

Abstract
As explained by the free drug theory, the unbound fraction of drug has long been thought to drive the efficacy of a molecule. Thus, the fraction unbound term, or fu, appears in equations for fundamental pharmacokinetic parameters such as clearance, and is used when attempting in vitro to in vivo extrapolation (IVIVE). In recent years though, it has been noted that IVIVE does not always yield accurate predictions, and that some highly protein bound ligands have more efficient uptake than can be explained by their unbound fractions. This review explores the evolution of fu terms included when implementing IVIVE, the concept of protein-facilitated uptake, and the mechanisms that have been proposed to account for facilitated uptake.
Keywords: Protein binding, Albumin-facilitated uptake, In vitro-in vivo extrapolation
Graphical Abstract
1. Introduction
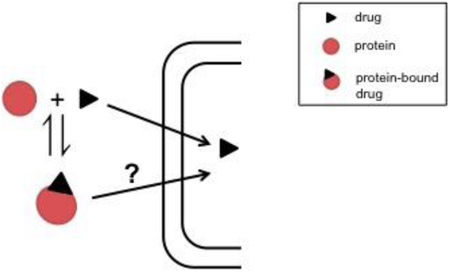
Beginning as early as 1949 with Goldstein’s review of the interactions between drugs and proteins, pharmaceutical scientists have recognized the importance of plasma protein binding (PPB) in pharmacokinetics and pharmacodynamics. Appearing in equations for several parameters including clearance and volume of distribution, it is one of the most fundamental properties in the field (Schmidt et al., 2010). The widely accepted free drug theory (FDT) (Fig. 1) explains that plasma protein binding is a rapid equilibrium process allowing a constant concentration of free drug, and in the absence of energy-dependent processes, this free drug concentration is the same on both sides of a membrane at steady state (Trainor, 2007). The other main principle of FDT is that only free drug can reach the site of action (or metabolism), and therefore the free drug concentration is what drives the pharmacological effect of a molecule (Smith et al., 2010).
Fig. 1: Free Drug Theory.

According to free drug theory only free drug can reach the site of action or metabolism, and at steady state, the unbound drug concentration is the same on both sides of the membrane.
While PPB is one of the most fundamental properties, it is often misinterpreted and compounds are wrongfully “optimized” based on protein binding measurements (Smith et al., 2010; Liu et al., 2014). Hueberger, Schmidt, and Derendorf (2013) nicely summarize the answer to the question they pose in their paper entitled, “When Is Protein Binding Important?”. In the final section they discuss the use of fu, the unbound fraction of drug, in the in vitro-in vitro extrapolation (IVIVE) of hepatic clearance. In honor of Dr. Derendorf’s retirement in this dedicatory issue, this review expands upon the questions surrounding protein binding, protein-facilitated uptake, and IVIVE.
1.1. Plasma Protein Binding
1.1.1. Major drug binding proteins
While there are several drug-binding components in plasma including lipoproteins and globulins, human serum albumin (HSA) and α−1-acid glycoprotein (AAG) have been the most extensively studied and are present in large enough amounts to have an effect on drug action.
HSA, a 66 kDa globular protein containing 585 amino acids including a large amount of charged residues, is present in the body at a relatively constant concentration of 600 μM (Quinlan et al., 2005). It is the most abundant protein in human plasma and accounts for 50% of total plasma protein content (Farrugia, 2010). Physiologically HSA cmbhelps maintain colloid osmotic pressure, and is capable of binding both endogenous ligands (such as fatty acids and bilirubin) as well as xenobiotics (Quinlan et al., 2005). Composed of 3 homologous domains (I-III) each with two sub-domains (A and B) (Quinlan et al., 2005), HSA has several low affinity binding sites and at least two high affinity drug binding sites (Sudlow site I and Sudlow site II) with a bias for binding acidic drugs (Sudlow et al., 1975). The protein undergoes different transitions depending on pH: the neutral-fast (N-F) transition between pH 5.0–3.5 that causes elongation, the fast-elongated (F-E) or acid expansion transition below pH 3.5 that causes further expansion, and the neutral-base (N-B) transition between pH 7.0–9.0 that causes enhanced binding at site I (Wanwimolruk and Birkett, 1982; Dockal et al., 2000).
AAG, a 38–48 kDa acidic protein containing 204 amino acids, can have more variable concentrations in the body. In healthy subjects it is typically present at 12–31 μM, however as an acute-phase protein synthesized in the liver, it can be as high as 60 μM in some disease states (Trainor, 2007). In other disease states, while levels of AAG are unchanged, its binding capacity is reduced (Israili and Dayton, 2001). AAG binding has also been shown to be dependent on age, gender, ethnicity, obesity, pregnancy, and diurnal changes (Bohnert and Gan, 2013). While multiple binding sites have been reported, only one appears to be important for drug binding, and it has a preference for basic and neutral drugs (Israili and Dayton, 2001). The lower baseline levels of AAG as well as its possibility to fluctuate can readily cause drug-binding effects.
The association and dissociation of the drug-protein complex is rapid (Schmidt et al., 2010) and at equilibrium can be described as:
where kon and koff are the association and dissociation rate constants, and the affinity of the drug for binding to the protein can be described by the association constant, ka, or its inverse, the dissociation constant, kd
| (1) |
1.1.2. Methods to measure plasma protein binding
While there are several methods used to measure protein binding and in depth reviews comparing them (Bohnert and Gan, 2013; Howard et al., 2010), the three most widespread methods measuring equilibrium binding in vitro will be briefly summarized here (Fig. 2).
Fig. 2: In vitro methods for measuring fu.

The fraction unbound can be determined with A) equilibrium dialysis B) ultrafiltration or C) ultracentrifugation.
The most commonly used technique to measure protein binding in the pharmaceutical industry is equilibrium dialysis (Fig. 2A). Using a device with two chambers separated by a semipermeable membrane, the protein-drug solution is added on one side, while buffer is added on the opposing side. When equilibrium is reached, the free fraction can be determined by measuring the total drug concentration in the protein chamber and the free drug concentration in the buffer chamber. Despite its ease of use, equilibrium dialysis still has disadvantages that must be considered including nonspecific binding to the membrane and the apparatus, volume shifts due to colloidal osmotic pressure, Gibbs-Donnan effects where charged particles near the membrane do not distribute evenly on both sides of the membrane, protein leakage across the membrane if the integrity of the membrane is compromised, and the need to determine the time required to reach equilibrium (Howard et al., 2010).
Other commonly used methods are ultrafiltration and ultracentrifugation. Ultrafiltration uses centrifugal force and a semipermeable membrane to separate a protein-free phase from a drug-protein solution (Fig. 2B). With this method the drug-protein solution is placed in the upper chamber of the two-chambered device and centrifugation (~2,000g) is used to move the unbound drug into the lower chamber (Bohnert and Gan, 2013). Here the total drug concentration is measured before centrifugation, and the free drug concentration is measured in the lower chamber at the end of the process. Similar to equilibrium dialysis, nonspecific binding to the membrane and the apparatus, Gibbs-Donnan effects, and protein leakage must be considered. In addition, more rigorous temperature and pH control are needed, and molecular sieving, where plasma water passes though the membrane faster than drug molecules, must be recognized (Howard et al., 2010).
Ultracentrifugation separates a drug-protein solution into several phases by using a high centrifugal force (~500,000g) for a long period of time (10–24hr) (Bohnert and Gan, 2013) (Fig. 2C). While this method avoids the membrane issues encountered with equilibrium dialysis and ultrafiltration, it has its own challenges. After centrifugation, three distinct layers are formed: a top layer containing very low-density lipoproteins and chylomicrons, a middle layer of free drug, and a bottom layer containing high-density molecules including albumin, AAG, and lipoproteins. To determine the free fraction, total drug concentration is measured before centrifugation and free drug concentration is measured from the middle layer at the end of the process. By having distinct layers, binding to specific proteins such as low-density lipoproteins vs. albumin can be determined (Brockman et al., 2015). The three layers can lead to the experimental difficulties since the top lipid layer must not be disrupted during sampling, and it is necessary to use the correct centrifugation parameters to ensure a truly protein-free middle layer (Nakai et al., 2004). Additionally, free drug can sediment depending on its shape, size, and the run temperature, while back diffusion of drugs from the protein-free layer is possible (Bohnert and Gan, 2013).
While these three methods are among the most commonly used, reviews summarize additional methods including gel filtration, chromatography, capillary electrophoresis, erythrocyte partitioning, surface plasmon resonance, and microdialysis (Howard et al., 2010; Bohnert and Gan, 2013). Despite advances in technology, there is still uncertainty in the fu values generated using these standard methodologies for highly bound drugs. Recent DDI guidelines reflect the low confidence in measured fu values as regulatory agencies decided that the lower limit should be 0.01, regardless of the actual measured value, to avoid false negative DDI predictions (Di et al., 2017). However, approximately one third of experimental drugs have high protein binding (≥ 99%), and using warfarin and itraconazole as examples, Di et al. (2017) show that fu ≤ 0.01 may be accurately measured using appropriate methods.
2. IVIVE and Protein Binding
Given that the current drug development process is expensive and time-consuming, (Parasrampuria et al., 2018), it is important to identify failures as early in the pipeline as possible to reduce inefficiency. When deciding which new chemical entities (NCE) to move forward, one of the most important pharmacokinetic parameters to consider is clearance as it is linked to drug exposure, half life, and dosing interval (Benet, 2010). Allometric scaling has been shown to be useful for drugs that are primarily cleared renally, however it is less useful for compounds cleared hepatically (Huh et al., 2011). Instead, in vitro - in vivo extrapolation is commonly used to predict the hepatic clearance of compounds and accurately understanding fu is crucial for these predictions.
The typical IVIVE process involves determining the intrinsic clearance (CLint) of a compound in vitro using microsomes or hepatocytes, which can then be scaled to an in vivo prediction using physiologically based scaling factors as well as a model of hepatic disposition such as the well-stirred model (Table 1). CLint is a measure of the body’s ability to remove drug in the absence of protein binding or blood flow limitations (Rowland et al., 1973; Wilkinson and Shand, 1975), and in vitro CLint is commonly measured by either uptake or substrate depletion assays (Houston, 1994; Chao et al., 2010). Rane et al. (1977) first suggested the use of in vitro uptake assays for hepatic clearance prediction and determined CLint under low substrate concentrations ([S] <<Km) using Eq. (2)
| (2) |
where Vmax represents the maximum velocity of metabolism and the Michaelis-Menten constant, km, is the concentration of drug at half Vmax (Rane et al., 1977). For substrate depletion assays, the metabolic rate parameter, k, can be determined from the slope of the linear regression of the log percentage of drug remaining versus time, and CLint can then be determined using Eq. (3)
| (3) |
where Vinc is the volume of the in vitro incubation (Obach et al., 1997; Obach, 1999).
Table 1:
Equations Commonly Used for IVIVE
| Model | Clearance Equation | |
|---|---|---|
| Traditional Well-Stirred | (5) | |
| Correction for binding in the incubation | (6) | |
| Correction for pH gradient | (10) | |
| Correction for pH gradient and PLR | (13) |
CLint,in vitro can then be scaled to CLint,in vivo (Eq. 4) using scaling factors (SF) that typically include 40 mg microsomal protein/g liver (Hakooz et al., 2006) or 120 million hepatocytes/g liver (Hallifax et al., 2010) as well as 21.4 g liver/kg bodyweight (Hallifax et al., 2010).
| (4) |
Following this scale-up, a model of hepatic disposition such as the well-stirred model (Rowland et al., 1973; Wilkinson and Shand, 1975) shown as Eq. (5),
| (5) |
(where QH is hepatic blood flow andfu,B is the fraction unbound in blood), the parallel tube model (Winkler et al., 1973), or the dispersion model (Roberts and Rowland, 1986) is commonly applied. It has been shown that clearance predictions are similar among the three models except for high clearance drugs, where the well-stirred model will cause underprediction (Chiba et al., 2009). However, our lab recently derived the theoretical basis for the extraction ratio and found that when organ clearance is calculated as the product of the extraction ratio (ER) and blood flow (Q) to the organ, it is only consistent with the well-stirred model (Benet et al. 2018). The paper goes on to explain why comparisons of the different models are not possible and IVIVE can only potentially work for the well-stirred model. Within the well-stirred model, it is important to note that total drug concentrations must be measured in blood and the fraction unbound is in reference to blood since the liver is capable of removing drug from both plasma and blood cells (Yang et al., 2007).
