

Abstract
The addition of a silicon-hydrogen or a boron-hydrogen bond across a carbon-carbon multiple bonds is a well-established method for the introduction of versatile silane and borane functional groups to base hydrocarbon feedstocks. Transition metal catalysis, historically with precious second- and third- row transition metals, has been used to broaden the scope of the hydrofunctionalization reaction, improve reaction rate and enhance selectivity. The anti-Markovnikov selectivity of platinum-catalyzed hydrosilylation of alkenes, for example, is an enabling synthetic technology in the multibillion-dollar silicones industry. Increased emphasis on sustainable catalytic methods and more economic processes has shifted focus to catalysis with more earth-abundant transition metals such as iron, cobalt and nickel. This review describes contemporary approaches and offers a contextual analysis of catalytic alkene hydrosilylation and hydroboration reactions using first-row transition metals. Emphasis is placed on defining advances in the field, what constitutes catalyst cost, safety, and important design features to enable precious metal-like reactivity, as well as new chemistry that is unique to first-row transition metals.
Modern chemistry finds its origins in a primeval drive to produce complex creations from simple raw materials where “state-of-the-art” could mean transforming sand into glass, tree bark into therapeutics, and even the unrelenting, albeit, futile attempts to transform base metals into gold.1 Modern society now relies on the transformation of abundant hydrocarbon feedstocks2 into functionalized building blocks that find application as medicines, flavors and fragrances, modern materials, fuels and fine chemicals. Alkenes and alkynes are among the most abundant and versatile whose supply is increasing due to the development of vast global natural gas deposits.3
Transition metal-catalyzed hydrosilylation4–6 and hydroboration7,8 reactions are among the most widely studied and widely employed transformations for the upgrading of commodity alkenes and alkynes, owing to the versatility and utility of the functionalized product. Both reactions fall into the broader category of hydrofunctionalization reactions, where a hydrogen atom and a functional group are added across the π-system of the substrate (FIG 1a). These processes are atom economical as both the hydrogen and functional group are retained in the product without formation of byproducts. Both metal-catalyzed hydrosilylation and hydroboration reactions trace their routes to hydrogenation processes and often times catalysts discovered for H2 addition to alkenes and alkynes are later applied to other types of hydrofunctionalization processes. These reactions are also mechanistically similar and involve related fundamental steps in a catalytic cycle (FIG 1b) including insertion of the unsaturated C-C bond into a M-X (X = H, SiR3, BR2) bond.
Figure 1 |. Olefin hydrosilylation versus olefin hydroboration: Motivations and mechanism.

a | Transition metal catalysis enable hydrosilylation and hydroboration of readily available olefins for use in commodity chemicals and fine chemicals, respectively. b | Transition metal-catalyzed hydrosilylation and hydroboration are mechanistically similar and may undergo alkene insertion into a metal hydride (Pathway A) or alkene insertion into either a metal silyl or metal boryl (Pathway B). For clarity, oxidative addition of H-FG and reductive elimination of the C-FG bond were not shown.
Despite their mechanistic similarities, the application of these processes differ significantly and impact how metrics for success in catalyst development should be evaluated. Alkene hydrosilylation is most commonly used as an industrial commodity chemical process to prepare commercial silicones; its application in fine chemical synthesis is less common. As such, the substrate scope, especially with respect to the silane reagent is relatively narrow and challenges are typically related to catalyst selectivity, activity and fidelity. Alkene hydroboration, by contrast, is often applied on relatively small scale and is most useful in the preparation of fine chemicals and intermediates in synthesis. Successful catalysts should function with a host of substrates and functional groups. The origin of the difference can be traced to the role of the added functional group – in hydrosilylation the silicon substituent is desired in the final product to achieve the targeted properties whereas in hydroboration, the boron group is typically transient and used in subsequent chemical transformations to form new carbon-carbon bonds or converted to other functional groups such as alcohols, amides, amines and halides. While it is true that organosilanes can also undergo additional functionalization, much like organoboron compounds, these transformations are often more challenging, requiring harsher reagents and conditions, consequently limiting their scope.9
The applications and utility of hydrosilylation and hydroboration reactions also determine the motivation for catalyst-controlled regioselectivity. In hydrosilyation, nearly all commercial silicones have the silicon substituent at the terminal position of the hydrocarbon chain and therefore minimizing competing Markovnikov addition is essential. By contrast, hydroboration catalysts that enable different regioselectivities are invaluable as this translates onto new disconnections in complex molecule settings. This review highlights and contextualizes many of the recent developments in state-of-the-art catalysts with earth-abundant metals for both hydrosilylation and hydroboration. Although many of the early motivations for these studies was focused on achieving precious metal-like reactivity with iron, cobalt and nickel, there is now little doubt that first-row metals, when placed in the appropriate coordination environment, can compete with more traditional precious metals in terms of activity and selectivity in hydrofunctionalization. More contemporary catalysts exploit the unique properties of the first-row metal or through ligand design, enable new chemistries previously unobserved in more traditional precious metal catalyzed processes. As these new catalysts are discovered, it is important to contextualize the outcomes in how the products are ultimately used.
ALKENE HYDROSILYLATION
Evolution and Perspectives on Industrial Hydrosilylation – Motivations for Contemporary Innovations.
Metal-catalyzed olefin hydrosilylation is one of the largest scale applications of homogeneous catalysis by value of product sold and is an enabling transformation in the silicones industry.4–6 Organosilicon products derived from olefin hydrosilylation are usually added to a diverse array of consumer products such as cosmetics,10 car tires,11 contact lenses,12 textiles5 and paints5 as performance enhancers. These compounds are also used as agricultural adjuvants10 and as release coatings for labels and tapes13 (BOX 1). For perspective on the enormous scale and impact of hydrosilylation, just one product, n-octyl-Si(OEt)3, a performance enhancer for masonry products and glass, is manufactured annually on a >6000 ton scale.5 It is also used as a penetrating sealer that dramatically improves weather resistance of highways, bridges, runways and airports.14 It is likely that every reader of this review has interacted with or used a product of hydrosilylation within the last 24 hours.
Box 1 |. Platinum-catalyzed hydrosilylation.
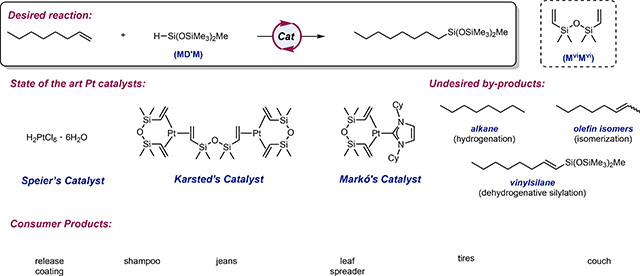
For the past five decades, the silicones industry has relied on precious metal catalysts, typically Pt, Rh, Ir and Ru, to promote hydrosilylation.15 Speier’s discovery that hexachloroplatinic acid, [H2PtCl6]•H2O, is an effective catalyst for alkene hydrosilylation16,17 was a transformative breakthrough in the field and initiated the widespread implementation of platinum catalysis in the silicones industry.6 Because of the long induction periods associated with this catalyst and its poor solubility in polysiloxane mixtures, Karstedt introduced divinyltetramethylsiloxane (MviMvi)-ligated platinum(0) catalysts that exhibited improved activity, selectivity as well as solubility in polysiloxane compositions and is now the most established, and widely used catalyst in the industry (BOX 1).6 Even with this important advance, the lability of the MviMvi ligands provides a pathway for the formation of platinum nanoparticles that give rise to unwanted side reactions, including alkene hydrogenation, isomerization and dehydrogenative silylation, resulting in increased product cost and waste as distillations are required to achieve the desired purity of the final product.18 Collodial platinum also contributes to discoloration of the product, reducing overall quality of the silicone.17 To eliminate formation of nanoparticles, Markó introduced strongly coordinating ligands in to the platinum catalysts and N-heterocyclic carbine-supported variants proved the most effective (BOX 1).19,20
In 2010, Troegel and Stohrer outlined the challenges in precious metal-catalyzed industrial hydrosilylation reactions and identified three major commercial applications of the process:15 (i) functional silanes, (ii) silicone release coatings and (ii) silicone rubbers. They noted that the attributes of the ideal catalyst that satisfy the quality requirements of each silicone are different for each application. For functional silanes, where the final product can be purified by distillation, the platinum catalyst can be recycled from the heavy organosilicon waste streams, and the contribution of catalyst cost to the final product is relatively low.21 The more significant metrics are the chemo- and regioselectivity of the process. In the case of silicone release coatings and rubbers, the morphology and viscosity of the product prohibits recovery of the platinum catalyst. In 2007 alone, it was estimated that the silicones industry consumed 5.6 metric tons of platinum.22 Clearly this is an application where more inexpensive catalysts would have an impact.
Catalysts with Earth-Abundant Transition Metals Catalysts for Alkene Hydrosilylation.
The economic consequences associated with platinum-catalyzed alkene hydrosilylation have long been recognized by the silicones industry. Other perhaps less obvious motivations to shift to more Earth-abundant metals include the price volatility associated with the precious metals market, the toxicity of heavy metals and the environmental consequences associated with mining elements of scarce abundance in the Earth’s crust.23,24,25 Contemporary developments in the discovery of first row transition metal catalysts for alkene hydrosilylation have recently been reviewed by Deng26 and Huang27 and will not be presented in detail here. To date, the majority of earth-abundant metal catalysts for alkene hydrosilylation use primary and secondary silanes. The resulting silanes and silicones are often not commercially relevant as residual Si-H bonds decrease the stability and hence utility of the final product. Accordingly, the focus of this review will be the evolution of pyridine(diimine) iron dinitrogen complexes and later generation α-diimine nickel complexes as pre-catalysts for alkene hydrosilylation. Furthermore, examples will be by and large, limited to reactions involving industrially relevant tertiary silanes and will feature important advances in the design and application of catalysts with earth-abundant metals and offer a critical analysis of advances in the field, important metrics for commercialization and unique chemistry unavailable with known precious metal catalysts.
Iron-catalyzed alkene hydrosilylation was first demonstrated by Nesmeyanov and coworkers where the authors reported that terminal olefins react with tertiary silanes in the presence of catalytic Fe(CO)5 at 100–140 °C to yield mixtures of hydrosilylation and dehydrogenative silylation products.28 Mechanistic studies by Wrighton established the intermediacy [Fe(CO)3] as the active species.29,30 This insight resulted in the development of a pre-catalyst initiation method by Grant and co-workers using sustained, near-ultraviolet pulsed irradiation to generate the catalytically active [Fe(CO)3] species on the nanosecond timescale.31 These findings inspired our laboratory to prepare reduced, aryl-substituted pyridine(diimine) (PDI) iron complexes iron complexes. The corresponding dihalide compounds, (ArPDI)FeX2, were well known pre-catalysts for ethylene polymerization,32 and, if the pyridine(diimine) is serving exclusively as a π-acceptor, [(PDI)Fe] is isolobal with [Fe(CO)3].33 Pyridine(diimine) ligands offer the advantage of ease of synthesis, as well as steric and electronic modularity34 with one-electron redox potentials spanning over 400 mV by 4-substitution on the central pyridine.35 Iron dinitrogen complexes were targeted as an unactivated, terminally bound N2 ligand was expected to be substitutionally labile, thus lowering the barrier for the ligand dissociation step required to access the active species.
In 2004, our laboratory reported the preparation and hydrosilylation activity of the 2,6-diisopropyl phenyl-substituted pyridine(diimine) iron dinitrogen complex, (iPrPDI)Fe(N2)2 (Complex 1 FIG 2).36 Terminal alkenes, such as 1-hexene and styrene underwent exclusive anti-Markovnikov hydrosilylation with PhSiH3 in the presence 0.3 mol % of 1 at 22 °C with no evidence for alkene isomerization (1-hexene) or dehydrogenative silylation. No reaction was observed when the tertiary silane, Et3SiH was used in place of PhSiH3. Complex 2, where the C-methyl imine substituents in 1 are replaced with phenyl groups, resulted in a more active hydrosilylation catalyst for 1-hexene using phenylsilane.35 However, when more hindered substrates such as cyclohexene are employed, 2 proved less active than 1 due to deactivation pathways involving η6 coordination of the phenyl and aryl substituents on the chelate to iron.37
Figure 2 |. Base metal catalysts for the hydrosilylation of commercially important substrates with rates and selectivities exceeding those associated with platinum.

Complexes 1-4 are symmetrical and the “Ar” group on the imine nitrogen is identical to the one depicted.
As platinum prices began to increase in the mid- to late 2000s, silicone manufacturers again became interested in more inexpensive alternatives to platinum. Nickel38 and palladium39 had been explored previously, and the activity and selectivity of these catalysts were inferior to platinum. Our 2004 publication36 attracted the attention of Momentive Performance Materials and in 2008, a highly productive academic-industrial collaboration was initiated.
