and were the first endogenous ligands identified for the cannabinoid receptors (7). Marnett and co-workers showed that endocannabinoid oxygenation by COX utilizes the Tyr-385–based radical mechanism employed for AA. Their work is reviewed in Ref. 8.
COX-1 and COX-2 are both targets of common nonspecific nonsteroidal anti-inflammatory drugs (nsNSAIDs), including ibuprofen and naproxen (Fig. 1). Aspirin is a nsNSAID that unlike other inhibitors covalently and irreversibly modifies COXs by acetylating Ser-530 and interfering with AA access to the COX active site (9–14). COX-2 “specific” inhibitors referred to as coxibs are more selective toward COX-2; however, some coxibs bind with high affinity to COX-1 and affect responses of COX-1 to nsNSAIDs and FAs. Most nsNSAIDs and coxibs are hydrophobic, many cause time-dependent COX inhibition, and each is metabolized at different rates. No one assay method provides a simple comparison of the specificities and potencies of COX inhibitors. A useful comparison based on in vitro whole-blood assays is described by Grosser et al. (15).
Each COX is associated with different biologies, but there is a level of functional complementarity (16, 17). Both COX isoforms are present on the luminal surface of the ER and associated inner membrane of the nuclear envelope (18–20). Importantly, COX-2 is also located in the Golgi apparatus where it is likely involved in PGE2 biosynthesis (21). COX-2 is also associated with lipid droplets (22). Differences between the subcellular locations of COX-1 and COX-2 have consequences in terms of the availability of substrates and agents that modulate COX activities (e.g. hydroperoxides and FAs that are not substrates).
Cyclooxygenase structure
COXs are sequence homodimers consisting of tightly associated monomers. Each monomer contains three domains: an N-terminal epidermal growth factor-like domain; a membrane-binding domain (MBD); and a globular C-terminal catalytic domain (Fig. 2) (23). The heme group is located near the surface of the catalytic domain, where it serves as a “functional bridge” between the POX and COX active sites. The MBD is composed of four short amphipathic helices, with an opening in its center. The amphipathic helices serve to anchor the homodimer to the surface of the membrane through the side chains of hydrophobic amino acids that protrude into the bilayer (24, 25). FAs are envisioned to enter the COX active site through the opening within the MBD. Cytosolic (c) PLA2 translocating to the surface of Golgi apparatus and the ER and nuclear envelope, depending on the cytosolic Ca2+ concentration (3, 26), is presumed to mobilize FA substrates that traverse the space between the monolayers of the bilayer entering methyl-end first into the pore formed by the MBD and from there into the COX active site.
Figure 2.
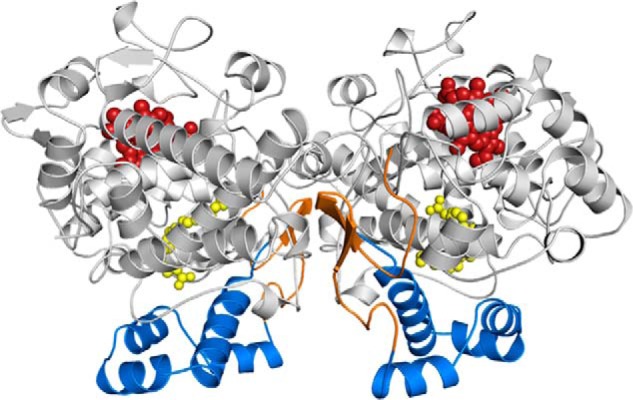
Structure of COX-2. Cartoon representation of the COX-2 biological dimer with AA (yellow spheres) bound within the COX channel in its productive (Ecat; left) and nonproductive (Eallo; right) poses (Protein Data Bank code 3HS5 (31)). The epidermal growth factor-like domain, MBD, and catalytic domains are colored orange, blue, and gray, respectively. The heme group is depicted as red spheres bound at the base of the POX active site.
Structural studies of FA binding to COX-1 and COX-2
At the atomic level, the interactions that govern FA binding, specificity, and catalysis by COX-1 have been well established (27–30), whereas the equivalent interactions for COX-2 have only recently come into focus (31, 32). In its catalytically productive conformation, AA is oriented in the COX channel of both COX-1 and COX-2 in an extended L-shaped conformation, with the carboxylate group located near the opening of the channel interacting with Arg-120 (Fig. 2). The ω-end of AA is located in a hydrophobic groove at the apex of the channel. In this orientation, C-13 of AA is optimally placed below Tyr-385, where the pro-S-hydrogen can be abstracted to initiate catalysis. The crystal structure of AA bound to COX-2 revealed AA in different conformations in the two monomers. The catalytically productive conformation of AA was observed in one monomer. The other monomer exhibited AA in a “nonproductive” pose, in which AA is bound inverted within the channel, with the carboxylate group stabilized by interactions with Tyr-385 and Ser-530 at the apex of the channel and the ω-end of AA directed toward the opening of the channel (31, 33).
Structure determinations have also characterized the binding of different FA substrates within the COX channel of COX-1 and COX-2 and provided insights into the molecular underpinnings responsible for the substrate selectivity observed for each isoform (30–32, 34). In general, these alternative FA substrates bind in extended L-shaped conformations, similar to that observed for AA, with their carboxylate groups located near Arg-120 and their ω-ends in the hydrophobic groove at the top of the channel. The resulting poses position C-13 (or C-11 in the case of linoleic acid and α-linolenic acid) below Tyr-385 for hydrogen abstraction, with the contacts and residues involved in the stabilization of the substrate within the channel highly conserved. Similar to that observed for AA, both nonproductive and productive poses are observed in each monomer for EPA bound in the COX channel of COX-2 (31).