Despite the common use of IVIVE, it is surprising that in vitro measures from human liver tissue cannot adequately predict in vivo human hepatic clearance (Bowman and Benet, 2016), and in vitro measures from rat liver tissue cannot adequately predict in vivo rat hepatic clearance (Wood et al., 2017). Looking at data from both human microsomes and hepatocytes, 66.5% of predictions fell more than 2-fold outside measured in vivo values, and prediction accuracy was comparable for both human microsome and hepatocyte data (Bowman and Benet, 2016). Wood et al. (2017) also saw similar results with approximately 75.0% of human microsome and hepatocyte data falling more than 2-fold outside measured in vivo values. Wood et al. (2017) summarize, “ultimately, the in vitro causes of under prediction are likely to be multifactorial,” some of which may include preparation process issues, cofactor depletion, and the possibility of extra-hepatic metabolism (Chiba et al., 2009).
We recently introduced a new term Rss, the ratio of the volume of distribution of drug in the whole liver to the volume of distribution of drug in the hepatocyte water at steady state (Sodhi and Benet, 2017). We suggest that poor IVIVE predictions may result in part from the use of a chemical approach, where the liver is considered to be homogenous organ, to predict a pharmacokinetic parameter. In pharmacokinetics the liver is a heterogeneous organ containing both aqueous and lipid components into which the drug can distribute, and this difference needs to be accounted for in the scale up of IVIVE by including Rss. However, for drugs that have an Rss value of close to 1.0, current metabolic IVIVE predictions may work.
Heuberger et al. (2013) discuss that one of the other major questions regarding IVIVE scale-up is which fu terms to include, which we elaborate on here.
2.1. Nonspecific Binding & fu,inc
As mentioned earlier, the widely accepted free drug theory says that only unbound drug can exert pharmacological effects, and thus when doing IVIVE the unbound drug concentrations in plasma, tissue, and assays should be considered (Faed, 1981). It has been well recognized that when inputting data into the well-stirred model, the fraction unbound in blood (fu,B) must be used, or the fraction unbound in plasma (fu,p) divided by the blood to plasma ratio (RB) (Yang et al., 2007). More recently the idea that the fraction unbound in the in vitro assay may need to be incorporated as well has begun to be implemented (Obach, 1996; Ito and Houston, 2005; Venkatakrishan et al., 2000; Ring et al., 2011).
Since nonspecific binding may occur in the in vitro system if drug binds to the incubation plate, to proteins in the assay media, or to proteins and lipids of microsomes or cells, another binding term, fu,inc, or the fraction of drug unbound in the incubation, can be included as shown in Eq. 6. As Obach (1996) explained, “Thus, most Km and Ki values reported in the literature that use impure in vitro systems are artifactual overestimates, because they are based on total substrate or inhibitor concentration added to the incubation (i.e., the nominal concentration) and not the free substrate or inhibitor available to bind to the enzyme.”
| (6) |
2.1.1. fuinc measured in vitro
The same assays for measuring PPB as mentioned previously (see section 1.1.2) can also be used for measuring fu,mic, the fraction unbound in a microsomal incubation, and fu,hep, the fraction unbound in a hepatocyte incubation. Equilibrium dialysis is again commonly used and to measure binding to microsomes, drug and microsomes without cofactors (NADPH/UDPGA) are added to one chamber, and buffer is added to the other (Obach, 1997). The value of fu,mic can be determined as the ratio of the concentration on the buffer side to the concentration on the microsome plus drug side. Unlike PPB measurements, volume shifts due to osmotic forces are not observed (Obach, 1997).
Measuring fu,hep is not quite as simple as excluding cofactors. Hepatocytes that have been deactivated at room temperature and subject to freeze/thaw cycles are commonly used to minimize the complication of simultaneous metabolism (Di et al., 2012). An alternative method is to use live hepatocytes preincubated with metabolic inhibitors such as 1-amino-benzotriazole and salicylamide for the assay, however equilibrium dialysis time vs. half-life of metabolism of the compounds must be considered (Austin et al., 2005). Furthermore, although the presence of specific, saturable binding sites did not appear to fit microsomal binding data (Austin et al., 2002), the test compound binding to hepatocytes could potentially be displaced by the inhibitors. Using only metabolic inhibitors also does not provide direct information about whether the measured binding is truly binding to the cell wall, or is actually intracellular accumulation from uptake. Despite the potential disadvantages, when comparing the use of live hepatocytes with inhibitors vs. dead hepatocytes for fu,hep measurements, overall there was no statistically significantly difference for a dataset of 17 compounds (Austin et al., 2005).
When running assays for both fuinc and CLint determinations, it is important to note that nonspecific binding increases as phospholipid concentration, microsomal protein concentration, or cell density increases (Kalvass et al., 2001; Margolis and Obach, 2003; Di et al., 2012). This is particularly relevant as microsomal protein concentrations can vary as much as 200-fold among laboratories (Kalvass et al., 2001). To try to avoid nonspecific binding, very low microsomal concentrations can be used; however under certain conditions such as investigating phase II metabolic reactions or intestinal metabolism, higher concentrations are needed (Gertz et al., 2008).
After proposing the incorporation of a fu,inc term for IVIVE (Obach, 1996), Obach (1999) tested the idea on data collected from 29 drugs in human microsomes. IVIVE prediction accuracy was examined when when only fu,B was included in the scale up (Fig. 3A), when both fu,B and fu,inc terms were used (Fig. 3B), and when no binding terms were used (under the assumption the values of fu,B and fu,inc would cancel) (Fig. 3C). However, as Kalvass et al. (2001) mention, “Since the free fraction in microsomes is determined, in part, by the choice of microsomal concentration in the incubation, equivalent free fractions between plasma and microsomes should be considered coincidental.” Obach (1999) reports that predictions were best for acidic compounds when both the fu,B and fu,inc binding terms were included and basic compounds when no fu terms were included, with which we agree. Obach (1999) also states that for neutral compounds, no binding terms gave better predictions, but we believe both methods were comparable in accuracy although no binding terms overpredicts and both binding terms underpredicts. Note in Fig. 3 that since AFE values on the y-axis are represented on a log scale, visual comparisons must be validated with numerical values. Riley et al. (2005) also saw similar trends with hepatocytes. Overall, predictions were best when both binding terms were included for acidic, basic, and neutral compounds. The worst accuracy occurred if no fu terms were included and in this scenario, predictions were very poor for acidic drugs and better for basic and neutral drugs where fu,inc may be large and cancel with fu,B.
Fig. 3: Accuracy of predictions from Obach (1999) using the well-stirred model.

The accuracy is examined using: A) the traditional model with fu,B only; B) both fu,B and fu,inc; and C) no binding terms assuming fu,B and fu,inc cancel. The shaded area highlights predictions falling within two fold of observed values.
2.1.2. fu,inc estimated in silico
Given that the experimental methods for measuring fu,inc are not high-throughput, in silico models for predicting the value have been proposed (Gao et al., 2010). Since it is believed that the phospholipid component is the primary contributor to nonspecific binding (Margolis and Obach, 2003), it makes sense that the extent of binding would increase with increasing lipophilicy as proposed by Austin et al. (2002).
Noting that basic compounds have enhanced binding over neutral and acidic compounds of similar lipophilicity (thought to be due to favorable electrostatic interactions between the protonated base and the phosphate groups), Austin et al. (2002) suggested using logP for basic compounds and logD7.4 for acidic and neutral compounds. Based on data from 37 compounds with fu,mic values of <0.9, Eq. (7) was developed to predict fu,inc based only on ionization and lipophilicy (r2 = 0.82). The equation was created based on both rat and human in vitro microsome binding data, but as Austin et al. (2002) mention, it has been shown that fu,mic is generally independent of species (Obach, 1997; Zhang et al., 2010)
| (7) |
where C is the microsomal protein concentration in mg/mL.
Other groups have built on this relationship and Hallifax and Houston (2006) proposed a quadratic equation, Eq. (8), (n=92, r2 = 0.75) for determining fu,mic. Using an expanded dataset of 127 compounds, no major differences were seen between the two equations for fu,mic predictions for low and high lipophilicity drugs. However, for intermediate drugs (logP/D values between 2.5 – 5.0) the Hallifax equation was more accurate (Gertz et al., 2008). For compounds with logP/D <0, where there is expected to be negligible interaction with microsomal protein, predictions from the Austin et al. (2002) equation are expected to be accurate, and it would be inappropriate to use the Hallifax and Houston (2006) equation due to the nonlinear nature (Gertz et al., 2008).
| (8) |
While these in silico predictions can be useful, Gertz et al. (2008) saw that minor variation in logP predictions could lead to high variations in fu,mic predictions, and in certain cases fu,mic should still be determined experimentally. However, they did find good agreement between using predicted logP values from software packages and experimental determinations. A similar equation, Eq. (9), was later proposed for fu,hep prediction (Austin et al., 2005; Kilford et al., 2008)
| (9) |
where VR is the ratio of cell volume to incubation volume and is 0.005 at a cell concentration of 106 cells/mL.
Groups continue to try to improve fu,inc predictions (Gao et al., 2008) and there are reviews that discuss the nuances of the methods more in detail (Poulin and Haddad, 2001; Gertz et al., 2008; Emoto et al., 2009; Gao et al., 2010). However, while many studies support the incorporation of fu,inc, there are still some that question its utility (Fagerholm, 2007). Even when both fu,inc and fu,B terms are incorporated in IVIVE, it is clear that there are still inconsistencies that need to be solved.
2.2. pH Difference and F1
Berezhkovskiy (2011) proposed adding an ionization factor, F1, to account for the difference in pH of extra- and intracellular water in hepatocytes (pH 7.4 vs. 7.0). F1 is defined as the ratio of the unbound, unionized (neutral) drug fractions in plasma and intracellular tissue water, and is added into the well-stirred model as a product of CLint as in Eq. (10). If the pH were the same or if the compound were neutral then F1 equals 1. For basic drugs where F1 >1, CL predictions will be higher (up to 6.3 fold for diprotic acids, 15 fold for triprotic acids) helping with IVIVE underprediction, and conversely for acidic drugs where F1 <1, CL predictions will be lower, helping with IVIVE overprediction. The largest difference in predictions is expected for low extraction ratio drugs where the prediction is directly proportional to F1.
| (10) |
where and is the neutral drug fraction in plasma (or the concentration of unbound neutral drug in plasma divided by the concentration of unbound drug in plasma) and is the neutral drug fraction in intracellular water. These ratios can be calculated as:
When measuring CLint with hepatocytes, if buffer with a pH of 7.4 is utilized, and assuming the intracellular pH is maintained at 7.0, there would be no need to account for F1 in doing IVIVE. However, the pH gradient appears to be disrupted as the fraction of buffer in the incubation is larger than the fraction of extracellular water in the liver (Berezhkovskiy et al., 2012). When measuring CLint with microsomes, it would always be necessary to account for F1 since the cellular integrity does not exist. A preliminary test of the method with microsomal data from 25 highly bound drugs did not completely cancel the underprediction seen, but showed improvement (Poulin et al. 2012b).