One challenge outlined for the pyridine(diimine) iron catalysts was activity with commercially relevant tertiary alkoxy- and siloxy-substituted silanes. Because decreasing the size of the 2,6-aryl substituents had previously been shown to increase the activity of pyridine(diimine) iron alkene hydrogenation catalysts,40 initial experiments were conducted with [(MePDI)Fe(N2)]2(μ2-N2)40 (Complex 3, FIG 2). Exclusive anti-Markovnikov hydrosilylation of 1-octene was observed with just ppm levels of 3. The hydrosilylation of 1-octene with MD’M with Karstedt’s catalyst (30 ppm, 72 °C) produces 80% yield of the desired linear octylsilane product along with products arising from olefin hydrogenation, isomerization and dehydrogenative silylation,19 demonstrating that iron, when in an appropriate coordination environment, can offer competitive if not superior, activity and selectivity to commercial platinum catalysts. Re-investigation of 1 with tertiary silanes, (Me3SiO)2MeSiH (MD’M), and (EtO)3SiH, and 1-octene also resulted in highly effective anti-Markovnikov hydrosilylation and established the increased reactivity of alkoxy-substituted silanes over purely alkylated ones.41
Figure 2 highlights the hydrosilylation activity of 3 for the preparation of commercial products. For example, the siloxane from 1-octene and MD’M is prepared on an approximately 6000 ton scale annually and is used to enhance cosmetic formulations and weatherproofing for masonry products, metal oxide and glass.5 It is notable that the iron catalyst 3, does not promote any observable side reactions such as alkene isomerization, hydrogenation or dehydrogenative silylation, which are commonly observed up to 20% overall with platinum (FIG 2).41 Performing the hydrosilylation of 1-octene with either (EtO)3SiH or Et3SiH required a slightly higher loading of 500 (rather than 200) ppm and the resulting product finds application as filler treatments14,42 and textile finish lubricants,43,44 respectively (FIG 2c,d). Again, exclusive anti-Markovnikov selectivity was observed, highlighting one of the advantages of the pyridine(diimine) iron catalysts. This exquisite selectivity was maintained with styrene and methyl capped polyethers (FIG 2b, FIG 2e), both notoriously challenging substrates that often undergo undesired, competing olefin isomerization and in the case of styrene, Markovnikov addition with commercial platinum catalysts.45 In the United States, the polyether products are manufactured on a scale of approximately 4 million pounds per year. All of these examples fall under the category of functional silanes,15 where the final product can be purified by distillation and therefore, the catalysts can be recovered, reprocessed and recycled. In these cases, superior selectivity is the more important metric than catalyst cost as energy intensive and costly distillation procedures can be obviated.
The crosslinking of silicone monomers to form silicone fluids is one of the largest applications of metal-catalyzed alkene hydrosilyation.15 These silicone products have excellent adhesion properties and clean release properties and find widespread application as linear coatings in labels, tapes and stamps.13,22 The gelatinous viscosity of the silicone product makes economic recovery of the platinum catalyst almost impossible, and, as a result, lost precious metal contributes to approximately 25–30% of the overall cost of product. This application is one that would clearly benefit from an inexpensive, earth-abundant metal catalyst. As will be detailed in a later section, many factors beyond the price of the metal contribute to catalyst cost.
Pyridine(diimine) iron catalyst 3 proved highly effective for the crosslinking of the two most common silicone fluids, SL6020 and SL6100 (the historical trade name of these materials is used for consistency), and the resulting crosslinked product exhibited nearly identical properties to commercial material made from platinum catalysis (FIG 2f).41 The rate of the iron-catalyzed hydrosilylation was on par with typical reaction conditions for commercial platinum-catalyzed cross-linking reactions. With 500 ppm of iron precatalyst 3, complete cross-linking was observed after 2 hours at room temperature. With platinum, catalyst lodings between 25 ppm to 150 ppm are employed at temperatures curing temperatures between 93 °C to 141 °C and curing times between 1.6 to 2.3 hours.46 Despite this tremendous breakthrough, several limitations of the iron-catalyzed reaction were identified. First, the silicone product had a slightly yellow discoloration, arising not from free residual iron, but rather from very small amounts of free pyridine(diimine), a strong chromophore. Secondly and perhaps more concerning, 3 is highly air sensitive. With many release coatings of this type, the platinum-catalyzed hydrosilylation is engineered to occur at a rate commensurate with the application. For example, in the synthesis of labels, the hydrosilylation occurs as the backing of the label is being prepared and in air. These essential features must be replicated for any next generation catalyst to displace platinum.
One advantage of pyridine(diimine)-based catalysts is their steric and electronic modularity. These properties were used to solve an interesting contemporary challenge in metal-catalyzed hydrosilylation – the selective hydrosilylation of 1,2,4-trivinylcyclohexane (TVCH).47 Commercial sources of this compound are typically composed of four stereoisomers. Addition of a [Si-H] unit of an alkoxysilane across the 4-alkene is desirable as two adjacent vinyl groups left intact would be available for subsequent functionalization with sulfur to crosslink into a rubber matrix for application in low rolling resistance tires. Karstedt’s and Speier’s catalysts yield mixtures of products from multiple silylations with poor regioselectivity, thereby generating considerable waste and requiring fractional distillation.4 Iron catalyst 1 proved highly active and selective for the single hydrosilylation of the 4-position of TVCH with both MD’M and (EtO)3SiH.47 Reducing the size of the 2,6-aryl substituents resulted in less selective catalysts, largely from disilylation. To improve activity, an electron- donating [NMe2] was introduced into the 4-position (Complex 4, FIG 2) of the pyridine while the 2,6-isopropyl aryl substituents were maintained for selectivity (FIG 2g).
Other tridentate ligands have also been explored to support iron catalysts for hydrosilylation. Iron dialkyl complexes of terpyridine, pyridine bis(oxazoline) and pyridine(diimines) were prepared and evaluated as precatalysts for the hydrosilylation of 1-octene with MD’M and (EtO)3SiH.48 Only (terpy)Fe(CH2SiMe3)2 (Complex 5, FIG 2) exhibited activity but required heating to 60 °C. Notably, this pre-catalyst also promoted the hydrosilylation of vinylcyclohexene oxide with MD’M without opening of the epoxide (FIG 2h). The product of this hydrosilylation reaction is sold commercially for use as high performance coatings for glass, wood, leather and plastics.49 This reactivity, to our knowledge, remains unique among first-row transition metal alkene hydrosilylation catalysts.
Since these initial discoveries, NNN-type pincers have been explored in iron-catalyzed alkene hydrosilylation. Nakazawa evaluated ketimine-type imino(bipyridine) ligands for the hydrosilylation of 1-octene with PhSiH3, Ph2SiH2, and PhMeSiH2 as well as alkoxysilanes using NaBEt3H in situ activation methods.50 High activities were observed, as was tolerance, to remote amino, chloride and thioether functional groups.
Catalyst Development: New Reactivity Modes and Overcoming Challenges to Implementation.
Reduced pyridine(diimine) iron complexes represent a milestone in both alkene hydrosilylation and catalysis with earth-abundant transition metals. These molecules were among the first to demonstrate that first-row transition metals can compete or even outperform precious metals in olefin hydrosilylation.36,41 Despite these advances, significant challenges remain for commercial implementation. The extreme air and moisture sensitivity of these compounds is the principal roadblock.
In situ activation was identified early on by the then Cornell-Momentive team as a strategy to use more easily handled pyridine(diimine) iron halide complexes with an appropriate reducing agent.51 In fact, our laboratory utilized this approach in our initial communication of the ironcatalyzed [2+2] cycloaddition of α,ω-dienes and demonstrated the effectiveness of NaBEt3H for this purpose.51 In 2010, the Cornell-Momentive team explored and patented a method using a variety of reductants including metal hydrides, alkoxides and alkylating agents to serve as activators for pyridine(diimine) iron-catalyzed hydrosilylation.52 This approach identified a host of successful promotors for the iron-catalyzed reaction but was abandoned as we found that any activator sufficiently reducing to generate active [(PDI)Fe] catalysts triggered disproportionation of alkoxy-substituted silanes to pyrrophoric SiH4!52 These methods and the associated safety hazards were well documented in the literature53,54,55,56 and generation of even small amounts of silane gas is unacceptable on an industrial scale and hence this route was abandoned. Nevertheless, this route has been widely applied57 including in high-throughput screening58 and was re-investigated and published by Thomas and coworkers in 2017.59 Again, interaction of KOtBu was definitively shown to generate SiH4 gas, which was observed in the 29Si NMR spectrum of the hydrosilylation mixture.59
Because of the extreme dangers associated with in situ activation using strong reductants, alternative strategies imparting air stability were explored. Efforts were expanded to pyridine(diimine)cobalt catalysts; Gibson and coworkers had demonstrated that preparation of alkyl derivatives was straightforward and modular,60 certainly more so than preparation of pyridine(diimine) iron dinitrogen compounds. The performance of this class of compounds along with bis(phosphine) cobalt derivatives in alkene hydrogenation demonstrated expanded functional group tolerance and perhaps hints of improved air stability.61
The pyridine(diimine) cobalt alkyl complex, (MesPDI)CoCH3 (Complex 7, FIG 3b), a chelate similar to one of the most active iron catalysts,41 promotes the dehydrogenative silylation of linear α-olefins with either MD’M or (EtO)3SiH to yield allysilanes with high selectivity (FIG 4a).62 Remarkably, internal alkenes produce the same products: allylsilanes with the silicone group located at the end of the hydrocarbon chain (FIG 4a-1 and FIG 4a-3). In each case, the starting olefin serves as the hydrogen acceptor and is converted to the corresponding alkane. Applying this reactivity to the crosslinking of silicone fluids allowed abundant alkenes to serve as functional equivalents of α,ω-dienes to tether two poly(dimethyl)-poly(methylhydrogen) siloxanes by a defined carbon chain length (FIG 4a-4). In the silicones industry, the applications of allylsilanes have not yet been fully explored; while dehydrogenative silylation has been known for decades, often mixtures of vinyl and allylsilanes are obtained. The exclusive allylic selectivity enabled by cobalt complex 7 enables the synthesis of these versatile products from readily-available starting materials and provides the opportunity to develop potentially unique applications. For example, allylsilanes were also demonstrated to be attractive building blocks for organic synthesis, being readily oxidized to allylic alcohols or serving as allyl transfer agents in carbon-carbon bond-forming reactions.63
Figure 3 |. Overcoming practical limitations in base metal catalyzed hydrosilylation.

a | In situ activation of bench stable iron dihalides with reducing agents and bases poses a safety hazard from base-catalyzed alkoxysilane disproportionation to form pyrophoric SiH4 gas | b Switching the metal from iron to cobalt allows more modular catalyst synthesis but also switches the reactivity to dehydrogenative sulylation c | Substrate-activated air-stable metal carboxylate precursors provides the advantage of easy pre-catalyst handling without the possibility of hazardous alkoxysilane disproportionation d | The use of commercially-available, inexpensive ligands for base metal precursors significantly decreases catalyst cost but at the expense of catalyst activity.
Figure 4 |. Pyridine(diimine) cobalt catalyzed dehydrogenative silylation.

a | Scope b | Mechanism c | Catalyst design principle for promoting hydrosilylation over dehydrogenative silylation.
