

Abstract
Neutrophils have a critical role in regulating the immune system. The immune system is compromised during chemotherapy, increasing infection risks and imposing a need for regular monitoring of neutrophil counts. Although commercial hematology analyzers are currently used in clinical practice for neutrophil counts, they are only available in clinics and hospitals, use large blood volumes, and are not available at the point of care (POC). Additionally, phlebotomy and blood processing require trained personnel, where patients are often admitted to hospitals when the infections are at late stage due to lack of frequent monitoring. Here, a reliable method is presented that selectively captures and quantifies white blood cells (WBCs) and neutrophils from a finger prick volume of whole blood by integrating microfluidics with high-resolution imaging algorithms. The platform is compact, portable, and easy to use. It captures and quantifies WBCs and neutrophils with high efficiency (>95%) and specificity (>95%) with an overall 4.2% bias compared to standard testing. The results from a small cohort of patients (N = 11 healthy, N = 5 lung and kidney cancer) present a unique disposable cell counter, demonstrating the ability of this tool to monitor neutrophil and WBC counts within clinical or in resource-constrained environments.
Keywords: cancer monitoring, lens-free shadow imaging, microfluidic, neutropenia, neutrophil, Poisson distribution
1. Introduction
Neutrophils are important elements of an innate immune system and their number in blood is a valuable indicator of general health status.[1] Particularly, low neutrophil count in blood (neutropenia, Table 1) is attributed to various medical factors including autoimmune deficiency, congenital genetic disorder, and drugs used in chemotherapy and is a serious health complication due to the effects of a compromised immune system.[1–6] In severe and prolonged neutropenia, patients are significantly susceptible to bacterial infections, which causes delays in chemotherapy cycle, limits the dose of chemotherapeutic drugs, and substantially increases healthcare expenditures.[7–11] Further, chemotherapy-induced neutropenia is also associated with mortality that accounts for ≈9.5% of the cancer-related deaths in the United States.[12–16]
Table 1.
Neutropenia classification.[48]
| Absolute neutrophil count, ANC, (no. of cells per μL of blood) | Grade | Risk of infection |
|---|---|---|
| ANC > 1500 | grade 1, normal | Mild |
| 1500 > ANC > 1000 | grade 2, neutropenia | Moderate |
| 1000 > ANC > 500 | grade 3, neutropenia | Severe |
| ANC < 500 | grade 4, severe neutropenia | Life-threatening |
In clinical settings, neutrophil counts are determined by performing a complete blood count (CBC) in hospitals and centralized laboratories with commercial hematology analyzers by skilled personnel using large volumes (up to 6 mL) of whole blood obtained by venipuncture.[17] Further, due to lack of frequent monitoring, neutropenic patients are often admitted to hospitals when the infections are in advanced stages or of prolonged duration since inflammatory immune response is minimal, and neutropenia may also develop asymptomatically until late stages.[5,7,13,16] In addition, minors and elderly cancer patients face critical constraints, including limited mobility for frequent clinic visits and invasive phlebotomy practices.
Currently, various microfluidic-based devices and techniques are utilized to capture and count cells from blood including fluorescence microscopy, CMOS-based holographic imaging, and impedance measurement (Table 2).[18–27] However, these systems have critical impediments such as low capture efficiency with high biases in measurements, need for multiple sample preprocessing steps (i.e., centrifugation), and costly read-out systems, limiting their applicability at point of care (POC). Although microfluidic immune-based capture methods to isolate white blood cell (WBC) subtypes have been demonstrated including CD4 cells and CD8 cells,[26] capture of neutrophils has been particularly challenging with limited specificity and efficiency.[27] Therefore, there is an urgent need for inexpensive, easy-to-use, and robust devices and techniques to monitor neutrophil counts at the POC for patients with cancer during chemotherapy.[28,29]
Table 2.
Comparison of current microfluidic-based cell counting methods.
| Capture method | Target cell | Antibody concentration | Quantification method | Blood sample | Cell count accuracy | Sample preparation | Reference |
|---|---|---|---|---|---|---|---|
| Immunocapturing | CD4+ T-cells | 10 μg mL−1 | Bright-field microscopy | Whole blood | High bias at >800 cells (μL−1) | Unprocessed | [18] |
| Hydrodynamic filtration/enrichment | WBCs | N/A | Microscopy/staining on glass | Diluted whole blood | High bias | Unprocessed | [19] |
| Microfluidic flow | WBC subgroups | N/A | Impedance spectroscopy fluorescence | Purified cell subtypes | Moderate | Multistep processing | [20] |
| Immunocapturing | CD4+ T-cells | 10 μg mL−1 | Chemiluminescence fluorescence | Whole blood | Moderate | Multistep processing | [21] |
| Microfluidic flow | WBC subgroups | N/A | Impedance spectroscopy | Lysed blood | Moderate high bias at high counts | Multistep processing | [22] |
| Microfluidic flow dry smear | WBCs/RBCs | N/A | CMOS imaging | Whole blood | High bias | Unprocessed | [23] |
| Microfluidic flow | WBCs | N/A | Fluorescence CMOS imaging | Diluted, labeled whole blood | N/A in low range | Fluorescence labeling | [24] |
| Microfluidic capturing | WBCs | N/A | Fluorescence CMOS imaging | Diluted, labeled whole blood | Moderate N/A in low range | Fluorescence labeling | [25] |
| Immunocapturing | CD4+ and CD8+ T-cells | N/A | Impedance spectroscopy | Whole lysed blood | Moderate (high bias) | Multistep processing after capturing | [26] |
| Immunocapturing | Neutrophils | N/A | Immunofluorescence imaging | Whole blood | Moderate (high bias) N/A in low range | Low | [27] |
| Our platform | Neutrophils | 100 μg mL−1 | CMOS imaging | Whole-blood dilution PBS/RBC lysis | High | Low | This study |
Here, we present a microfluidic platform integrated with a lens-free shadow imaging system to capture and quantify neutrophils accurately using a high-resolution image algorithm[30] from a finger prick volume of whole blood (5.5 μL). The platform is compact, portable, inexpensive, disposable, easy to use, and can capture neutrophils and WBCs with over 95% efficiency. The specificity of the device is over a wide range of cell counts, including neutropenic regime (<2000 cells μL−1) at high accuracy with overall 4.2% bias in between measurements. Further, data from a small cohort patients (N = 11 healthy, N = 5 lung and kidney cancer) showed that the platform also provides neutrophil counts from clinical samples reliably as compared to the clinical gold standard (i.e., hematology analyzer count). Such a system could potentially reduce costs associated with hospitalization and eliminate impediments associated with current clinical practices, offering an improved quality of life.
