

Abstract
A series of emodin analogues have been demonstrated to exhibit potent antiproliferative activity in three human epidermal growth factor receptor 2 (HER2)-overexpressing cell lines. However, in docking simulations, not all of these emodin analogues docked into the HER2 protein binding site. As the epidermal growth factor receptor (EFGR) and HER2 proteins are members of the ErbB family, the present study aimed to determine whether these anthraquinone derivatives exhibit potent antitumour bioactivity due to their inhibition of EGFR protein. Two 2D quantitative structure-activity relationship (QSAR) models, applied using multiple linear regression and a support vector machine, indicated seven representative molecular descriptors of anthraquinone derivatives associated with their antitumour activities. Molecular docking simulation indicated the possible docking poses of binding in the EGFR kinase domain. Two 3D-QSAR models performed by comparative force field analysis and comparative similarity indices analysis indicated the favoured and disfavoured fields for four physicochemical parameters (steric and hydrophobic properties, and hydrogen bond donor and acceptor), which may further improve the antitumour properties. These results demonstrate the benefits of further investigations on the development of lead compounds with improved anticancer bioactivity.
Keywords: epidermal growth factor receptor, multiple linear regression, support vector machine, molecular docking simulation, comparative force field analysis, comparative similarity indices analysis
Introduction
The ErbB family of proteins consists of four receptor tyrosine kinases: ErbB1/human epidermal growth factor receptor (HER)1/epidermal growth factor receptor (EGFR), ErbB2/HER2, ErbB3/HER3 and ErbB4/HER4 (1). The overexpression or overactivity of EGFR has been linked to a number of types of cancer, including lung cancer, colon cancer, glioblastoma, and head and neck squamous cell carcinoma (2–5).
A previous study demonstrated that a series of analogues of emodin, which is an anthraquinone derivative, exhibited potent antiproliferative activity in three HER2-overexpressing cell lines, FaDu, HSC3 and OECM1 (6). Following molecular docking simulation, the results revealed that not all of these compounds were able to dock into the binding site of the HER2 protein. However, a number of anthraquinone derivatives, including the emodin analogues of the previous study, docked into the binding site of the EGFR protein during a docking simulation, and the majority of these compounds exhibited similar docking poses within the EGFR kinase domain. EGFR and HER2 are members of the ErbB protein family, with the FaDu and OECM1 cell lines being EGFR+/HER2+ cell lines, and HSC3 being an EGFR+/EGR2− cell line. It has been indicated that emodin preferentially suppresses the phosphorylation activities of HER-2/neu, compared with EGFR, but also that emodin is also able to suppress the EGF-induced tyrosine phosphorylation of EGFR at high concentrations (7). In addition, chrysophanic acid, which is a natural anthraquinone, has been demonstrated to exhibit antiproliferative activity by inhibiting the EGF-induced phosphorylation of EGFR and suppressing the activation of downstream signalling molecules (8). As the 38 compounds in the previous study had superior antiproliferative activity in the FaDu cell line than the other two cell lines, the present study aimed to determine whether these anthraquinone derivatives exhibit potent antiproliferative activity in EGFR-overexpressing cell lines rather than HER2-overexpressing cell lines by assessing the antiproliferative activity of the FaDu cell line.
In the present study, a number of quantitative structure-activity relationship (QSAR) models were applied in order to identify the association between the functional groups of anthraquinone derivatives and their antitumour functions. The docking simulation indicated the possible docking poses of anthraquinone derivatives in the EGFR kinase domain.
Materials and methods
Data collection
A total of 38 anthraquinone derivatives were collected as described previously (Fig. 1; Table I) (6). All 38 compounds were drawn using ChemBioOffice 2010 v12.0 (http://www.cambridgesoft.com/services/), and each compound was prepared using the Prepare Ligand protocol in Discovery Studio v2.5 (DS2.5) (Accelrys Software, Inc., San Diego, CA, USA.) to modify its ionization to the physiological state.
Figure 1.
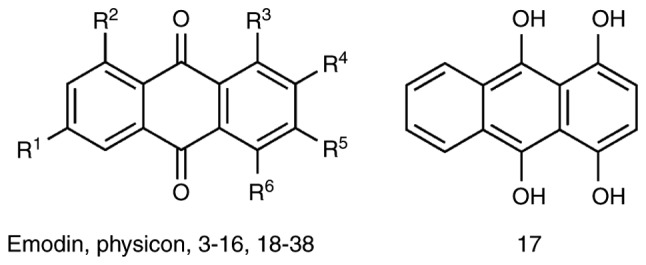
Chemical scaffolds of the anthraquinone derivatives.
