

Abstract
Background & Aims:
Acetaminophen (APAP) induced hepatotoxicity is a leading cause of acute liver failure worldwide. It is well established that the liver damage induced by acetaminophen exhibits diurnal variation. However, the detailed mechanism for the hepatotoxic variation is not clear. Here we aimed to determine the relative contributions of gut microbiota in modulating the diurnal variation of hepatotoxicity induced by APAP.
Methods:
Male Balb/C mice were treated with or without antibiotics and orally administrated a single dose of APAP (300 mg/kg) at ZT0 (when the light is on-start of resting period) and ZT12 (when the light is off-start of active period).
Results:
In agreement with previous findings, hepatic injury was markedly enhanced at ZT12 compared with ZT0. Interestingly, upon antibiotics treatment, ZT12 displayed protection against APAP hepatotoxicity similar to ZT0. Moreover, mice that received the cecal content from ZT12 showed more severe liver damage than mice that received the cecal content from ZT0. 16S sequencing data revealed significant differences in the cecal content between ZT0 and ZT12 in the compositional level. Furthermore, metabolomic analysis showed that the gut microbial metabolites were also different between ZT0 and ZT12. Specifically, the level of 1-phenyl-1,2-propanedione (PPD) was significantly higher at ZT12 than ZT0. Treatment with PPD alone did not cause obvious liver damage. However, PPD synergistically enhanced APAP induced hepatic injury in vivo and in vitro. Finally, we found Saccharomyces cerevisiae, which could reduce intestinal PPD levels, was able to markedly alleviate APAP induced liver damage at ZT12.
Conclusions:
The gut microbial metabolite, 1-phenyl-1,2-propanedione was responsible, at least in part, for the diurnal variation of hepatotoxicity induced by APAP by decreasing GSH levels.
Keywords: acetaminophen, gut microbiota, metabolomics, acute liver injury
Lay summary:
Acetaminophen (APAP) induced acute liver failure due to over dose is a leading public health problem. APAP induced liver injury exhibited diurnal variation, in particular, it causes more severe liver damage when taken at night compared with that in the morning. Here we showed that gut microbial metabolite, 1-phenyl-1,2-propanedione (PPD) is involved in the rhythmic hepatotoxicity induced by APAP by depleting hepatic GSH levels. Our data suggest gut microbiota may be a potential target for reducing APAP induced acute liver injury.
Graphical Abstract
Introduction
Acetaminophen (APAP) is a widely used analgesic and antipyretic drug [1,2]. However, severe hepatotoxicity occurs upon taken excessive doses is the leading cause for acute liver failure (ALF) in the Western world [3,4]. APAP is absorbed from the intestine and processed by liver [5]. When taken at a low dose, APAP is mainly eliminated by sulfation and glucuronidation, and the toxicity pathway makes only a small contribution and mainly detoxified by glutathione (GSH) [6–8]. With elevation of APAP concentration in hepatocytes, a greater portion of APAP can be metabolized into N-acetyl-p-benzoquinone imine (NAPQI) by cytochrome P450s, mainly by CYP2E1 [9,10]. NAPQI is detoxified by conjugation with hepatic GSH. However, if GSH is exhausted, the generated reactive metabolite accumulates and exerts toxic effects initiated by covalent binding to macromolecules and causing mitochondrial dysfunction, ultimately leading to hepatocyte necrosis and acute liver failure [11–13].
Based on numerous clinical and preclinical studies, it is well established that APAP induced acute liver injury exhibits diurnal variation [14,15]. Generally, APAP dosing at night leads to more severe liver damage than in the morning. Although several pieces of evidence pointed out that the variations of hepatic metabolic genes expression and hepatic GSH level were involved in the fluctuation [14,15], the detailed mechanism responsible for the diurnal variation is not fully understood. Exploring the potential and novel driver for the hepatotoxic rhythmicity is important for understanding APAP induced ALF. Accumulating evidence points to gut microbiota being involved in this process. Indeed, intestinal microbiota undergoes diurnal oscillations both at the compositional and functional level [16]. Gut microbiota has been demonstrated to regulate many extraintestinal organ diseases, including liver injury. For instance, intestinal bacteria translocate into liver and promote hepatic inflammation during CCl4-induced fibrosis [17]. Moreover, gut microbial-generated saturated fatty acid exerts protective effects against alcohol induced liver damage [18]. However, the detailed contributions of gut microbiota to drug induced liver damage and failure remain elusive. In the present study, we explored the direct connection between gut microbiota and APAP induced diurnal variation of hepatotoxicity, and the underlying mechanism by identifying the potential gut microbiota derived molecule that acts on the liver to influence the susceptibility to APAP induced liver injury.