2. Results
2.1. Optimization of Experimental Variables to Improve the Cell Capture Efficiency
To evaluate the neutrophil capture efficiency in microfluidic channels, we utilized three experimental variables: flow rate, antibody concentration, and blood incubation time (refer to the Experimental Section for further details of materials and methods) (Figure 1A–F, and Figure S1–S3, Supporting Information). Here, we initially investigated the effect of flow rate on capture efficiency for neutrophils in the microfluidic channels using three different flow rates: 5 μL min−1, 20 μL min−1, and hand pipetting (≈200 μL min−1), keeping antibody concentration constant at 50 μg mL−1. We calculated the captured efficiencies (Equation (1)) as 62% ± 3%, 52% ± 5%, and 32% ± 9% for these flow rates, respectively (Figure 2A) Then, keeping the flow rate constant (5 μL min−1), we evaluated three anti-CD66b antibody concentrations, namely, 25, 50, and 100 μg mL−1, and we observed the capture efficiencies 54% ± 7%, 69% ± 2%, and 99% ± 2%, respectively (Figure 2B). To evaluate the effect of incubation time on capture efficiency, we incubated diluted blood in microfluidic channels for 5 and 20 min, and compared our results with blood without incubation. The capture efficiency was calculated as 70% ± 4%, 99% ± 2%, and 44% ± 9%, respectively (Figure 2C). The results presented the highest capture efficiency for immune cells in a microfluidic channel for 5 μL min−1 flow rate with 100 μg mL−1 antibody concentration and 20 min of incubation time (Figure 2D). In these experiments, we initially load the channels with samples under flow, then incubated the devices without active flow. Finally, we run wash solution with the initial flow rate that varies in each optimization step to remove unbound cells.
Figure 1.
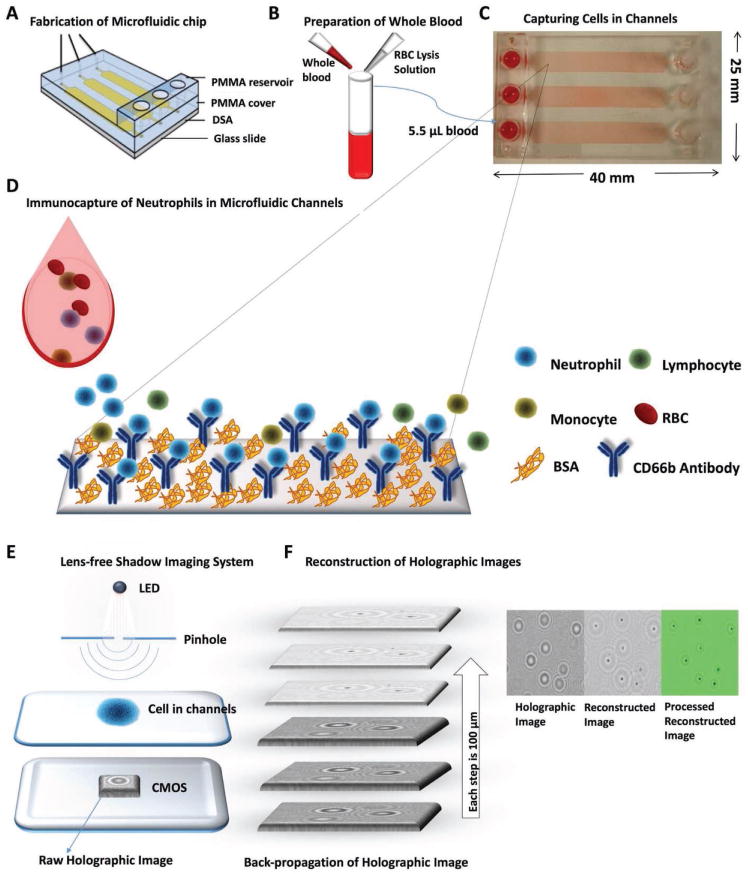
Lens-free shadow imaging platform for capturing and quantification of neutrophils to monitor neutropenia at the POC. A) PMMA, DSA, and microscope glass slide are assembled to fabricate the microfluidic chip with channels (height: 50 μm). B) Finger prick volume of whole blood is diluted with PBS/RBC lysis buffer in 1:5. C) Blood was withdrawn with a syringe pump (flow rate 5 μL min−1) into microfluidic channels. D) A biotinylated antibody specific to CD45 and CD66 cell surface marker binds to white blood cells and neutrophils, respectively, on the channel surfaces. The remaining unbound cells were washed away with PBS. The channel surfaces were also coated with BSA to prevent nonspecific binding. E) Captured neutrophils are then imaged using a lens-free shadow imaging system. The images were then processed with the ImageJ software to quantify the cell number. F) The patterns formed holographic images on CMOS sensor and reconstructed using an in-house written image processing algorithm by back-propagation method for every 100 μm distance starting from the raw image. The algorithm resolves the diffraction patterns and places a dot at the center of each holographic set. These images were further processed to improve quality.
Figure 2.
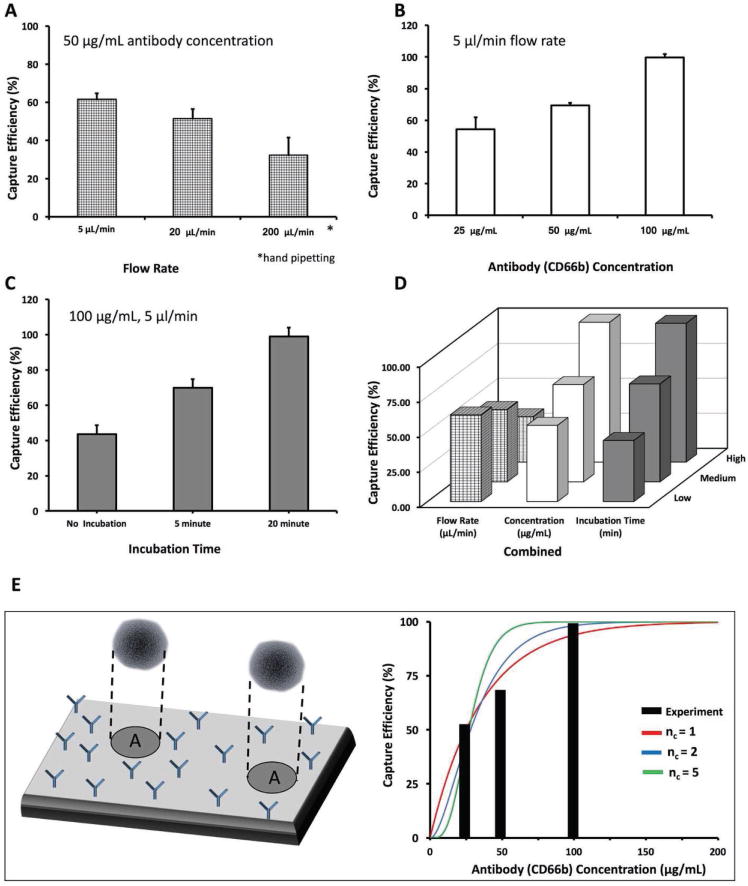
Optimization of the experimental variables. To define the optimum conditions for cell capture, we examined the effect of three interrelated variables: flow rate, capture antibody concentration, and incubation time of blood. A) To optimize the flow rate, we kept the antibody concentration constant and varied the flow rate. We observed that the highest capture efficiency was achieved with 5 μL min−1 flow rate. B) For optimizing antibody concentration, we kept the flow rate constant at 5 μL min−1, and varied the antibody concentration. We observed that the highest capture efficiency was achieved using 100 μg mL−1. C) For optimizing the incubation time, we kept the antibody concentration at 100 μg mL−1, the flow rate at 5 μL min−1, and used three different time periods. D) A graph of all these three parameters together, showing that the highest capture efficiency was achieved with 20 min incubation time, 5 μL min−1 flow rate, and 100 μg mL−1 antibody concentration. E) Results of a theoretical model that uses a Poisson distribution of cells within the microfluidic channel surface, captured by varying number of surface antibodies (anti-CD66b antibodies). The model suggests that the 2–5 binding sites per neutrophil is enough to achieve the observed collection efficiencies and fits the form of the experimental efficiency curve.