There are subtle differences associated with the binding of different FA substrates to COX-1 and COX-2. With COX-2 there is not a functional requirement for a direct interaction between the substrate carboxylate group and Arg-120, and COX-2 has a larger COX channel volume. This conformational freedom is the major reason that COX-2 has a more promiscuous substrate preference than COX-1 (32). In the case of COX-1, an interaction between the carboxylate end of the substrate and Arg-120 is required for binding and catalysis. With different substrates having different chain lengths and degrees of unsaturation, this leads to suboptimal alignments of C-13 and Tyr-385. As a result, there is inefficient hydrogen abstraction and oxygenation of these substrates (35). Conversely, binding of different FA substrates within the COX channel of COX-2 is driven by coordinated interactions between the FAs and multiple residues lining the channel, rather than by a single Arg-120 determinant (32). Comparisons made between the conformations of AA and different FA substrates bound in the COX channel of COX-2 show significant differences in the positions of the carboxylate ends of these substrates, similar to that observed in COX-1. However, the lack of a requirement for binding to Arg-120 in COX-2 results in the proper insertion of the ω-end of the FA in the hydrophobic groove, which leads to optimal alignment of carbon 13 with Tyr-385.
Structural studies of endocannabinoid binding to COX-2
The X-ray crystal structure of COX-2 in complex with 1-AG revealed the molecular details of how endocannabinoid substrates bind within the COX channel (36). (2-AG undergoes acyl migration in biological buffers resulting in the conversion of 2-AG to 1-AG.) 1-AG binds to both monomers of the dimer. In each monomer, the glycerol moiety is located near the opening of the channel with the ω-end of the acyl chain inserted into the hydrophobic groove. However, subtle differences in the conformations of 1-AG in each monomer were observed. In one monomer, the ω-end of 1-AG does not fully insert into the hydrophobic groove, resulting in the misalignment of carbon 13 for hydrogen abstraction. Conversely, the ω-end of 1-AG bound in the opposite monomer binds deep in the hydrophobic groove resulting in the optimal alignment of carbon 13 below Tyr-385 for hydrogen abstraction. This conformation mimics the productive pose of AA. Thus, 1-AG binds in a nonproductive conformation in one monomer and in a productive conformation in the other monomer, analogous to that observed for AA binding to COX-2. Movement of the side chain of Leu-531 near the opening of the channel provides the conformational flexibility required to accommodate the bulkier 2,3-dihydroxypropyl moiety and facilitate productive binding.
Cyclooxygenases functioning as conformational heterodimers
COX-1 has been known to be a sequence homodimer since 1977 (37). Until about 10 years ago, it was widely believed that each COX monomer comprising a dimer operated independently. This dogma was based on enzyme kinetics (38), measurements of the stoichiometry of heme binding (39), and aspirin acetylation (40). Additionally, and as noted above, structural studies with COX-1 and later COX-2 showed the enzymes to be composed of identical protein monomers.4
Evidence contradictory to the view that COX monomers function independently was first published in the mid 1980s. Kulmacz and Lands (41) showed that there is one high-affinity site for heme per ovine (ov) COX-1 dimer and later that complete COX inhibition occurs at a ratio of one nsNSAID molecule per dimer (42). They suggested that the two COX-1 subunits are “distinct.” One early COX-2 structure showed AA positioned in different orientations in the two COX sites (33). Finally, pharmacological studies by Rimon and co-workers (43, 44) found that coxibs interfere with the inhibition of COX-1 by some nsNSAIDs without affecting COX-1 activity toward AA. However, these various bits of information, being difficult to explain, were largely ignored.
It is now clear that COXs operate in solution as conformational heterodimers each composed of an allosteric (Eallo) and a catalytic (Ecat) monomer that function cooperatively as illustrated in Fig. 3. In the case of COX-2, the Eallo/Ecat pair is quite stable suggesting that it is formed and stabilized during the folding and processing of COX-2 (13). It is essential to understand that multiple interactions can occur involving each COX isoform with numerous substrate and nonsubstrate FAs and 2-AG and many nsNSAIDs and coxibs. Obviously, this makes for considerable complexity. In the remainder of this JBC Review, we highlight only some aspects of what has been observed to date.
Figure 3.
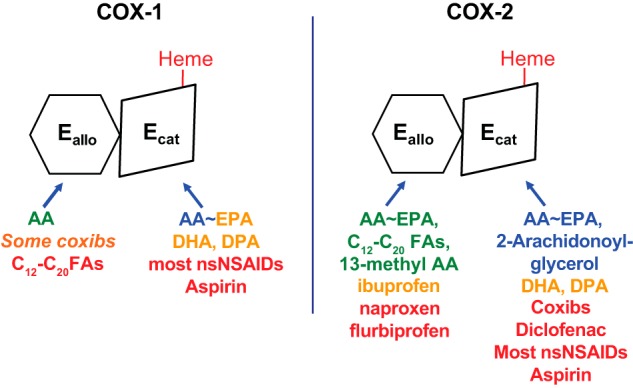
Isoform-specific interactions of COX substrates, nonsubstrate FAs, and COX inhibitors with huCOX-1 and huCOX-2. Each COX isoform functions as a conformational heterodimer composed of an allosteric (Eallo) and a catalytic (Ecat) subunit. The individual subunits of human COXs differ both in their affinities for different ligands and in their responses to binding of the ligands. Efficient COX substrates are shown in blue in the approximate order of their catalytic efficiencies. FAs that are inefficient COX substrates can interfere with prostaglandin formation, typically by competing with AA for the Ecat; these are shown in orange (e.g. EPA, DHA, and DPA). Ligands shown in green allosterically stimulate COX activity. Ligands shown in red interfere with COX activity either allosterically by binding Eallo or competitively by binding Ecat. Ligands that bind Eallo can also affect responses to COX inhibitors. For example, nonsubstrate FAs bound to Eallo of huCOX-1 increase the rate of aspirin acetylation, whereas celecoxib (in orange) bound to Eallo of huCOX-1 can interfere with aspirin action. This figure and the legend are adapted from Ref. 64.