Materials and methods
Animals
Seven- to eight-week-old male specific pathogen free Balb/C mice were used (unless otherwise stated). Mice were gavaged with 300mg/kg APAP dissolved in warm phosphate buffered saline (PBS) at ZT0 (8:00 AM) or ZT12 (8:00 PM). For antibiotics treatment, mice received vancomycin (100 mg/kg), neomycin sulfate (200 mg/kg), metronidazole (200 mg/kg) and ampicillin (200 mg/kg) intragastrically once daily for 3 days. Mice were not fasted before APAP treatment and were sacrificed at 24 hours after APAP treatment. All mice had free access to food and water and were maintained in a temperature-controlled colony room on a 12:12-h light/dark cycle (8:00 AM, light on; 8:00 PM, light off). All experimental procedures were in compliance with the National Institutes of Health guidelines and were approved by the local Animal Care and Use Committee of the Southern Medical University.
Metabolomic analysis
The non-targeted metabolomics was described previously [19,20]. Briefly, UPLC-Q-TOF/MS (ACQUITY UPLC I-Class, Waters) and ESI-QTOF/MS (Xevo G2-S Q-TOF, Waters) were used. The chromatographic column is the ACQUITY UPLC HSS T3 Column (100Å, 1.8 μm, 2.1 mm × 50 mm, 1/pkg [186003538]). Mobile phase A contained 0.1% formic acid water, and mobile phase B contained 0.1% formic acid acetonitrile. The gradient elution was conducted as follows: 0–1 min, 99% A and 1% B; 1–2 min, 80% A and 20% B; 2–3 min, 50% A and 50% B; 3–7 min, 20% A and 80% B; 7–9 min, 1% A and 99% B; 9–10 min, 99% A and 1% B. The injection volume was 2 μl, and the flow rate was 0.4 mL/min. The capillary and cone voltages were set to 3 kV and 30 V, respectively. The acquisition rate was set to 0.2 s with a 0.01−s inter-scan delay. The nebulization gas was set to 800 L/h at a temperature of 500°C, the cone gas was set to 60 L/h, and the source temperature was set to 120°C. The UPLC-Q-TOF/MS data were processed and analyzed using MarkerLynx (Waters) in the Masslynx software (version 4.1) for peak detection and alignment. Multivariate statistical analysis was performed using the EZinfo software.
For targeted metabolomic analysis (PPD measurement), samples were extracted ultrasonically in methanol for 10 min, followed by filtration and the analysis was performed by gas chromatography-mass spectrometry (GC/MS, Finnigan) with TG WAX column (30m × 0.25mm × 0.25μm). The column temperature was conducted as follows: 100°C (2min)—20°C/min—200°C (2min), and the flow rate was 1 mL/min. The split ratio was 40:1. The analysis was performed using a system equipped with an EI source. The Single-ion scanning mode was used to improve sensibility and the fragment of m/z = 105 was collected for quantification.
Fecal microbiota transplantation (FMT)
Fecal microbiota transplantation was performed according to the modified method described previously [21,22]. In short, 6–8-week-old male Balb/C mice received antibiotics (described above) intragastrically once daily for 5 days to deplete the gut microbiota. The cecal content of the donor mice (ZT0 group, ZT12 group) was collected and resuspended in PBS at 0.125g/ml. An amount of 0.15 ml was administered to mice by gavage at ZT0. After 3 days, mice were gavaged with 300mg/kg APAP and were sacrificed at 24 hours after APAP treatment.
For further details regarding the materials used, please refer to the CTAT table and supplementary information.
Results
Dependence of the diurnal variation of APAP induced acute liver injury on gut microbiota
Mice were treated with non-absorbable antibiotics (ABX) continuously for 3 days to explore the contribution of intestinal microbiota to APAP induced diurnal variation of hepatotoxicity. After ABX administration, mice were treated with a single oral dose of APAP at ZT0 and ZT12, respectively. In agreement with previous findings, APAP did not reveal obvious hepatotoxic effects when administrated at ZT0; however, the same dose of APAP resulted in significant liver damage at ZT12 as evidenced by plasma aminotransferases elevation and necrotic area after 24h APAP administration (Fig. 1A–C). ABX treatment almost completely abolished the ALT/AST elevation and the livers from ABX treated mice displayed much less necrosis compared with control animals. Furthermore, the hepatic expression of inflammatory factors was similarly decreased with by ABX consistent with less liver injury (Fig. 1D, Supplementary Fig. 1).
Figure 1:

The diurnal variation of APAP induced hepatotoxicity was dependent on gut microbiota. Balb/C mice were treated with APAP for 24 hours with or without antibiotics pretreatment. (A) Plasma ALT, AST levels (n=6–12). (B) Representative gross liver appearance. (C) Hepatic HE staining and quantification of necrotic area (n=6–12). (D) mRNA levels of key cytokines and chemokines in the liver (n=6–12). (E) Mice received antibiotics intragastrically once daily for 5 days followed by receiving the cecal content from the donor mice (ZT0 or ZT12 group) once daily for 3 days. Mice were then gavaged with 300mg/kg APAP at ZT0 and were sacrificed 24 hours after APAP treatment. HE staining (n=6–12). (F) mRNA levels of key cytokines and chemokines in the liver (n=6–12). ZT0 = 8am. ZT12 = 8pm. The results are expressed as the mean ± S.E.M. Two-tailed Student’s t-test was used for statistical evaluation. *p<0.05. Scale bar: 100 μm. N.S.: non-siginificance.