2.2. Modeling of Neutrophil Capture in a Microfluidic Channel
To evaluate the experimental findings, we utilized a Poisson model of attachment and detachment of cells (Figure 2E). Given that even at relatively slow perfusion speeds, the majority of neutrophils will be suspended in the bulk of the microfluidic device and few will be able to bind, we assume that no binding will happen during the initial flow-in or final washing steps. We also consider how quickly neutrophils will settle to the chip surface and determine whether at the 20 min incubation time, all neutrophils are able to reach the active surface of the device. On the active surface of the device, we consider that there will be some number of adhesion points, due to the presence of the antibodies. It is unknown how many of these there will be, due to many experimental variables, including rate of antibody binding to the surface, self-binding, and diffusion from the bulk to the surface. To quantify this, we assume that the number of active binding sites is proportional to the concentration of antibodies.
To determine the capture rate, we randomly distribute the binding sites onto a target surface, and then randomly distribute neutrophils onto the surface. If the neutrophil surface area includes nc or greater binding sites, it is considered permanently captured, while if it has fewer, it is considered lost. We repeat this across many trials to determine the overall capture rate. The results are shown in Figure 2E (obtained using Equation (4)) where the model fits the experimental data well, and suggests at the order of 2–5 binding sites per neutrophil is enough to achieve the observed collection efficiencies.
2.3. Cell Capture from Whole Blood in Different Buffer Solutions with Various Dilutions
To evaluate and compare the capture efficiencies in different buffer solutions and concentrations, we utilized various dilutions of whole blood in PBS (i.e., undiluted whole blood, 1:2, 1:5, 1:10, 1:20, and 1:30 diluted whole blood solutions) and in red blood cell (RBC) lysis buffer (1:5 diluted whole blood solution) (Figure S4, Supporting Information). We observed that capture efficiency was ≈30% for unprocessed whole blood in all cases. This low efficiency was attributed to overwhelming number of red blood cells present in blood (there are ≈1000 RBCs present in whole blood for every WBC). To validate this negative interference of RBCs on WBC capturing in microfluidic channels, we compared the number of captured cells in whole blood with RBC depleted whole blood. We observed that the number of captured cells was 11 267 in whole blood while the number was significantly increased to 35 634 in RBC depleted whole blood (Figure S5, Supporting Information) sample. To avoid this negative interference, we then diluted blood samples in PBS and observed that the absolute number of captured cells decreased in higher dilutions (i.e., 4875 cells in 1:2 PBS diluted solution and 1251 cells in 1:30 diluted solution) (Figure 3A) as the result of dilution. In contrast, capture efficiency increased in solutions with higher dilution ratio (i.e., 26% capture efficiency in 1:2 dilution whereas >95% capture efficiency in 1:30 dilution). In addition, we also evaluated the capture efficiency by diluting blood samples using RBC lysing buffer (no processing such as centrifugation). Further, the capture efficiency for 1:5 diluted sample was recorded as higher than 95% (Figure 3B). We observed capture efficiencies higher than 95% with PBS buffer at 1:30 dilution and with RBC lysing buffer at 1:5 dilution. In addition, with PBS-diluted samples, we observed nonspecific RBC binding in the channels in post-washing steps, however with the RBC lysing buffer the negative interference (nonspecific binding) was eliminated due to ruptured RBCs. In addition, we also evaluated the capture efficiency of cells in a control channel, which do not have any antibodies on the glass surface (all the other steps are similarly performed). This channel provided no significant fluorescent signal for quantitative comparison as seen in Figure S4F (Supporting Information), which suggests that the nonspecific binding of the targeted cells (neutrophils and WBCs) was not present.
Figure 3.
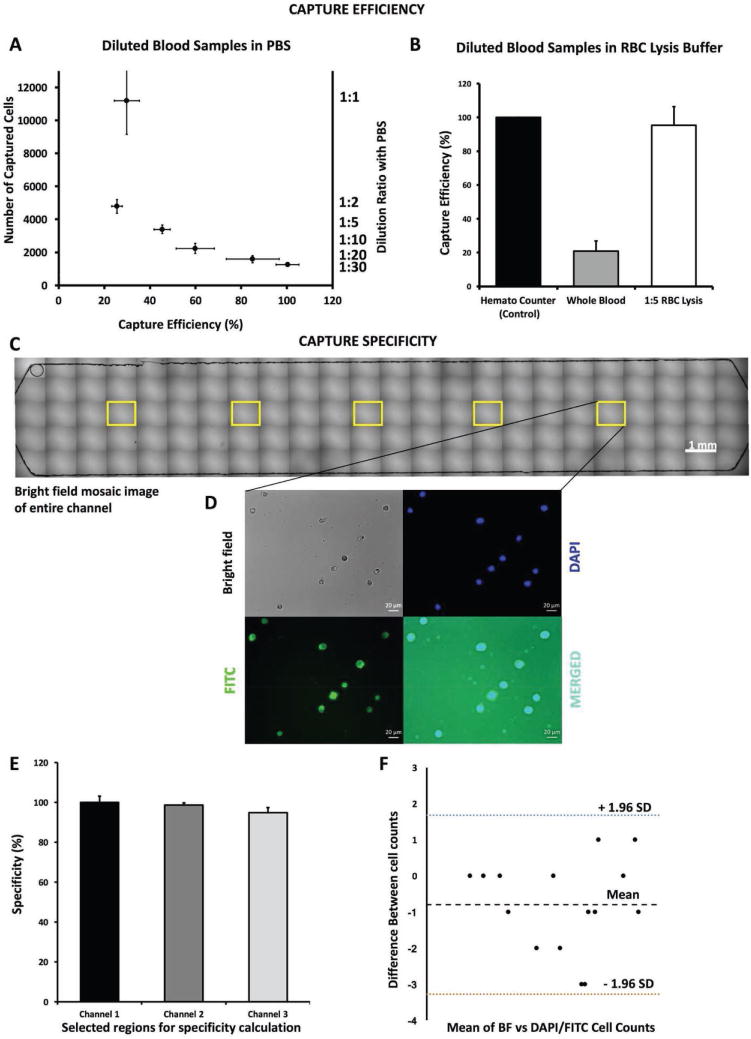
Effect of different buffer solutions with varying dilutions on cell capture efficiency. To evaluate the effect of different buffers on cell capture efficiencies, we utilized PBS and RBC lysis buffer solutions for dilution of whole blood. A) In PBS, we used six sample solutions diluted with PBS (undiluted whole blood (WB)). We observed that the number of captured cells decreased with higher dilutions, while the capture efficiency increased with higher dilutions, and the highest efficiency was achieved at 1:30 dilution (>95%). We also observed that the lowest efficiency was obtained when undiluted whole blood samples were used and this was attributed to significantly high number of RBCs present in blood, thus preventing white blood cells to bind to the functionalized surface. B) To assess the effect of RBC lysis, we compared undiluted whole blood and whole blood diluted 1:5 in lysis buffer. C) To evaluate the specificity of the captured cells, we calculated the number of captured neutrophils, compared them using microscopy images taken from five randomly assigned regions within each microfluidic channel (yellow labeled regions of interests), and created from mosaic of 120 microscopy images. D) We used bright field light (gray), DAPI fluorescence (DNA stain, blue), and FITC fluorescence microscopy (anti-CD66b antibody staining, green) images and merged them to see overlapping cells for specificity determination. E) We obtained an average specificity percent higher than 95% in all regions after comparing the number of the cells in each imaging modality. F) We then used the Bland–Altman method to analyze the difference in variation between imaging techniques and we found HLoA as 1.68%, LLoA as −3.28%, and mean as −0.8%.