Evidence for half-site COX-2 activity
In studies instigated by the work of Rimon and co-workers (43, 44), we prepared and characterized several COX-2 heterodimers composed of a COX-inactive (Mutant) and a COX-active (Native) monomer (45). Hybrid dimers of this type are designated Mutant/Native human (hu) COX-2 to denote their subunit compositions. One of the heterodimers investigated was G533A/Native huCOX-2. A homodimeric G533A/G533A huCOX-2 mutant lacks COX activity but retains POX activity, suggesting that the mutant homodimer is structurally intact. We expected that were the monomers to act independently, the G533A/Native huCOX-2 would have 50% of native COX activity and 100% of the native POX activity. However, G533A/Native huCOX-2 and Native/Native huCOX-2 had almost the same COX activities and POX activities (45). This finding suggested but did not prove that the Native/Native huCOX-2 homodimer was displaying half-site COX activity.5
Interactions of huCOX-2 and huCOX-2 with AA versus EPA
The first indication that half-site functioning relates to allosteric regulation came upon comparing the oxygenation of two COX substrates AA and EPA alone and together with COX-1 versus COX-2 (Fig. 3) (46). The ω-3 “fish oil” FA EPA was known to be a poorer substrate than AA of both COX-1 and COX-2 (47, 48). Under optimal conditions COX-1 oxygenates EPA at about one-tenth the rate of AA to form primarily PGH3 (46). COX-2 oxygenates EPA at about 45% the rate of AA to form PGH3 along with comparable amounts of monohydroxy-FAs (49). We found EPA to be a good inhibitor of AA oxygenation by COX-1 (46); however, only very high concentrations of EPA inhibited AA oxygenation by COX-2. Our most surprising observation was that despite AA and EPA having similar Km values with COX-2 when tested individually, COX-2 preferentially oxygenates AA when EPA and AA are tested together (46). We only later realized that EPA bound to Eallo of COX-2 promotes AA (versus EPA) oxygenation by Ecat.
The results of studies comparing AA and EPA led us to examine the effects of numerous other FAs on COXs. These studies have indicated that common saturated and monounsaturated “nonsubstrate” FAs as well as polyunsaturated substrate FAs each interact differently with the two different subunits of each isoform (Fig. 3). Different nsNSAIDs and coxibs bind COX subunits with different affinities and effects. Moreover, the interactions of FAs with COXs not only modulate COX activities but also the responses to COX inhibitors. We presume these effects are physiologically and pharmacologically important. The overall response in terms of net COX activity provides a readout of the FA composition and concentration (i.e. “FA tone”) of the environment in which each isoform operates. As noted above, COX-1 functions in the ER, whereas COX-2 likely functions primarily in the Golgi apparatus at least in PGE2 synthesis (21).
Interactions of huCOX-1 and huCOX-2 with nonsubstrate FAs
Direct evidence for co-interactions of two different FAs with two different COX subunits came from examining the effects that common FAs that are not COX substrates (nonsubstrate FAs) have on COX-1 and COX-2 (Fig. 3) (50–54). We summarize studies with AA and palmitic acid (PA) interacting with huCOX-2 as an example (Fig. 4, A and B). We found that PA can enhance COX-2–mediated oxygenation of AA by more than 2-fold (50, 51, 54). This is likely attributable to a small decrease in Km and a small increase in Vmax values (50, 53, 54). The 2-fold stimulatory effect of PA is seen at high PA/AA ratios when PA occupies a significant fraction of the COX sites of Eallo (Fig. 4B). It turns out that AA itself binds Eallo with high affinity but AA bound to Eallo is not oxygenated (Fig. 4A) (51, 52).6 AA competes with and displaces PA from Eallo of COX-2, and at high AA/PA ratios the stimulatory effect of PA is eliminated. Yet even very high PA concentrations fail to inhibit AA oxygenation by COX-2. These outcomes are a consequence of a 20-fold difference in the Kd values of AA and PA for binding to Eallo (i.e. 0.26 and 7.5 μm, respectively)6 and a relative inability of PA to bind Ecat of COX-2 (i.e. Kd of >50 μm, well above the critical micelle concentration of PA (51)) (Fig. 4). Analysis of the X-ray crystal structure of PA in complex with COX-2 reveals that PA is bound to only one monomer of the dimer, with its carboxylate group located near the opening of the channel and interacting with Arg-120 (51). PA also fails to interfere with cross-linking between the COX-2 subunits, a characteristic of ligands that bind only one of the two monomers (50, 54). PA is the most effective of all common nonsubstrate FAs in activating COX-2, but other saturated and monounsaturated nonsubstrate FAs bind with similar affinities to Eallo of COX-2 (51)
Figure 4.
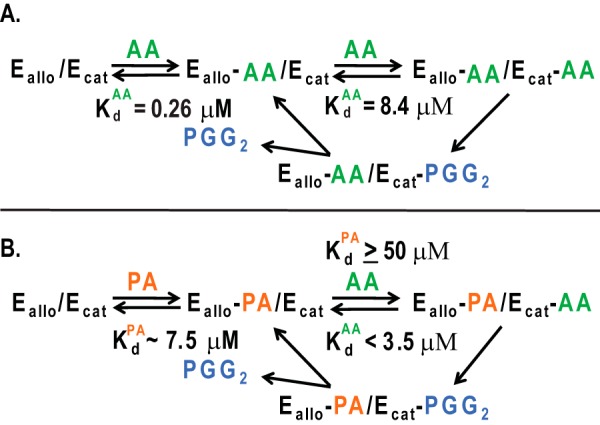
Model for allosteric interactions between monomers of the COX-2 homodimer caused by the binding of FAs. Ecat is the catalytic monomer to which heme is bound, whereas Eallo is the allosteric monomer that does not bind heme. A, binding of AA to Eallo and Ecat of huCOX-2 and subsequent oxygenation of AA. The Kd value for Ecat is the Km for AA (8.4 μm) in the absence of other FAs. The Kd value for AA binding to Eallo was determined to be 0.26 μm.6 B, binding of AA to Eallo and Ecat in the presence of PA. We make the following assumptions: (a) that the Kd value for binding of AA to the Ecat site is equal to the Km value in the presence of PA (i.e. < 3.5 μm), and (b) that the Kd value for PA binding to the Ecat site is high (≥50 μm) because PA does not inhibit AA oxygenation by huCOX-2 even at low concentrations of AA. We estimate that the Kd values for the binding of PA and stearic acid to the Eallo site are both ∼7.5 μm.5 Note that the Kd value for PA binding to Eallo is 30-fold that for AA binding to Eallo so that at all relevant FA concentrations AA and PA will compete for binding to Eallo, and the effect of PA will be determined by the PA/AA ratio. This figure and the legend are modified from Ref. 51.