To further clarify the contribution of gut microbiota to APAP induced liver injury, we performed fecal microbiota transplantation (FMT). The cecal content from ZT0 and ZT12 was collected and orally administrated to the recipients whose gut microbiota had been depleted with ABX. Microbiome 16s analysis showed that the clusters between ZT0 donor and ZT0 recipient, ZT12 donor and ZT12 recipient were overlapped, suggesting similar fecal microbiota between donor and recipient. (Supplementary Fig. 2). More importantly, as revealed in Fig. 1E, livers from mice that received ZT12 cecal content displayed significantly increased necrotic area compared with mice that received ZT0 content upon APAP administration at ZT0. Similarly, the hepatic cytokines/chemokines expression was also markedly enhanced in ZT12 transplanted mice (Fig. 1F). The antibiotic pretreatment did not protect the mice against APAP after ZT12 microbiota transfer, which rules out a direct inhibitory effect of antibiotics in the liver. These data, together with ABX experiment, clearly demonstrated the APAP hepatotoxic diurnal variation is dependent on gut microbiota.
In particular, intestinal microbiota from ZT12 could serve as a determinant to promote APAP induced acute liver injury. Although the necrotic area in the ZT12 to ZT0 fecal transplant was less than ZT12, this may reflect incomplete reconstitution of the microflora or other factors which might contribute to resistance of ZT0 to injury.
Intestinal microbiota exhibit diurnal variation
To explore the underlying mechanism by which gut microbiota modulate APAP induced hepatotoxicity, we next examined the diurnal variation of the compositional intestinal microbiota by analyzing 16s rRNA. Total bacterial load in the cecum exhibited non-significant trend toward higher levels in ZT12 compared with ZT0 (Supplementary Fig. 3A). Moreover, Firmicutes/Bacteroidetes ratio, which reflects the overall microbial composition in the gut, was significantly decreased in ZT12 versus ZT0 (Fig. 2A). Specifically, at the phyla level, increased abundance of Bacteroidetes and decreased abundance of Actinobacteria were observed in the cecal content of ZT12 versus ZT0 (Fig. 2B, Supplementary Fig. 3B). Moreover, in the genus level, the abundance of Lactobacillus and Enterorhabdus were enriched in ZT0 whearas Pseudoflavonifractor, Alistipes, Bacteroides, Barnesiella and Rikenella were enriched in ZT12 (Supplementary Fig. 3C,D). PCoA analysis also revealed the separation of two clusters of ZT0 and ZT12 (Fig. 2C). Our previous FMT experiment revealed that the plots between ZT0 recipients and ZT12 recipients were also separated, further demonstrated the microbial composition was transferable by transplantation (Supplementary Fig. 2). These results indicated that the composition of the gut microbiota displayed diurnal variation.
Figure 2:

Diurnal variation in both composition and metabolomics of gut microbiota. (A) Relative Firmicutes/Bacteroidetes ratio in the cecum (n=6–12). (B) The relative abundance of bacteria at the phylum level in the cecum (n=6–12). (C) Scatter plots of unweighted uniFrac PCoA for the microbial composition in the cecal content (n=8). (D) Scatter plots of PCA for the microbioal metabolomics in the ceal content (n = 6). (E) Volcano plot of the metabolomic analysis (n=6). (F) PPD levels in cecal content and liver at ZT0 and ZT12 (n=5–6). The results are expressed as the mean ± S.E.M. Two-tailed Student’s t-test was used for statistical evaluation. *p<0.05.
We next evaluated the gut microbial functional level between ZT0 and ZT12 by detecting the bacterial metabolites using untargeted metabolomic analysis. As shown in Fig. 2D, the metabolic profile was completely shifted in ZT12 compared with ZT0 as evidenced by PCA analysis which was widely used in metabolomics analysis. Furthermore, several metabolites were enriched either in ZT0 or ZT12 (Fig. 2E). Short chain fatty acids (SCFAs) and some long chain fatty acids like C15:0 and C17:0 were important bacteria-derived metabolites [18]. Although we did not find significant difference in the levels of C15:0 and C17:0, some SCFAs were different between ZT0 and ZT12. Specifically, C3:0 (Propionate) was higher while Iso-C5:0 (Isovalerate) showed increase trend in ZT12 compared with ZT0. (Supplementary Fig. 4). One compound, 1-phenyl-1,2-propanedione (PPD) which stood out as exhibiting potential reaction with hepatocytes based on its structure. Cecal PPD levels were around 2-fold higher in ZT12 than ZT0 based on metabolomics analysis (Fig. 2F). In addition, other than in the cecum, PPD concentration was also significantly elevated in liver of ZT12 than ZT0 (Fig. 2F). These data suggested gut microbial metabolite was also shifted with the diurnal variation in the microbiota.
To further confirm the PPD was generated from intestinal microbiota, we treated mice with ABX and found the PPD was dramatically decreased compared with controls at ZT12 (Fig. 3A). Furthermore, we isolated several intestinal bacteria from human stool and cultured them in vitro, and then analyzed the PPD level in the medium. As presented in Fig. 3B and Supplementary Fig. 5, some strains like E. coli, Citrobacter freundii, Clostridium difficile and Enterococcus faecalis generated PPD. However, interestingly, the Lactobacillus casei and Bacteroides thetaiotaomicron, which had been reported to exhibit beneficial effects on host [23,24], did not produce PPD. These data further demonstrated intestinal microbiota could generate PPD. Therefore, one may conclude that PPD level in the colon and liver is a potential biomarker of bacteria which increase the risk of APAP toxicity.