3. Protein Facilitated Uptake
Baker and Bradley (1966) were first to suggest violations of the free drug theory and that hepatic uptake may occur directly from the albumin-drug complex, not just from free drug. Later studies also noted that highly protein bound ligands had more efficient hepatic uptake than could be accounted for by just their unbound concentrations. This phenomenon became known as albumin-mediated uptake. The idea gained traction in the 1980’s when single-pass liver perfusion studies with various ligands including taurocholate (Forker and Luxon, 1981), rose bengal (Forker and Luxon, 1983), oleate (Weisiger and Ma, 1987), and warfarin (Tsao et al., 1988) demonstrated the facilitated uptake. More recently it has been noted that as fu,p decreases, underprediction with traditional IVIVE increases (Baker and Parton, 2007). This trend may also be seen with data from human hepatocytes compiled by Wood et al. (2017) (Fig. 4). While only 9 drugs with fu,p values ranging from 0.001–0.01 were included, the AFE may be higher in this range. Accordingly, some have tried using total rather than free drug concentrations in clearance equations to eliminate the underprediction (Obach, 1999; Riley et al., 2005), which conceptually supports the idea of albumin-mediated uptake. When conducting in vitro studies, groups have also found that adding HSA or plasma to microsome and hepatocyte incubations can cause decreases in Km values and improved IVIVE results (Blitzer and Lyons, 1985; Ludden et al., 1997; Shibata et al., 2002; Blanchard et al., 2004; Blanchard et al., 2006; Skaggs et al., 2006; Baker and Parton, 2007; Rowland et al., 2008; Wattanachai et al., 2011; Gill et al., 2012; Wattanachai et al., 2015; Mao et al., 2018). Poulin et al. (2016) have nicely summarized several of these studies. When proteins are added to the incubations, the drug uptake rates decrease less than would be expected with the decrease in unbound drug concentrations.
Fig. 4: IVIVE prediction error and fu,p.

Examining the accuracy of predictions from human hepatocytes taken from Wood et al. (2017), as fu,p decreases to the 0.001–0.01 range, average fold error (AFE) may increase. If AFE <1, the reciprocal is plotted.
While there are now several examples suggesting that perhaps total drug, not unbound drug, can drive hepatic uptake and clearance and should be considered when doing IVIVE, the mechanism explaining why has not yet been agreed upon. We review here the state of past and present hypotheses (Fig. 5).
Fig. 5: Hypotheses to explain albumin-facilitated uptake.

A) Presence of an albumin receptor where uptake can occur due to direct uptake of unbound ligand or after specific interaction of the albumin-ligand complex with its receptor; B) Rate-limiting dissociation where free ligand uptake is faster than ligand dissociation from albumin; C) Rate-limiting diffusion of ligand through the UWL where the slow diffusion of unbound ligand is supplemented with the diffusion of more soluble bound ligand; D) Conformational change where uptake occurs from the direct uptake of unbound ligand in plasma or after a conformational change of the albumin-ligand complex due to cell membrane binding catalyzing the release of drug; E) Ionic interactions with the cell membrane where the diffusional distance for unbound ligand is decreased; and F) Transporter-induced protein binding shift where a high affinity transporter may strip ligand from the ligand-drug complex.
3.1. Specific Albumin Receptor on Hepatocyte Surface
The earliest hypothesis to explain albumin-mediated uptake was that there is an albumin-receptor on the hepatocyte cell surface (Fig. 5A). Oleate was one of the first ligands used to suggest this (Weisiger et al., 1981). When increasing [14C]oleate concentration but keeping bovine albumin concentration constant, oleate uptake increased linearly relative to concentration in the perfused rat liver. However, when increasing both oleate and albumin concentration (1:1 so the unbound oleate concentration is constant), there was a saturable process, where albumin appeared to be acting as a competitive inhibitor resulting in a plot similar to Fig. 6. 125I-albumin was then used to evaluate the possibility of albumin binding to hepatocytes and there appeared to be a single high-affinity binding site specific for albumin. Of the wide variety of potential displacement proteins tested, including several known to have hepatocyte surface receptors, only other albumin molecules (human and rat) significantly displaced the bovine albumin (Ockner et al., 1983). It was later further shown that there appears to be no species specificity for the potential hepatocyte-albumin interaction (Reed and Burrington, 1989). Based on the Stokes radius of albumin and assuming that all sites are occupied, it was estimated that somewhere between 1 to 8% of the total hepatocyte surface is occupied by the albumin-receptor complex (Ockner et al., 1983), and a dissociation constant of albumin binding to hepatocytes was estimated (Table 2). Weisiger et al. (1981) justified their hepatocyte receptor hypothesis by explaining that if only the 0.1% of free oleate accounted for uptake, the dissociation from the oleate-albumin complex would need to be extremely rapid to explain the higher extraction, but the half-time for the dissociation of oleate from albumin is actually significantly longer than the time required for blood to pass through the liver.
Fig. 6: Saturation vs. linear results.

An example of a saturation curve (solid line) that is seen in several studies when the concentrations of albumin and ligand are varied (at a fixed 1:1 ratio). This is in contrast to when the concentration of ligand is varied at a fixed albumin concentration and uptake is linear (dashed line). Saturation is suggested to occur for instance when free albumin is competing with the ligand-albumin complex for receptors (section 3.1), or when the rate limiting transport step shifts from ligand dissociation to influx or metabolism (section 3.2).
Table 2:
Estimated dissociation constants (Kd) for albumin binding to hepatocyte cell surface
| Kd (μM) | # Sites/Cell | Methodology | Source |
|---|---|---|---|
| 25 ± 7 | 10 (± 3) × 106 | 125I-labeled bovine albumin binding to rat hepatocytes (ligand free), 20°C, 30 min. | Weisiger et al., 1981 |
| 53 ± 13 | 10.4 (± 1.9) × 106 | 131I-labeled rose bengal in perfused rats, various concentrations of BSA, fit to their kinetic model | Forker & Luxon, 1983 |
| 157 ± 47 | Warfarin in perfused rats, 37°C, fit to facilitated dissociation kinetic model (Eq. 11) | Tsao et al., 1988 | |
| 2.5 ± 1.7 | 2.2 (± 1.3) × 106 | 125I-labeled monomeric albumin binding to rat hepatocytes, 4°C, 30 min. | Wright et al., 1987 |
| 4.4 ± 1.8 | 7.4 (± 2.3) × 106 | 125I-labeled monomeric albumin binding to rat hepatocytes, 37°C, 30 min | Wright etal., 1987 |
| 1.9 ± 1.0 | 3.9 (± 3.0) × 106 | 125I-bovine albumin binding to rat hepatocytes, 20°C | Reed and Burrington, 1989 |
| 1.1 ± 0.5 | 2.0 (± 1.1) × 106 | 125I-rat albumin binding to rat hepatocytes, 20°C | Reed and Burrington, 1989 |
An even earlier paper also suggested the potential role of an albumin receptor on the hepatocyte cell surface, but through a different mechanism. Bloomer et al. (1973) saw that when the IgG fraction of goat anti-human albumin was added to a solution of [14C]bilirubin and human albumin, [14C]bilirubin was completely recovered in the supernatant. This suggested that bilirubin can be easily separated from albumin if albumin reacts with another macromolecule, and led to their hypothesis that if albumin receptors in the membrane interact with albumin at bilirubin binding sites, more bilirubin could be separated from albumin as it passes the hepatocyte surface, leading to higher unbound bilirubin diffusion.
While early studies suggested the possibility of a membrane protein with high affinity for albumin, others collected data that were not consistent with the hypothesis. Stremmel et al. (1983) examined the binding of 125I-labeled rat albumin to rat liver plasma membranes attempting to characterize the proposed hepatocyte albumin receptor. Running incubations for 30 minutes at various temperatures and various plasma membrane and albumin concentrations, there was no evidence of specific binding to the membrane. For instance, using 5 pmol of albumin and 10 mg of membrane protein, only 2.3% of the incubated albumin was recovered in the membrane pellet, and after two washes, only 0.09% remained, indicating that most of the albumin was trapped within the pellet, or loosely associated as it was easily removed with washing. Adding excess unlabeled albumin did not cause inhibition of binding, heat denaturation of the membranes caused no binding changes, and the amount of albumin binding to rat erythrocyte ghosts was the same as to the liver plasma membrane. These investigators employed additional methodologies and used ultraviolet irradiation to prove that the failure to observe binding was not due to a rapid dissociation rate, and ran affinity chromatography with solubilized membrane proteins over albumin-agarose gels and did not find one with high albumin affinity. Stremmel et al. (1983) went on to suggest that perhaps there are less specific interactions between the liver cell surface and albumin-ligand complex instead of a specific hepatocytic albumin receptor.
Similar to Stremmel et al. (1983), Stollman et al. (1983) did not find evidence for the interaction of albumin with a hepatocyte receptor. Perfusing rat livers with a protein-free fluorocarbon medium, Stollman et al. (1983) measured the uptake of [3H]bilirubin with either 125I-albumin, 125I-ligandin (an intracellular protein known to bind bilirubin with high affinity), or free with a [14C]sucrose reference and found the same uptake across the three conditions. Furthermore, after injecting [3H]bilirubin with 125I-albumin and with [14C]sucrose and seeing no delay in 125I-albumin transit compared to that of [14C]sucrose, they concluded that the off-rate of albumin from a receptor would have to be very rapid, which would be unusual.
The specific albumin receptor theory would also not be able to account for the enhanced clearance seen for ligands bound to other proteins such as β-lactoglobulin, (Nunes et al., 1988; Burczynski et al., 1990). Finally, Reed and Burrington (1989) examined the interactions of two fragments of albumin with rat hepatocytes, and calculating similar dissociation constants in both cases, they concluded that since there does not seem to be a specific site on albumin that interacts with hepatocytes, receptor recognition seems unlikely.
3.2. Rate-Limiting Dissociation of Ligand from the Albumin-Ligand Complex
To further explore the determinants of hepatic uptake and the possibility of an albumin receptor, Weisiger et al. (1984) evaluated the role of bovine albumin on sulfobromophthalein (BSP) uptake in skates. Believing that since skates naturally lack albumin they would not have evolved an albumin receptor, the goal was to see if a different kinetic behavior occurred. Using a single-pass perfused liver model, two different steps in the uptake process were determined. For fixed albumin concentrations, as total BSP concentration increased, linear saturation kinetics were present suggesting that the rate-limiting step in these situations does not involve albumin. This was called the “intrinsic” uptake step. When the concentration of albumin and BSP were varied at a fixed molar ratio, the uptake rates did not correlate with the estimated equilibrium concentrations in the perfusate, and the saturation kinetics seen occurred at uptake velocities too small to saturate the intrinsic uptake step. The data collected from skates were similar to that from rats with oleate (Weisiger et al., 1981), however since skates have no reason to have an albumin receptor, an alternate explanation for the results was needed. In this case, the rate-limiting step was hypothesized to be the spontaneous dissociation of BSP from albumin, going back to the traditional assumption that clearance only occurs for free ligand (Fig. 5B). If free BSP clearance is faster than BSP can be replenished by dissociation from albumin, equilibrium would not actually be present in the sinusoid, and the dependence of clearance on the bound ligand concentration is explained. As the albumin concentration increases, reassociation of free BSP to albumin can start to occur instead of only BSP clearance, and when binding equilibrium is established the uptake then becomes limited by the intrinsic uptake step.
Weisiger et al. (1984) explain that although similar kinetics were seen in rats, this dissociation limited model would not explain the findings since the BSP uptake rate is more rapid in rats and exceeds the rate of spontaneous dissociation. Weisiger and Ma (1987) saw that the removal rate for oleate in perfused rat liver with dilute albumin solutions is similar to the spontaneous dissociation rate measured in vitro, supporting the dissociation-limited model in rats for this ligand. If the albumin receptor model were to hold true, the removal rate could be much higher since it is not limited by the rate of spontaneous dissociation.
To further explore this, van der Sluijs et al. (1987) measured dibromosulfophthalein (DBSP) uptake in rat liver perfused with native albumin vs. lactosylated albumin. After demonstrating that DBSP had similar protein binding to the lactosylated albumin as to native albumin, a 40% decrease in the hepatic uptake rate constant for the lactosylated albumin was found. These investigators also conducted rapid filtration experiments and saw that the dissociation rate constant of DBSP from lactosylated albumin was half that from albumin and concluded that the decreased off-rate could explain the decreased hepatic uptake, providing further evidence for the idea of dissociation-limited uptake.