Unlike huCOX-2, huCOX-1 is uniformly inhibited, as opposed to being stimulated or unaffected by common nonsubstrate FAs (52, 54). Maximal inhibition of 55% occurs at a nonsubstrate FA/AA ratio of 20 with PA, oleic acid, and stearic acid. There is little nonsubstrate FA specificity compared with that observed with huCOX-2 where PA is 2-fold more effective than stearic acid in augmenting huCOX-2 (54).
The fact that the level of inhibition of COX-1 by nonsubstrate FAs plateaus indicates that inhibition by nonsubstrate FAs does not involve competition with AA binding to Ecat. Additionally, nonsubstrate FAs can displace unreacted [14C]AA from Eallo of huCOX-1 suggesting that inhibition by nonsubstrate FAs occurs allosterically via Eallo of COX-1.6 Marnett and co-workers (53) observed that another FA 13-Me-AA does not significantly affect AA oxygenation by ovCOX-1 but that 13-Me-AA does enhance 2-AG oxygenation by COX-1. Thus, 13-Me-AA can also bind Eallo of COX-1 (Fig. 3). The X-ray crystal structure of 13-Me-AA bound to COX-2 reveals that 13-Me-AA binds to both monomers of the dimer and in a nonproductive conformation with its carboxylate bound near Tyr-385 at the apex of the COX channel.
Interactions of COX-1 versus COX-2 with substrate FAs
Nine ω-3 and ω-6 FAs having 18, 20, or 22 carbons were compared as substrates for huCOX-1 and huCOX-2 (Fig. 3) (54). AA and DHLA are the best substrates for both isoforms. Based on Km measurements, the Kd values for AA binding to Ecat of COX-1 and COX-2 are indistinguishable (∼8 and 4 μm, respectively). The Kd values for AA binding to Eallo of COX-1 and COX-2 are also similar (∼0.26 and 0.35 μm, respectively).6
Seven substrate FAs (excepting AA and DHLA) are modest inhibitors of COX-1 (45 ± 5% inhibition) when tested at a FA/AA ratio of five. Nonsubstrate FAs somewhat augment the inhibitory effects of EPA and docosahexaenoic acid (DHA; 22:6ω3) on AA oxygenation by COX-1. This suggests that in solution (as opposed to under crystallization conditions)4 none of these FAs efficiently binds Eallo of COX-1 (54).
Only the C-22 polyunsaturated FAs adrenic acid (22:4ω6), docosapentaenoic acid (DPA; 22:5ω3), and DHA are modest inhibitors of AA oxygenation by COX-2 (50, 54). EPA and AA both bind to Eallo of COX-2 with similar affinities (Kd ∼0.25 μm) (54); DHLA was not tested but likely has a high affinity for Eallo. Other C-18 and C-22 polyunsaturated FA substrates bound Eallo very poorly (Kd ≥25 μm).6
Structural comparisons of the interactions of COX-2 with nonsubstrate and substrate FAs
Detailed structural comparisons between the two monomers with AA and EPA bound in productive versus nonproductive conformations (31) or with PA bound to a single monomer (51) did not uncover significant structural deviations that could be related to the molecular basis for allosteric regulation by FAs. The observation of different conformations of AA and EPA within monomers suggests that there is a difference between the COX sites of the dimer at some point during protein crystallization and is consistent with the half-site functioning of COX-2 in solution.4
Based on studies of COX-2 mutants, it is unclear whether the nonproductive conformation of AA is functionally significant in solution. In a Y385F/Native huCOX-2 heterodimer about 90% of the enzyme is of the form Y385F (Eallo)/Native (Ecat) and the Km for AA is the same as native COX-2 (13); this heterodimer is effectively inhibited by nsNAIDs and coxibs that function via Eallo (55). Accordingly, Y385F/Native huCOX-2 can be used as a platform to make and test individual mutations in Eallo versus Ecat. An R120A substitution in Eallo generates Y385F R120A/Native huCOX-2 in which the Vmax is reduced by two-thirds compared with that of Y385F/Native huCOX-2 but without affecting the Km value for AA. Importantly, [14C]AA does not bind to Eallo of a Y385F R120A/Native huCOX-2, and PA does not activate this mutant. Furthermore, COX inhibitors that function via Eallo fail to inhibit Y385F R120A/Native huCOX-2. These results and studies of related mutants suggest to us that FAs and allosteric COX inhibitors bring about their effects by binding to Arg-120 of Eallo and affecting in different ways the positioning of the downstream loop involving residues 126–129 (12, 45, 55, 56). This loop interacts with the partner Ecat subunit (12, 50, 56). More broadly, the results indicate that the function of FA binding to Eallo of COX-2 is to relieve a tonic inhibition of Ecat by Eallo (55).
Interactions with COX inhibitors, nsNSAIDs and coxibs
Most COX inhibitors exhibit competitive inhibitory kinetic behavior when tested in standard inhibitor assays (i.e. when inhibitor and substrates AA and O2 are combined, enzyme is then added and initial rates are measured). However, many COX inhibitors also cause time-dependent enzyme inhibition. This general phenomenon is only captured when enzyme is preincubated for several minutes with an inhibitor (42, 57, 58). Time-dependent inhibition involves inhibitor binding to a COX site followed by time-dependent structural changes. Many of these changes are slowly reversible (42, 59–61). This time-dependent property of inhibitors has therapeutic consequences. Time-dependent inhibitors do not covalently modify COXs. The exception is aspirin, which acetylates Ser-530 of Ecat (9–14).
The findings that inhibitors bind one of the two COX sites of a dimer with a higher affinity (42, 45, 61) and that aspirin acetylates only one monomer (i.e. Ecat) (12, 13, 49) raises the question of whether an inhibitor is competing with AA binding to Eallo (Kd ∼0.25 μm) or Ecat (Kd ∼5–10 μm) or both. There are examples of several different scenarios (Fig. 3).
COX inhibitor acting time-dependently via Eallo or Ecat
At least two nsNSAIDs, naproxen and flurbiprofen, are time-dependent inhibitors (52, 55, 62) that appear to act via Ecat of COX-1 (52) but via Eallo of COX-2 (Fig. 3) (13, 55).