Figure 3:

Production of PPD by human intestinal bacteria. (A) Cecal PPD level at ZT12 after antibiotics treatment at ZT12 (n=3). The results are expressed as the mean ± S.E.M. Two-tailed Student’s t-test was used for statistical evaluation. *p<0.05. (B) Single bacteria strains were isolated from human stool and cultured in vitro. The PPD chromatograms (GC/MS) for the medium of specific bacteria strain.
PPD synergistically enhanced APAP induced liver damage in vivo and in vitro
To further link the APAP induced diurnal variation of hepatotoxicity to gut microbial metabolite variation, we next focused on the pathophysiological effect of PPD, namely does PPD actually contribute to the liver injury. APAP and PPD alone did not cause obvious liver injury in ZT0. However, mice that were co-treated with PPD and APAP exhibited increased liver damage as evidenced by plasma ALT level (Fig. 4A) and hepatic necrosis (Fig. 4B). The level of hepatic PPD after addition of exogenous PPD (Fig. 4C) was comparable to the ZT12 level (Fig. 2F). Hepatic cytokine expression confirmed the phenotype (Fig. 4D). We next determined the effects of PPD in vitro by isolating primary mouse hepatocytes. Similar to animal experiments, PPD and APAP alone lead to minimal cell toxicity while co-treatment with PPD and APAP caused increased cell damage as monitored by PI staining (Fig. 4E). These data clearly demonstrated that PPD could synergistically enhance APAP induced liver injury in vivo and in vitro. As noted above, we cannot exclude that other products of the microbiome at ZT0 contribute to resistance so that the extent of necrosis by the combination of APAP and PPD is dampened compared to ZT12 APAP, or other bacterial products at ZT12 contribute to worse toxicity.
Figure 4:

PPD synergistically enhanced APAP induced liver injury. Mice were treated with APAP or PPD at ZT0 for 24h. (A) Plasma ALT levels (n=5–9). (B) HE staining and necrotic area quantification (n=5–9). (C) Hepatic PPD level after PPD treatment (n=3). (D) mRNA levels of key cytokines and chemokines in the liver (n=5–9). (E) Primary cultured hepatocytes were treated with APAP, PPD or both for 6 hours, PI staining and quantification (n=3). The results are expressed as the mean ± S.E.M. Two-tailed Student’s t-test was used for statistical evaluation. *p<0.05. Scale bar: 100 μm.
PPD directly depletes hepatic GSH
To examine the mechanism by which microbiota and PPD influence APAP induced liver damage, we monitored several key pathways involved in the hepatotoxicity caused by APAP. First, APAP absorption as characterized by the peak plasma APAP concentration and the area under curve (AUC) after APAP administration was not significantly altered upon ABX treatment at ZT12 (Supplementary Fig. 6) excluding the possibility of a microbiota mediated enterohepatic cycling of APAP due to deconjugation in the gut as recently described with tacrine which exhibits a biphasic plasma profile [25]. Additionally, we found CYP2E1 protein level was also not affected by ABX treatment, indicating gut microbiota may not influence APAP bioactivation (Supplementary Fig. 7). Then we found that the PCNA level between control and ABX after 24hr APAP administration was comparable, suggesting gut microbiota was also not involved in hepatocyte regeneration (Supplementary Fig. 8). However, significantly reduced p-JNK level was observed in ABX treated mice compared with controls at ZT12 after 1h APAP administration (Fig. 5A). Furthermore, in the absence of APAP, decreased hepatic reduced GSH level was observed in ZT12 compared with ZT0, while ABX treatment partially but significantly recovered GSH level in ZT12 (Fig. 5B). However, no diurnal variation in glutamate cysteine ligase-catalytic subunit (GCL-c) expression (ZT0 versus ZT12) was observed (Supplementary Fig. 9), indicating that enzymatic GSH synthesis capacity may not explain the phenotype of hepatic GSH levels. Moreover, hepatic GSSG/GSH ratio was highest at ZT12 whereas ABX could significantly reduce GSSG/GSH ratio (Supplementary Fig. 10A). Since higher hepatic GSH synthesis was closely associated with more food intake (substrate availability), we next monitored the food consumption with ABX treatment. There was no difference in food intake during day phase (ZT0-ZT12, Supplementary Fig. 11A) but the mice receiving ABX consumed less during night phase (ZT12-ZT0, Supplementary Fig. 11B). In addition, the ratio of food intake during day and night phase was comparable between control and ABX groups (Supplementary Fig. 11C). These results indicated the modulation of GSH by ABX at ZT12 was independent of food consumption. This does not exclude the likelihood that consumption of diet containing GSH precursor amino acids contribute to the diurnal variation but superimposed is an effect of the microbiome on GSH levels. These data suggested the augmented liver damage at ZT12 than ZT0 may result from lowered hepatic GSH level and gut microbiota may modulate GSH which influences the susceptibility to APAP induced hepatotoxicity. Although the starting GSH level was approximate 35% higher after ABX at ZT12, the GSH fell to comparable low levels 1 hour after APAP (Supplementary Fig. 12) at the sublethal dose of APAP. The extra GSH may have been enough to handle the bulk of NAPQI generated.