It should be noted that these early in vitro measures of the dissociation rate constant have limitations (Burczynski and Luxon, 1995). If a solid-phase acceptor is used, where it is assumed that the free ligand is diffusing and binding to the acceptor, there could be direct collisional exchange between albumin and the acceptor causing ligand transfer as well, overestimating the spontaneous dissociation rate constant. If stop-flow fluorescence is used, where the conformation change in albumin is measured, if the change is slower than the dissociation rate, the rate constant could be underestimated.
3.3. Rate-Limiting Diffusion of Ligand Through the Unstirred Water Layer
Another explanation for albumin-facilitated uptake is related to the idea of an unstirred water layer (UWL) in the space of Disse, or the space that separates sinusoidal lining cells from hepatocytes. The idea of an UWL is common with intestinal absorption where rates of highly permeable compounds are known to have an upper limit (Komiya et al., 1980). Given that the space of Disse contains a matrix of fibrillar material and hepatocytes have microvilli and adherent water film (Ichikawa et al., 1992), groups began exploring the potential role that an UWL may play in the liver.
Bass and Pond (1988) created a “pseudofacilitation” model, returning to the idea that uptake occurs only from the unbound fraction of ligand. This model is reviewed in detail by Burczynski and Luxon (1995). The hypothesis is that ligands undergoing cellular uptake can be rate limited by the UWL adjacent to the cell surface or can be rate limited by permeability through the membrane itself (Weisiger et al., 1989b). For ligands with high membrane permeability that are rate limited by the UWL, a concentration gradient will develop within the UWL in the absence of protein (Burczynski and Luxon, 1995). With the addition of protein, traditionally binding causes a decrease in the diffusion rate of the protein-ligand complex as compared to free ligand (Amidon et al., 1982; Weisiger et al., 1989b). However, for highly lipophilic ligands, which have limited diffusional flux across aqueous barriers, the presence of protein promotes aqueous solubility (Weisiger et al., 1989b; Burczynski and Luxon, 1995). The slow diffusion of unbound ligand is therefore supplemented with the diffusion of more soluble bound ligand. The albumin-ligand complex will try to replenish the depleted unbound ligand near the cell surface and restore equilibrium. As a result there is decreased diffusional distance for the unbound ligand and the unbound concentration driving uptake is increased (Burczynski and Luxon, 1995). When the bound ligand concentration is much higher than that of the free ligand, the average diffusional flux of the bound ligand can be greater than that of the free, making the flux appear to only depend on the bound concentration (Weisiger et al., 1989b) (Fig. 5C).
Ichikawa et al. (1992) explored the uptake of ligands with various permeabilites in perfused rat liver and isolated rat hepatocytes. Using highly-permeable diazepam and taurocholate in the perfusion study, as BSA concentrations increased and free fraction decreased, the extraction ratio did not greatly change, showing albumin-mediated transport of the compounds. Tolbutamide and salicylate, intermediate permeability compounds, also exhibited some albumin-mediated transport as their extraction ratios decreased to one-third when the free fractions were decreased to one-tenth. With cefodizime, a compound with low permeability, the extraction ratio decreased as the free fraction decreased, leading to the conclusion that albumin-mediated transport was not observed, and supporting the hypothesis.
Ichikawa et al. (1992) point out that it is possible that slow dissociation from albumin (see section 3.2) could also play a role. However when investigating perfused livers vs. hepatocytes, the highly permeable compounds had a lower influx clearance in the perfused livers. It is argued that if the dissociation-limited transport were to play a large role, the influx clearances should be similar, since koff should be the same in both systems. The rate-limiting diffusion theory can explain the saturation kinetics seen in earlier studies (Weisiger et al., 1981) as the increase in unbound clearance reaches a maximum, which is determined by the product of the effective membrane permeability and total surface area (Pond et al., 1992).
However, others believe that the UWL cannot fully explain albumin-facilitated uptake. Although the use of polyethylene sheeting has been questioned (Schwab and Goresky, 1991), Burczynski et al. (1989) found that while palmitate clearance with hepatocytes was about 7 fold faster than with polyethylene, the codiffusion of bound and free palmitate to the cell surface could only account for about 20% of the facilitated clearance observed. Furthermore, Pond et al. (1992) tested the pseudofacilitation model (Bass and Pond, 1988) on [3H]palmitic acid uptake data generated in hepatocytes finding a high dependence on parameter estimate selection. Their experimental results agree with the theoretical model predictions if a reported low equilibrium association constant was used (15 μM−1); however if higher values also reported were used (62 and 94 μM−1), the measured unbound clearance exceeded the model predictions.
Comparing the uptake of oleate and BSP with albumin in isolated perfused rat liver vs. hepatocyte suspensions, Nunes et al. (1988) measured the same kinetics in both experiments, concluding that the facilitated uptake observed was not dependent on the intact lobule characteristics or diffusion barrier in the space of Disse present only in the perfused livers. Similarly, Blitzer and Lyons (1985) still saw taurocholate facilitated uptake in rat basolateral liver plasma membrane vesicles, which were vigorously mixed during the experiments and for which the UWL effects were expected to be minimal.
3.4. Interactions with the Hepatocyte Cell Surface (not albumin receptor)
As these alternative hypotheses continued arising, more work was simultaneously conducted concerning the putative hepatocyte receptor hypothesis. As Stremmel et al. (1983) suggested, instead of looking for a specific albumin receptor, the focus shifted to more general interactions that could be occurring at the liver cell surface with the albumin-ligand complex catalyzing ligand dissociation.
3.4.1. Conformational change
Using rose bengal, Forker and Luxon (1983) explained that while the albumin-ligand complex may interact with the cell surface, since albumin itself is not removed (Nilsson and Berg, 1977), free ligand must ultimately be what is interacting with the transport carrier, and must interact without mixing with the pool of free ligand in the extracellular fluid. They suggested that the binding of the complex to the cell surface may lead to a conformational change in albumin reducing its binding affinity for the ligand and/or presenting the ligand in a favorable location for uptake (Fig. 5D) (Forker et al., 1982). They also added an important corollary that the sites on the cell surface would have a similar affinity for albumin whether or not ligand is bound. This then suggests that the interaction at the cell surface is not ligand specific, and the affinity of albumin for the surface is independent of ligand concentration. Creating a kinetic model where the total rate of ligand removal is proportional to the mass of free ligand plus the mass that is bound to the cell surface as albumin-ligand complexes, they fit their perfusion data and compared the calculated values to those obtained by Weisiger et al. (1981) with ligand-free albumin and hepatocytes (Table 2). The similar results supported their kinetic model and Forker and Luxon concluded that albumin binding to the cell surface explained how bound ligand, in this case rose bengal, in another perfusion study, taurocholate (Forker and Luxon, 1981), and in a study using rat liver cell monolayers, palmitate (Fleischer et al., 1986) is available for hepatic uptake.
3.4.1.1. Facilitated-dissociation model
Tsao et al. (1988) also suggested this hypothesis with warfarin where the interaction between the cell surface and warfarin-BSA complex (Kd = 157 μM, Table 2), would induce a conformational change in albumin and decrease the binding affinity of warfarin. Having previously run perfusion studies in normal rats and analbuminemic rats and seeing albumin-mediated uptake of warfarin in both cases led to the conclusion that a specific albumin receptor on the cell surface may not be necessary for the uptake (Tsao et al., 1986). A “facilitated-dissociation” kinetic model was developed to include both the uptake of unbound drug and the uptake of drug from the albumin-drug complex after a conformational change. Assuming that the dissociation of warfarin from albumin is much faster than the perfusion or hepatic uptake rate and thus, warfarin binding to albumin is at equilibrium, and assuming that the uptake of unbound warfarin is linear compared to warfarin concentration, the equation for the uptake rate of drug is:
| (11) |
where C0 is the total concentration of ligand, pm and PB,influx are the permeability clearances for unbound ligand and unbound ligand dissociated from the drug-ligand complex respectively, while fu is the fraction of unbound ligand in the extracellular fluid expressed as
where n is the number of binding sites on albumin, kd is the dissociation constant of ligand and λ is the fraction of albumin bound to the surface of hepatocytes (assuming both unbound albumin and ligand-bound albumin compete for the same binding sites on the surface) expressed as
where Bmax is the capacity of albumin binding sites on the surface of hepatocytes and Kd,m is the dissociation constant of bound albumin from the hepatocyte surface (Tsao et al., 1988; Miyauchi et al., 2018).
Based on their model, Tsao et al. (1988) simulated the contributions of the unbound and bound drug. At low albumin concentrations, the increase in albumin increases the albumin-bound warfarin, which facilitates the uptake of bound warfarin, but at high albumin concentrations, the increase in albumin leads to competition of free albumin and albumin-bound warfarin for the liver cell surface binding sites, and warfarin uptake is inhibited.
Twenty years later Miyauchi et al. (2018) revisited the facilitated dissociation model finding it could accurately predict the uptake of two organic anion transporting polypeptide substrates, 1-anilino-8-napthalene sulfonate (ANS) in primary cultured rat hepatocytes, and pitavastatin in isolated human hepatocytes. They performed a curve-fitting exercise based on their experimental data escalating the albumin concentration in the incubation medium and upon finding the calculated line fit well, the authors concluded that the enhancement of clearance can be accurately predicted by the facilitated-dissociation model.
3.4.1.2. Acidic microenvironment
Burczynski et al. (1997) and others point out that the acidic microenvironment of the hepatocyte may also play a role since it has been shown to decrease the albumin binding of anthracyclines and long-chain fatty acids.
At physiological pH, the negatively charged groups on the hepatocyte cell surface attract positively charged ions to try to maintain electroneutrality (Burczynski and Luxon, 1995). The presence of H+ ions then lowers the pH of the environment near the hepatocyte that can modulate albumin conformation changes through the Neutral-Base and Neutral-Fast transitions where ligands can be released from the proteins.
Using absorption and electron spin resonance spectroscopy, Horie et al. (1988) showed albumin undergoes conformational changes through interaction with hepatocellular membranes, as well as other membrane types, similar to those seen in the Neutral-Base and Neutral-Fast transitions of albumin. However, Foker and Ghiron (1988) point out that that the nitroxide spin label used could react with the 59 free amino groups or the single available SH group of albumin, so the labeling may not be site specific and the results cannot be considered definitive.
3.4.2. Ionic interactions between the cell surface and albumin-ligand complex
An alternative theory for the role that the hepatocyte cell surface plays in facilitated uptake is that ionic interactions can occur between the hepatocyte plasma membrane and the protein-ligand complex. This would then decrease the diffusional distance for the unbound ligand and provide more unbound ligand to the cell surface for uptake (Burczynski et al., 1997) (Fig. 5E). When this hypothesis was suggested, most studies in the field had used albumin as a binding protein and although β-lactoglobulin was tested (Burczynski et al., 1990), it has a similar isoelectric point (pI) to albumin, and no difference in uptake would be expected between the two.
Burczynski et al. (1997) investigated the potential of the ionic interaction first by measuring [3H]-palmitate clearance for binding proteins with different pIs: AAG (pI=2.7), albumin (pI=4.9), and lysozyme (pI=11.0). The clearance with the basic lysozyme was 6.3 fold greater than with the acidic AAG and 3.2 fold greater than with albumin. This agrees with the hypothesis that the net positive charge of lysozyme would be expected to be attracted to the negatively charged groups on the membrane surface. Burczynski et al. (1997) also examined the uptake with 0.23 μM albumin vs. 2.3 mM lysozyme plus 0.23 μM albumin where the unbound palmitate fraction would be expected to be lower, and found that the clearance was statistically higher with the lysozyme plus albumin, further supporting the hypothesis. In a subsequent paper they went on to chemically modify albumin (though maleylation, succinylation, and cationization) to have pIs between 2.0–8.6 to exclude the possibility of conformational and binding site differences between proteins (Burczynski et al., 2001). After showing that the dissociation rate constants were not statistically significant from each other, and finding that [3H]-palmitate clearance significantly increased by 0.27 units with a unit increase in pI, they concluded that the ionic interactions hypothesis holds true.