Time-independent inhibitors
Ibuprofen and mefenamic acid are considered to be time-independent inhibitors of AA oxygenation by both COX-1 (52) and COX-2 (51, 63). With COX-2, ibuprofen appears to bind both monomers of COX-2 to completely inhibit AA oxygenation (51, 63), but ibuprofen has a slightly higher affinity for Eallo than Ecat of huCOX-2 (Fig. 3) (64). Curiously, ibuprofen inhibition of huCOX-1 becomes time-dependent in the presence of nonsubstrate FAs (52). Double-tryptophan murine COX-2 mutants at positions 89/90 or 89/119 enhance the time dependence of ibuprofen and mefenamic acid (65). (R)-Profens are substrate-selective inhibitors of 2-AG oxygenation by COX-2 (61). Unlike AA, 2-AG binds very poorly to Eallo.
Some coxibs preferentially bind Eallo of COX-1 but Ecat of COX-2
Celecoxib preferentially inhibits AA oxygenation by COX-2 versus COX-1. However, celecoxib binds with higher affinity to Eallo of COX-1 than Ecat of COX-2 (12) (Fig. 3). Celecoxib binding to Eallo of COX-1 does not inhibit AA oxygenation (presumably because AA can readily displace celecoxib from Eallo), but celecoxib binding does interfere with the actions of some COX-1 inhibitors, including aspirin that act via Ecat of COX-1 (12). Celecoxib and rofecoxib as well as diclofenac and indomethacin cause time-dependent inhibition of COX-2 that appears to involve their interactions with Ecat (13).
Effects of FAs on interactions of PGHSs with nsNSAIDs and coxibs
Nonsubstrate FAs can either inhibit or potentiate the actions of nsNSAIDs or coxibs. Potentiation of the effect of a COX inhibitor by a nonsubstrate FA suggests that the COX inhibitor functions via Ecat (e.g. celecoxib, diclofenac, and indomethacin with COX-2 (13, 51)). Nonsubstrate FAs interfere with COX-2 inhibition by agents that act via Eallo of COX-2 (e.g. naproxen and flurbiprofen). Furthermore, inhibitory responses to naproxen and flurbiprofen are attenuated in Y385F S121P/Native (55) and Y385F R120A/Native huCOX-2 (13)—heterodimers in which interactions with Arg-120 of Eallo are abrogated. Conversely, the inhibitory response to diclofenac, which acts via Ecat, is not affected in these mutants, although the inhibitory response to ibuprofen inhibition is partially attenuated (55).
Acknowledgments
This JBC Review is written to honor Dr. Herbert Tabor on the occasion of his 100th birthday. W. L. S. was privileged to have served with him from 2000 to 2015 as a JBC Associate Editor and to recall Herb's favorite phrase: “And I'm sure you will all agree….” (Fig. 5).
Figure 5.

Herb Tabor and colleagues at the JBC Associate Editor meeting in San Francisco, February 10, 2010. Back (left to right): Vince Hascall, Jerry Lingrel, Jim Stull, Ken Neet, David Russell, Chuck Samuel, Fred Guengerich, Jim Siedow, John Exton, Bob Lehman, Bob Simoni, and George Carman. Front (left to right): Norma Allewell, Bill Smith, Linda Spremulli, Tom Vanaman, Herb Tabor, Dale Benos, Judy Bond, Joel Gottesfeld, Martha Fedor, and Xiao-Fan Wang.
This work was supported by National Institutes of Health Grants GM 68848 (to W. L. S.), CA 130810 (to W. L. S.), HL 117798 (to W. L. S.), GM 077176 (to M. G. M.), and GM 115386 (to M. G. M.). This JBC Review is part of a collection honoring Herbert Tabor on the occasion of his 100th birthday. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
It is now known that COXs only crystallize in a reasonable amount of time in a symmetric form with both monomers occupied with a heme group and a COX ligand (56).
Subsequent studies indicate that G533A/Native huCOX-2 folds such that G533A is the allosteric subunit (Eallo) and Native is the catalytic subunit (Ecat) (51).
The binding of FAs to Eallo versus Ecat can be estimated by measuring the levels of unreacted [14C] AA, which is that bound to Eallo, after incubations performed at high enzyme to substrate concentrations (e.g. using 1 μm [14C]AA with 0.1 to 2 μm COX (51)).
- PGHS
- prostaglandin endoperoxide H synthase
- 2-AG
- 2-arachidonoylglycerol
- 1-AG
- 1-arachidonoylglycerol
- 13-Me-AA
- 13-methylarachidonic acid
- AA
- arachidonic acid
- AEA
- anandamide
- COX
- cyclooxygenase
- DHA
- docosahexaenoic acid
- DHLA
- dihomo-γ-linolenic acid
- DPA
- docosapentaenoic acid
- EPA
- eicosapentaenoic acid
- FA
- fatty acid
- hu
- human
- MBD
- membrane binding domain
- nsNSAIDs
- nonspecific nonsteroidal anti-inflammatory drugs
- ov
- ovine
- PA
- palmitic acid
- PG
- prostaglandin
- POX
- peroxidase
- ER
- endoplasmic reticulum.