Mechanistic studies were conducted to uncover the origin of dehydrogenation and the allylic selectivity.62 This understanding was also sought after to alter the selectivity from dehydrogenative silylation to hydrosilylation. The data support a pathway involving formation of a cobalt-silyl following activation of the cobalt alkyl complex (FIG 4b). Selective 2,1-insertion of the alkene followed by β-hydrogen elimination directed away from the large silyl substituent releases the allyl silane and generates a cobalt-hydride. Insertion of the substrate alkene and hydrogen transfer to the alkyl by the silane generates the observed alkane and accounts for the hydrogen balance in catalytic reaction. We reasoned that interception of the cobalt alkyl with silane following insertion into the putative Co-Si bond rather than β-hydrogen elimination would yield hydrosilylation product (FIG 2c). Indeed Grant and Brookhart observed a similar pathway with [Cp*Co]-derived catalysts.64
Pyridine(diimine) cobalt complexes were synthesized with smaller N-imine substituents (FIG 3b, FIG 4c) to switch between dehydrogenative silylation and hydrosilylation.65 Introduction of a methyl group on the N-imine position was effective as the cobalt alkyl complex promoted the hydrosilylation of 1-octene with (EtO)3SiH with no detectable allyl silane.65
Other ligand platforms on cobalt also promote hydrosilylation over dehydrogenative silylation. Weix, Holland and coworkers reported air-stable cobalt ß-diketiminate pre-catalysts that when activated with alkyl lithiums, Grignard reagents or KOtBu promote the hydrosilylation of 1-hexene with PhSiH3.66 However, the use of the air-sensitive, isolated pre-catalysts were required for the same transformation using (EtO)3SiH to prevent alkoxysilane disproportionation by the added activator. Fout and co-workers also reported air-sensitive, reduced cobalt nitrogen compounds as effective pre-catalysts for the hydrosilylation of alkenes with industrially relevant tertiary silanes.67
With appropriate ligands on cobalt identified to promote hydrosilylation over dehydrogenative silylation, strategies were explored to develop air-stable catalysts without using external activators that promote the disproportionation of the silane reagent.65 Cobalt(II) carboxylates are air-stable and one of the most inexpensive sources of cobalt and were therefore attractive for preparing precursors for catalysis. Our laboratory first demonstrated that Co(OAc)2 in combination with phosphines generated active catalysts for hydroboration and limited C(sp2)-H borylation.68 The borane reagent served not only as a substrate for the catalytic reaction but also as an activator to reduce the cobalt to the active form. We reasoned that silanes may serve a similar role and allow the use of air stable pyridine(diimine) cobalt(II) carboxylates in alkene hydrosilylation (FIG 3c). During the course of our investigations, Nagashima reported an identical approach for the activation of cobalt(II) and iron(II) carboxylates for the hydrosilylation of styrene and other terminal alkenes with tertiary silanes using isocyanides as the ligands (FIG 3c).69
The N-methylated pyridine(diimine) cobalt(II) acetate, (MeAPDI)Co(OAc)2 (9, FIG 3c) was synthesized in a straightforward fashion and is bench stable.65 This complex was an effective pre-catalyst for the hydrosilylation of 1-octene with (EtO)3SiH but heating to 80 °C was required to likely overcome the poor solubility of the cobalt compound. The elevated temperature reduced the selectivity of hydrosilylation to 87:13 with allylsilane accounting for the balance of the mixture. These compounds were readily prepared from the free pyridine(diimine) and cobalt(II) carboxylate and were characterized as S = 3/2 derivatives with both κ1 and κ2 carboxylate ligands. More soluble carboxylates such as 2-ethylhexanoate increased both the activity and selectivity of the hydrosilylation reaction and exposure of the solid catalyst to air for 18 hours produced no erosion of catalyst performance. Pyridine(bisoxazoline) and its carbon analog, TFAPDI, were also effective catalysts (Complex 6, FIG 2). This family of catalysts was effective for a range of alkenes and tertiary silanes. The hydrosilylation of allyl glycidyl ether with (OEt)3SiH was carried out on a 10-gram scale with just 50 ppm Co (FIG 2i).65 The resulting epoxy functional silane from this hydrosilylation reaction is a commercial product used as an adhesion promoter for caulks, sealants, adhesives and coatings.70
Both (MeAPDI)Co(EH)2 (Complex 10, FIG 3c) and (TFAPDI)Co(EH)2 (Complex 6, FIG 2) (EH = 2-ethylhexanoate) were effective for the cross linking of siloxane polymers.65 These studies highlight the tradeoff between catalyst activity and cost. The ligand for the N-methylated precursor, MeAPDI, is prepared in a single step from inexpensive methylamine and 2,6-diacetylpyridine.65 The corresponding cobalt(II) pre-catalyst was less efficient for the silicone crosslinking reaction, requiring 55 ppm of (MeAPDI)Co(EH)2 and heating to 80 °C and the resulting product was slightly discolored due to the relatively high metal-ligand loading. By contrast, TFAPDI is relatively challenging to prepare through a low yielding, three-step synthetic sequence from commercially available materials. However, the resulting cobalt precatalyst, (TFAPDI)Co(EH)2 (6, FIG 2), is more active for silicone crosslinking, operating at ambient temperature at 1 ppm loading.65 As such, the resulting silicone is colorless and is indistinguishable from the material made from platinum catalysis. These results highlight an important point regarding catalyst cost: when ligands are involved, synthesis of the ligand and associated purification usually contributes significantly more to overall cost than the difference associated with the transition metals. Protecting the metal with simple, substitutionally labile ligands such as an alkene that can be removed by hydrosilylation obviates the need for custom ligand synthesis and greatly reduces ligand cost (FIG 3d). Divinyltetramethylsiloxane (MviMvi) is used as a supporting ligand for Karstedt’s catalyst. Iron71 and nickel38 analogs have been prepared but promote dehydrogenative silylation thus limiting their utility. Nagashima and coworkers have developed η2-Si-H containing ligands for protection and activtation of carbonyl iron complexes and studied their application in C-H functionalization of indoles72 and hydrosilylation of 1-octene with MD’M, albeit with reduced activity to the pyridine(diimine) iron dinitrogen compounds.73 For instance, the hydrosilylation of 1-octene with MD’M requires 3 mol% of Nagashima’s iron catalyst (FIG 3d, right) at 80 °C to furnish 75% yield of the desired product after 3 hours of reaction time,73 while with pyridine(diimine) catalyst 3, only 200 ppm (0.05 mol%) is required to achieve full conversion at room temperature after 15 minutes of reaction time (FIG 2a).41
The pyridine(diimine) and related NNN-supported cobalt(II) dihalide and bis(carboxylate) pre-catalysts were subsequently studied by RajanBabu and coworkers for the hydrosilylation of terminal alkenes and dienes with PhSiH3, PhSiH2, PhSi(Me)H2.74 For the halide complexes, the relatively dangerous NaBEt3H activation52 procedure was used. Liu and Deng recently extended the silane activation procedure to N-heterocyclic carbene-supported cobalt(II) complexes and demonstrated reduction to cobalt(I) and high activity hydrosilylation catalysis with 1-octene and other alkenes and (EtO)3SiH. However, the authors did not comment on the air-stability of the cobalt(II) bis(amide) precatalysts.75
The periodic relationship between platinum and nickel has long inspired the search for hydrosilylation catalysts based on the less expensive and more terrestrially abundant first row metal. Common nickel precursors such as Ni(COD)2, Ni(acac)2 as well as the analogue of Karstedt’s catalyst, Ni2{[(CH=CH)-SiMe2]2O}3 all promote significant dehydrogenative silylation.38 More recent efforts using various ligand designs resulted in improvements in selectivity, principally with aryl silanes.6 Most notably, Hu and coworkers described a nickel pincer catalyst for the hydrosilylation of a range of alkenes with Ph2SiH2.76 While significant advances,38,76 selective anti-Markovnikov hydrosilylation of commercially relevant alkene-silane combinations remained by and large elusive.
Because of the success with cobalt(II) carboxylates, initial catalyst evaluation studies were conducted with the corresponding nickel(II) carboxylates. Aryl- and alkyl-substituted α-diimine ligands were selected due to their low cost, ease of synthesis and ability to generate isoelectronic nickel catalysts to the pyridine(diimine) iron complexes. Combination of iPrDI with hydrocarbon soluble nickel carboxylate precursors produced highly active and anti-Markovnikov selective catalysts for the hydrosilylation of 1-octene with tertiary alkoxy- and siloxy-substituted silanes.77 Application of the method of continuous variations and stoichiometric studies identified the formally Ni(I) hydride dimer, [(iPrDI)NiH]2 as the nickel compound, which was previously synthesized by Yang and coworkers,78 resulting from activation and reduction of the carboxylate ligands. Crystallographic metrical parameters and DFT calculations on [(iPrDI)NiH]2 and the corresponding monomer, (iPrDI)NiH are consistent with a nickel(II) center with a oneelectron-reduced α-diimine ligand. The rate law (rate = kobs[Ni]1/2[1-octene][(EtO)3SiH]) and deuterium labeling studies establish a pathway involving fast and reversible alkene insertion followed by turnover limiting carbon-silicon bond formation.77 This mechanism involving nickel(II) intermediates containing one-electron reduced α-diimines is distinct from the well-established Chalk-Harrod mechanism operative with platinum catalysts (FIG 5).79,80
Figure 5 |. Proposed mechanism for alkene hydrosilylation with α-diimine nickel complexes.

The nickel-catalyzed hydrosilylation of 1-octene with triethoxysilane was scaled to 10 grams with 96% yield of n-octyl silane with >98% anti-Markovnikov selectivity using 1 mol% of nickel(2-EH)2 (R = 2-ethylhexanoate) and 1 mol% of the iPrDI ligand. This hydrosilylation can be carried out with 2.5 ppm of Karstedt’s catalyst to furnish 93.7% of the desired product after 1 hour of reaction time, albeit with a higher temperature of 70 °C.81 Silicone cross linking was also successful at ppm loadings and a clear and colorless product was obtained from air-stable precursors.77 Thus, the α-diimine nickel catalyst solved many of the practical challenges associated with earth-abundant catalysts: inexpensive ligands, air stable precursors and activity and anti-Markovnikov selectivity that is on par or superior to platinum catalysts.
Phosphine-ligated nickel(II) complexes were reported to catalyze the hydrosilylation of alkenes and alkynes with chlorosilanes at high temperatures, although de-chlorination of the silane is observed in some cases.82 In addition to well-defined, molecular precursors, heterogeneous nickel catalysts have also been explored for alkene and diene hydrosilylation. Lappert and coworkers reported Ziegler-type conditions involving the activation of Ni(acac)2 with AlEt3 for the hydrosilylation of 1,3-dienes and terminal acetylenes.77 More recently Shimida and coworkers described a similar protocol involving Ni(acac)2 and related derivatives activated with NaBEt3H for 1,3-diene hydrosilylation.84 Amorphous mixtures containing reduced nickel and various salts were generated from nickel(II) alkoxides and tertiary silanes and shown to be active for the hydrosilylation of 1-decene and other terminal and internal alkenes with HSi(OMe)3.85 Special care should be exercised with this silane due to its known ability to undergo disproportionation to SiH4 even when simply stored in a glass bottle.53
Opportunities and Outlook in Alkene Hydrosilylation with Earth-Abundant Transition Metals.
The past decade has witnessed transformative advances in the area of alkene hydrosilylation catalyzed by Earth-abundant first row transition metals. Given the scale and practical importance of transition metal catalysis in the silicones industry, attempts have been made over several decades to find more inexpensive alternatives to platinum.28 Early generation catalysts were plagued by impractical or challenging modes of catalyst activation, poor selectivity and low activity. The introduction of potentially redox-active and non-innocent33 pyridine(diimine) provided the first examples of iron catalysts that could compete with precious metals. As commercial interest in these catalysts grew, significant obstacles to implementation were identified. Catalyst cost extends far beyond the price of the transition metal; ligand synthesis and most importantly ease of handling are essential factors if Earth-abundant transition metals are ever to displace platinum. Complexes that have multistep ligand synthesis, require specialized starting materials or require handling in a glovebox will ultimately be “more expensive” than simple sources of platinum in the current market. Perhaps if the volatile precious metal market soars, interest will increase in more synthetically advanced or fragile catalysts.
Scores of metal-ligand combinations and a variety of catalyst activation modes have been evaluated for hydrosilylation reactions since the initial reports of the pyridine(diimine) iron catalysts.36,41 The diversity of structures that support active catalysts with iron, cobalt and nickel for this reaction is remarkable. Despite this progress, substantial challenges remain. Much is often made of substrate scope; while alkene hydrosilylation may evolve into a valuable method in synthesis,27 the primary application of the reaction is in the silicones industry. Many important commercial reactions have yet to be successfully addressed with first row transition metal catalysts. Chlorosilanes are widely used in platinum-catalyzed hydrosilylation but are incompatible with almost all known non-platinum catalysts.5 Likewise the hydrosilylation of vinyl chloride or allylic halides is also problematic.5 Recent advances in iron-catalyzed hydrogen isotope exchange,86 cobalt-catalyzed C-H borylation87,88 and alkene hydrogenation61 suggest that catalysts with broader functional group tolerance may be within reach.
The discovery of first-row transition metal catalysts for alkene hydrosilylation also offers the opportunity for mechanistic pathways distinct from platinum catalysts. While early carbonyl complexes of iron and cobalt likely operate by Chalk-Harrod or modified Chalk-Harrod pathways,64 complexes bearing redox active ligands have been shown to offer new pathways. Oxidative addition reactions, for example, operate by cooperative metal-ligand redox events.89 Understanding these processes and exploiting one-electron chemistry as a distinct feature of first row transition metal catalysis may ultimately offer catalyst design principles unavailable with heavier transition elements.
ALKENE AND TERMINAL ALKYNE HYDROBORATION
Uncatalyzed and Catalyzed Alkene Hydroboration.