3.4.2.1. fu,p-adjusted
Specifically citing the ionic interactions hypothesis, Poulin et al. (2012b) hypothesized that the whole-liver fu may be larger than fu,p in vivo and an adjusted fu term, fu,p-adjusted should be utilized. The authors proposed using a plasma-to-whole-liver-concentration ratio (PLR) of plasma binding proteins to correct for the difference between extracellular protein binding and liver protein binding and also built in the ionization factor, F1, mentioned previously (see section 2.2) to create Eq. 12
| (12) |
Poulin et al. (2012b) reported that Eq. (12) is applicable for HSA-bound drugs. For drugs bound to AAG fu,p-adjusted is not used and only the F1 correction as in Eq. (11) is employed since AAG levels are lower than HSA in plasma, and previous studies suggested that facilitated uptake is greater with HSA than with AAG (Qin et al., 1994; Bilello et al., 1996).
To estimate the PLR value for HSA, they note that the levels of HSA in the intracellular liver are negligible compared to the levels in the interstitial space and plasma, so the PLR is really a concentration ratio of the proteins in the plasma vs. interstitial space, and they assumed a homogenized distribution of HSA in accordance with the well-stirred model assumptions. For humans the PLR was estimated to be 13.3. The same value can be used for rat and monkey, but for dog, a value of 8.5 is used due to the greater volume of interstitial fluid (Poulin et al., 2012a).
| (13) |
Adding the new term to the well-stirred model, Eq. (13), and applying it to data generated in plasma-free microsomal incubations for 25 highly bound compounds, Poulin et al. (2012b) found no systematic over- or underprediction, an AFE close to unity, and the best predictions for bases.
Hallifax and Houston (2012) conducted a similar evaluation of methods using a larger dataset of 107 drugs, finding that the Poulin fu,p-adjusted method was the least biased (AFE for hepatocytes =1.3, for microsomes = 1.7) compared to the Berezhokovkiy F1 method or the conventional method with fu,B. They raise some concerns about the method though, showing that if a hepatocytosolic pH of 7.2 instead of 7.0 had been used (given the range of 7.0–7.4 reported in the literature), or if a PLR of 133 instead of 13.3 were used, there would be overprediction for acids. Hallifax and Houston (2012) ultimately conclude that the fu,p-adjusted method may not offer significant improvement over a simple empirical correction. However, using a simple empirical correction factor based on AFE, or using one based on a regression analysis (Sohlenius-Sternbeck et al., 2012; Yamagata et al., 2017), requires analysis of an in vivo dataset and is dependent on compound selection in that dataset.
Poulin and coworkers have continued to validate their model showing it performs better than using an empirical correction (Poulin et al., 2012a) and performs well for hepatocytes (Poulin and Haddad, 2013; 2015), and when combining metabolism with transporter and permeability data (Poulin, 2013).
The PLR values mentioned earlier are for protein-free incubations, where it is assumed that protein-facilitated uptake is not occurring, and “the CLint determined in vitro should represent only a measure of the hepatic uptake of the free drug moiety by contrast to the in vivo condition in liver where the bound drug moiety is also assumed to be available for uptake” (Da-Silva et al., 2018). To apply this methodology to different experimental procedures with varied albumin concentrations, the PLR value can be changed to the actual concentration ratio of HSA in the buffer/plasma/perfusate and the organ material, and if an assay has albumin at a similar level to that in vivo, the PLR should be 1 (Poulin and Haddad, 2015). This has recently been confirmed to improve prediction accuracy for naproxen and bisphenol A in isolated perfused rat livers with different albumin concentrations (Bounakta et al., 2018; Poulin et al., 2017) and in the HepatoPac® system with 25 compounds (Da-Silva et al., 2018).
Recently, the fu,p-adjusted model was compared to the facilitated dissociation model (Poulin and Haddad, 2018) by considering the data for ANS and pitavastatin from Miyauchi et al. (2018). While specific input parameters are required for the facilitated-dissociation as in Eq. 11), such as the relative interaction capacity, dissociation constant, number of binding sites, and albumin concentration, the Poulin fu,p-adjusted model (Eq. 12) requires less input parameters. The two models were shown to both improve IVIVE, and to be conceptually and mathematically equivalent, particularly for pitavastatin. However, for ANS, which had a lower capacity of interaction with the membrane, the fu,p-adjusted model overestimated the unbound CLint in albumin in vivo since the model assumes each interaction between the albumin-ligand complex and cell surface would lead to facilitated uptake.
3.5. Transporter Induced Protein Binding Shift
Most recently, we have proposed a transporter-induced protein-binding shift as a mechanism of facilitated uptake (Bowman et al., 2017). Many of the previous hypotheses were developed before the transporter field emerged, and it is now clear that transporters can violate the principles of free drug theory as uptake transporters can elevate the intracellular free concentration significantly above that in plasma (Giacomini et al., 2010). It has also been noted that highly protein bound compounds that are substrates of transporters often have the poorest IVIVE predictions (Soars et al., 2007). Our hypothesis is that high affinity binding to cell membrane proteins such as OATPs may be able to change the equilibrium of the nonspecific binding between drugs and plasma proteins. If a highly protein bound drug has a higher affinity for a transporter than for albumin, or the suggested hepatocyte cell surface (Table 2), the transporter may be able to strip the drug from the protein before the drug dissociates itself and is at binding equilibrium (Fig. 5F). This would mean that protein binding is not restricting the access of these compounds and using fu values measured at equilibrium in static assays (see section 1.1.2) would yield incorrect results. Using statins, known substrates of the OATP1B1 transporter, our preliminary results show an expected increase in affinity (decrease in measured unbound Km) for uptake in serum vs. protein-free buffer incubations with highly bound drugs, and minimal change in unbound Km values for drugs with low binding for which the transporter induced shift does not need to occur.
Recent data in the literature also agree with our hypothesis although the authors offer different explanations. Examining the previously mentioned IPRL work by Poulin et al. (2017) and Bounakta et al. (2018), for the highly protein bound bisphenol A, the unbound Km value decreases dramatically with the addition of albumin, and shows less of a decrease for naproxen which has lower protein binding (Table 3). Miyauchi et al. (2018) tested the uptake of 1-anilino-8-naphthalene sulfonate (ANS) in rat hepatocytes with bovine serum albumin and pitavastatin in human hepatocytes with HSA. A facilitated uptake process was seen for both compounds, but there was a more dramatic increase in the unbound uptake for pitavastatin, which has a higher affinity for OATP, than for ANS which has a relatively lower affinity for Oatp. The authors hypothesized, “the higher the affinity for the transporter, the more effective is the albumin-mediated enhancement.” Furthermore, when examining the uptake of highly bound new chemical entity, compound A, which was shown to be an OATP1B3 substrate, the unbound clearance increased with increased HSA concentration in HEK293 cells where the mechanisms hypothesized in hepatocytes may not be present, but overexpression of the OATP transporters is (Fukuchi et al., 2017). Ongoing work in our laboratory will continue to test to validity of this hypothesis.
Table 3:
Data from Poulin et al. (2017) and Bounakta et al. (2018)
| Compound | fu,p | Km [ALB]= 0 g/L |
Km [ALB]=30 g/L |
[ALB]=30 g/L |
Fold Dif unbound Km |
|---|---|---|---|---|---|
| Bisphenol A | 0.045 | 13.4 | 3.5 | 0.16 | 83.8 |
| Naproxen | 0.12 | 98.9 | 174.4 | 20.9 | 4.73 |
3.6. Alternative Explanations, Cells, Proteins
The theories mentioned above have gained the most traction and are frequently cited today. Over the years other possibilities have been mentioned, but often deemed less likely. One early suggestion was that there may be direct transfer of ligand from plasma albumin to hepatic intracellular binding proteins (Bloomer et al., 1973). For this to happen, the intracellular proteins would need to be in close proximity to the plasma albumin and have a high enough affinity for the ligand. A similar hypothesis is direct transfer where collisional exchanges (random or from ionic interactions) between the ligand-protein complex and cell membrane could cause direct transfer of the ligand to the cell without dissociation in the extracellular fluid (Wootan et al., 1993; Zucker et al., 1995; Burczynski and Luxon, 1995).
Suggesting an alternative explanation for the putative albumin receptor, Reed and Burrington (1989) hypothesized that there are two populations of albumin. One is a “surface” population that binds to the container surface and undergoes a conformation change that causes the molecules to have higher affinity for the hepatocyte surface, and can then transfer directly between the container wall and cell wall. The other population is the native population, which binds with lower affinity to the cell surface.
Another hypothesis is that albumin affects the hepatocyte membrane potential and/or fluidity (Tsao et al., 1988; Forker and Luxon, 1995). Using perfused rat livers and a microelectrode, Weisiger et al. (1989a) showed that depolarization of cells decreased oleate uptake, while hyperpolarization increased uptake. This could explain the increase in clearance seen in the presence of albumin for the non-protein bound antipyrine (Øie and Fiori, 1985). Another idea that has been mentioned proposes the endocytosis of the protein-drug complex (Stremmel et al., 1983; Poulin et al., 2012b).
No matter which is ultimately the correct mechanism, it is clear that the unbound clearance of many ligands is enhanced as the concentration of plasma protein is increased. This protein-facilitated uptake has been shown to occur in other organs and cell types besides the liver/hepatocytes including myocytes (Hütter et al., 1984a,b; Rauch et al., 1987; Sorrentino et al., 1989, Elmadhoun et al., 2001), adipocytes (Sorrentino et al., 1989), proximal tubules (Besseghir et al., 1989), perfused kidney (Taft and Sweeney, 1995), brain (Pardridge et al., 1983), and human embryonic kidney cells overexpressing OATP1B1 and 1B3 (Fukuchi et al. 2017), as well as inert material including polyethylene (Burczynski, et al. 1989) and n-decane (Weisiger et al., 1989b). Facilitated uptake may also be considered for nanoparticles bound to albumin as they are used to deliver drugs to tumor cells more effectively, and perhaps the complex also interacts with cells (Saptarshi et al., 2013; Poulin et al., 2016). Facilitated uptake has been shown not only for ligands bound to albumin, but also to proteins such as β-lactoglobulin (Nunes et al., 1988; Burczynski et al., 1990) and ligandin (Stollman et al., 1983).
4. Conclusions
This review explores the use of the fu parameter in IVIVE both in terms of the traditional scale up where only unbound drug is believed to undergo uptake, and in terms of protein-facilitated uptake, where highly protein bound ligands have more efficient uptake than can be accounted for by just their unbound concentrations. While the addition of fu,inc and/or F1 to the well-stirred model has sound reasoning and is commonly being implemented now, the mechanisms behind protein-facilitated uptake and how to kinetically describe this process have yet to be agreed upon. Two of the popular models, the fu,p-adjusted model (see section 3.4.2.1) and the FDM (see section 3.4.1.1) were recently compared and found to be complementary (Poulin and Haddad, 2018). Since fu,p and pKa values can be estimated in silico, the fu,p-adjusted model is suitable for early stage drug discovery. (However, plasma protein binding assays must still be conducted to determine the major binding protein, and how to implement fractional binding has yet to be determined). While the FDM provides more specific results, more experimental data are needed before making it suitable for use in the later stages of drug discovery. Regardless of which mechanism or combination of mechanisms is ultimately correct, it is clear that steady state uptake of highly bound ligands is lower in the presence of proteins, but higher than predicted by traditional equilibrium binding. Additional studies and understanding of these mechanisms are needed to ultimately improve IVIVE and the drug development process.