References
- 1. Tsai A. L., and Kulmacz R. J. (2010) Prostaglandin H synthase: resolved and unresolved mechanistic issues. Arch. Biochem. Biophys. 493, 103–124 10.1016/j.abb.2009.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smith W. L., Urade Y., and Jakobsson P. J. (2011) Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 111, 5821–5865 10.1021/cr2002992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Leslie C. C. (2015) Cytosolic phospholipase A2: physiological function and role in disease. J. Lipid Res. 56, 1386–1402 10.1194/jlr.R057588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Murakami M., Yamamoto K., Miki Y., Murase R., Sato H., and Taketomi Y. (2016) The roles of the secreted phospholipase A2 gene family in immunology. Adv. Immunol. 132, 91–134 10.1016/bs.ai.2016.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hamberg M., and Samuelsson B. (1967) Oxygenation of unsaturated fatty acids by the vesicular gland of sheep. J. Biol. Chem. 242, 5344–5354 [PubMed] [Google Scholar]
- 6. Liu Y., and Roth J. P. (2016) A revised mechanism for human cyclooxygenase-2. J. Biol. Chem. 291, 948–958 10.1074/jbc.M115.668038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rouzer C. A., and Marnett L. J. (2008) Non-redundant functions of cyclooxygenases: oxygenation of endocannabinoids. J. Biol. Chem. 283, 8065–8069 10.1074/jbc.R800005200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rouzer C. A., and Marnett L. J. (2011) Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem. Rev. 111, 5899–5921 10.1021/cr2002799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DeWitt D. L., el-Harith E. A., Kraemer S. A., Andrews M. J., Yao E. F., Armstrong R. L., and Smith W. L. (1990) The aspirin and heme-binding sites of ovine and murine prostaglandin endoperoxide synthases. J. Biol. Chem. 265, 5192–5198 [PubMed] [Google Scholar]
- 10. Lecomte M., Laneuville O., Ji C., DeWitt D. L., and Smith W. L. (1994) Acetylation of human prostaglandin endoperoxide synthase-2 (cyclooxygenase-2) by aspirin. J. Biol. Chem. 269, 13207–13215 [PubMed] [Google Scholar]
- 11. Loll P. J., Picot D., and Garavito R. M. (1995) The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nat. Struct. Biol. 2, 637–643 10.1038/nsb0895-637 [DOI] [PubMed] [Google Scholar]
- 12. Rimon G., Sidhu R. S., Lauver D. A., Lee J. Y., Sharma N. P., Yuan C., Frieler R. A., Trievel R. C., Lucchesi B. R., and Smith W. L. (2010) Coxibs interfere with the action of aspirin by binding tightly to one monomer of cyclooxygenase-1. Proc. Natl. Acad. Sci. U.S.A. 107, 28–33 10.1073/pnas.0909765106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dong L., Sharma N. P., Jurban B. J., and Smith W. L. (2013) Pre-existent asymmetry in the human cyclooxygenase-2 sequence homodimer. J. Biol. Chem. 288, 28641–28655 10.1074/jbc.M113.505503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lucido M. J., Orlando B. J., Vecchio A. J., and Malkowski M. G. (2016) Crystal structure of aspirin-acetylated human cyclooxygenase-2: insight into the formation of products with reversed stereochemistry. Biochemistry 55, 1226–1238 10.1021/acs.biochem.5b01378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grosser T., Fries S., and FitzGerald G. A. (2006) Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J. Clin. Invest. 116, 4–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li X., Mazaleuskaya L. L., Yuan C., Ballantyne L. L., Meng H., Smith W. L., FitzGerald G. A., and Funk C. D. (2018) Flipping the cyclooxygenase (Ptgs) genes reveals isoform-specific compensatory functions. J. Lipid Res. 59, 89–101 10.1194/jlr.M079996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li X., Mazaleuskaya L. L., Ballantyne L. L., Meng H., FitzGerald G. A., and Funk C. D. (2018) Differential compensation of two cyclooxygenases in renal homeostasis is independent of prostaglandin-synthetic capacity under basal conditions. FASEB J. 32, 5326–5337 10.1096/fj.201800252R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rollins T. E., and Smith W. L. (1980) Subcellular localization of prostaglandin-forming cyclooxygenase in Swiss mouse 3T3 fibroblasts by electron microscopic immunocytochemistry. J. Biol. Chem. 255, 4872–4875 [PubMed] [Google Scholar]
- 19. Otto J. C., and Smith W. L. (1994) The orientation of prostaglandin endoperoxide synthases-1 and -2 in the endoplasmic reticulum. J. Biol. Chem. 269, 19868–19875 [PubMed] [Google Scholar]
- 20. Spencer A. G., Woods J. W., Arakawa T., Singer I. I., and Smith W. L. (1998) Subcellular localization of prostaglandin endoperoxide H synthases-1 and -2 by immunoelectron microscopy. J. Biol. Chem. 273, 9886–9893 10.1074/jbc.273.16.9886 [DOI] [PubMed] [Google Scholar]
- 21. Yuan C., and Smith W. L. (2015) A cyclooxygenase-2-dependent prostaglandin E2 biosynthetic system in the Golgi apparatus. J. Biol. Chem. 290, 5606–5620 10.1074/jbc.M114.632463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Accioly M. T., Pacheco P., Maya-Monteiro C. M., Carrossini N., Robbs B. K., Oliveira S. S., Kaufmann C., Morgado-Diaz J. A., Bozza P. T., and Viola J. P. (2008) Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 68, 1732–1740 10.1158/0008-5472.CAN-07-1999 [DOI] [PubMed] [Google Scholar]
- 23. Picot D., Loll P. J., and Garavito R. M. (1994) The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature 367, 243–249 10.1038/367243a0 [DOI] [PubMed] [Google Scholar]
- 24. Otto J. C., and Smith W. L. (1996) Photolabeling of prostaglandin endoperoxide H synthase-1 with 3-trifluoro-3-(m-[125I]iodophenyl)diazirine as a probe of membrane association and the cyclooxygenase active site. J. Biol. Chem. 271, 9906–9910 10.1074/jbc.271.17.9906 [DOI] [PubMed] [Google Scholar]