Figure 5:

PPD directly depleted GSH. (A) Mice were treated with or without ABX followed by APAP administration for 1h at ZT0 and ZT12, respectively. Hepatic p-JNK, JNK protein levels (n = 3). (B) Hepatic reduced GSH levels after antibiotics treatment (n=8–12). (C) Mice were treated with or without PPD for 30 min followed by APAP administration for 1h at ZT0. Hepatic p-JNK, JNK protein levels (n = 3). (D) Reduced GSH levels in the livers after PPD treatment for 1 hour at ZT0 (n=8–12). (E) Reduced GSH levels in primary hepatocyte after PPD treatment for 6 hours (n=8–12). (F) APAP adducts levels in the livers after PPD treatment for 1 hour at ZT0 (n = 3). The results are expressed as the mean ± S.E.M. Two-tailed Student’s t-test was used for statistical evaluation. *p<0.05. N.S.: non-siginificance.
We then focused on the potential effect of PPD on GSH levels and related hepatic pathophysiologic changes. PPD synergistically enhanced hepatic p-JNK expression after 1h APAP treatment (Fig. 5C). Moreover, as presented in Fig. 5D and Supplementary Fig. 10B, PPD treatment modestly decreased reduced GSH and importantly enhanced GSSG/GSH ratio in vivo. Similar results were observed in primary cultured hepatocyte (Fig. 5E, Supplementary Fig. 10C). In agreement with GSH findings, PPD + APAP treatment markedly enhanced APAP adducts proteins compared with APAP administration alone in liver, which is consistent with the augmented p-JNK activation and hepatotoxicity observed in PPD and APAP co-treated mice (Fig. 5F). Though these data indicate that PPD might directly deplete GSH in the liver, the extent of GSH depletion from PPD and reversal by ABX are relatively modest but may be sufficient to change the threshold and dose of APAP required to generate APAP adducts and activate p-JNK. It is alternatively possible that the redox change in GSSG/GSH ratio induced by PPD was a key in sensitization to enhanced toxicity. We further determined the effects of N-Acetyl-L-cysteine (NAC) and SP600125 on the cytotoxicity. NAC could restore GSH depletion induced by APAP administration while SP600125 could inhibit JNK activation. We found both NAC and SP600125 were able to markedly rescue the cell death induced by APAP and PPD co-treatment (Supplementary Fig. 13), indicating oxidative stress and JNK activation were involved in the enhanced toxicity of APAP and PPD co-treatment.
Saccharomyces cerevisiae (Yeast) could decrease intestinal PPD and protect liver from APAP induced hepatotoxicity
Since intestinal PPD level was important for APAP induced liver damage at ZT12, we determined whether reduced PPD level in the gut may be beneficial for the hepatotoxicity caused by APAP. Indeed, biotransformation of PPD by Saccharomyces cerevisiae was widely used in industry especially in beer production, specifically, yeast could reduce the PPD’s carbonyls into hydroxyl [26]. We administrated Saccharomyces cerevisiae orally to mice and found the PPD levels in the cecal content were significantly decreased (Fig. 6A). Interestingly, yeast administration markedly attenuated APAP induced liver damage at ZT12 as evidenced by lowered plasma ALT, AST level and reduced necrotic area (Fig. 6B–D). Although we cannot exclude other beneficial effects of Saccharomyces cerevisiae, these results clearly revealed that yeast could ameliorate APAP induced liver injury as accompanied by PPD reduction.
Figure 6:

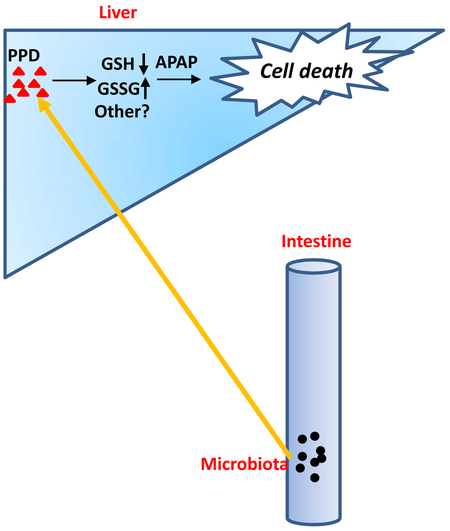
The protective effect of Saccharomyces cerevisiae (Yeast) against APAP induced hepatotoxicity was accompanied with diminished PPD concentration in the cecum. Mice were gavaged with yeast at ZT10 for 3 days followed by APAP administration for 24h at ZT12. (A) Cecal PPD level and representative chromatograms (n=5–9). (B) Plasma ALT, AST levels (n=5–9). (C) Representative liver photography. (D) HE staining and necrotic area quantification (n=5–9). (E) Working model of this work: Gut microbiota generated more PPD at ZT12 and more PPD translocated into liver and synergistically enhanced hepaic damage induced by APAP through a mechanism that depended on PPD-induced GSH depletion, redox change or possibly other unknown mechanisms. The results are expressed as the mean ± S.E.M. Two-tailed Student’s t-test was used for statistical evaluation. *p<0.05. Scale bar: 100 μm.