5. Acknowledgements
CMB was supported by the National Science Foundation Graduate Research Fellowship Program [Grant 1144247]; LZB is a member of the UCSF Liver Center supported by NIH Grant [P30 DK026743].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References
- Amidon GE, Higuchi WI, Ho NF, 1982. Theoretical and experimental studies of transport of micelle-solubilized solutes. J. Pharm. Sci 71, 77–84. [DOI] [PubMed] [Google Scholar]
- Austin RP, Barton P, Cockroft SL, Wenlock MC, Riley RJ, 2002. The influence of nonspecific microsomal binding on apparent intrinsic clearance, and its prediction from physicochemical properties. Drug Metab. Dispos 30, 1497–1503. [DOI] [PubMed] [Google Scholar]
- Austin RP, Barton P, Mohmed S, Riley RJ, 2005. The binding of drugs to hepatocytes and its relationship to physicochemical properties. Drug Metab. Dispos 33, 419–425. [DOI] [PubMed] [Google Scholar]
- Baker KJ, Bradley SE, 1966. Binding of sulfobromophthalein (BSP) sodium by plasma albumin. Its role in hepatic BSP extraction. J. Clin. Invest 45, 281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Parton T, 2007. Kinetic determinants of hepatic clearance: plasma protein binding and hepatic uptake. Xenobiotica. 37:1110–1134. [DOI] [PubMed] [Google Scholar]
- Bass L, Pond SM, 1988. The puzzle of rates of cellular uptake of protein-bound ligand In: Pecile A, Rescigno A (Eds.), Pharmacokinetics: Mathematical and Statistical Approaches to Metabolism and Distribution of Chemicals and Drugs. Plenum Press, London: pp. 241–265. [Google Scholar]
- Benet LZ, 2010. Clearance (née Rowland) concepts: a downdate and an update. J. Pharmacokinet. Pharmacodyn 37, 529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benet LZ, Liu S, Wolfe AR, 2018. The universally unrecognized assumption in predicting drug clearance and organ extraction ratio. Clin. Pharmacol. Ther 103, 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezhkovskiy LM, 2011. The corrected traditional equations for calculation of hepatic clearance that account for the difference in drug ionization in extracellular and intracellular tissue water and the corresponding corrected PBPK equation. J. Pharm. Sci 100, 1167–1183. [DOI] [PubMed] [Google Scholar]
- Berezhovskiy LM, Liu N, Halladay JS, 2012. Consistency of the novel equations for determination of hepatic clearance and drug time course in liver that account for the difference in drug ionization in extracellular and intracellular tissue water. J. Pharm. Sci 101, 516–518. [DOI] [PubMed] [Google Scholar]
- Besseghir K, Mosig D, Roch-Ramel F, 1989. Facilitation by serum albumin of renal tubular secretion of organic anions. 256, F475–484. [DOI] [PubMed] [Google Scholar]
- Bilello JA, Bilello PA, Stellrecht K, Leonard J, Norbeck DW, Kempf DJ, Robins T, Drusano GL, 1996. Human serum alpha 1 acid glycoprotein reduces uptake, intracellular concentration, and antiviral activity of A-80987, an inhibitor of the human immunodeficiency virus type 1 protease. Antimicrob. Agents Chemother 40, 1491–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard N, Hewitt NJ, Silber P, Jones H, Coassolo P, Lavé T, 2006. Prediction of hepatic clearance using cryopreserved human hepatocytes: a comparison of serum and serum-free incubations. J. Pharm. Pharmacol 58, 633–641. [DOI] [PubMed] [Google Scholar]
- Blanchard N, Richert L, Notter B, Delobel F, David P, Coassolo P, Lavé T, 2004. Impact of serum on clearance predictions obtained from suspensions and primary cultures of rat hepatocytes. Eur. J. Pharm. Sci 23, 189–199. [DOI] [PubMed] [Google Scholar]
- Blitzer BL, Lyons L, 1985. Enhancement of Na+-dependent bile acid uptake by albumin: direct demonstration in rat basolateral liver plasma membrane vesicles. Am. J. Physiol 249, G34–38. [DOI] [PubMed] [Google Scholar]
- Bloomer JR, Berk PD, Vergalla J, Berlin NI, 1973. Influence of albumin on the hepatic uptake of unconjugated bilirubin. Clin. Sci. Mol. Med 45, 505–516. [DOI] [PubMed] [Google Scholar]
- Bohnert T, Gan LS, 2013. Plasma protein binding: from discovery to development. J. Pharm. Sci 102, 2953–2994. [DOI] [PubMed] [Google Scholar]
- Bounakta S, Bteich M, Mantha M, Poulin P, Haddad S, 2018. Predictions of bisphenol A hepatic clearance in the isolated perfused rat liver (IRPL): impact of albumin binding and of co-administration with naproxen. Xenobiotica. 48, 135–147. [DOI] [PubMed] [Google Scholar]
- Bowman CM, Benet LZ, 2016. Hepatic clearance predictions from in vitro-in vivo extrapolaton and the Biopharmaceutics Drug Disposition Classification System. Drug Metab. Dispos 44, 1731–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman CM, Okochi H, Benet LZ, 2017. The effect of protein binding on the interaction between transporters and highly protein bound drugs. Poster presented at: The International Pharmaceutical Federation Pharmaceutical Sciences World Congress, Stockholm, Sweden, May 2017. [Google Scholar]
- Brockman AH, Oller HR, Moreau B, Kriksciukaite K, Bilodeau MT, 2015. Simple method provides resolution of albumin, lipoprotein, free fraction, and chylomicron to enhance the utility of protein binding assays. J. Med. Chem 58, 1420–1425. [DOI] [PubMed] [Google Scholar]
- Burczynski FJ, Cai Z-S, Moran JB, Forker EL, 1989. Palmitate uptake by cultured hepatocytes: albumin binding and stagnant layer phenomena. Am. J. Physiol 257, G584–593. [DOI] [PubMed] [Google Scholar]
- Burczynski FJ, Luxon BA, 1995. Is there facilitated uptake of fatty acids by theliver? Interpretation and analysis of experimental data. Can. J. Physiol. Pharmacol 73, 409–420. [DOI] [PubMed] [Google Scholar]
- Burczynski FJ, Moran JB, Cai Z-S, Forker EL, 1990. β-Lactoglobulin enhances the uptake of free palmitate by hepatocyte monolayers: the relative importance of diffusion and facilitated dissociation. Can. J. Physiol. Pharmacol 68, 201–206. [DOI] [PubMed] [Google Scholar]
- Burczynski FJ, Wang GQ, Elmadhoun B, She YM, Roberts MS, Standing KG, 2001. Hepatocyte [3H]-palmitate uptake: effect of albumin surface charge modification. Can. J. Physiol. Pharmacol 79, 868–875. [PubMed] [Google Scholar]
- Burczynski FJ, Wang GQ, Hnatowich M, 1997. Effect of binding protein surface charge on palmitate uptake by hepatocyte suspensions. Br. J. Pharmacol 120, 1215–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao P, Uss AS, Cheng KC, 2010. Use of intrinsic clearance for prediction of human hepatic clearance. Expert Opin. Drug Metab. Toxicol 6, 189–198. [DOI] [PubMed] [Google Scholar]
- Chiba M, Ishii Y, Sugiyama Y, 2009. Prediction of hepatic clearance in human from in vitro data for successful drug development. AAPS J. 11, 262–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da-Silva F, Boulenc X, Vermet H, Compigne P, Gerbal-Chaloin S, Daujat-Chavanieu M, Klieber S, Poulin P, 2018. Improving prediction of metabolic clearance using quantitative extrapolation of results obtained from human hepatic micropatterned cocultures model and by considering the impact of albumin binding. J. Pharm. Sci 107, 1957–1972. [DOI] [PubMed] [Google Scholar]
- Di L, Keefer C, Scott DO, Strelevitz TJ, Chang G, Bi YA, Lai Y, Duckworth J, Fenner K, Troutman MD, Obach RS, 2012. Mechanistic insights from comparing intrinsic clearance values between human liver microsomes and hepatocytes to guide drug design. Eur. J. Med. Chem 57, 441–448. [DOI] [PubMed] [Google Scholar]
- Di L, Breen C, Chambers R Eckley ST, Fricke R, Ghosh A, Harradine P, Kalvass JC, Ho S, Lee CA, Marathe P, Perkins EJ, Qian M, Tse S, Yan Z, Zamek-Gliszczynski MJ, 2017. Industry perspective on contemporary protein-binding methodologies: considerations for regulatory drug-drug interaction and related guidelines on highly bound drugs. J. Pharm. Sci 106, 3442–3452. [DOI] [PubMed] [Google Scholar]
- Dockal M, Carter DC, Rüker F, 2000. Conformational transitions of the three recombinant domains of human serum albumin depending on pH. 275, 3042–3050. [DOI] [PubMed] [Google Scholar]
- Elmadhoun B, Wang GQ, Kirshenbaum LA, Burczynski FJ, 2001. Palmitate uptake by neonatal rat myocytes and hepatocytes. Role of extracellular protein. Eur. J. Biochem 268, 3145–3153. [DOI] [PubMed] [Google Scholar]
- Emoto C, Murayama N, Rostami-Hodjegan A, Yamazaki H, 2009. Utilization of estimated physicochemical properties as an integrated part of predicting hepatic clearance in the early drug-discovery stage: impact of plasma and microsomal binding. Xenobiotica. 39, 227–235. [DOI] [PubMed] [Google Scholar]
- Faed EM, 1981. Protein binding of drugs in plasma, interstitial fluid and tissues: effect on pharmacokinetics. Eur. J. Clin. Pharmacol 21, 77–81. [DOI] [PubMed] [Google Scholar]
- Fagerholm U, 2007. Prediction of human pharmacokinetics—evaluation of methods for prediction of hepatic metabolic clearance. J. Pharm. Pharmacol 59, 803–828. [DOI] [PubMed] [Google Scholar]
- Farrugia A, 2010. Albumin usage in clinical medicine: tradition or therapeutic? Transfus. Med. Rev 24, 53–63. [DOI] [PubMed] [Google Scholar]
- Fleischer AB, Shurmantine WO, Luxon BA, Forker EL, 1986. Palmitate uptake by hepatocyte monolayers. Effect of albumin binding. J. Clin. Invest 77, 964–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forker EL, Ghiron C, 1988. ESR, albumin, and the riddle of organic anion uptake by the liver. Am. J. Physiol 254, G463–464. [DOI] [PubMed] [Google Scholar]
- Forker EL, Luxon BA, 1981. Albumin helps mediate removal of taurocholate by rat liver. J. Clin. Invest 67, 1517–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forker EL, Luxon BA, 1983. Albumin-mediated transport of rose bengal by perfused rat liver. Kinetics of the reaction at the cell surface. J. Clin. Invest 72, 1764–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forker EL, Luxon BA, Snell M, Shurmantine WO, 1982. Effect of albumin binding on the hepatic transport of rose bengal: surface-mediated dissociation of limited capacity. J. Pharmacol. Exp. Ther 223, 342–347. [PubMed] [Google Scholar]
- Fukuchi Y, Toshimoto K, Mori T, Kakimoto K, Tobe Y, Sawada T, Asaumi R, Iwata T, Hashimoto Y, Nunoya K-I, Imawaka H, Miyauchi S, Sugiyama Y, 2017. Analysis of nonlinear pharmacokinetics of a highly albumin-bound compound: contribution of albumin-mediated hepatic uptake mechanism. J. Pharm. Sci 106, 2704–2714. [DOI] [PubMed] [Google Scholar]
- Gao H, Steyn SJ, Chang G, Lin J, 2010. Assessment of in silico models for fraction of unbound drug in human liver microsomes. Expert Opin. Drug Metab. Toxicol 6, 533–542. [DOI] [PubMed] [Google Scholar]
- Gao H, Yao L, Mathieu HW, Zhang Y, Maurer TS, Troutman MD, Scott DO, Ruggeri RB, Lin J, 2008. In silico modeling of nonspecific binding to human liver microsomes. Drug Metab. Dispos 36, 2130–2135. [DOI] [PubMed] [Google Scholar]