- 25. Spencer A. G., Thuresson E., Otto J. C., Song I., Smith T., DeWitt D. L., Garavito R. M., and Smith W. L. (1999) The membrane binding domains of prostaglandin endoperoxide H synthase-1 and -2: peptide mapping and mutational analysis. J. Biol. Chem. 274, 32936–32942 10.1074/jbc.274.46.32936 [DOI] [PubMed] [Google Scholar]
- 26. Leslie C. C., Gangelhoff T. A., and Gelb M. H. (2010) Localization and function of cytosolic phospholipase A2α at the Golgi. Biochimie 92, 620–626 10.1016/j.biochi.2010.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bhattacharyya D. K., Lecomte M., Rieke C. J., Garavito M., and Smith W. L. (1996) Involvement of arginine 120, glutamate 524, and tyrosine 355 in the binding of arachidonate and 2-phenylpropionic acid inhibitors to the cyclooxygenase active site of ovine prostaglandin endoperoxide H synthase-1. J. Biol. Chem. 271, 2179–2184 10.1074/jbc.271.4.2179 [DOI] [PubMed] [Google Scholar]
- 28. Rieke C. J., Mulichak A. M., Garavito R. M., and Smith W. L. (1999) The role of arginine 120 of human prostaglandin endoperoxide H synthase-2 in the interaction with fatty acid substrates and inhibitors. J. Biol. Chem. 274, 17109–17114 10.1074/jbc.274.24.17109 [DOI] [PubMed] [Google Scholar]
- 29. Malkowski M. G., Ginell S. L., Smith W. L., and Garavito R. M. (2000) The X-ray crystal structure of prostaglandin endoperoxide H synthase-1 complexed with arachidonic acid. Science 289, 1933–1937 10.1126/science.289.5486.1933 [DOI] [PubMed] [Google Scholar]
- 30. Thuresson E. D., Malkowski M. G., Lakkides K. M., Rieke C. J., Mulichak A. M., Ginell S. L., Garavito R. M., and Smith W. L. (2001) Mutational and X-ray crystallographic analysis of the interaction of dihomo-γ-linolenic acid with prostaglandin endoperoxide H synthases. J. Biol. Chem. 276, 10358–10365 10.1074/jbc.M009378200 [DOI] [PubMed] [Google Scholar]
- 31. Vecchio A. J., Simmons D. M., and Malkowski M. G. (2010) Structural basis of fatty acid substrate binding to cyclooxygenase-2. J. Biol. Chem. 285, 22152–22163 10.1074/jbc.M110.119867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vecchio A. J., Orlando B. J., Nandagiri R., and Malkowski M. G. (2012) Investigating substrate promiscuity in cyclooxygenase-2: the role of Arg-120 and residues lining the hydrophobic groove. J. Biol. Chem. 287, 24619–24630 10.1074/jbc.M112.372243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kiefer J. R., Pawlitz J. L., Moreland K. T., Stegeman R. A., Hood W. F., Gierse J. K., Stevens A. M., Goodwin D. C., Rowlinson S. W., Marnett L. J., Stallings W. C., and Kurumbail R. G. (2000) Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature 405, 97–101 10.1038/35011103 [DOI] [PubMed] [Google Scholar]
- 34. Malkowski M. G., Thuresson E. D., Lakkides K. M., Rieke C. J., Micielli R., Smith W. L., and Garavito R. M. (2001) Structure of eicosapentaenoic and linoleic acids in the cyclooxygenase site of prostaglandin endoperoxide H synthase-1. J. Biol. Chem. 276, 37547–37555 10.1074/jbc.M105982200 [DOI] [PubMed] [Google Scholar]
- 35. Malkowski M. G. (2017) in Encyclopedia of Inorganic and Bioinorganic Chemistry (Scott R. A., ed) pp. 1–18, John Wiley & Sons Ltd., Chichester, UK [Google Scholar]
- 36. Vecchio A. J., and Malkowski M. G. (2011) The structural basis of endocannabinoid oxygenation by cyclooxygenase-2. J. Biol. Chem. 286, 20736–20745 10.1074/jbc.M111.230367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Van der Ouderaa F. J., Buytenhek M., Nugteren D. H., and Van Dorp D. A. (1977) Purification and characterisation of prostaglandin endoperoxide synthetase from sheep vesicular glands. Biochim. Biophys. Acta 487, 315–331 10.1016/0005-2760(77)90008-X [DOI] [PubMed] [Google Scholar]
- 38. Swinney D. C., Mak A. Y., Barnett J., and Ramesha C. S. (1997) Differential allosteric regulation of prostaglandin H synthase 1 and 2 by arachidonic acid. J. Biol. Chem. 272, 12393–12398 10.1074/jbc.272.19.12393 [DOI] [PubMed] [Google Scholar]
- 39. Roth G. J., Machuga E. T., and Strittmatter P. (1981) The heme-binding properties of prostaglandin synthetase from sheep vesicular gland. J. Biol. Chem. 256, 10018–10022 [PubMed] [Google Scholar]
- 40. Van Der Ouderaa F. J., Buytenhek M., Nugteren D. H., and Van Dorp D. A. (1980) Acetylation of prostaglandin endoperoxide synthetase with acetylsalicylic acid. Eur. J. Biochem. 109, 1–8 10.1111/j.1432-1033.1980.tb04760.x [DOI] [PubMed] [Google Scholar]
- 41. Kulmacz R. J., and Lands W. E. (1984) Prostaglandin H synthase. Stoichiometry of heme cofactor. J. Biol. Chem. 259, 6358–6363 [PubMed] [Google Scholar]
- 42. Kulmacz R. J., and Lands W. E. (1985) Stoichiometry and kinetics of the interaction of prostaglandin H synthase with anti-inflammatory agents. J. Biol. Chem. 260, 12572–12578 [PubMed] [Google Scholar]
- 43. Rosenstock M., Danon A., Rubin M., and Rimon G. (2001) Prostaglandin H synthase-2 inhibitors interfere with prostaglandin H synthase-1 inhibition by nonsteroidal anti-inflammatory drugs. Eur. J. Pharmacol. 412, 101–108 10.1016/S0014-2999(00)00931-6 [DOI] [PubMed] [Google Scholar]
- 44. Burde T., and Rimon G. (2002) On the interaction of specific prostaglandin H synthase-2 inhibitors with prostaglandin H synthase-1. Eur. J. Pharmacol. 453, 167–173 10.1016/S0014-2999(02)02450-0 [DOI] [PubMed] [Google Scholar]
- 45. Yuan C., Rieke C. J., Rimon G., Wingerd B. A., and Smith W. L. (2006) Partnering between monomers of cyclooxygenase-2 homodimers. Proc. Natl. Acad. Sci. U.S.A. 103, 6142–6147 10.1073/pnas.0601805103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wada M., DeLong C. J., Hong Y. H., Rieke C. J., Song I., Sidhu R. S., Yuan C., Warnock S., Schmaier A. H., Yokoyama C., Smyth E. M., Wilson S. J., FitzGerald G. A., Garavito R. M., Sui de X., Regan J. W., and Smith W. L. (2007) Specificities of enzymes and receptors of prostaglandin pathways with arachidonic acid and eicosapentaenoic acid derived substrates and products. J. Biol. Chem. 282, 22254–22266 10.1074/jbc.M703169200 [DOI] [PubMed] [Google Scholar]