Hydroboration, a landmark discovery reported in 1956 by H. C. Brown,90 opened new opportunities in organic synthesis, particularly with respect to the regioselective hydrofunctionalization of alkenes and alkynes. With highly reactive boranes such as diborane (B2H6), borane-THF (BH3•THF), and commonly used alkyboranes such as 9-borabicyclo[3.3.1]nonane (9-BBN), these reactions are highly exergonic and typically do not require a catalyst.91 Examples with BH3•SMe2 and 9-BBN applied to complex molecule synthesis are numerous and include Lonomycin A,92 an antibiotic, and Nebivolol,93 a blood pressure lowering agent, respectively. In these examples, the intermediate trialkylboranes were immediately oxidized without isolation due to their high reactivity, extreme air-sensitivity and incompatibility with purification by chromatography. Dialkoxyboranes such as catecholborane (HBCat) and pinacolborane (HBPin), the latter being most common, offered improved stability and handling but are sluggish to add to alkenes and alkynes and often do not undergo hydroboration in the absence of catalysts due to the diminished electrophilicity of the boron atoms.94 (BOX 2)
Box 2 |. Transition metal catalyzed hydroboration enables new opportunities for fine chemical synthesis.
Uncatalyzed alkene hydroboration is widely used in the synthesis of complex molecules due to its predictable anti-Markovnikov selectivity and incredibly high efficiency. However, the resulting trialkylborane products from uncatalyzed hydroborations are often air-sensitive and hard to purify, precluding their isolation. Transition metal catalysts allow the synthesis of air-stable, easy to handle alkylboronate esters which can streamline further reactivity studies. Moreover, transition metals have the ability to exert control on chemoselectivity, regioselectivity as well as enantioselectivity, which can enable new opportunities for fine chemical synthesis.
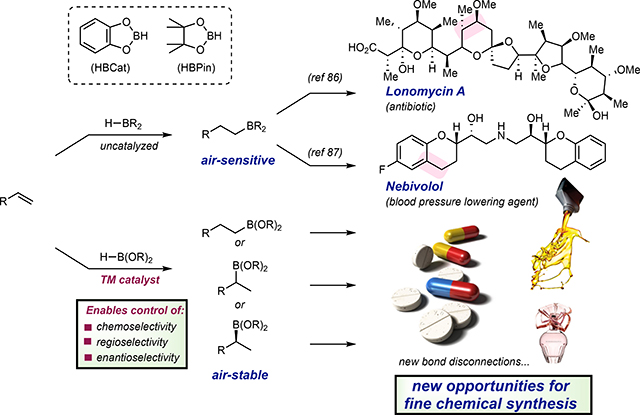
The discovery of transition metal catalysts for alkene and alkyne hydroboration with stabilized dialkoxy-substituted boranes is attractive as the resulting alkylboronate ester products are air- and bench-stable and compatible with chromatographic purification. Perhaps more importantly, the opportunity for transition metal catalysis to impart chemo-, regio- and enantioselectivity,95,96,97 coupled with the immensely rich reactivity of organoboron compounds make transition metal-catalyzed hydroboration an enabling tool for the synthesis of fine chemicals that may find promising applications in pharmaceutical, petroleum and fragrance industries (BOX 2). While many may view alkene and alkyne hydroboration as a mature area and perhaps even a solved problem, this section of the review will highlight the many new opportunities to access desirable organoboronate building blocks that open new avenues in synthesis.
Overview of Precious Metal-Catalyzed Alkene Hydroboration.
Much like hydrosilylation, considerable effort has been devoted to precious metal-catalyzed alkene hydroboration and a brief overview is given to provide the necessary context for advances with Earth-abundant transition metals. Männing and Nöth’s seminal report describing the application of (Ph3P)3RhCl to alkene hydroboration98 with HBPin initiated extensive investigations on next generation catalysts to improve reaction scope, applications and to understand the mechanism. Comprehensive reviews have been published on this topic91,94,95,96 and only selected findings that illustrate the state-of-the-art in the field and highlight the unmet needs of the process and the opportunities for catalysts with Earth-abundant transition metal are discussed.
Common reactivity patterns observed with rhodium and iridium catalysts are presented in FIG 6 (a-f). Terminal alkenes undergo hydroboration to yield linear alkylboronate esters98,99 (FIG 6a) while with vinylarenes methods to obtain linear99 and branched products100 (FIG 6b) including enantioselective variants101 are known. Hydroboration of 1,2-disubstituted linear alkenes such as 4-hexene proceed with internal selectivity,102 and examples of isomerization-hydroboration of these types of alkenes to yield terminally-functionalized products have been reported (FIG 6c).99,103,104 This process was shown to be capricious with (Ph3P)3RhCl99,105 and later attributed to partially oxidized rhodium.106 Diastereoselective, directed hydroborations to yield the 1,3-anti isomer (FIG 6d) as well as the 1,3-syn isomer (FIG 6e) have been reported with rhodium97 and iridium,97 respectively, with cyclic monosubstituted cyclohexenes. These directed hydroborations were effective with acyclic alkenes as well (FIG 6f).97
Figure 6 |. Hydroboration reactivity of precious metal catalysts.

Precious metal catalyzed hydroboration of a | terminal alkenes, b | vinyl arenes, and c | acylic 1,2-disubstituted alkenes. Directed hydroborations of cyclic monosubstituted cyclohexenes furnishing the d | 1,3-anti isomer and the e | 1,3-syn isomer f | directed hydroboration of acyclic internal alkenes g | (E)-selective and f | (Z)-selective hydroboration of terminal alkynes.
Hydroboration of Alkenes with Earth-Abundant, First-Row Transition Metal Complexes.
Alkene Hydroboration with Iron and Cobalt.
Despite the potential mechanistic similarity, examples of alkene hydroboration with first-row transition metals have lagged behind hydrosilylation, likely due to the economic motivations for replacement of precious metals with the latter. Ritter and coworkers pioneered this area with the selective 1,4-hydroboration of 1,3-dienes with HBPin promoted by an in situ-activated iminopyridine iron catalyst.107 The catalytic method was chemoselective for dienes but alkenes proved unreactive; the low affinity of the reduced iron complex for the latter type of substrate was offered as the rationale for the lack of turnover.107 Given the established reactivity of pyridine(diimine) and terpyridine iron complexes in alkene hydrogenation108 and hydrosilylation,48,65 it was unsurprising that these classes of catalyst could be extended to hydroboration. Huang and coworkers were first to publish a demonstration of the method109 by adaptation of an in situ activation protocol initially reported by Bouwkamp and coworkers for pyridine(diimine) iron-catalyzed [2+2] cycloaddition.51 Among the iron complexes with tridentate ligands evaluated, Milstein’s bipyridyl phosphine ligand110 was identified as optimal. The cobalt variant of this catalyst, again using NaBHEt3 activation, with improved activity and the same alkene scope was published one year later by the same group.111
While these catalysts were important in demonstrating the utility of iron and cobalt for the hydroboration of α-olefins with improved efficiency when compared with rhodium and iridium catalysts known at the time, the reported method was inactive for internal alkenes representing a limitation compared to known precious metal catalysts. Contemporary with Huang’s report, our laboratory reported that (ArPDI)Fe(N2)2 (Ar = 2,6-iPr2-C6H3; 2,4,6-Me3-C6H2) served as single-component catalysts for alkene hydroboration with HBPin.112 The well-defined iron catalysts proved superior to in situ activation methods as internal alkenes such as 4-octene underwent facile hydrofunctionalization. The isolated iron dinitrogen compounds enabled the exclusive formation of the terminal product, 1-octylboronate ester, from the hydroboration of 4-octene with HBPin.112 Such an isomerization-hydroboration sequence with 4-octene, similar to what has been observed with precious metal catalysts99,103,104 was subsequently reported by other laboratories following our report using (N-phosphinoamidate)cobalt catalysts,113 an alkylated amide-derived N,N,N-Fe(II) complex,114 and NHC-ligated iron carbonyl compounds.115 Cyclic olefins such as cyclohexene and cyclooctene were also efficiently hydroborated to furnish the 2° alkylboronate ester products. The pyridine(diimine) iron pre-catalysts, likely because of the large aryl substituents, have proven ineffective for the hydroboration of tri- and tetrasubstituted alkenes.
As with the alkene hydrosilylation, the air-sensitivity and challenges associated with the preparation of pyridine(diimine) iron dinitrogen compounds inspired the search for more accessible alternatives. Pyridine(diimine)cobalt alkyl compounds,60 (ArPDI)CoCH3, have proven more straightforward to prepare and offer a range of chelate modifications unavailable with the corresponding iron dinitrogen compounds.116 The second generation 4-pyrrolidinyl-substituted cobalt complex 11, promoted the isomerization-hydroboration of hindered, essentially unactivated tri- and tetra-substituted alkenes (FIG 7a).116 Even though this cobalt complex contains the same aryl substituents as in the (ArPDI)Fe(N2)2 complexes described above, the presence of the electron-donating pyrrolidinyl group at the 4-position likely increases the hydricity of the cobalt-hydride. For hindered tri- and tetra-substituted alkenes, the turnover-limiting step is likely alkene insertion and the electron-donating pyrrolidinyl group lowers the overall barrier for alkene insertion and thus enabling access to these challenging alkene substrates. The results of these studies illustrate the benefits of the readily synthesized, modular cobalt alkyl pre-catalysts. With the analogous pyridine(diimine) iron dinitrogen compounds, derivatives have not been easily accessed and the synthetic chemistry is a bottleneck for creating a diverse pre-catalyst library.
Figure 7 |. New reactivity enabled by base metal catalysts.

Remote hydrofunctionalization in the hydroboration of a | hindered tri- and tetra-substituted alkenes, b | internal alkenes containing a reactive functional group c | Accessing branched selectivity in activated substrates via controlled thermodynamic alkene isomerization followed by hydroboration d | Synthesis of homobenzyltriboronate esters via double dehydrogenative borylation followed by hydroboration e | Enantioselective hydroboration of activated 1-substituted vinylarenes and f | unactivated 1,1-disubstituted alkenes g/h | Enantioselective hydroboration/cyclization of 1,6-enynes i | (Z)-selective hydroboration of terminal alkynes with a mechanism distinct from its precious metal counterparts j | Unique alkynylboronate insertion mechanism was leveraged to develop a protocol involving 1,1-diboration of terminal alkynes k | Selective Markovnikov addition of unactivated terminal alkenes and l | unactivated 1,2-disubstituted alkenes are unmet needs in alkene hydroboration chemistry.
Exclusive formation of the terminally functionalized alkyl boronate ester as the product provides a convenient method for the hydrofunctionalization of remote positions independent of the position of the starting C=C bond (FIG 7a).116 This reactivity has not been reported with precious metal alkene hydroboration catalysts and highlights the new reactivity enabled by first row transition metals. The high activity of these cobalt catalysts enabled chemoselective isomerization-hydroboration of an internal alkene containing an ester, retaining the carbonyl group (FIG 7b). Deuterium labeling experiments as well as stoichiometric experiments on the cobalt pre-catalyst support a mechanism wherein a cobalt hydride undergoes reversible olefin insertion and β–hydride elimination to furnish a primary alkyl cobalt complex that is intercepted by HBPin to yield the terminal alkylboronate ester product (FIG 8a).116 The observation of deuterium throughout the alkyl chain establishes a reversible insertion and β-hydrogen elimination sequence, where interception of cobalt secondary alkyl intermediates with HBPin, either by σ-bond metathesis or oxidative addition-reductive elimination, is kinetically unfavorable. Once the cobalt migrates to the primary position of the alkyl chain, carbon-boron bond formation occurs and accounts for the preference for terminal selectivity in hydroboration.116 It is likely that for internal olefins, the turnover-limiting step is olefin isomerization. These observations are supported by the relatively poor performance of this class of catalyst toward cyclic alkenes such as cyclohexene and cyclooctene. When considering substrate scope, most would consider olefins of this type trivial as compared to more hindered tri- and tetrasubstituted alkenes. The reticence of pyridine(diimine) cobalt catalysts to form secondary carbon-boron bonds, likely due to the presence of the large aryl substituents, is the origin of this effect. It is important to note, however, that the (ArPDI)Fe(N2)2 compounds, which contain the same large aryl substituents, can effectively catalyze the hydroboration of these cyclic internal olefins presumably due to the decreased steric penalty in the interception of iron-secondary alkyl intermediates by HBPin by virtue of the larger atomic radius of iron than cobalt. This olefin isomerization-functionalization strategy was applied in the synthesis of linear alpha olefins (LAOs),117 which find use as high-value chemical intermediates in the petrochemical industry,118 from internal olefins. Using cobalt complex 7, chain-walking isomerization-hydroalumination followed by thermolysis allows the net transformation of internal olefins to LAOs.117
Figure 8 |. Proposed mechanisms in base metal catalyzed alkene and terminal alkyne hydroboration explaining new reactivity.

a | Proposed mechanism for remote hydrofunctionalization via isomerization-hydroboration b | Alkynylboronate insertion into a cobalt hydride as the origin of (Z)-selectivity in the hydroboration of terminal alkynes with pre-catalyst 16.