- Gertz M, Kilford PJ, Houston JB, Galetin A, 2008. Drug lipophilicity and microsomal protein concentration as determinants in the prediction of the fraction unbound in microsomal incubations. Drug Metab. Dispos 36, 535–542. [DOI] [PubMed] [Google Scholar]
- Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, Hoffmaster KA, Ishikawa T, Keppler D, Kim RB, Lee CA, Niemi M, Polli JW, Sugiyama Y, Swaan PW, Ware JA, Wright SH, Yee SW, Zamek-Gliszczynski MJ, Zhang L, 2010. Membrane transporters in drug development. Nat. Rev. Drug Discov 9, 215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill KL, Houston JB, Galetin A, 2012. Characterization of in vitro glucurondation clearance of a range of drugs in human kidney microsomes: comparison with liver and intestinal glucuronidation and impact of albumin. Drug Metab. Dispos 40, 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein A 1949. The interactions of drugs and plasma proteins. J. Pharmacol. Exp. Ther 95, 102–165. [PubMed] [Google Scholar]
- Hakooz N, Ito K, Rawden H, Gill H, Lemmers L, Boobis AR, Edwards RJ, Carlile DJ, Lake BG, Houston JB, 2006. Determination of a human hepatic microsomal scaling factor for predicting in vivo drug clearance. Pharm. Res 23, 533–539. [DOI] [PubMed] [Google Scholar]
- Hallifax D, Foster JA, Houston JB, 2010. Prediction of human metabolic clearance from in vitro systems: retrospective analysis and prospective view. Pharm. Res 27, 2150–2161. [DOI] [PubMed] [Google Scholar]
- Hallifax D, Houston JB, 2006. Binding of drugs to hepatic microsomes: comment and assessment of current prediction methodology with recommendation for improvement. Drug Metab. Dispos 34, 724–726. [DOI] [PubMed] [Google Scholar]
- Hallifax D, Houston JB, 2012. Evaluation of hepatic clearance prediction using in vitro data: emphasis on fraction unbound in plasma and drug ionisation using a database of 107 drugs. J. Pharm. Sci 101, 2645–2652. [DOI] [PubMed] [Google Scholar]
- Heuberger J, Schmidt S, Derendorf H, 2013. When is protein binding important? J. Pharm. Sci 102, 3458–3467. [DOI] [PubMed] [Google Scholar]
- Horie T, Mizuma T, Kasai S, Awazu S, 1988. Conformational change in plasma albumin due to interaction with isolated rat hepatocyte. Am. J. Physiol 254, G465–470. [DOI] [PubMed] [Google Scholar]
- Houston JB, 1994. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharmacol 47, 1469–1479. [DOI] [PubMed] [Google Scholar]
- Howard ML, Hill JJ, Galluppi GR, McLean MA, 2010. Plasma protein binding in drug discovery and development. Comb. Chem. High Throughput Screen 13, 170–187. [DOI] [PubMed] [Google Scholar]
- Huh Y, Smith DE, Feng MR, 2011. Interspecies scaling and prediction of human clearance: comparison of small- and macro-molecule drugs. Xenobiotica. 41, 972–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hütter JF, Piper HM, Spieckermann PG, 1984a. Myocardial fatty acid oxidation: evidence for an albumin-receptor-mediated membrane transfer of fatty acids. Basic Res. Cardiol 79, 274–282. [DOI] [PubMed] [Google Scholar]
- Hütter JF, Piper HM, Spieckermann PG, 1984b. Kinetic analysis of myocardial fatty acid oxidation suggesting an albumin receptor mediated uptake process. J. Mol. Cell. Cardiol 16, 219–226. [DOI] [PubMed] [Google Scholar]
- Ichikawa M, Tsao SC, Lin T-H, Miyauchi S, Sawada Y, Iga T, Hanano M, Sugiyama Y, 1992. ‘Albumin-mediated transport phenomenon’ observed for ligands with high membrane permeability: effect of the unstirred water layer in the Disse’s space of rat liver. J. Hepatol 16, 38–49. [DOI] [PubMed] [Google Scholar]
- Israili ZH, Dayton PG, 2001. Human alpha-1-glycoprotein and its interactions with drugs. Drug Metab. Rev 33, 161–235. [DOI] [PubMed] [Google Scholar]
- Ito K, Houston JB, 2005. Prediction of human drug clearance from in vitro and preclinical data using physiologically based and empirical approaches. Pharm. Res 22, 103–112. [DOI] [PubMed] [Google Scholar]
- Kalvass JC, Tess DA, Giragossian C, Linhares MC, Maurer TS, 2001. Influence of microsomal concentration on apparent intrinsic clearance: implications for scaling in vitro data. Drug Metab. Dispos 29, 1332–1336. [PubMed] [Google Scholar]
- Kilford PJ, Gertz M, Houston JB, Galetin A, 2008. Hepatocellular binding ofdrugs: correction for unbound fraction in hepatocyte incubations using microsomal binding or drug lipophilicity data. Drug Metab. Dispos 36, 1194–1197. [DOI] [PubMed] [Google Scholar]
- Komiya I, Park JY, Kamani A, Ho NFH, Higuchi WI, 1980. Quantitative mechanistic studies in simultaneous fluid flow and intestinal absorption using steroids as model solutes. Int. J. Pharm 4, 249–262. [Google Scholar]
- Liu X, Wright M, Hop CECA, 2014. Rational use of plasma protein and tissue binding data in drug design. J. Med. Chem 57, 8238–8248. [DOI] [PubMed] [Google Scholar]
- Ludden LK, Ludden TM, Collins JM, Pentikis HS, Strong JM, 1997. Effect of albumin on the estimation, in vitro, of phenytoin Vmax and Km values: implications for clinical correlation. J. Pharmacol. Exp. Ther 282, 391–396. [PubMed] [Google Scholar]
- Mao J, Doshi U, Wright M, Hop CECA, Li AP, Chen Y, 2018. Prediction of the pharmacokinetics of pravastatin as an OATP substrate using plateable human hepatocytes with human plasma data and PBPK modeling. CPT Pharmacometrics Syst. Pharmacol 7, 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis JM, Obach RS, 2003. Impact of nonspecific binding to microsomes and phospholipid on the inhibition of cytochrome P4502D6: implications for relating in vitro inhibition data to in vivo drug interactions. Drug Metab. Dispos 31, 606–611. [DOI] [PubMed] [Google Scholar]
- Miyauchi S, Masuda M, Kim S-J, Tanaka Y, Lee K-R, Iwakado S, Nemoto M, Sasaki S, Shimono K, Tanaka Y, Sugiyama Y, 2018. The phenomenon of albumin-mediated hepatic uptake of organic anion transport polypeptide substrates: prediction of the in vivo uptake clearance from the in vitro uptake by isolated hepatocytes using a facilitated-dissociation model. Drug Metab. Dispos 46, 259–267. [DOI] [PubMed] [Google Scholar]
- Nakai D, Kumamoto K, Sakikawa C, Kosaka T, Tokui T, 2004. Evaluation of the protein binding ratio of drugs by a micro-scale ultracentrifugation method. J. Pharm. Sci 93, 847–854. [DOI] [PubMed] [Google Scholar]
- Nilsson M, Berg T, 1977. Uptake and degradation of formaldehyde-treated 125I-labelled human serum albumin in rat liver cells in vivo and in vitro. Biochim. Biophys. Acta 497, 171–182. [DOI] [PubMed] [Google Scholar]
- Nunes R, Kiang C-L, Sorrentino D, Berk PD, 1988. ‘Albumin-receptor’ uptake kinetics do not require an intact lobular architecture and are not specific for albumin. J. Hepatol 7, 293–304. [DOI] [PubMed] [Google Scholar]
- Obach RS, 1996. The importance of nonspecific binding in in vitro matrices, its impact on enzyme kinetic studies of drug metabolism reactions, and implications for in vitro-in vivo correlations. Drug Metab. Dispos 24, 1047–1049. [PubMed] [Google Scholar]
- Obach RS, 1997. Nonspecific binding to microsomes: impact on scale-up of in vitro intrinsic clearance to hepatic clearance as assessed through examination of warfarin, imipramine, and propranolol. Drug Metab. Dispos 25, 1359–1369. [PubMed] [Google Scholar]
- Obach RS, 1999. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos 27, 1350–1359. [PubMed] [Google Scholar]
- Obach RS, Baxter JG, Liston TE, Silber BM, Jones BC, MacIntyre F, Rance DJ, Wastall P, 1997. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther 283, 46–58. [PubMed] [Google Scholar]
- Ockner RK, Weisiger RA, Gollan JL, 1983. Hepatic uptake of albumin-bound substances: albumin receptor concept. Am. J. Physiol 245, G13–18. [DOI] [PubMed] [Google Scholar]
- Øie S, Fiori F, 1985. Effects of albumin and alpha-1 acid glycoprotein on elimination of prasozin and antipyrine in the isolated perfused rat liver. J. Pharmacol. Exp. Ther 636–640. [PubMed] [Google Scholar]
- Parasrampuria DA, Benet LZ, Sharma A, 2018. Why drugs fail in late stages of development: case study analyses from the last decade and recommendations. AAPS J. doi: 10.1208/s12248-018-0204-y. [DOI] [PubMed] [Google Scholar]
- Pardridge WM, Sakiyama R, Fierer G, 1983. Transport of propranolol and lidocaine through the rat blood-brain barrier. Primary role of globulin-bound drug. J. Clin. Invest 71, 900–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond SM, Davis CKC Bogoyevitch MA, Gordon RA, Weisiger RA, Bass L, 1992. Uptake of palmitate by hepatocyte suspensions: facilitation by albumin? Am. J. Physiol 262, G883–894. [DOI] [PubMed] [Google Scholar]
- Poulin P, 2013. Prediction of total hepatic clearance by combining metabolism, transport, and permeability data in the in vitro-in vivo extrapolation methods: emphasis on apparent fraction unbound in liver for drugs. J. Pharm. Sci 102, 2085–1095. [DOI] [PubMed] [Google Scholar]
- Poulin P, Haddad S, 2011. Microsome composition-based model as a mechanistic tool to predict nonspecific binding of drugs in liver microsomes. J. Pharm. Sci 100, 4501–4517. [DOI] [PubMed] [Google Scholar]
- Poulin P, Haddad S, 2013. Toward a new paradigm for the efficient in vitro-in vivo extrapolation of metabolic clearance in humans from hepatocyte data. J. Pharm. Sci 102, 3239–3251. [DOI] [PubMed] [Google Scholar]
- Poulin P, Haddad S, 2015. Albumin and uptake of drugs in cells: additional validation exercises of a recently published equation that quantifies the albumin-facilitated uptake mechanism(s) in physiologically based pharmacokinetic and pharmacodynamic modeling research. J. Pharm. Sci 104, 4448–4458. [DOI] [PubMed] [Google Scholar]
- Poulin P, Haddad S, 2018. Extrapolation of the hepatic clearance of drugs in the absence of albumin in vitro to that in the presence of albumin in vivo: comparative assessment of 2 extrapolation models based on the albumin-mediated hepatic uptake theory and limitations and mechanistic insights. J. Pharm. Sci doi: 10.1016/j.xphs.2018.03.012. [DOI] [PubMed] [Google Scholar]
- Poulin P, Bteich M, Haddad S, 2017. Supplemental analysis of the prediction of hepatic clearance of binary mixtures of bisphenol A and naproxen determined in an isolated perfused rat liver model to promote the understanding of potential albumin-facilitated hepatic uptake mechanism. J. Pharm. Sci 106, 3207–3214. [DOI] [PubMed] [Google Scholar]
- Poulin P, Burczynski FJ, Haddad S, 2016. The role of extracellular binding proteins in the cellular uptake of drugs: impact on quantitative in vitro-to-in vivo extrapolations of toxicity and efficacy in physiologically based pharmacokinetic-pharmacodynamic research. J. Pharm. Sci 105, 497–508. [DOI] [PubMed] [Google Scholar]
- Poulin P, Hop CECA, Ho Q, Halladay JS, Haddad S, Kenny JR, 2012a. Comparative assessment of in vitro-in vivo extrapolation methods used for predicting hepatic metabolic clearance of drugs. J. Pharm. Sci 101, 4308–4326. [DOI] [PubMed] [Google Scholar]