- 47. Culp B. R., Titus B. G., and Lands W. E. (1979) Inhibition of prostaglandin biosynthesis by eicosapentaenoic acid. Prostaglandins Med. 3, 269–278 10.1016/0161-4630(79)90068-5 [DOI] [PubMed] [Google Scholar]
- 48. Liu W., Cao D., Oh S. F., Serhan C. N., and Kulmacz R. J. (2006) Divergent cyclooxygenase responses to fatty acid structure and peroxide level in fish and mammalian prostaglandin H synthases. FASEB J. 20, 1097–1108 10.1096/fj.05-5273com [DOI] [PubMed] [Google Scholar]
- 49. Sharma N. P., Dong L., Yuan C., Noon K. R., and Smith W. L. (2010) Asymmetric acetylation of the cyclooxygenase-2 homodimer by aspirin and its effects on the oxygenation of arachidonic, eicosapentaenoic, and docosahexaenoic acids. Mol. Pharmacol. 77, 979–986 10.1124/mol.109.063115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yuan C., Sidhu R. S., Kuklev D. V., Kado Y., Wada M., Song I., and Smith W. L. (2009) Cyclooxygenase allosterism, fatty acid-mediated cross-talk between monomers of cyclooxygenase homodimers. J. Biol. Chem. 284, 10046–10055 10.1074/jbc.M808634200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dong L., Vecchio A. J., Sharma N. P., Jurban B. J., Malkowski M. G., and Smith W. L. (2011) Human cyclooxygenase-2 is a sequence homodimer that functions as a conformational heterodimer. J. Biol. Chem. 286, 19035–19046 10.1074/jbc.M111.231969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zou H., Yuan C., Dong L., Sidhu R. S., Hong Y. H., Kuklev D. V., and Smith W. L. (2012) Human cyclooxygenase-1 activity and its responses to COX inhibitors are allosterically regulated by nonsubstrate fatty acids. J. Lipid Res. 53, 1336–1347 10.1194/jlr.M026856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kudalkar S. N., Nikas S. P., Kingsley P. J., Xu S., Galligan J. J., Rouzer C. A., Banerjee S., Ji L., Eno M. R., Makriyannis A., and Marnett L. J. (2015) 13-Methylarachidonic acid is a positive allosteric modulator of endocannabinoid oxygenation by cyclooxygenase. J. Biol. Chem. 290, 7897–7909 10.1074/jbc.M114.634014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dong L., Zou H., Yuan C., Hong Y. H., Kuklev D. V., and Smith W. L. (2016) Different fatty acids compete with arachidonic acid for binding to the allosteric or catalytic subunits of cyclooxygenases to regulate prostanoid synthesis. J. Biol. Chem. 291, 4069–4078 10.1074/jbc.M115.698001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dong L., Yuan C., Orlando B. J., Malkowski M. G., and Smith W. L. (2016) Fatty acid binding to the allosteric subunit of cyclooxygenase-2 relieves a tonic inhibition of the catalytic subunit. J. Biol. Chem. 291, 25641–25655 10.1074/jbc.M116.757310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sidhu R. S., Lee J. Y., Yuan C., and Smith W. L. (2010) Comparison of cyclooxygenase-1 crystal structures: cross-talk between monomers comprising cyclooxygenase-1 homodimers. Biochemistry 49, 7069–7079 10.1021/bi1003298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Smith W. L., and Lands W. E. (1971) Stimulation and blockade of prostaglandin biosynthesis. J. Biol. Chem. 246, 6700–6702 [PubMed] [Google Scholar]
- 58. Rome L. H., and Lands W. E. (1975) Structural requirements for time-dependent inhibition of prostaglandin biosynthesis by anti-inflammatory drugs. Proc. Natl. Acad. Sci. U.S.A. 72, 4863–4865 10.1073/pnas.72.12.4863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Percival M. D., Ouellet M., Vincent C. J., Yergey J. A., Kennedy B. P., and O'Neill G. P. (1994) Purification and characterization of recombinant human cyclooxygenase-2. Arch. Biochem. Biophys. 315, 111–118 10.1006/abbi.1994.1478 [DOI] [PubMed] [Google Scholar]
- 60. Laneuville O., Breuer D. K., Dewitt D. L., Hla T., Funk C. D., and Smith W. L. (1994) Differential inhibition of human prostaglandin endoperoxide H synthases-1 and -2 by nonsteroidal anti-inflammatory drugs. J. Pharmacol. Exp. Ther. 271, 927–934 [PubMed] [Google Scholar]
- 61. Duggan K. C., Hermanson D. J., Musee J., Prusakiewicz J. J., Scheib J. L., Carter B. D., Banerjee S., Oates J. A., and Marnett L. J. (2011) (R)-Profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2. Nat. Chem. Biol. 7, 803–809 10.1038/nchembio.663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Duggan K. C., Walters M. J., Musee J., Harp J. M., Kiefer J. R., Oates J. A., and Marnett L. J. (2010) Molecular basis for cyclooxygenase inhibition by the non-steroidal anti-inflammatory drug naproxen. J. Biol. Chem. 285, 34950–34959 10.1074/jbc.M110.162982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Prusakiewicz J. J., Duggan K. C., Rouzer C. A., and Marnett L. J. (2009) Differential sensitivity and mechanism of inhibition of COX-2 oxygenation of arachidonic acid and 2-arachidonoylglycerol by ibuprofen and mefenamic acid. Biochemistry 48, 7353–7355 10.1021/bi900999z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dong L., Zou H., Yuan C., Hong Y. H., Uhlson C. L., Murphy R. C., and Smith W. L. (2016) Interactions of 2-O-arachidonylglycerol ether and ibuprofen with the allosteric and catalytic subunits of human COX-2. J. Lipid Res. 57, 1043–1050 10.1194/jlr.M067512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Blobaum A. L., Xu S., Rowlinson S. W., Duggan K. C., Banerjee S., Kudalkar S. N., Birmingham W. R., Ghebreselasie K., and Marnett L. J. (2015) Action at a distance: mutations of peripheral residues transform rapid reversible inhibitors to slow, tight binds of cyclooxygenase-2. J. Biol. Chem. 290, 12793–12803 10.1074/jbc.M114.635987 [DOI] [PMC free article] [PubMed] [Google Scholar]