2.4. Specificity Evaluation for Neutrophil Capturing
To determine the capture specificity for neutrophils in microfluidic channels, we acquired microscopic images of the entire channel using bright-field (BF) and fluorescence microscopy (i.e., DAPI as marker for all WBCs, FITC for CD66b-positive neutrophils). We evaluated five randomly selected, equidistant regions of interest in each channel (Figure 3C,D, and Figure S6, Supporting Information) (number of channels: N = 3, number of regions: n = 5) and counted the captured cells in the microscopy images. The capture specificity for neutrophils was calculated using Equation (2). We observed that in each microfluidic channel, the neutrophils were captured with a significantly high specificity of 98.02% ± 2.72% indicating robustness of surface functionalization and experimental procedure (Figure 3E). We then used the Bland–Altman method to analyze the difference in counts between BF and other imaging modalities and found that the higher limit of agreement (HLoA) was 1.68% and lower LoA (LLoA) was −3.28% with mean of −0.8%.[31]
2.5. Proof of Concept for a Lens-Free Shadow Imaging System Integrated with Microfluidic Device
To evaluate the efficiency, accuracy, and integration performance of the lens-free shadow imaging system with the microfluidic device, we compared BF microscopy and DAPI fluorescence images with holographic and reconstructed lens-free shadow images of the captured cells in the channels (Figure 4A–D). The images taken from five randomly selected regions in the channels indicated highly overlapping cell morphology in the lens-free shadow imaging system, as compared to fluorescence and BF microscopy images (Figure 4 insets). We obtained average cell specificity of 98.01% ± 3.52% (N = 5, number of regions, for n = 3, imaging modalities, for all three imaging mechanisms) in all these cases (Figure 4E). We also showed the difference variation in between these counts using Bland–Altman graph and found a mean of number difference at −4.7 (Figure 4F). These results showed that the microfluidic device, integrated with the lens-free shadow imaging system, can be utilized for capturing and quantification of neutrophils from whole blood with high efficiency and accuracy.
Figure 4.
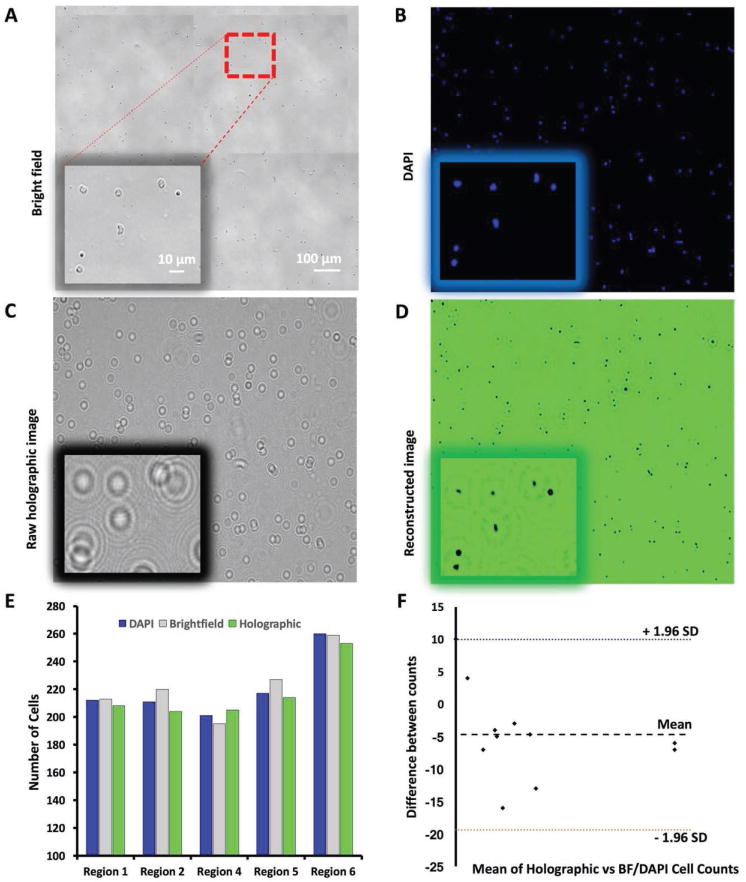
Evaluation of lens-free shadow imaging system integrated with the microfluidic device. A) To evaluate the capture specificity of the lens-free shadow imaging system, we obtained a bright-field image of the entire channel with a 10× microscope objective, and identified five random regions of interest for cell counting, and compared the results using all four imaging techniques. The red-dashed square shows the actual region that is in the insets of each image taken with 40× magnification. B) An image of the one of randomly selected five regions taken with a DAPI filter (blue). C) A holographic image obtained from the same region with the lens-free shadow imaging system. D) The reconstructed image produced by the deconvolution of the holographic image of the same region using the angular spectrum method and the back-propagation algorithm. E) We calculated the number of cells in each of the five regions using bright field microscopy, fluorescent microscopy, and the lens-free reconstructed system images, and obtained a comparable number count in each region. We compared the cell counts between reconstructed images obtained via DAPI staining and bright field microscopy, and used their count ratio as specificity. F) We then used the Bland–Altman method to analyze the cell count differences between imaging techniques. We found HLoA as 9.96, LLoA as −19.39, and mean of −4.7.
2.6. Evaluation of Microfluidic Device Performance and Validation of the Platform with Clinical Samples
We analyzed the performance of the platform in comparison to the gold standard method (i.e., hematology analyzer count using the Beckman Coulter Ac⋅T-diff Hematology Analyzer) using healthy control and cancer patient blood samples (Figure 5A). We observed that our device performed comparably with the commercially available hematology analyzer in capturing and quantifying of WBCs and neutrophils (based on CD45 or CD66b surface antigen capture, respectively), with a linear regression R2 value of 0.99 (N = 11, number of devices, n = 1,2, and 3, number of channels) (Figure 5B). We then utilized the platform using clinical samples from kidney and lung cancer patients to capture and quantify neutrophils and observed a correlation with a R2 value of 0.91 (N = 5, number of devices, n = 1,2, and 3, number of channels) (Figure 5C). We also compared the methods using a student t-test and found no significant difference between them (for p-value threshold set at 0.05, t = 0.36). When we analyzed all the measurements using each channel, we observed that the platform overall provided very reliable measurements that are in good correlation with the analyzer (R2 = 0.97 for N = 36 channels). We also found that the platform has the capability to measure the neutrophil counts in the neutropenic regime (less than 2000 cells) obtained with diluted samples (Figure 5D). We then utilized the accuracy parameter described in the manual of the analyzer (Beckman-Coulter Ac.T-diff hematology analyzer), which is ±300 cells (μL−1) of blood or ±5% bias in between measurements for WBCs and neutrophils. The microfluidic device, integrated with the lens-free shadow imaging system, could capture and quantify the neutrophils and WBCs from blood samples with comparable efficiency, with 241.1 cells at ±4.2% bias (Figure 5E). In addition, we also obtained high counts of immune cells (about 10 000 and more cells) that can be attributed to infections in blood. These results indicate that the platform can potentially be used with high accuracy, comparable with commercially available hematology blood counters, using significantly lower volumes of blood in comparison.
Figure 5.