- Poulin P, Kenny JR, Hop CECA, Haddad S, 2012b. In vitro-in vivo extrapolation of clearance: modeling hepatic metabolic clearance of highly bound drugs and comparative assessment with existing calculation methods. J. Pharm. Sci 101, 838–851. [DOI] [PubMed] [Google Scholar]
- Qin M, Nilsson M, Øie S, 1994. Decreased elimination of drug in the presence of alpha-1-acid glycoprotein is related to a reduced hepatocyte uptake. J. Pharmacol. Exp. Ther 269, 1176–1181. [PubMed] [Google Scholar]
- Quinlan GJ, Martin GS, Evans TW, 2005. Albumin: biochemical properties and therapeutic potential. Hepatology. 41, 1211–1219. [DOI] [PubMed] [Google Scholar]
- Rane A, Wilkinson GR, Shand DG, 1977. Prediction of hepatic extraction ratio from in vitro measurement of intrinsic clearance. J. Pharmacol. Exp. Ther 200, 420–424. [PubMed] [Google Scholar]
- Rauch B, Bode C, Piper HM, Hütter JF, Zimmermann R, Braunwell E, Hasselbach W, Kübler W, 1987. Palmitate uptake in calcium tolerant, adult rat myocardial single cells—evidence for an albumin mediated transport across sarcolemma. J. Mol. Cell. Cardiol 19, 159–166. [DOI] [PubMed] [Google Scholar]
- Reed RG, Burrington CM, 1989. The albumin receptor effect may be due to a surface-induced conformational change in albumin. J. Biol. Chem 264, 9867–9872. [PubMed] [Google Scholar]
- Riley RJ, McGinnity DF, Austin RP, 2005. A unified model for predicting human hepatic, metabolic clearance from in vitro intrinsic clearance data in hepatocytes and microsomes. Drug Metab. Dispos 33, 1304–1311. [DOI] [PubMed] [Google Scholar]
- Ring BJ, Chien JY, Adkison KK, Jones HM, Rowland M, Jones RD, Yates JWT, Ku MS, Gibson CR, He H, Vuppugalla R, Marathe P, Fischer V, Dutta S, Sinha VK, Björnsoon T, Lavé T, Poulin P, 2011. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 3: comparative assessment of prediction methods of human clearance. J. Pharm. Sci 100, 4090–4110. [DOI] [PubMed] [Google Scholar]
- Roberts MS, Rowland M, 1986. A dispersion model of hepatic elimination: 1. formulation of the model and bolus considerations. J. Pharmacokinet. Biopharm 14, 227–260. [DOI] [PubMed] [Google Scholar]
- Rowland A, Elliot DJ, Knights KM, Mackenzie PI, Miners JO, 2008. The “albumin effect” and in vitro-in vivo extrapolation: sequestration of long-chain unsaturated fatty acids enhances phenytoin hydroxylation by human liver microsomal and recombinant cytochrome P450 2C9. Drug Metab. Dispos 36, 2008. [DOI] [PubMed] [Google Scholar]
- Rowland M, Benet LZ, Graham GG, 1973. Clearance concepts in pharmacokinetics. J. Pharmacokinet. Biopharm 1, 123–136. [DOI] [PubMed] [Google Scholar]
- Saptarshi SR, Duschl A, Lopata AL, 2013. Interaction of nanoparticles with proteins: relation to bio-reactivity of the nanoparticle. J. Nanotechnol 11, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S, Gonzalez D, Derendorf H, 2010. Significance of protein binding in pharmacokinetics and pharmacodynamics. J. Pharm. Sci 99, 1107–1122. [DOI] [PubMed] [Google Scholar]
- Schwab AJ, Goresky CA, 1991. Free fatty acid uptake by polyethylene: what can one learn from this? Am. J. Physiol 261, G896–906. [DOI] [PubMed] [Google Scholar]
- Shibata Y, Takahashi H, Chiba M, Ishii Y, 2002. Prediction of hepatic clearance and availability by cryopreserved human hepatocytes: an application of serum incubation method. Drug Metab. Dispos 30, 892–896. [DOI] [PubMed] [Google Scholar]
- Skaggs SM, Foti RS, Fisher MB, 2006. A streamlined method to predict hepatic clearance using human liver microsomes in the presence of human plasma. J. Pharmacol. Toxicol. Methods 53, 284–290. [DOI] [PubMed] [Google Scholar]
- Smith DA, Di L, Kerns EH, 2010. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat. Rev. Drug Discov 9, 929–939. [DOI] [PubMed] [Google Scholar]
- Soars MG, McGinnity DF, Grime K, Riley RJ, 2007. The pivotal role of hepatocytes in drug discovery. Chem. Biol. Interact 168, 2–15. [DOI] [PubMed] [Google Scholar]
- Sodhi JS, Benet LZ, 2017. Impact of drug distribution within the liver on measurement of metabolic elimination rate constants (k) and clearance (CL). Poster presented at: The International Pharmaceutical Federation Pharmaceutical Sciences World Congress, Stockholm, Sweden, May 2017. [Google Scholar]
- Sohlenius-Sternbeck AK, Jones C, Ferguson D, Middleton BJ, Projean D, Floby E, Bylund J, Afzelius L, 2012. Practical use of the regression offset approach for the prediction of in vivo intrinsic clearance from hepatocytes. Xenobiotica. 42, 841–853. [DOI] [PubMed] [Google Scholar]
- Sorrentino D, Robinson RB, Kiang C-L, Berk PD, 1989. At physiologic albumin/oleate concentrations oleate uptake by isolated hepatocytes, cardiac myocytes, and adipocytes is a saturable function of the unbound oleate concentration. Uptake kinetics are consistent with the conventional theory. J. Clin. Invest 84, 1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stollman YR, Gärtner U, Theilmann L, Ohmi N, Wolkoff AW, 1983. Hepatic bilirubin uptake in the isolated perfused rat liver is not facilitated by albumin binding. J. Clin. Invest 72, 718–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremmel W, Potter BJ, Berk PD, 1983. Studies of albumin binding to rat liver plasma membranes: implications for the albumin receptor hypothesis. Biochim. Biophys. Acta 756, 20–27. [DOI] [PubMed] [Google Scholar]
- Sudlow G, Birkett DJ, Wade DN, 1975. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol 11, 824–832. [PubMed] [Google Scholar]
- Taft DR, Sweeney KR, 1995. The influence of protein binding on the elimination of acetazolamide by the isolated perfused rat kidney: evidence of albumin-mediated tubular secretion. J. Pharmacol. Exp. Ther 274, 752–760. [PubMed] [Google Scholar]
- Trainor GL, 2007. The importance of plasma protein binding in drug discovery. Expert Opin. Drug Discov 2, 51–64. [DOI] [PubMed] [Google Scholar]
- Tsao SC, Sugiyama Y, Sawada Y, Iga T, Hanano M, 1988. Kinetic analysis of albumin-mediated uptake of warfarin by perfused rat liver. J. Pharmacokinet. Biopharm 16, 165–181. [DOI] [PubMed] [Google Scholar]
- Tsao SC, Sugiyama Y, Sawada Y, Nagase S, Iga T, Hanano M, 1986. Effect of albumin on hepatic uptake of warfarin in normal and analbuminemic mutant rats: analysis by multiple indicator dilution method. J. Pharmacokinet. Biopharm 14, 51–64. [DOI] [PubMed] [Google Scholar]
- van der Sluijs P, Postema B, Meijer DKF, 1987. Lactosylation of albumin reduces uptake rate of dibromosulfophthalein in perfused rat liver and dissociation rate from albumin in vitro. Hepatology. 7, 688–695. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan K, von Moltke LL, Obach RS, Greenblatt DJ, 2000. Microsomal binding of amitriptyline: effect on estimation of enzyme kinetic parameters in vitro. J. Pharmacol. Exp. Ther 293, 343–350. [PubMed] [Google Scholar]
- Wanwimolruk S, Birkett DJ, 1982. The effects of N-B transition of human serum albumin on the specific drug-binding sites. Biochim. Biophys. Acta 709, 247–255. [DOI] [PubMed] [Google Scholar]
- Wattanachai N, Polasek TM, Heath TM, Uchaipichat V, Tassaneeyakul W, Tassaneeyakul W, Miners JO, 2011. In vitro-in vivo extrapolation of CYP2C8-catalyzed paclitaxel 6α-hydroxylation: effects of albumin on in vitro kinetic parameters and assessment of interindividual variability in predicted clearance. Eur. J. Clin. Pharmacol 67, 815–824. [DOI] [PubMed] [Google Scholar]
- Wattanachai N, Tassaneeyakul W, Tassaneeyakul W, 2015. The effects of bovine serum albumin on kinetic characterization of human liver microsomal CYP2C19 and CYP2E1 activities. Thai J. Pharmacol 37, 27–40. [Google Scholar]
- Weisiger RA, Ma WL, 1987. Uptake of oleate from albumin solutions by rat liver. Failure to detect catalysis of the dissociation of oleate from albumin by an albumin receptor. J. Clin. Invest 79, 1070–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisiger R, Gollan J, Ockner R, 1981. Receptor for albumin on the liver cell surface may mediate uptake of fatty acids and other albumin-bound substances. Science. 211, 1048–1051. [DOI] [PubMed] [Google Scholar]
- Weisiger RA, Fitz JG, Scharschmidt BR 1989a. Hepatic oleate uptake. Electrochemical driving forces in intact rat liver. J. Clin. Invest 83, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisiger RA, Pond SM, Bass L, 1989b. Albumin enhances unidirectional fluxes of fatty acid across a lipid-water interface: theory and experiments. Am. J. Physiol 257, G904–916. [DOI] [PubMed] [Google Scholar]
- Weisiger RA, Zacks CM, Smith ND, Boyer JL, 1984. Effect of albumin binding on extraction of sulfobromophthalein by perfused elasmobranch liver: evidence for dissociation-limited uptake. Hepatology. 4, 492–501. [DOI] [PubMed] [Google Scholar]
- Wilkinson GR, Shand DG, 1975. A physiologic approach to hepatic drug clearance. Clin. Pharmacol. Ther 18, 377–390. [DOI] [PubMed] [Google Scholar]
- Winkler K, Keiding S, & Tystruup N, 1973. In: Paumgartner G & Preisig R (Eds.) The Liver: Quantitative Aspects of Structure and Functions. S. Karger, Basel, pp. 44–155. [Google Scholar]
- Wood FL, Houston JB, Hallifax D, 2017. Clearance prediction methodology needs fundamental improvement: trends common to rat and human hepatocytes/microsomes and implications for experimental methodology. Drug Metab. Dispos 45, 1178–1188. [DOI] [PubMed] [Google Scholar]
- Wootan MG, Bernlohr DA, Storch J, 1993. Mechanism of fluorescent fatty acid transfer from adipocyte fatty acid binding protein to membranes. Biochemistry. 32, 8622–8627. [DOI] [PubMed] [Google Scholar]
- Wright TL, Lysenko N, Ockner RK, Weisiger RA, 1987. Interaction of natural and synthetic albumin polymers with hepatocytes. Hepatology. 7, 294–301. [DOI] [PubMed] [Google Scholar]
- Yamagata T, Zanelli U, Gallemann D, Perrin D, Dolgos H, Petersson C, 2017. Comparison of methods for the prediction of human clearance from hepatocyte intrinsic clearance for a set of reference compounds and an external evaluation set. Xenobiotica. 47, 741–751. [DOI] [PubMed] [Google Scholar]
- Yang J, Jamei M, Yeo KR, Rostami-Hodjegan A, Tucker GT, 2007. Misuse of the well-stirred model of hepatic drug clearance. Drug Metab Dispos. 35, 501–502. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yao L, Lin J, Gao H, Wilson TC, Giragossian C, 2010. Lack of appreciable species differences in nonspecific microsomal binding. J. Pharm. Sci 99, 3620–3627. [DOI] [PubMed] [Google Scholar]
- Zucker SD, Goessling W, Gollan JL, 1995. Kinetics of bilirubin transfer between serum albumin and membrane vesicles. Insight into the mechanism of organic anion delivery to the hepatocyte plasma membrane. J. Biol. Chem 270, 1074–1081. [DOI] [PubMed] [Google Scholar]