Discussion
Gut microbiota was recently found to exhibit circadian rhythm, at both the compositional and functional level. Such variation is believed to influence host pathophysiological status. For example, high fat diet leads to aberrant microbial oscillations which results in the disruption of host circadian system, finally promoting obesity development [27]. Although the mediators from microbiota to modulate host response are not fully understood, metabolites are believed to be the key factor. We previously reported that chronic alcohol feeding would diminish the capacity of gut microbiota to synthesize saturated long chain fatty acids. Consequently, gut barrier was markedly disrupted and bacteria/bacterial products translocated into liver [18]. In addition, gut microbiota metabolize choline from diet to trimethylamine (TMA) which can be further oxidized in liver to trimethylamine-N-oxide (TMAO). TMAO may promote atherosclerosis development [28,29]. Indeed, gut microbiota is able to generate a great number of metabolites for their survival and many of these compounds are likely to be metabolized by the liver. Hence it is possible for gut microbiota to modulate hepatotoxicity after drug administration.
The diurnal variation in susceptibility to APAP toxicity has been known for some time. While our studies were being conducted, another group published work on the diurnal variation to gut microbiome and our findings are in agreement [30]. Although they showed a diurnal susceptibility to APAP hepatotoxicity, the mechanism was not determined. We demonstrated that antibiotic treatment protected against APAP toxicity at ZT12 and cecal content transfer from ZT12 (time of susceptibility) converted resistance to susceptibility to APAP toxicity in ZT0 mice. Most investigations in the field have recognized that fasting increases susceptibility of APAP, presumably by decreasing the GSH pool size. Indeed, though not removed from food, mice have very low food consumption during day-light phase. However, our finding that the ABX treatment protected and ZT12 flora sensitized to APAP which could not be explained in food consumption, suggested that diet restricted food consumption was insufficient to explain our findings. Indeed, ABX treatment significantly increased ZT12 GSH levels despite lower food intake during night. Though not increased to the GSH levels at ZT0, the increase might be expected to require an increased acute dose of APAP to overcome the GSH protection. Therefore, although decreased diet intake in day-light phase is likely to contribute to lower GSH, a microbiota dependent effect is also contributing and making the liver more susceptible to the standard nonlethal but toxic dose of APAP that we used. Probably because this dose is right at the threshold for toxicity. Of note, although diurnal oscillation of the core circadian genes is the same in rodents and human, their functional output (sleep/wake cycle) is opposite and thus more severe liver toxicity at ZT12 (during active phase) in mice is not similar to more severe liver toxicity when APAP is taken in the evening
We next pursued the mechanisms for the ZT0 versus ZT12 toxicity and ABX protection by systematically assessing the early upstream pathway in APAP toxicity. We found no significant differences in absorption in a time course of blood APAP, no differences in CYP2E1 and GCL-c levels in ZT0 versus ZT12. However, we found that the different microbiome at ZT12 vs. ZT0 was producing an increase of some metabolites, including PPD. We focused on this compound because: (1) the level in the intestine exhibited diurnal variation similar to APAP toxic variation; (2) the host does not produce this small compound; (3) the dicarbonyl structure is highly possible to be processed by liver. PPD was found increased at ZT12, not only in the cecum but also in the liver, and the concentration of PPD at ZT12 was reduced by ABX treatment. We then demonstrated that exogenous PPD alone lead to modest GSH depletion in the liver and cells which potentiated APAP toxicity. The likelihood the PPD could directly form an adduct with GSH to an extent sufficient to deplete GSH is implausible considering the excess of hepatic GSH relative to PPD. However, combined with the large GSH depletion by the borderline toxic dose of APAP, sufficient NAPQI was made to increase adducts and activate JNK. In addition, the quinone like structure of PPD acting as a redox cycling compound generating ROS and altering to the redox state (GSSG/GSH) may have contributed. Importantly, the moderately lower GSH levels induced by PPD brings the GSH pool closer to APAP induced depletion, favoring NAPQI availability, i.e. less detoxification due to lower GSH pool size rather than increased production of NAPQI. Once GSH is depleted and covalent binding has occurred, increased ROS are released from mitochondria which then activates ASK1 leading to JNK activation and a sustained p-JNK→mitochondria→ROS→ASK1→JNK activation loop [31–36]. In the APAP model, this high level of ROS production in mitochondria ultimately leads to MPT mediated necrosis [37]. Importantly, the administration of PPD along with APAP enhanced covalent binding and sustained JNK activation accounting for PPD potentiating APAP toxicity. Clearly PPD can explain the effect of the microbiome on APAP toxicity but more work is needed to explore other possible actions of PPD or other bacterial products (Fig. 6E).