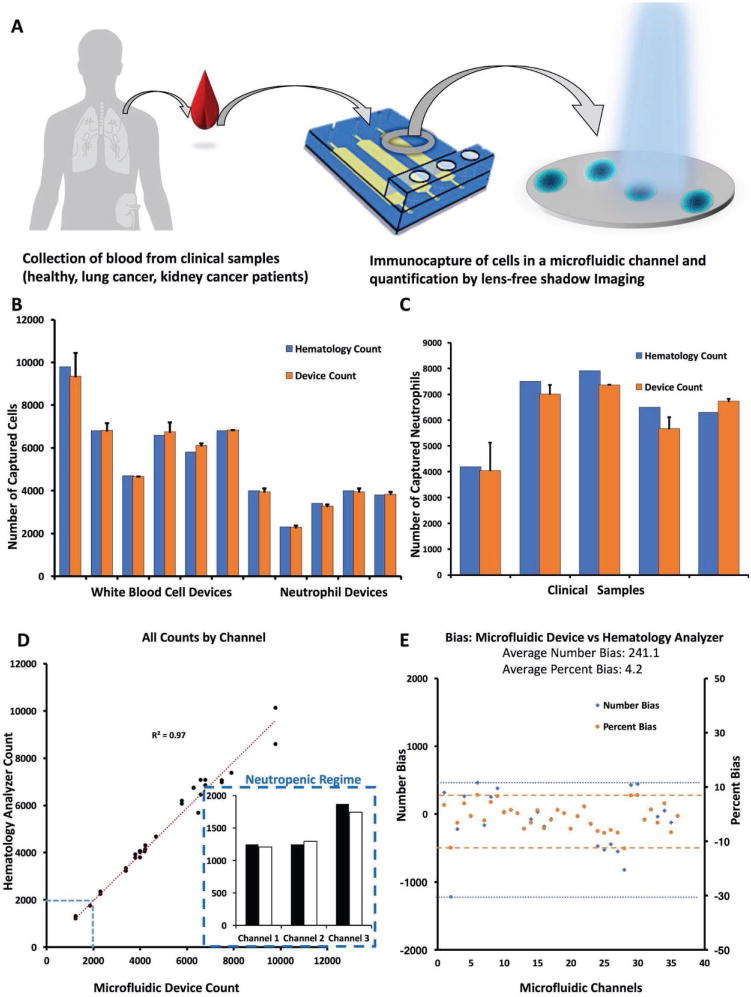
Performance evaluation of the platform and validation with clinical samples. We evaluated the platform capacity and performance using healthy control samples and with samples from patients with lung and kidney cancer and compared with the Beckman–Coulter Ac⋅T-diff Hematology Analyzer. A) The experiment was performed with WBCs, as well as neutrophils from whole blood obtained from healthy individuals and cancer patients. 20 μL of whole blood was diluted with 1:5 RBC lysis buffer, withdrawn and incubated in the microfluidic channels. The cells of interest were captured in the channels using capture antibodies (CD45 antibody for WBCs and CD66b for neutrophils). B) We compared the number of WBCs and neutrophils from healthy controls using the platform and gold standard commercial hematology analyzer, and they provided a linear correlation with R2 of 0.99 (N = 11, number of health sample devices, n = 1, 2, and 3, number of channels). C) We then compared the neutrophil count from cancer patients using lens-free shadow imaging system and hematology analyzer. The clinical samples provided a good correlation with R2 of 0.91 (N = 5, number of patient sample devices, n = 1, 2, and 3, number of channels in each device). We also compared the methods using a t-test and found no significant difference between them (for p-value threshold set at 0.05, t = 0.36). D) We then compared all the microfluidic device counts with hematology analyzer using measurements from each channel. We found that the platform has significantly comparable performance with R2 value of 0.97 (n = 36). The platform was also able to measure the cell counts that were in the neutropenic regime, as shown in the inset (these counts were obtained from 1:20 and 1:30 diluted blood samples). E) We then calculated the overall average bias in the number of cells and found it to be ±241.1 cells with an average of 4.2%, which is comparable to the gold standard (the accuracy error margin in commercial analyzer is ±5%, and 300 cells in the device manual).
3. Discussion
Neutropenia poses a significant health risk to patients with cancer, particularly during chemotherapy due to suppressed immune mechanisms.[14,16,32] Therefore, regular monitoring of patient health status carries utmost importance in environments, where resources are limited, especially for neonatal, pediatric, and elderly patients (particularly those with small veins) for which frequent clinic visits and phlebotomy are burdensome and discouraging. Here, we presented a microfluidic device integrated with a lens-free shadow imaging platform, which is a versatile tool for common hematology blood counts performed at clinics, particularly with a potential at the POC. The platform provided reliable results, with accuracy comparable to currently used hematology analyzers. Additionally, lens-free shadow imaging systems have been utilized for cell imaging in various studies (Table 2).[30,33–37] Here, one of the advantages of using this system is to image a significantly large area of interest—since it has a wide field of view of ≈30 mm2—as compared to regular light microscopes.[34] In addition, the commercial analyzers are bulky, expensive, and requires continuous supply of buffers for operation and regular maintenance, which increases the overall cost. The presented platform is relatively inexpensive and uses disposable polymeric materials.
Although immuno-capturing cells from whole blood using microfluidic technology have been studied earlier, low capture efficiencies with high bias between measurements have been reported. These devices use blood volumes larger than their microfluidic channel volume, which results in large variations in cell count measurements.[18,26,27,38,39] These variations may be attributed to high abundance of RBCs in whole blood, and we have shown that this challenge can be overcome by using dilutions with buffers to achieve higher capture efficiency. Specifically, we have shown that the capture efficiency increases significantly with higher dilutions in a PBS buffer.
In current clinical practice, neutrophils and WBCs are counted using sophisticated and expensive devices (such as FACS and hematology analyzers) with labor intensive and invasive blood handling processes commonly performed in centralized laboratories by trained personnel. Further, these devices require costly and highly technical calibration and maintenance during operation. These requirements limit their utilization at the POC, particularly for cancer patients, where frequent clinical visits and blood draws are needed. To address these challenges, our platform presents various improvements including processing of low sample volumes, easy-to-use cell counting, a portable and relatively inexpensive system, and minimally invasive blood collection.
Microfluidic devices provide various advantages over conventional plate-based microwells for diagnostic tests such as blood separation and cell count.[40] Such advantages include, precise control of sample volume with minuscule amount, wide range of geometries mimicking the biological environment, and ability to expose the biological moieties to shear stress using flow thus mimicking their natural environment.[41,42]
Microfluidic technologies can also create technical challenges such as bubble formation, preservation of the devices during transportation to remote areas and storage in these locations, and requirement of an active flow mechanism to wash away the unbound blood components. These processes involve a few manual operations and the use of syringe pumps that may interfere their utilization for POC applications. However, these challenges are usually in part addressed during commercialization for mass production since we envision that a commercialized version of the platform will be fully automated to minimize manual handling. In addition, there are practical innovations that further help address these problems, such as the use of trehalose to increase shelf-life for on-chip immune chemistry, and utilization of externally actuated reservoirs to replace microfluidic pumps, enabling translation.[43] Therefore, the technical challenges present in similar platforms for POC applications can be minimized with these improvements. In the future, these devices integrated with portable platforms have significant promise for broad applications in mobile healthcare applications and can be utilized in detection and diagnosis of various diseases and disorders at the POC.[44–46]
4. Experimental Section
Experimental Procedure
The experimental procedure was performed in four steps: i) the fabrication of the microfluidic device, ii) the functionalization of the glass surface, iii) immunocapturing of total WBCs and neutrophils using the device, and iv) quantification the captured neutrophils using a lens-free shadow imaging platform.