Despite the improved activity of 11, neat conditions and elevated temperatures were required for the hydroboration of hindered tri and tetrasubstituted olefins.116 To improve catalyst activity, cobalt compounds with bidentate α–diimine ligands were evaluated with the hypothesis that the smaller steric profile of the cobalt catalyst and the open coordination site may improve overall activity by lowering the barrier for coordination of the sterically hindered alkene. As anticipated, α–diimine cobalt allyl complex 12, (iPrDI)Co(η3-C3H5), was shown to be exceptionally active for challenging tri-, tetra- and geminally substituted alkenes, surpassing the catalytic activity of complex 11 (FIG 7a).119 However, hydroboration with pre-catalyst 12 was sluggish with styrenes and diene substrates such as limonene and this catalytic inhibition was attributed to the formation of inactive (iPrDI)Co(η6-arene) and (iPrDI)Co(η3-allyl) complexes, respectively. This highlights that while revealing an open coordination site in the pre-catalyst results in more facile alkene coordination, it also promotes deleterious catalyst deactivation pathways. These factors should be taken into consideration in future catalyst design.
Complementary synthetic and catalytic methods are desirable to access different products from the same starting materials through catalyst control. To achieve this objective, we sought cobalt complexes that would chain walk in the “opposite” direction, namely away from the terminus of an alkyl chain to internal positions. We sought to leverage the known alkene isomerization activity of the cobalt hydride phosphine complex 13, (PPh3)CoH(N2),120 a compound originally prepared by Sacco and Rossi, and couple it to carbon-boron bond formation. A method was developed where pre-mixing the alkene with 13 to promote isomerization to thermodynamically preferred internal positions followed by addition of HBPin resulted in isolation of benzyl boronate esters from alkenyl arenes with up to 10 methylene linkers between the olefin and the arene (FIG 7c).68 This method also proved successful for the synthesis 1,1-diboronate esters from α,ω-dienes.68
(Z)-selective hydroboration of terminal alkynes: mechanism and elucidation of the origin of selectivity.
Metal-catalyzed hydroboration of terminal alkynes presents an opportunity to control the stereo- and regiochemistry of the addition of the boron to the C≡C bond. A large number of transition metal catalysts are known to prepare the thermodynamically preferred (E)-vinylboronate ester.121–125 The prevalence of metal-hydrides and their ability to insert alkynes through hydrometallation accounts for the abundance of catalysts for this transformation (FIG 6g).126 Methods to synthesize the isomeric (Z)-vinylboronate esters, however, are relatively scarce and usually require multistep routes that are often difficult to control because of competing isomerization to the corresponding (E) isomer.127
Two precious metal catalysts have been reported to promote the (Z)-selective hydroboration of terminal alkynes. In 2000, Miyaura and co-workers published phosphine-ligated rhodium and iridium pre-catalysts and found that NEt3 and catecholborane provided higher (Z)-selectivity compared to the more commonly used pinacolborane (HBPin).128 An additional diol exchange step is required to generate more air- and silica-stable (Z)-vinylpinacolboronate esters. Twelve years later, Leitner and coworkers published the application of a nonclassical ruthenium hydride complex, [Ru(PNP)(H)2(H2)] to the (Z)-selective hydroboration of terminal alkynes with HBPin at - 15 °C.129 In both of these reports, the intermediacy of metal vinylidine species130 were proposed to account for the observed (Z)-selectivity (FIG 6h).
Many additional possible regio- and stereoselectivity patterns are available for the addition of borane (HBR2) and diborane (R2B-BR2) to C≡C bonds. To date, precious metal catalysts have been lacking in accessing many of these patterns. Given the value of various types of vinylboronates as building blocks for synthesis, there is a tremendous opportunity for first-row transition metals that extends well beyond potential cost and environmental advantages to access unique structural types.
In 2015, our laboratory reported that the cyclohexyl substituted pyridine(diimine) cobalt methyl complex, (CyAPDI)CoCH3 (16, FIG 7), promoted the (Z)-selective hydroboration of 1-alkynes using HBPin.131 High yields and diastereoselectivities were obtained with 3 mol% of the cobalt pre-catalyst for the valuable (Z)-vinylboronate esters (FIG 7i). A range of alkyne substituents was tolerated in the cobalt-catalyzed reaction including: protected propargyl ethers, propargyl and phthalamide substituents. This protocol obviates the need for diol exchange128 and low temperatures129 reported for the corresponding rhodium and ruthenium catalysts, respectively, to achieve high (Z)-selectivity.
Deuterium-labelling experiments with both deuterated alkyne and borane as well as stoichiometric experiments with the pre-catalyst and isolated intermediates were used to rule out the metal-vinylidene pathway invoked with precious metal catalysts. The data for the cobalt-catalyzed process are consistent with formation of cobalt acetylide complex obtained from interaction of 16 and the terminal alkyne. This compound engages with HBPin to form a hydrido cobalt alkynylboronate intermediate that undergoes insertion to form a vinyl cobalt product. Protonation with the terminal alkyne releases the (Z)-vinylboronate ester and regenerates the cobalt acetylide (FIG 8b). Stereodefined syn-hydrometallation of the alkynylboronate accounts for the observed (Z)-selectivity – a mechanism that is distinct from the precious metal catalysts.131
Performing the terminal alkyne hydroboration procedure with the aryl-substituted pyridine(diimine) cobalt complex (iPrPDI)CoCH3 (17, FIG 7) resulted in exclusive isolation of the (E)-vinylboronate ester product. The relative rates of reaction of 16 and 17 with terminal alkynes and HBPin accounts for the divergent selectivity. Aryl-substituted 17 engages HBPin to form the known cobalt hydride, (iPrPDI)CoH which undergoes alkyne insertion and reaction with borane to yield the observed (E)-vinylboronate product. With the (Z)-selective catalyst, the cobalt alkyl reacts faster with terminal alkynes obviating formation of a cobalt hydride and suppressing the (E)-selective pathway. The deuterium labeling experiments in the hydroboration of 1-octyne with DBPin using 16 and 17 as the pre-catalysts where 1,1 and 1,2 DBPin addition to the alkyne were observed, respectively, are consistent with this proposal. The origin of these relative rate differences is currently not understood. One notable difference between two classes of catalyst are their geometric preferences. With 17, the large aryl groups enforce planar geometry while with 16, the cyclohexyl groups are bent below the idealized plane of the pyridine(diimine) ligand and favor more distorted, pseudo tetrahedral geometries. Compounds with alkyl in place of aryl imine substituents are also more electron-rich. Delineation of the role of these effects on catalytic performance and selectivity is an interesting area for future study.
Since this initial publication, Kirchner reported iron(II) polyhydride pincer complexes as effective pre-catalysts for (Z)-selective terminal alkyne hydroboration.132 Vinylidene intermediates were proposed to account for the observed (Z)-selectivity, although no evidence for this proposal was presented.
In the mechanism depicted in FIG 8b, both the [H] and [BPin] components of the borane are transferred to the terminal carbon of the alkyne, suggesting that other classes of terminal alkyne 1,1-difunctionalization reactions should also be possible. Indeed, 16 is an effective pre-catalyst for the 1,1-diboration of alkynes with B2Pin2 to yield 1,1-diboryl alkenes (FIG 7j).133 Unsymmetrical diboron reagents such as BPin-BDan (Dan = naphthalene-1,8-diaminato) were also effective and yielded stereochemically pure, unsymmetric 1,1-vinyldiboronates. This enabled the synthesis of stereodefined trisubstituted alkenes, addressing a fundamentally important problem in organic synthesis. This 1,1-difunctionalization method was ultimately applied to the synthesis of tiagabine, an epilepsy medication.133 This is yet another example where the new mechanisms available to base metal catalysts not only provide more robust reaction conditions and alternatives in explaining reaction outcomes, but also provide a blueprint in the design and development of new catalytic reactions.
Beyond Pyridine(diimines): New Ligand Designs.
Achiral Alkene Hydroboration.
New catalysts based on iron,134–136 cobalt,137–139 nickel,140 manganese141 and even magnesium142 have been since been discovered for alkene hydroboration following the initial reports described above. Effective Earth-abundant transition metal pre-catalysts have been reported with a diverse array of ligands, including phosphine-iminopyridines,134 β-diketiminates,135 alkoxy-tethered NHCs136 (iron), CCC-type NHCs pincers137,139 and bipyridine metal organic frameworks (MOFs)138 (cobalt), phosphine-NHCs (nickel),140 terpyridines (manganese),141 as well as tris-(benzimidazolyl)methyl ligands on magnesium.142 Although no new reactivity patterns were disclosed with these examples, they nonetheless exemplify the applicability of different first row transition metals and diverse ligand architectures available for olefin hydroboration.
Using the same Milstein-type PNN ligands for cobalt in alkene hydroboration,111 Huang and coworkers reported a new and interesting reactivity pattern utilizing olefin hydroboration.143 In this method, two sequential dehydrogenative borylations of a vinylarene followed by hydroboration of the resulting 1,1-diboryl vinylarene with the HBPin generated in situ allowed the selective synthesis of 1,1,1-triboronates using vinyl arenes and B2Pin2 as starting materials (FIG 7d).143 This chemistry demonstrates the unique reactivity available to base metal catalysts by unlocking novel mechanisms.
Enantioselective Alkene Hydroboration.
In 2014, Huang144 and Lu145 published consecutive reports on the cobalt-catalyzed enantioselective hydroboration of notoriously challenging 1,1-disubstituted aryl alkenes146 using iminopyridine-oxazoline (IPO) ligands (FIG 7e). The reported enantioselectivities exceed those obtained previously with iridium.147 The C1 symmetric148 IPO ligand used in these studies contains a 2,6-diisopropylphenyl protecting element and an oxazoline chiral element. A similar strategy was previously used by our laboratory for asymmetric hydrogenation of α-substituted styrenes using C1 symmetric PDI cobalt alkyl pre-catalysts,108 where the same large 2,6-diisopropylphenyl protecting element was used to prevent formation of inactive bis(chelate) metal complexes149,150 and the chiral element is derived from commercially available chiral alkylamines. This ligand design was further applied to the enantioselective hydroboration-hydrogenation of internal alkynes using chiral imidazoline iminopyridine ligands (IIP), where the oxygen in the IPO ligand was replaced by an N-phenyl group.151 In this protocol, regioselective syn-hydroboration of the alkyne furnishes a vinylboronate that further undergoes enantioselective hydrogenation to yield enantioenriched chiral secondary organoboronates. Despite the demonstrated success of these chiral tridentate ligands in transferring their chiral information to pro-chiral alkenes, multi-step synthetic procedures are required, limiting rapid evaluation of derivatives. Recently, Ge and coworkers reported the use of chiral bisphosphine-ligated cobalt pre-catalysts, previously shown to be effective in asymmetric hydrogenation reactions,61 for the enantioselective hydroboration-cyclization of 1,6-enynes with HBPin (FIG 7g, 7h).152 These chiral bisphosphine ligands have also been utilized in copper catalysis where bisphosphine-ligated copper catalysts were shown to be highly effective in the enantioselective hydroboration of unactivated 1,1-disubstituted alkenes,153 overcoming the limitation of the (IPO)cobalt catalysts where high enantioselectivities could only be obtained in activated styrene derivatives (FIG 7f).144,145
Conclusions and Outlook in Alkene and Alkyne Hydroboration with Earth-Abundant Transition Metals.
Over the past seven years, base metal-catalyzed alkene hydroboration has attracted considerable attention and has become a vibrant field of research as evidenced by a number of significant breakthroughs and associated publications. Catalysts with Earth-abundant metals have proven, in some cases, to outperform their precious metal counterparts in terms of activity and selectivity but more importantly in several cases enable new bond disconnections that were not otherwise possible. These findings clearly demonstrate that controlling selectivity in hydroboration is far from a solved problem and many frontiers remain. It is therefore not surprising that the recent research efforts in the field are still being directed toward the discovery of new catalyst platforms for this transformation. Many of these new catalysts reported for hydroboration contain a tridentate ligand and a first row transition metal in low oxidation state or conversely in a high oxidation state in combination with an appropriate reductant such as NaHBEt3 or a Grignard reagent. These examples provide valuable proof-of-concept for the viability of diverse ligand and metal combinations for olefin hydroboration.
With a few exceptions, the base metal catalyzed reports either exhibit poor functional group compatibility or do not give details to what types of substrates were unsuitable for the pre-catalyst. Because olefin hydroboration finds the most potential in fine chemical synthesis, demonstration of exquisite regio- and chemoselectivity in a complex molecule containing an alkene as well as a suite of other functional groups will constitute an important advance in the field. There have been examples where chemoselective alkene hydroboration was observed with terminal alkenes containing different functional groups such as amino groups, esters, epoxides, halides, ketones, etc, 139 however, when more hindered alkenes are used, the catalysts are often poisoned by these functional groups. These observations demonstrate an important point related to reporting the functional group tolerance associated with a catalyst; often times this is merely a measure of relative rates. Increasing the reactivity of an olefin, in this case using terminal alkenes, will increase the “tolerance” of the reaction to functionality. Special care should be taken when interpreting such results.