Female mice are usually resistant to APAP induced hepatotoxicity [8]. Therefore male mice were mostly used to report the diurnal variation of APAP induced acute liver injury [14,15]. However, we found the female mice that received male ZT12 feces exhibited increased necrosis compared with female mice that received male ZT0 feces (Supplementary Fig. 14), indicating that gut microbiota may also influence the diurnal variation of liver damage induced by APAP in female mice, though the microbiome alone makes a very modest contribution to the resistance of female mice to APAP.
Another important finding of this study is that oral administration of Saccharomyces cerevisiae could significantly attenuate APAP induced liver damage at ZT12. The reason we choose Saccharomyces cerevisiae was due to the fact that live yeast have been widely used in industries such as beer production to reduce the carbonyl on PPD to hydroxyl [26]. In our study, such biocatalysis alleviated the hepatic burden of metabolizing PPD and prevented the effect on GSH level. This finding suggests the translational significance of the current study, i.e. Saccharomyces cerevisiae pretreatment may reduce the risk of APAP induced ALF, when applied to populations at enhanced risk like chronic alcoholism or starvation. Of note, further work remains to be done to exclude additional mechanisms for the protection by Saccharomyces cerevisiae.
In conclusion, we provide a novel mechanism for the diurnal variation of APAP induced acute liver damage mediated by gut microbial functional oscillations.
Supplementary Material
Highlights.
The diurnal variation of APAP induced acute liver injury is dependent on gut microbiota.
Gut microbiota derived 1-phenyl-1,2-propanedione (PPD) played the key role in the diurnal variation of APAP induced hepatotoxicity.
PPD participated in liver damage development by directly depleting hepatic GSH level.
Acknowledgments
Financial Support
This study was supported in part by the National Science Foundation of China (31500952), the Natural Science Funds for Distinguished Young Scholar of Guangdong province (2016A030306043) and the award of Young Pearl Scholars of Guangdong province to PC. The Grant of NSFC-Guangdong Joint Foundation of China (U1601225), National Natural Science Foundation of China (81372030) and Key Scientific and Technological Program of Guangzhou City (201607020016) to JY. National Institute of Health grants (R01DK067215, P30DK048522) and its analytic/metabolic/instrumentation core to NK.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: All authors have no potential conflicts of interest to declare.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher’s website.
References:
- 1.Sheehan WJ, Mauger DT, Paul IM, Moy JN, Boehmer SJ, Szefler SJ, et al. Acetaminophen versus ibuprofen in young children with mild persistent asthma. N Engl J Med 2016;375(7):619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu Y, Zhang C, Chen YH, Wang H, Zhang ZH, Chen X, et al. Immature mice are more susceptible than adult mice to acetaminophen-induced acute liver injury. Sci Rep 2017;7:42736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larsen FS, Wendon J. Understanding paracetamol-induced liver failure. Intensive Care Med 2014;40:888–890. [DOI] [PubMed] [Google Scholar]
- 4.Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005;42:1364–1372. [DOI] [PubMed] [Google Scholar]
- 5.Schäfer C, Schröder KR, Höglinger O, Tollabimazraehno S, Lornejad-Schäfer MR. Acetaminophen changes intestinal epithelial cell membrane properties, subsequently affecting absorption processes. Cell Physiol Biochem 2013;32(2):431–447. [DOI] [PubMed] [Google Scholar]
- 6.Possamai LA, McPhail MJ, Khamri W, Wu B, Concas D, Harrison M, et al. The role of intestinal microbiota in murine models of acetaminophen-induced hepatotoxicity. Liver Int 2015;35(3):764–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kučera O, Endlicher R, Rychtrmoc D, Lotková H, Sobotka O, Červinková Z. Acetaminophen toxicity in rat and mouse hepatocytes in vitro. Drug Chem Toxicol 2017;40(4):448–456. [DOI] [PubMed] [Google Scholar]
- 8.Du K, Williams CD, McGill MR, Jaeschke H. Lower susceptibility of female mice to acetaminophen hepatotoxicity: Role of mitochondrial glutathione, oxidant stress and c-jun N-terminal kinase. Toxicol Appl Pharmacol 2014;281(1):58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Rongen A, Välitalo PA, Peeters MY, Boerma D, Huisman FW, van Ramshorst B, et al. Morbidly obese patients exhibit increased CYP2E1-mediated oxidation of acetaminophen. Clin Pharmacokinet 2016;55(7):833–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaplowitz N, Win S, Than TA, Liu ZX, Dara L. Targeting signal transduction pathways which regulate necrosis in acetaminophen hepatotoxicity. J Hepatol 2015;63(1):5–7. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J, Min RWM, Le K, Zhou S, Aghajan M, Than TA, et al. The role of MAP2 kinases and p38 kinase in acute murine liver injury models. Cell Death Dis 2017;8(6):e2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim YH, Hwang JH, Kim KS, Noh JR, Choi DH, Kim DK, et al. Metformin ameliorates acetaminophen hepatotoxicity via Gadd45β-dependent regulation of JNK signaling in mice. J Hepatol 2015;63(1):75–82. [DOI] [PubMed] [Google Scholar]