Microfluidic Device Fabrication
To fabricate the microfluidic devices, poly(methyl methacrylate) (PMMA) (McMaster-Carr, Atlanta, GA, USA) and 50 μm thick double-sided adhesive polymer film (DSA, iTapestore, Scotch Plains, NJ, USA) were cut with laser cutter (Versa VLS2.3, Scottsdale, AZ, USA) with channel length of 25 mm × 4 mm width and 50 μm height (Figure 1A, and Figure S1, Supporting Information). Glass slides (VWR International, Radnor, PA, USA) were attached to PMMA layer using DSA to form microfluidic channels. PMMA layer provided an outlet and a reservoir for blood inlet.
Surface Functionalization
Glass slides (24 mm × 40 mm) were cleaned in absolute ethanol (Sigma Aldrich, St. Louis, MO, USA) solution under sonication for 15 min. The slides were then rinsed with deionized water and dried under nitrogen gas. Dried glass slides were treated with O2 plasma (IoN 3 MHz, PVA Tepla, Corona, CA, USA) for 2 min. Plasma-treated glass slides were immediately transferred into a solution of 3-mercaptopropyltrimethoxysilane (3-MPTS) (4% in ethanol, Sigma Aldrich, St. Louis, MO, USA) and incubated for 45 min at room temperature for silanization of the surface. Then, slides were rinsed with 1 mL of ethanol and dried at room temperature. These surface-treated slides were attached to PMMA layer with DSA to form the channels (Figure 1A). Channels were first washed with 100 μL phosphate-buffered saline (PBS) (pH 7.4, Life Technologies, Grand Island, NY, USA). Gamma-maleimidobutyryl-oxysuccinimide ester (GMBS) (30 μL, 223 × 10−6 M in dimethyl sulfoxide (DMSO), Sigma Aldrich, St. Louis, MO, USA) diluted with PBS (6 × 10−6 M final) was then introduced into channels and incubated for 45 min at room temperature in dark. After channels were washed twice with 100 μL of PBS, 40 μL of neutravidin (0.2 mg mL−1 in PBS, Sigma Aldrich, St. Louis, MO, USA) was introduced to the channels and incubated overnight at 4 °C. Channels were then washed twice with PBS (100 μL) again to remove unbound molecules. Chips were functionalized with 50 μL of 100 μg mL−1 biotinylated cell-specific antibodies to capture cells of interest. Biotinylated human anti-CD45 antibody (monoclonal mouse IgG1 Clone # 2D1, BAM1430 R&D Systems (Minneapolis, MN, USA)) was used to capture all white blood cells regardless of their subtype, meanwhile biotinylated human anti-CEACAM-8/anti-CD66-b antibody (polyclonal sheep IgG, E. coli-derived recombinant human CEACAM-8/CD66b Gln35-His141 Accession # P31997, BAF4246, R&D Systems (Minneapolis, MN, USA)) was used to capture neutrophils from whole blood (collected in EDTA blood collection tubes, Stanford Blood Center, Palo Alto, CA, USA, and Stanford Cancer Institute, Stanford, CA, USA) (Figure S2, Supporting Information). Finally, bovine serum albumin (BSA) (Sigma Aldrich, St. Louis, MO, USA) solution was used (3% in PBS) as a blocking agent and was incubated for an hour at room temperature to prevent nonspecific binding of cells to channel surfaces; channels were then washed with PBS.
Immune-Cell Capture
Microfluidic chips were connected to a syringe pump (Harvard Apparatus, Holliston, MA, USA) to control the flow rate inside channels using plastic tubing (Cole-Parmer, Vernon Hills, IL, USA). To eliminate the overwhelming number of RBCs and prevent their interference on the surface, whole blood was initially diluted in 1:5 ratio with either PBS or RBC lysing buffer solution (Thermo Fisher Scientific Waltham, MA, USA) (Figure 1B). Then blood samples were introduced into reservoirs of the chips on the inlets of the channels and withdrawn at 5 μL min−1 flow rate for 5.5 μL (Figure S1, Supporting Information) to fill the entire channel (Figure 1C). Then, blood sample was incubated for 20 min for the cells of interest to settle down to the glass substrate. At this step, only neutrophils were captured using anti-CD66b antibodies, while other subtypes of WBCs were washed away with PBS (Figure 1D). After the channels were washed with 100 μL of PBS again, the cells were stained with 2-phenylindole-4′,6-dicarboxamidine dihydrohydrochloride (DAPI) (Life Technologies Co., Carlsbad, CA, USA) to label nuclei of the entire white blood cell population and fluorescein isothiocyanate (FITC) (Life Technologies Co., Carlsbad, CA, USA) conjugated anti-CD66b antibody to label neutrophils for 30 min at room temperature in a dark environment. After staining, channels were washed again with 100 μL PBS. Then, chips were inspected under fluorescence microscope (Zeiss Observer Z1, Carl Zeiss Microscopy, Thornwood, NY, USA) and lens-free shadow imaging system (Figure 1E). Fluorescence microscopy was used only for the validation studies of cell capture. Once the desired efficiencies of capture and specificity were achieved, these do not need: to be used since the rest of the experiments (particularly patient samples) was performed with the lens-free shadow imaging system. The captured cells were quantified using two parameters: i) capture efficiency: the percentage of captured cells as compared to the actual expected number obtained by hematology analyzer and ii) specificity: the ratio of the cells of interest to all the cells that were captured. These are defined as:
| (1) |
| (2) |
We also performed the same neutrophil capturing procedure with clinical samples obtained from kidney and lung cancer patients (obtained from Prof. Alice Fan and Prof. Heather Wakelee at Stanford University School of Medicine, Division of Oncology). Blood samples were collected from patients at the Stanford Cancer Institute after informed consent per Stanford University Institutional Review Board approved protocols (IRB:12597).
Lens-Free Shadow Imaging System
The captured cells were quantified in microfluidic channels using a lens-free shadow imaging system. Briefly, lens-free shadow imaging platform was realized by utilizing angular spectrum method (ASM).[33,34,47] In ASM, light diffracting from an object is expanded into a superposition of complex plane waves. The samples are illuminated by a partially coherent illumination system, and light diffracted from the edges of the cells forms diffraction patterns as hologram shadows onto the surface of a CMOS-based imaging sensor. To obtain the original image, ASM-based back-propagation technique was performed. In this method, the raw holographic image is multiplied with the transfer function of light in glass, and an inverse fast Fourier transform is then performed at every 100 μm distance in the negative propagation direction. At a certain distance, an image with resolvable features is achieved and a deconvolved image is reconstructed (Figure 1F, and Figure S3, Supporting Information). The distortion and newly formed holographic patterns can be observed by further back propagating from this threshold z value. The reconstructed image provides sharp features at the figure center of each diffraction pattern, which corresponds to captured neutrophil cells in the microfluidic channel.