One of the unsolved problems in alkene hydroboration chemistry is the selective Markovnikov addition of unactivated (non-styrenyl) α-olefins (FIG 7k). An ingenious solution to this problem involves a formal hydroboration with a diboron reagent and a proton source has been reported recently154 using systems developed by Hoveyda and co-workers,155,156 including an enantioselective variant by Aggarwal.157 However, the exquisite control of regioselectivity in the hydroboration of unsymmetric 1,2-disubstituted olefins still remains to be achieved (FIG 7l). More modular platforms that enable expedited evaluation of metal/ligand combinations are needed in order to possibly tackle this challenging problem.
As outlined in this review, catalysts with Earth-abundant metals offer complementary reactivity from their precious metal counterparts. However, the mechanistic underpinnings of this divergence in reactivity are lacking and further research is needed to pinpoint the factors that account for the regioselectivity differences between precious and base metal systems. Such a first principles understanding can form a fundamental framework where chemists can leverage the intrinsic differences between these classes of catalysts beneficial for the invention and development of useful catalytic transformations that extend beyond alkene hydroboration.
Acknowledgements
The authors thank Princeton University for financial support. J. V O. acknowledges the Howard Hughes Medical Institute International Student Research Fellowship and the 2016 Harold W. Dodds Honorific Fellowship (awarded by the Graduate School at Princeton University).
References
- 1).Principe LM in The Secrets of Alchemy (University of Chicago Press, 2013). [Google Scholar]
- 2).2017. World Energy Outlook: https://www.eia.gov/outlooks/ieo/pdf/0484(2017).pdf
- 3).Johnson J Global Energy Markets in Turmoil, International Energy Agency Says. Chem. Eng. News 95, 15 (2017). [https://cen.acs.org/articles/95/i46/Global-energy-markets-turmoil-International.html] [Google Scholar]
- 4).Marciniec B Catalysis by transition metal complexes of alkene silylation – recent progress and mechanistic implications. Coord. Chem. Rev 249, 2374–2390 (2005). [Google Scholar]
- 5).Marciniec B, Maciejewski H, Pietraszok C & Pawluc P in Advances in Silicone Science (ed. Matisons J) vol 1 (Springer, 2009). [Google Scholar]
- 6).Nakajima Y & Shimada S Hydrosilylation reactions of olefins: recent advances and perspectives. RSC Adv. 5, 20603–20616 (2015). [Google Scholar]
- 7).Vogels CM & Westcott SA Recent advances in organic synthesis using transition metal-catalyzed hydroborations. Curr. Org. Chem 9, 687–699 (2005). [Google Scholar]
- 8).Burgess K & Ohlmeyer MJ Transition-metal promoted hydroboration of alkenes, emerging methodology for organic transformations. Chem. Rev 91, 1179–1191 (1991). [Google Scholar]
- 9).Komiyama T, Minami Y & Hiyama T Recent advances in transition-metal-catalyzed synthetic transformations of organosilicon reagents. ACS Catalysis 7, 631–651 (2017). [Google Scholar]
- 10).Pukhnarevitch VB, Lukevics E, Kopylova LI & Voronkov M Perspectives of Hydrosilation (Institute for Organic Synthesis, Riga, Latvia, 1992). [Google Scholar]
- 11).Herzig C US Patent 6,265,497; 2001.
- 12).Friedman G, Sperry P & Brossas J US Patent 5,166,298; 1992.
- 13).Lewis LN, Stein J, Gao Y, Colborn RE & Hutchins G Platinum catalysts used in the silicones industry. Platin. Met. Rev 41, 66–75 (1997). [Google Scholar]
- 14).Momentive Performance Materials, Silquest* A-137 Technical Data Sheet. 2011, HCD-10164.
- 15).Troegel D & Stohrer J Recent advances and actual challenges in late transition metal catalyzed hydrosilylation of olefins from an industrial point of view. Coord. Chem. Rev 255, 1440–1459 (2011). [Google Scholar]
- 16).Speier JL, Webster JA & Barnes GH The Addition of Silicon Hydrides to Olefinic Double Bonds. Part II. The Use of Group VIII Metal Catalysts. J. Am. Chem. Soc 79, 974–979 (1957). [Google Scholar]
- 17).Lewis LN & Lewis N Platinum-catalyzed hydrosilylation-colloid formation as the essential step. J. Am. Chem. Soc 108, 7228–7231 (1986). [Google Scholar]
- 18).Stein J, Lewis LN, Gao L & Scott RA In situ determination of the active catalyst in hydrosilylation reactions using highly reactive Pt(0) catalyst precursors. J. Am. Chem. Soc 121, 3693–3703 (1999). [Google Scholar]
- 19).Markó IE et al. Selective and efficient platinum(0)-carbene complexes as hydrosilylation catalysts. Science 298, 204–206 (2002). [DOI] [PubMed] [Google Scholar]
- 20).Berthon-Gelloz G, Schumers J-M, Lucaccioni F, Tinant B, Wouters J, Markó IE Expedient, direct synthesis of (L)Pt(0)(1,6-diene) complexes from H2PtCl6. Organometallics 26, 5731–5734 (2007). [Google Scholar]
- 21).Bai H In situ platinum recovery and color removal from organosilicon streams. Ind. Eng. Chem. Res 51, 16457–16466 (2012). [Google Scholar]
- 22).Holwell AJ Optimised technologies are emerging which reduce platinum usage in silicone curing. Platin. Met. Rev 52, 243–246 (2008). [Google Scholar]
- 23).Chirik PJ & Weighardt K Radical ligands confer nobility on base-metal catalysts. Science 327, 794–795 (2010). [DOI] [PubMed] [Google Scholar]
- 24).Chirik PJ Iron- and cobalt-catalyzed alkene hydrogenation: catalysis with both redox-active and strong field ligands. Acc. Chem. Res 48, 1687–1695 (2015). [DOI] [PubMed] [Google Scholar]
- 25).Fürstner A Iron catalysis in organic synthesis: a critical assessment of what it takes to make this base metal a multitasking champion. ACS Cent. Sci 2, 778–789 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Sun J & Deng L Cobalt complex-catalyzed hydrosilylation of alkenes and alkynes. ACS Catal. 6, 290–300 (2016). [Google Scholar]
- 27).Du X & Huang Z Advances in base-metal-catalyzed alkene hydrosilylation. ACS Catal. 7, 1227–1243 (2017). [Google Scholar]
- 28).Nesmeyanov AN, Freidlina R. Kh., Chukovskaya EC, Petrova RG & Belyavsky AB Addition, substitution, and telomerization reactions of olefins in the presence of metal carbonyls or colloidal iron. Tetrahedron 17, 61–68 (1962). [Google Scholar]
- 29).Schroeder MA & Wrighton MS Pentacarbonyliron(0) photocatalyzed reactions of trialkylsilanes with alkenes. J. Organomet. Chem 128, 345–358 (1977). [Google Scholar]
- 30).Mitchener JC & Wrighton MS Photogeneration of Very Active Homogeneous Catalysts Using Laser Light Excitation of Iron Carbonyl Precursors. J. Am. Chem. Soc 103, 975–977 (1981). [Google Scholar]
- 31).Whetten RL, Fu KJ & Grant ER Pulsed-laser photocatalytic isomerization and hydrogenation of olefins. J. Am. Chem. Soc 104, 4270–4272 (1982). [Google Scholar]
- 32).Small BL, Brookhart M & Bennett AMA Highly active iron and cobalt catalysts for the polymerization of ethylene J. Am. Chem. Soc 120, 4049–4050 (1998). [Google Scholar]
- 33).Chirik PJ Preface: forum on redox-active ligands. Inorg. Chem 50, 9737–9740 (2011). [DOI] [PubMed] [Google Scholar]
- 34).Gibson VC, Redshaw C & Solan GA Bis(imino)pyridines: surprisingly reactive ligands and a gateway to new families of catalysts. Chem. Rev 107, 1745–1776 (2007). [DOI] [PubMed] [Google Scholar]
- 35).Darmon JM; Turner ZR; Lobkovsky E & Chirik PJ Electronic effects in 4-substituted bis(iminopyridines) and the corresponding reduced iron compounds. Organometallics 31, 2275–2285 (2012).22675236 [Google Scholar]
- 36).Bart SC, Lobkovsky E & Chirik PJ Preparation and Molecular and Electronic Structures of Iron(0) Dinitrogen and Silane Complexes and Their Application to Catalytic Hydrogenation and Hydrosilylation. J. Am. Chem. Soc 126, 13794–13807 (2004). [DOI] [PubMed] [Google Scholar]
- 37).Archer AM, Bouwkamp MW, Cortez M, Lobkovsky E & Chirik PJ Arene coordination in bis(imino)pyridine iron complexes: identification of catalyst deactivation pathways in iron-catalyzed hydrogenation and hydrosilation. Organometallics 25, 4269–4278 (2006). [Google Scholar]
- 38).Meciejewski H, Marciniec B & Kownacki I Catalysis of hydrosilylation part XXXIV. High catalytic efficiency of the nickel equivalent of Karstedt catalyst [{Ni(η-CH2=CHSiMe2)2O}2{μ-CH2=CHSiMe2)2O}]. J. Organomet. Chem 597, 175–181 (2000). [Google Scholar]
- 39).LaPointe AM, Rix FC & Brookhart M Mechanistic studies of palladium(II)-catalyzed hydrosilylation and dehydrogenative silation reactions. J. Am. Chem. Soc 119, 906–917 (1997). [Google Scholar]
- 40).Russel SK, Darmon JM, Lobkovsky E & Chirik PJ Synthesis of aryl-substituted bis(imino)pyridine iron dinitrogen complexes. Inorg. Chem 49, 2782–2792 (2010). [DOI] [PubMed] [Google Scholar]
- 41).Tondreau AM et al. Iron Catalysts for Selective Anti-Markovnikov Alkene Hydrosilylation Using Tertiary Silanes. Science 335, 567–570 (2012). [DOI] [PubMed] [Google Scholar]
- 42).Plueddemann E in Silane Coupling Agents (New York: ) ed. 2 (Plenum Press, 1991). [Google Scholar]
- 43).Petrea RD and Schuette RL US Patent 5,288,416; 1994.
- 44).Plonsker L US Patent 4,932,976; 1990.
- 45).Sprengers JW, de Greef M, Duin MA & Elsevier CJ Stable platinum(0) catalysts for catalytic hydrosilylation of styrene and synthesis of [Pt(Ar-bian)(η2-alkene)] complexes. Eur. J. Inorg. Chem 2003, 3811–3819 (2003). [Google Scholar]
- 46).Momentive Performance Materials, SilForce SL6000 Technical Data Sheet. 2016, HCD-10896.
- 47).Atienza CCH et al. High Selectivity Bis(imino)pyridine Iron Catalysts for the Hydrosilylation of 1,2,4-Trivinylcyclohexane. ACS Catal. 2, 2169–2172 (2012). [Google Scholar]
- 48).Tondreau AM et al. Synthesis, electronic structure, and alkene hydrosilylation activity of terpyridine and bis(imino)pyridine iron dialkyl complexes. Organometallics 31, 4886–4893 (2012). [Google Scholar]
- 49).Momentive Performance Materials, CoatOSil* 1770 Technical Data Sheet. 2011, HCD-10012.
- 50).Toya Y, Hayasakai K & Nakazawa H Hydrosilylation of olefins catalyzed by iron complexes bearing ketamine type iminobipyridine ligands. Organometallics 36, 1727–1735 (2017). [Google Scholar]
- 51).Bouwkamp MW, Bowman AC, Lobkovsky E & Chirik PJ Iron-catalyzed [2π+2π] cycloaddition of α,ω-dienes: the importance of redox-active supporting ligands. J. Am. Chem. Soc 128, 13340–13341 (2006). [DOI] [PubMed] [Google Scholar]
- 52).Chirik PJ, Tondreau AM, Delis JGP, Lewis KM, Weller KJ & Nye A US Patent 13/302,236; 2010.