- 13.Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N. C-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 2006;131:165–178. [DOI] [PubMed] [Google Scholar]
- 14.Kakan X, Chen P, Zhang J. Clock gene mPer2 functions in diurnal variation of acetaminophen induced hepatotoxicity in mice. Exp Toxicol Pathol 2011;63(6):581–585. [DOI] [PubMed] [Google Scholar]
- 15.Kim YC, Lee SJ. Temporal variation in hepatotoxicity and metabolism of acetaminophen in mice. Toxicology 1998;128(1):53–61. [DOI] [PubMed] [Google Scholar]
- 16.Thaiss CA, Zeevi D, Levy M, Zilberman-Schapira G, Suez J, Tengeler AC, et al. Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell 2014;159(3):514–529. [DOI] [PubMed] [Google Scholar]
- 17.Fouts DE, Torralba M, Nelson KE, Brenner DA, Schnabl B. Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J Hepatol 2012;56(6):1283–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen P, Torralba M, Tan J, Embree M, Zengler K, Stärkel P, et al. Supplementation of saturated long-chain fatty acids maintains intestinal eubiosis and reduces ethanol-induced liver injury in mice. Gastroenterology 2015;148(1):203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker A, Pfitzner B, Neschen S, Kahle M, Harir M, Lucio M, et al. Distinct signatures of host-microbial meta-metabolome and gut microbiome in two C57BL/6 strains under high-fat diet. ISME J 2014;8(12):2380–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han H, Xiao H, Zhang K, Lu Z. Impact of 4-epi-oxytetracycline on the gut microbiota and blood metabolomics of Wistar rats. Sci Rep 2016;6:23141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gregory JC, Buffa JA, Org E, Wang Z, Levison BS, Zhu W et al. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J Biol Chem 2015; 290:5647–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schuijt TJ, Lankelma JM, Scicluna BP, de Sousa e Melo F, Roelofs JJ, de Boer JD, et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut 2016;65(4):575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagnerberger S, Spruss A, Kanuri G, Stahl C, Schröder M, Vetter W, Bischoff SC, et al. Lactobacillus casei Shirota protects from fructose-induced liver steatosis: a mouse model. J Nutr Biochem 2013;24(3):531–538. [DOI] [PubMed] [Google Scholar]
- 24.Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host–microbial relationships in the intestine. Science 2001;291:881–884. [DOI] [PubMed] [Google Scholar]
- 25.Yip LY, Aw CC, Lee SH, Hong YS, Ku HC, Xu WH, et al. The liver-gut microbiota axis modulates hepatotoxicity of tacrine in the rat. Hepatology. In Press. doi: 10.1002/hep.29327. [DOI] [PubMed] [Google Scholar]
- 26.Lourenço E Rodrigues JAR, Moran PJS. Preparation of an (−)-ephedrine intermediate through asymmetric reduction of 1-phenyl-1,2-propanedione by anaerobically pre-treated baker’s yeast. J Mol Catal B-Enzym 2004;29:37–40. [Google Scholar]
- 27.Leone V, Gibbons SM, Martinez K, Hutchison AL, Huang EY, Cham CM, et al. Effects of diurnal variation of gut microbes and high-fat feeding on host circadian clock function and metabolism. Cell Host Microbe 2015;17(5):681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 2013;19(5):576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen ML, Yi L, Zhang Y, Zhou X, Ran L, Yang J, et al. Resveratrol attenuates trimethylamine-N-Oxide (TMAO)-induced atherosclerosis by regulating TMAO synthesis and bile acid metabolism via remodeling of the gut microbiota. MBio 2016;7(2):e02210–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thaiss CA, Levy M, Korem T, Dohnalová L, Shapiro H, Jaitin DA, et al. Microbiota diurnal rhythmicity programs host transcriptome oscillations. Cell 2016;167(6):1495–1510. [DOI] [PubMed] [Google Scholar]
- 31.Win S, Than TA, Han D, Petrovic LM, Kaplowitz N. C-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J Biol Chem 2011;286: 35071–35078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma M, Gadang V, Jaeschke A. Critical role for mixed-lineage kinase 3 in acetaminophen-induced hepatotoxicity. Mol Pharmacol 2012;82:1001–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakagawa H, Maeda S, Hikiba Y, Ohmae T, Shibata W, Yanai A et al. Deletion of apoptosis signal-regulating kinase 1 attenuates acetaminophen-induced liver injury by inhibiting c-Jun N-terminal kinase activation. Gastroenterology 2008;135: 1311–1321. [DOI] [PubMed] [Google Scholar]
- 34.Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem 2008;283:13565–13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kon K, Kim JS, Jaeschke H, Lemasters JJ. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 2004;40:1170–1179. [DOI] [PubMed] [Google Scholar]
- 36.Saito C, Lemasters JJ, Jaeschke H. c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicol Appl Pharmacol 2010;246:8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han D, Dara L, Win S, Than TA, Yuan L, Abbasi SQ et al. Regulation of drug-induced liver injury by signal transduction pathways: critical role of mitochondria. Trends Pharmacol Sci 2013;34:243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.