Cell-Capture Model
In the model, a group of antibodies was assumed to be randomly distributed on the surface of the chip. Cells were then placed down on the surface of the channel, and determined if they have enough antibodies nearby to stay stuck in the channel when it is washed. It is considered that some constant fraction, φ, of the antibodies in the channel will end up bound to the surface. Given an antibody mass m, a channel height h, and assuming a cell binding neighborhood of area A, one can write the total expected number of antibodies that a cell will interact with as λ = Ahρ/φ, where ρ is the antibody concentration in the fluid (Figure 2E). Since the antibodies are randomly distributed, a Poissonian sampling of the surface is used and assumed that any cell with more than some critical number of bound antibodies, nc, will stay in the channel. This distribution is given by:
| (3) |
The total number of bound neutrophils in the channel is the sum of all cells that have nc or more attached antibodies. Dividing by the total number of trials, a bound fraction is obtained given by the cumulative distribution function of the Poisson distribution, namely:
| (4) |
where Γ is the Gamma function.
Supplementary Material
Acknowledgments
U.D. acknowledges partial support from NIH Grant Nos. R01 DE024971, U54CA199075, U54EB015408, and NSF CBET 1464673. E.T. and J.K. acknowledge partial support from NSF CBET 1309933. The authors would like to thank Yusuf Yesil, Sherry Zhou, and Dr. Sencer Ayas for their contribution during the experiments, analysis, and drawing of Figure 1B and 4D and thoughtful discussions. H.I. developed the study, performed the experiments, analyzed the data, wrote the manuscript; J.L.K. and E.T. performed the theoretical analysis, wrote the modeling section in manuscript, read and corrected the manuscript; M.O.O. performed patient sample experiments; M.O., helped with data analysis and manuscript writing, H.C.T. performed the initial study and started the experiments; C.R.H., Y.I., T.J.M., J.S.P., N.G.D., H.W., and A.F. provided and contributed with clinical samples, details of blood components, and clinical interpretation and relevance of results; U.D. conceived the study, analyzed the data, wrote manuscript, and supervised the overall study.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
U.D. is a founder of and has an equity interest in: i) DxNow Inc., a company that is developing microfluidic and imaging technologies for point-of-care diagnostic solutions, ii) Koek Biotech, a company that is developing microfluidic IVF technologies for clinical solutions, and iii) Levitas Inc., a company focusing on developing products for liquid biopsy. U.D.’s interests were viewed and managed in accordance with the conflict of interest policies.
Contributor Information
Dr. Hakan Inan, Demirci Bio-Acoustic-MEMS in Medicine (BAMM) Laboratory, Stanford University School of Medicine Department of Radiology, Canary Center at Stanford for Cancer Early Detection, 3155 Porter Drive, Palo Alto, CA 94304, USA.
James L. Kingsley, Department of Physics, Worcester Polytechnic Institute, 100 Institute Road, Worcester, MA 01609-2280, USA
Dr. Mehmet O. Ozen, Demirci Bio-Acoustic-MEMS in Medicine (BAMM) Laboratory, Stanford University School of Medicine Department of Radiology, Canary Center at Stanford for Cancer Early Detection, 3155 Porter Drive, Palo Alto, CA 94304, USA.
Prof. Huseyin Cumhur Tekin, Department of Bioengineering, Izmir Institute of Technology, 35100 Urla, Izmir, Turkey
Dr. Christian R. Hoerner, Department of Medicine, Division of Oncology, Stanford University School of Medicine, Stanford, CA 94305, USA
Yoriko Imae, Stanford Cancer Institute, Stanford, CA 94305, USA.
Thomas J. Metzner, Stanford Cancer Institute, Stanford, CA 94305, USA
Jordan S. Preiss, Stanford Cancer Institute, Stanford, CA 94305, USA
Dr. Naside Gozde Durmus, Department of Biochemistry, Stanford University, CA 94305, USA
Prof. Mehmet Ozsoz, Independent Scholar, 35100, 6500/1 Sokak, No:8F, Karsiyaka/Izmir, Turkey
Prof. Heather Wakelee, Department of Medicine, Division of Oncology, Stanford University School of Medicine, Stanford, CA 94305, USA Stanford Cancer Institute, Stanford, CA 94305, USA.
Prof. Alice C. Fan, Department of Medicine, Division of Oncology, Stanford University School of Medicine, Stanford, CA 94305, USA Stanford Cancer Institute, Stanford, CA 94305, USA.
Prof. Erkan Tüzel, Department of Physics, Worcester Polytechnic Institute, 100 Institute Road, Worcester, MA 01609-2280, USA
Prof. Utkan Demirci, Demirci Bio-Acoustic-MEMS in Medicine (BAMM) Laboratory, Stanford University School of Medicine Department of Radiology, Canary Center at Stanford for Cancer Early Detection, 3155 Porter Drive, Palo Alto, CA 94304, USA; Department of Electrical Engineering (by courtesy), Stanford University, Stanford, CA 94305, USA.
References
- 1.Dale DC, Cottle TE, Fier CJ, Bolyard AA, Bonilla MA, Boxer LA, Cham B, Freedman MH, Kannourakis G, Kinsey SE, Davis R, Scarlata D, Schwinzer B, Zeidler C, Welte K. Am J Hematol. 2003;72:82. doi: 10.1002/ajh.10255. [DOI] [PubMed] [Google Scholar]
- 2.Gibson C, Berliner N. Blood. 2014;124:1251. doi: 10.1182/blood-2014-02-482612. [DOI] [PubMed] [Google Scholar]
- 3.Hsieh MM, Everhart JE, Byrd-Holt DD, Tisdale JF, Rodgers GP. Ann Intern Med. 2007;146:486. doi: 10.7326/0003-4819-146-7-200704030-00004. [DOI] [PubMed] [Google Scholar]
- 4.Newburger PE, Dale DC. Semin Hematol. 2013;50:198. doi: 10.1053/j.seminhematol.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boxer LA. Hematology Am Soc Hematol Educ Program. 2012;2012:174. doi: 10.1182/asheducation-2012.1.174. [DOI] [PubMed] [Google Scholar]
- 6.Boxer L, Dale DC. Semin Hematol. 2002;39:75. doi: 10.1053/shem.2002.31911. [DOI] [PubMed] [Google Scholar]
- 7.Lyman GH, Abella E, Pettengell R. Crit Rev Oncol/Hematol. 2014;90:190. doi: 10.1016/j.critrevonc.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 8.Lyman GH, Poniewierski MS, Crawford J, Dale DC, Culakova E. Blood. 2015;126:2089. [Google Scholar]
- 9.Cameron D. Br J Cancer. 2009;101:S18. doi: 10.1038/sj.bjc.6605272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chindaprasirt J, Wanitpongpun C, Limpawattana P, Thepsuthammarat K, Sripakdee W, Sookprasert A, Wirasorn K. Asian Pac J Cancer Prev. 2013;14:1115. doi: 10.7314/apjcp.2013.14.2.1115. [DOI] [PubMed] [Google Scholar]