- 53).Ryan JW Redistribution and Reduction Reactions of Alkoxysilanes. J. Am. Chem. Soc 84, 4730–4734 (1962). [Google Scholar]
- 54).Buchwald SL Silane Disproportionation Results in Spontaneous Ignition. Chem. Eng. News 71, 2 (1993). [http://pubs.acs.org/cen/safety/19930329.html] [Google Scholar]
- 55).Berk SC & Buchwald SL An Air-Stable Catalyst System for the Conversion of Esters to Alcohols. J. Org. Chem 57, 3751–3753 (1992). [Google Scholar]
- 56).Buslov I, Keller SC & Hu X Alkoxy Hydrosilanes as Surrogates of Gaseous Silanes for Hydrosilation of Alkenes. Org. Lett 18, 1928–1931 (2016). [DOI] [PubMed] [Google Scholar]
- 57).Greenhalgh MD, Frank DJ & Thomas SP Iron-catalyzed chemo-, region-, and stereoselective hydrosilylation of alkenes and alkynes using a bench-stable iron(II) precatalyst. Adv. Synth. Catal 356, 584–590 (2014). [Google Scholar]
- 58).Brandstadt K, Cook S, Nguyen BT, Surgenor A, Taylor R, Tzou M US Patent 61/536,799; 2011
- 59).Docherty JH, Peng J, Dominey AP & Thomas SP Activation and discovery of earth-abundant metal catalysts using sodium tert-butoxide. Nat. Chem 9, 595–600 (2017). [DOI] [PubMed] [Google Scholar]
- 60).Gibson VC, Tellman KP, Humphries MJ & Wass DF Bis(imino)pyridine cobalt alkyl complexes and their reactivity towards ethylene: a model system for β-hydrogen chain transfer. Chem. Commun 0, 2316–2317 (2002). [DOI] [PubMed] [Google Scholar]
- 61).Friedfeld MR, Shevlin M, Hoyt JM, Krska SW, Tudge MT & Chirik PJ Cobalt precursors for high-throughput discovery of base metal asymmetric alkene hydrogenation catalysts. Science 342, 1076–1080 (2013). [DOI] [PubMed] [Google Scholar]
- 62).Atienza CCH et al. Bis(imino)pyridine Cobalt-Catalyzed Dehydrogenative Silylation of Alkenes: Scope, Mechanism, and Origins of Selective Allylsilane Formation. J. Am. Chem. Soc 136, 12108–12118 (2014). [DOI] [PubMed] [Google Scholar]
- 63).McAtee JR, Martin SES, Ahneman DT, Johnson KA & Watson DA Preparation of allyl and vinyl silanes by the palladium-catalyzed silylation of terminal olefins: a silyl-Heck reaction. Angew. Chem. Int. Ed 51, 3663–3667 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64).Brookhart M & Grant BE Mechanism of a Cobalt(III)-Catalyzed Olefin Hydrosilation Reaction: Direct Evidence for a Silyl Migration Pathway. J. Am. Chem. Soc 115, 2151–2156 (1993). [Google Scholar]
- 65).Schuster CH, Diao T, Pappas I & Chirik PJ Bench-Stable, Substrate-Activated Cobalt Carboxylate Pre-Catalysts for Alkene Hydrosilylation with Tertiary Silanes. ACS Catal. 6, 2632–2636 (2016). [Google Scholar]
- 66).Chen C, Hecht MB, Kavara A, Brennesel WB, Mercado BQ, Weix DJ, & Holland PL Rapid, regioconvergent, solvent-free alkene hydrosilylation with a cobalt catalyst. J. Am. Chem. Soc 137, 13244–13247 (2015). [DOI] [PubMed] [Google Scholar]
- 67).Ibrahim AD, Entsminger SW, Zhu L and Fout AR A Highly Chemoselective Cobalt Catalyst for the Hydrosilylation of Alkenes using Tertiary Silanes and Hydrosiloxanes. ACS Catal. 6, 3589–3593 (2016). [Google Scholar]
- 68).Scheuermann ML, Johnson EJ & Chirik PJ Alkene isomerization–hydroboration promoted by phosphine–ligated cobalt catalysts. Org. Lett 17, 2716–2719 (2015). [DOI] [PubMed] [Google Scholar]
- 69).Noda D, Tahara A, Sunada Y & Nagashima H Non-Precious-Metal Catalytic Systems Involving Iron or Cobalt Carboxylates and Alkyl Isocyanides for Hydrosilylation of Alkenes with Hydrosiloxanes. J. Am. Chem. Soc 138, 2480–2483 (2016). [DOI] [PubMed] [Google Scholar]
- 70).Momentive Performance Materials, Silquest* A-1871 Technical Data Sheet. 2011, HCD-10053.
- 71).Marciniec B, Kownacka A, Kownacki I, HoffmNN M & Taylor R Hydrosilylation vs. dehydrogenative silylation of styrene catalyzed by iron(0) carbonyl complexes with multivinylsilicon ligands- mechanistic implications. J. Organomet. Chem 791, 58–65 (2015). [Google Scholar]
- 72).Sunada Y, Soejima H & Nagashima H Disilaferracycle dicarbonyl complex containing weakly coordinated η2-(H-Si) ligands: application to C-H functionalization of indoles and arenes. Organometallics 33, 5936–5939 (2014). [Google Scholar]
- 73).Sunada Y, Tsutsumi H, Shigeta K, Yoshida R, Hashimoto T & Nagashima H Catalyst design for iron-promoted reductions: an iron-disilyl-dicarbonyl complex bearing weakly coordinating η2-(H-Si) moieties. Dalton Trans. 42, 16687–16692 (2013). [DOI] [PubMed] [Google Scholar]
- 74).Raya B, Jing S, Balasanthiran V & RajanBabu TV Control of selectivity through synergy between catalysts, silanes, and reaction conditions in cobalt-catalyzed hydrosilylation of dienes and terminal alkenes. ACS Catal. 7, 2275–2283 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75).Liu Y & Deng L Mode of activation of cobalt(II) amides for catalytic hydrosilylation of alkenes with tertiary silanes. J. Am. Chem. Soc 139, 1798–1801 (2017). [DOI] [PubMed] [Google Scholar]
- 76).Buslov I, Becouse J, Mazza S, Montandon-Clerc M & Hu X Chemoselective Alkene Hydrosilylation Catalyzed by Nickel Pincer Complexes. Angew. Chem. Int. Ed 54, 14523–14526 (2015). [DOI] [PubMed] [Google Scholar]
- 77).Pappas I, Treacy S & Chirik PJ Alkene Hydrosilylation Using Tertiary Silanes with α-Diimine Nickel Catalysts. Redox-Active Ligands Promote a Distinct Mechanistic Pathway from Platinum Catalysts. ACS Catal. 6, 4105–4109 (2016). [Google Scholar]
- 78).Dong Q, Zhao Y, Su Y, SU J-H, Wu B & Yang X-J Synthesis and reactivity of nickel hydride complexes of an α-diimine ligand. Inorg. Chem 51, 13162–13170 (2012). [DOI] [PubMed] [Google Scholar]
- 79).Roy AK & Taylor RB The first alkene-platinum-silyl complexes: lifting the hydrosilation mechanism shroud with long-lived precatalytic intermediates and true Pt catalysts. J. Am. Chem. Soc 124, 9510–9524 (2002). [DOI] [PubMed] [Google Scholar]
- 80).Meister TK, Riener K, Gigler P, Stohrer J, Herrmann WA & Kühn FE Platinum catalysis revisited- unraveling principles of catalytic olefin hydrosilylation. ACS Catal. 6, 1274–1284 (2016). [Google Scholar]
- 81).Roy A; Boyer J; Koller J; Jenkins D; Trotto A; Kewis K Patent Application WO2015192029 A1; 2014.
- 82).Kiso Y, Kumada M, Tamao K & Umeno M Silicon Hydrides and Nickel Complexes I. Phosphine-Nickel(II) Complexes as Hydrosilylation Catalysts. J. Organomet. Chem 50, 297–310 (1973). [Google Scholar]
- 83).Lappert MF, Nile TA & Takahashi S Homogeneous catalysis:II. Ziegler systems as catalysts for hydrosilylation. J. Organomet. Chem 72, 425–439 (1974). [Google Scholar]
- 84).Srinivas V, Nakajima Y, Ando W, Sato K & Shimada S Bis(acetylacetonato)Ni(II)/NaHBEt3-catalyzed hydrosilylation of 1,3-dienes, alkenes and alkynes. J. Organomet. Chem 809, 57–62 (2016). [Google Scholar]
- 85).Buslov I, Song F, & Hu X An easily accessed nickel nanoparticle catalyst for alkene hydrosilylation with tertiary silanes. Angew. Chem. Int. Ed 55, 12295–12299 (2016). [DOI] [PubMed] [Google Scholar]
- 86).Yu RP, Hesk D, Rivera N, Pelczer I & Chirik PJ Iron-catalyzed tritiation of pharmaceuticals. Nature 529, 195–199 (2016). [DOI] [PubMed] [Google Scholar]
- 87).Obligacion JV, Semproni SP & Chirik PJ Cobalt-catalyzed C-H borylation. J. Am. Chem. Soc 136, 4133–4136 (2014). [DOI] [PubMed] [Google Scholar]
- 88).Obligacion JV, Bezdek MJ & Chirik PJ C(sp2)-H borylation of fluorinated arenes using an air-stable cobalt precatalyst: electronically enhanced site selectivity enables synthetic opportunities. J. Am. Chem. Soc 139, 2825–2832 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89).Darmon JM et al. Oxidative addition of carbon-carbon bonds with a redox-active bis(imino)pyridine iron complex. J. Am. Chem. Soc 134, 17125–17137 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90).Brown HC & Subba Rao BC A new powerful reducing agent-sodium borohydride in the presence of aluminum chloride and other polyvalent metal halides. J. Am. Chem. Soc 78, 2582–2588 (1956). [Google Scholar]
- 91).Carroll A-M, O’ Sullivan TP & Guiry PJ The development of enantioselective rhodium-catalyzed hydroboration of olefins. Adv. Synth. Catal 347, 609–631 (2005). [Google Scholar]
- 92).Evans DA, Ratz AM; Huff BE & Sheppard GS Total synthesis of the polyether antibiotic Lonomycin A (Emericid). J. Am. Chem. Soc 117, 3448–3467 (1995). [Google Scholar]
- 93).Volpicelli R et al. Process for preparing Nebivolol. U.S. Patent 8,258,323, 2012.