- 11.Kuderer NM, Dale DC, Crawford J, Cosler LE, Lyman GH. Cancer. 2006;106:2258. doi: 10.1002/cncr.21847. [DOI] [PubMed] [Google Scholar]
- 12.Schwartzberg LS. Clin Cornerstone. 2006;8:S5. doi: 10.1016/s1098-3597(06)80053-0. [DOI] [PubMed] [Google Scholar]
- 13.Lyman GH, Michels SL, Reynolds MW, Barron R, Tomic KS, Yu J. Cancer. 2010;116:5555. doi: 10.1002/cncr.25332. [DOI] [PubMed] [Google Scholar]
- 14.Crawford J, Dale DC, Lyman GH. Cancer. 2004;100:228. doi: 10.1002/cncr.11882. [DOI] [PubMed] [Google Scholar]
- 15.Dinan MA, Hirsch BR, Lyman GH. J Natl Compr Cancer Network. 2015;13:e1. doi: 10.6004/jnccn.2015.0014. [DOI] [PubMed] [Google Scholar]
- 16.Lyman GH, Lyman CH, Agboola O. Oncologist. 2012;10:427. doi: 10.1634/theoncologist.10-6-427. [DOI] [PubMed] [Google Scholar]
- 17.Blann A. Nurs Times. 2014;110:16. [PubMed] [Google Scholar]
- 18.Cheng X, Irimia D, Dixon M, Sekine K, Demirci U, Zamir L, Tompkins RG, Rodriguez W, Toner MA. Lab Chip. 2007;7:170. doi: 10.1039/b612966h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamada M, Seki M. Lab Chip. 2005;5:1233. doi: 10.1039/b509386d. [DOI] [PubMed] [Google Scholar]
- 20.Holmes D, Pettigrew D, Reccius CH, Gwyer JD, van Berkel C, Holloway J, Davies DE, Morgan H. Lab Chip. 2009;9:2881. doi: 10.1039/b910053a. [DOI] [PubMed] [Google Scholar]
- 21.Wang Z, Chin SY, Chin CD, Sarik J, Harper M, Justman J, Sia SK. Anal Chem. 2010;82:36. doi: 10.1021/ac902144w. [DOI] [PubMed] [Google Scholar]
- 22.Han X, Van Berkel C, Gwyer J, Capretto L, Morgan H. Anal Chem. 2012;84:1070. doi: 10.1021/ac202700x. [DOI] [PubMed] [Google Scholar]
- 23.Seo S, Isikman SO, Sencan I, Mudanyali O, Su TW, Bishara W, Erlinger A, Ozcan A. Anal Chem. 2010;82:4621. doi: 10.1021/ac1007915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu H, Mavandadi S, Coskun AF, Yaglidere O, Ozcan A. Anal Chem. 2011;83:6641. doi: 10.1021/ac201587a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu H, Sencan I, Wong J, Dimitrov S, Tseng D, Nagashima K, Ozcan A. Lab Chip. 2013;13:28. doi: 10.1039/c3lc41408f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watkins NN, Hassan U, Damhorst G, Ni H, Vaid A, Rodriguez W, Bashir R. Sci Transl Med. 2013;5:214ra170. doi: 10.1126/scitranslmed.3006870. [DOI] [PubMed] [Google Scholar]
- 27.Kotz KT, Xiao W, Miller-Graziano C, Qian W, Russom A, Warner EA, Moldawer LL, De A, Bankey PE, Petritis BO, Camp DG, Rosenbach AE, Goverman J, Fagan SP, Brownstein BH, Irimia D, Xu W, Wilhelmy J, Mindrinos MN, Smith RD, Davis RW, Tompkins RG, Toner M. Nat Med. 2010;16:1042. doi: 10.1038/nm.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma S, Zapatero-Rodríguez J, Estrela P, O’Kennedy R. Biosensors. 2015;5:577. doi: 10.3390/bios5030577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song Y, Huang YY, Liu X, Zhang X, Ferrari M, Qin L. Trends Biotechnol. 2014;32:132. doi: 10.1016/j.tibtech.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sobieranski AC, Inci F, Tekin HC, Yuksekkaya M, Comunello E, Cobra D, von Wangenheim A, Demirci U. Light: Sci Appl. 2015;4:e346. doi: 10.1038/lsa.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bland JM, Altman D. Lancet. 1986;327:307. [PubMed] [Google Scholar]
- 32.Sanmatías S, Clemente M, Giner-Bosch V, Giner V. Ann Oncol. 2011;22:181. doi: 10.1093/annonc/mdq332. [DOI] [PubMed] [Google Scholar]
- 33.Roy M, Seo D, Oh S, Chae Y, Nam MH, Seo S. Diagnostics. 2016;6:17. doi: 10.3390/diagnostics6020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gurkan UA, Moon S, Geckil H, Xu F, Wang S, Lu TJ, Demirci U. Biotechnol J. 2011;6:138. doi: 10.1002/biot.201000427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang X, Khimji I, Gurkan UA, Safaee H, Catalano PN, Keles HO, Kayaalp E, Demirci U. Lab Chip. 2011;11:2535. doi: 10.1039/c1lc20236g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roy M, Lee J, Jin G, Seo S, Nam MH. Proc 2013 Int Conf Emerging Trends in Communication, Control, Signal Processing and Computing Applications, IEEE-C2SPCA. IEEE; Piscataway, NJ, USA: 2013. [Google Scholar]
- 37.Moon S, Keles HO, Kim YG, Kuritzkes D, Demirci U. Proc 31st Annual Int Conf IEEE Engineering in Medicine and Biology Society: Engineering the Future of Biomedicine, EMBC. IEEE; Minneapolis, MN, USA: 2009. pp. 6376–6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moon SJ, Gurkan UA, Blander J, Fawzi WW, Aboud S, Mugusi F, Kuritzkes DR, Demirci U. PLoS One. 2011;6:e21409. doi: 10.1371/journal.pone.0021409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hassan U, Watkins NN, Reddy B, Damhorst G, Bashir R. Nat Protoc. 2016;11:714. doi: 10.1038/nprot.2016.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inan H, Wang S, Inci F, Baday M, Zangar R, Kesiraju S, Anderson KS, Cunningham BT, Demirci U. Sci Rep. 2017;7:3322. doi: 10.1038/s41598-017-02672-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Halldorsson S, Lucumi E, Gómez-Sjöberg R, Fleming RMT. Biosens Bioelectron. 2015;63:218. doi: 10.1016/j.bios.2014.07.029. [DOI] [PubMed] [Google Scholar]
- 42.El Assal R, Gurkan UA, Chen P, Juillard F, Tocchio A, Chinnasamy T, Beauchemin C, Unluisler S, Canikyan S, Holman A, Srivatsa S, Kaye KM, Demirci U. Sci Rep. 2016;6:39144. doi: 10.1038/srep39144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Asghar W, Yuksekkaya M, Shafiee H, Zhang M, Ozen MO, Inci F, Kocakulak M, Demirci U. Sci Rep. 2016;6:21163. doi: 10.1038/srep21163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shafiee H, Asghar W, Inci F, Yuksekkaya M, Jahangir M, Zhang MH, Durmus NG, Gurkan UA, Kuritzkes DR, Demirci U. Sci Rep. 2015;5:8719. doi: 10.1038/srep08719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inan H, Poyraz M, Inci F, Lifson MA, Baday M, Cunningham BT, Demirci U. Chem Soc Rev. 2017;46:366. doi: 10.1039/c6cs00206d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lifson MA, Ozen MO, Inci F, Wang S, Inan H, Baday M, Henrich TJ, Demirci U. Adv Drug Delivery Rev. 2016;103:90. doi: 10.1016/j.addr.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim SB, Bae H, Koo KI, Dokmeci MR, Ozcan A, Khademhosseini A. J Lab Autom. 2012;17:43. doi: 10.1177/2211068211426695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Common Terminology Criteria for Adverse Events (CTCAE), Version 4.0, DCTD, CTI, NIH, DHHS. National Institute of Cancer; 2009. (NIH publication #09-7473). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.