- 94).Beletskaya I & Pelter A Hydroborations catalyzed by transition metal complexes. Tetrahedron 53, 4957–5026 (1997). [Google Scholar]
- 95).Vogels CM & Westcott SA Recent advances in organic synthesis using transition metal-catalyzed hydroborations. Curr. Org. Chem 9, 687–699 (2005). [Google Scholar]
- 96).Burgess K & Ohlmeyer MJ Transition-metal promoted hydroboration of alkenes, emerging methodology for organic transformations. Chem. Rev 91, 1179–1191 (1991). [Google Scholar]
- 97).Evans DA, Fu GC & Hoveyda AH Rhodium(I)- and iridium(I)-catalyzed hydroboration reactions: scope and synthetic applications. J. Am. Chem. Soc 114, 6671–6679 (1992). [Google Scholar]
- 98).Männig D & Nöth H Catalytic hydroboration with rhodium complexes. Angew. Chem. Int. Ed. Engl 24, 878–879 (1985). [Google Scholar]
- 99).Yamamoto Y, Fujikawa R, Umemoto T & Miyaura N Iridium-catalyzed hydroboration of alkenes with pinacolborane. Tetrahedron 60, 10695–10700 (2004). [Google Scholar]
- 100).Burgess K et al. Reactions of catecholborane with Wilkinson’s catalyst: implications for transition metal-catalyzed hydroborations of alkenes. J. Am. Chem. Soc 114, 9350–9359 (1992). [Google Scholar]
- 101).Hayashi T; Matsumoto Y & Ito Y Catalytic asymmetric hydroboration of styrenes. J. Am. Chem. Soc 111, 3426–3428 (1989). [Google Scholar]
- 102.) Evans DA, Fu GC & Anderson BA Mechanistic study of the rhodium(I)- catalyzed hydroboration reaction. J. Am. Chem. Soc 114, 6679–6685 (1992). [Google Scholar]
- 103).Pereira S & Srebnik M Transition metal-catalyzed hydroboration of and CCl4 addition to alkenes. J. Am. Chem. Soc 118, 909–910 (1996). [Google Scholar]
- 104).Lata CJ & Crudden CM Dramatic effect of Lewis acids on the rhodium-catalyzed hydroboration of olefins. J. Am. Chem. Soc 132, 131–137 (2010). [DOI] [PubMed] [Google Scholar]
- 105).Pereira S & Srebnik M A study of hydroboration of alkenes and alkynes with pinacolborane catalyzed by transition metals. Tetrahedron Lett. 37, 3283–3286 (1996). [Google Scholar]
- 106).Hadebe SW & Robinson RS Microwave mediated rhodium-catalysed hydroboration of trans-4-octene with pinacolborane. Tetrahedron Lett. 47, 1299–1302 (2006). [Google Scholar]
- 107).Wu JY, Moreau B & Ritter T Iron-catalyzed 1,4-hydroboration of 1,3-dienes. J. Am. Chem. Soc 131, 12915–12917 (2009). [DOI] [PubMed] [Google Scholar]
- 108).Monfette S; Turner ZR; Semproni SP & Chirik PJ Enantiopure C1-Symmetric Bis(imino)pyridine Cobalt Complexes for Asymmetric Alkene Hydrogenation. J. Am. Chem. Soc 134, 4561–4564 (2012). [DOI] [PubMed] [Google Scholar]
- 109).Zhang L, Peng D, Leng X & Huang Z Iron-catalyzed, atom-economical, chemo- and regioselective alkene hydroboration with pinacolborane. Angew. Chem. Int. Ed 52, 3676–3680 (2013). [DOI] [PubMed] [Google Scholar]
- 110).Balaraman E, Gnanaprakasam B; Shimon LJW & Milstein D Direct hydrogenation of amides to alcohols and amines under mild conditions. J. Am. Chem. Soc 132, 16756–16758 (2010). [DOI] [PubMed] [Google Scholar]
- 111).Zhang L, Zuo Z, Leng X & Huang Z A cobalt-catalyzed alkene hydroboration with pinacolborane. Angew. Chem. Int. Ed 53, 2696–2700 (2014). [DOI] [PubMed] [Google Scholar]
- 112).Obligacion JV & Chirik PJ Highly selective bis(imino)pyridine iron-catalyzed alkene hydroboration. Org. Lett 15, 2680–2683 (2013). [DOI] [PubMed] [Google Scholar]
- 113).Ruddy AJ, Sydora OL, Small BL, Stradiotto M & Turculet L (N-phosphinoamidate)cobalt-catalyzed hydroboration: alkene isomerization affords terminal selectivity. Chem. Eur. J 20, 13918–13922 (2014). [DOI] [PubMed] [Google Scholar]
- 114).Tseng TK-N, Kampf JW & Szymczak NK Regulation of iron-catalyzed olefin hydroboration by ligand modifications at a remote site. ACS Catal. 5, 411–415 (2015). [Google Scholar]
- 115).Zheng J, Sortais J-B & Darcel C [(NHC)Fe(CO)4] efficient pre-catalyst for selective hydroboration of alkenes. ChemCatChem 6, 763–766 (2014). [Google Scholar]
- 116).Obligacion JV & Chirik PJ Bis(imino)pyridine cobalt-catalyzed alkene isomerization-hydroboration: a strategy for remote hydrofunctionalization with terminal selectivity. J. Am. Chem. Soc 135, 19107–19110 (2013). [DOI] [PubMed] [Google Scholar]
- 117).Weliange NM, McGuinness DS, Gardiner MG & Patel J Cobalt-bis(imino)pyridine complexes as catalysts for hydroalumination-isomerization of internal olefins. Dalton. Trans 45, 10842–10849 (2016). [DOI] [PubMed] [Google Scholar]
- 118).de Klerk A et al. Linear α-olefins from linear internal olefins by a boron-based continuous double-bond isomerization process. Ind. Eng. Chem. Res 46, 400–410 (2007). [Google Scholar]
- 119).Palmer WN, Diao T, Pappas I & Chirik PJ High-activity cobalt catalysts for alkene hydroboration with electronically responsive terpyridine and α–diimine ligands. ACS Catal. 5, 622–626 (2015). [Google Scholar]
- 120).Sacco A & Rossi M Hydride and nitrogen complexes of cobalt. Chem. Commun 316–316 (1967). [Google Scholar]
- 121).Pereira S & Srebnik M A study of hydroboration of alkenes and alkynes with pinacolborane catalyzed by transition metals. Tetrahedron Lett. 37, 3283–3286 (1996). [Google Scholar]
- 122).Lee et al. Stereoselective hydroboration of diynes and triyne to give products containing multiple vinylene bridges: a versatile application to fluorescent dyes and light-emitting copolymers. Organometallics 23, 4659–4575 (2004). [Google Scholar]
- 123).Nielson BM & Bielawski CW Photoswitchable metal-mediated catalysis: remotely tuned alkene and alkyne hydroborations. Organometallics 32, 3121–3128 (2013). [Google Scholar]
- 124).Pereira S & Srebnik M Hydroboration of alkynes with pinacolborane catalyzed by HZrCp2Cl. Organometallics 14, 3127–3128 (1995). [Google Scholar]
- 125).He X & Hartwig JF True metal-catalyzed hydroboration with titanium. J. Am. Chem. Soc 118, 1696–1702 (1996). [Google Scholar]
- 126).Otsuka S & Nakamura A Acetylene and allene complexes: their implication in homogeneous catalysis. Adv. Organomet. Chem 14, 245–283 (1976). [Google Scholar]
- 127).Hermann WA & Prinz M Applied Homogeneous Catalysis with Organometallic Compounds 2nd edn, 1119–1124 (2002). [Google Scholar]
- 128).Ohmura T, Yamamoto Y & Miyaura N Rhodium- or iridium-catalyzed trans-hydroboration of terminal alkynes giving (Z)-1-alkenylboron compounds. J. Am. Chem. Soc 122, 4990–4991 (2000). [Google Scholar]
- 129).Gunanathan C, Hölscher M, Pan F & Leitner W Ruthenium catalyzed hydroboration of terminal alkynes to Z-vinylboronates. J. Am. Chem. Soc 134, 14349–14352 (2012). [DOI] [PubMed] [Google Scholar]
- 130).Bruneau C & Dixneuf PH Metal vinylidenes and allenylidenes in catalysis: applications in anti-Markovnikov additions to terminal alkynes and alkene metathesis. Angew. Chem. Int. Ed 45, 2176–2203 (2006). [DOI] [PubMed] [Google Scholar]
- 131).Obligacion JV, Neely JM, Yazdani AN, Pappas I & Chirik PJ Cobalt catalyzed Z-selective hydroboration of terminal alkynes and elucidation of the origin of selectivity. J. Am. Chem. Soc 137, 5855–5858 (2015). [DOI] [PubMed] [Google Scholar]
- 132).Gorgas N, Alves LG, Stöger B, Martins AM, Veriros LF & Kirchner K Stable, yet highly reactive nonclassical iron(II) polyhydride pincer complexes: Z-selective dimerization and hydroboration of terminal alkynes. J. Am. Chem. Soc 139, 8130–8133 (2017). [DOI] [PubMed] [Google Scholar]
- 133).Krautwald S, Bezdek MJ & Chirik PJ Cobalt-catalyzed 1,1-diboration of terminal alkynes: scope, mechanism, and synthetic applications. J. Am. Chem. Soc 139, 3868–3875 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134).Gilbert-Wilson R, Chu W-Y & Rauchfuss TB Phosphine-iminopyridines as platforms for hydrofunctionalization of alkenes. Inorg. Chem 54, 5596–5603 (2015). [DOI] [PubMed] [Google Scholar]
- 135).Espinal-Viguri M; Woof CR & Webster RL Iron-catalyzed hydroboration: unlocking reactivity through ligand modulation. Chem. Eur. J 22, 11605–11608 (2016). [DOI] [PubMed] [Google Scholar]
- 136).MacNair AJ; Millet CRP; Nichol GS; Ironmonger A & Thomas SP Markovnikov-selective, activator-free iron-catalyzed vinylarene hydroboration. ACS Catal. 6, 7217–7221 (2016). [Google Scholar]
- 137).Reilly SW; Webster CE; Hollis K & Valle HU Transmetallation from CCC-NHC pincer Zr complexes in the synthesis of air-stable CCC-NHC pincer Co(III) complexes and iniatial hydroboration trials. Dalton Trans. 45, 2823–2828 (2016). [DOI] [PubMed] [Google Scholar]
- 138).Zhang T; Manna K & Lin W Metalorganic frameworks stabilize solution-inaccesible cobalt catalysts for highly efficient broad-scope organic transformations. J. Am. Chem. Soc 138, 3241–3249 (2016). [DOI] [PubMed] [Google Scholar]
- 139).Ibrahim AD, Entsminger SW & Fout AR Insights into a chemoselective cobalt catalyst for the hydroboration of alkenes and nitriles. ACS Catal. 7, 3730–3734 (2017). [Google Scholar]
- 140).Touney EE; Van Hoveln R; Buttke CT; Freidberg MD; Guzei IA & Schomaker JM Heteroleptic nickel complexes for the Markovnikov-selective hydroboration of styrenes. Organometallics 35, 3436–3439 (2016). [Google Scholar]
- 141).Zhang G; Zeng H; Wu J; Yin Z; Zheng S & Fettinger JC Highly selective hydroboration of alkenes, ketones and aldehydes catalyzed by a well-defined manganese complex. Angew. Chem. Int. Ed 55, 14369–14372 (2016). [DOI] [PubMed] [Google Scholar]
- 142).Rauch M; Ruccolo S & Parkin G Synthesis, Structure, and Reactivity of a Terminal Magnesium Hydride Compound with a Carbatrane Motif, [TismPriBenz]MgH: A Multifunctional Catalyst for Hydrosilylation and Hydroboration. J. Am. Chem. Soc 139, 13264–13267 (2017). [DOI] [PubMed] [Google Scholar]
- 143).Zhang L & Huang Z Synthesis of 1,1,1-tris(boronates) from vinylarenes by Co-catalyzed dehydrogenative borylations-hydroboration. J. Am. Chem. Soc 137, 15600–15603 (2015). [DOI] [PubMed] [Google Scholar]
- 144).Zhang L, Zuo Z, Wan X & Huang Z Cobalt-catalyzed enantioselective hydroboration of 1,1-disubstituted aryl alkenes. J. Am. Chem. Soc 136, 15501–15504 (2014). [DOI] [PubMed] [Google Scholar]
- 145).Chen J, Xi T & Lu Z Iminopyridine oxazoline iron catalyst for asymmetric hydroboration of 1,1-disubstituted aryl alkenes. Org. Lett 16, 6452–6455 (2014). [DOI] [PubMed] [Google Scholar]
- 146).Masamune S; Kim B; Petersen JS; Sato T & Veenstra SJ Organoboron compounds in organic synthesis. 1. Asymmetric hydroboration. J. Am. Chem. Soc 107, 4549–4551 (1985). [Google Scholar]
- 147).Mazet C & Gérard D Highly regio- and enantioselective catalytic asymmetric hydroboration of α-substituted styrenyl derivatives. Chem. Commun 47, 298–300 (2011). [DOI] [PubMed] [Google Scholar]
- 148).Bianchini et al. Oligomerisation of ethylene to linear α-olefins by CS and C1-symmetric [2,6-bis(imino)pyridyl]iron and –cobalt dichloride complexes. Eur. J. Inorg. Chem 1620–1631 (2003). [Google Scholar]
- 149).Tondreau AM; Darmon JM; Wile BM; Floyd SK; Lobkovsky E & Chirik PJ Enantiopure pyridine bis(oxazoline) “pybox” and bis(oxazoline) “box” iron dialkyl complexes: comparison to bis(imino)pyridine compounds and application to catalytic hydrosilylation of ketones. Organometallics 28, 3928–3940 (2009). [Google Scholar]
- 150).Wile BM et al. Reduction chemistry of aryl- and alkyl-substituted bis(imino)pyridine iron dihalide compounds: molecular and electronic structures of [(PDI)2Fe] derivatives. Inorg. Chem 48, 4190–4200 (2009). [DOI] [PubMed] [Google Scholar]
- 151).Guo J, Cheng B, Shen X & Lu Z Cobalt-catalyzed asymmetric sequential hydroboration/ hydrogenation of internal alkynes. J. Am. Chem. Soc 139, 15316–15319 (2017). [DOI] [PubMed] [Google Scholar]
- 152).Yu S, Wu C & Ge S Cobalt-catalyzed asymmetric hydroboration/cyclization of 1,6-enynes with pinacolborane. J. Am. Chem. Soc 139, 6526–6529 (2017). [DOI] [PubMed] [Google Scholar]
- 153).Jang WJ, Song SM, Moon JH, Lee JY & Yun J Copper-catalyzed enantioselective hydroboration of unactivated 1,1-disubstituted alkenes. J. Am. Chem. Soc 139, 13660–13663 (2017). [DOI] [PubMed] [Google Scholar]
- 154).Kerchner HA & Montgomery J Synthesis of secondary and tertiary alkylboranes via formal hydroboration of terminal and 1,1-disubstituted alkenes. Org. Lett 18, 5760–5763 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155).Jang H; Zhugralin AR; Lee Y & Hoveyda AH Highly selective methods for synthesis of internal (α-) vinylboronates through efficient NHC-Cu-catalyzed hydroboration of terminal alkynes. Utility in chemical synthesis and mechanistic basis for selectivity. J. Am. Chem. Soc 133, 7859–7871 (2011). [DOI] [PubMed] [Google Scholar]
- 156).Corberán R; Mszar NW & Hoveyda AH NHC-Cu-catalyzed enantioselective hydroboration of acyclic and exocyclic 1,1-disubstituted aryl alkenes. Angew. Chem. Int. Ed 50, 7079–7082 (2011). [DOI] [PubMed] [Google Scholar]
- 157).Smith JR; Collins BSL; Hesse MJ; Graham MA; Myers EL & Aggarwal VK Enantioselective Rhodium(III)-Catalyzed Markovnikov Hydroboration of Unactivated Terminal Alkenes. J. Am. Chem. Soc 139, 9148–9151 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]