Table I.
pIC50 values of anthraquinone derivatives in the FaDu cell line.
| Compound | R1 | R2 | R3 | R4 | R5 | R6 | pIC50 |
|---|---|---|---|---|---|---|---|
| Emodin | −OH | −OH | −OH | −H | −CH3 | −H | 4.33 |
| Physicona | −OMe | −OH | −OH | −H | −CH3 | −H | 3.46 |
| 3 | −OCOMe | −OCOMe | −OCOMe | −H | −COOH | −H | 3.93 |
| 4 | −OH | −OH | −OH | −H | −COOH | −H | 3.96 |
| 5 | −OCOMe | −OCOMe | −OCOMe | −H | −CH3 | −H | 4.48 |
| 6 | −OCOEt | −OCOEt | −OCOEt | −H | −CH3 | −H | 4.45 |
| 7a | −OCOPr | −OCOPr | −OCOPr | −H | −CH3 | −H | 4.34 |
| 8 | −OCOBu | −OCOBu | −OCOBu | −H | −CH3 | −H | 4.37 |
| 9 | −OMe | −OMe | −OMe | −H | −CH3 | −H | 4.41 |
| 10 | −H | −H | −NH2 | −NH2 | −H | −H | 3.63 |
| 11 | −H | −H | −H | −COOH | −H | −H | 3.79 |
| 12a | −H | −H | −H | −CH3 | −H | −H | 4.06 |
| 13a | −H | −H | −H | −SO3Na | −H | −H | 3.51 |
| 14 | −H | −H | −SO3Na | −H | −H | −H | 2.64 |
| 15 | −H | −H | −OH | −H | −H | −OH | 4.83 |
| 16a | −H | −H | −OH | −OH | −H | −OH | 4.39 |
| 17a | – | – | – | – | – | – | 5.14 |
| 18 | −H | −H | −NHMe | −H | −H | −NHMe | 3.04 |
| 19 | −H | −SO3K | −SO3K | −H | −H | −H | 3.74 |
| 20a | −H | −OH | −OH | −H | −H | −H | 4.83 |
| 21 | −H | −OH | −H | −H | −H | −OH | 4.42 |
| 22 | −OH | −H | −H | −OH− | −H | −H | 3.91 |
| 23 | −H | −H | −OH | −OH | −H | −H | 3.77 |
| 24a | −NH2 | −H | −H | −NH2 | −H | −H | 4.38 |
| 25 | −H | −H | −H | −OH | −NH2 | −H | 4.49 |
| 26 | −H | −H | −H | −NH2 | −H | −H | 4.44 |
| 27 | −H | −H | −H | −Et | −H | −H | 4.01 |
| 28a | −H | −H | −NH2 | −H | −H | −NH2 | 3.81 |
| 29 | −H | −NH2 | −H | −H | −H | −NH2 | 4.95 |
| 30 | −H | −H | −NHMe | −H | −H | −H | 4.23 |
| 31 | −H | −H | −NH2 | −CH3 | −H | −H | 4.25 |
| 32 | −H | −H | −NH2 | −H | −H | −H | 4.40 |
| 33 | −H | −H | −OH | −H | −H | −H | 4.40 |
| 34 | −H | −H | −H | −OH | −H | −H | 3.16 |
| 35 | −H | −OCOMe | −OCOMe | −H | −H | −H | 4.65 |
| 36 | −H | −OCOEt | −OCOEt | −H | −H | −H | 4.17 |
| 37a | −H | −OCOPr | −OCOPr | −H | −H | −H | 4.48 |
| 38 | −H | −OCOBu | −OCOBu | −H | −H | −H | 4.40 |
Test set. IC50, half maximal inhibitory concentration; pIC50, −log(IC50).
As described previously (6), the efficacy of antitumour activity was determined using a modified MTT method, and the half maximal inhibitory concentration (IC50) value was calculated by linear regression analysis (9). The FaDu cells (3,000 cells/well) were seeded into 96-well plates with a vehicle (dimethyl sulfoxide) or various concentrations of 38 test compounds using the dose range of 0–100 µM for 72 h at 37°C (5% CO2). The MTT (5 µg/ml) was added after 70 h of incubation. Subsequently, 40% dimethylformamide and 20% sodium dodecyl sulphate in H2O was treated as a solubilisation buffer and added into the wells to dissolve the violet formazan precipitation overnight at 37°C. A microplate reader (Molecular Devices, LLC, Sunnyvale, CA, USA) was used to detect the absorbance at 570 nm, and the linear regression analysis was performed to calculate the IC50 value. The IC50 value of the 38 test compounds for FaDu cells was found to lie between 7.3 µM (compound 17) and 2,285 µM (compound 14).
The X-ray crystal structures of the wild-type and T790M/L858R (TMLR) mutant EGFR kinase domains were obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (PDB ID, 5CAV and 5CAS, respectively; http://www.rcsb.org) (10) for use in the molecular docking simulation. The protein preparation of each EGFR kinase domain was performed using the Prepare Protein protocol in DS2.5 to remove water atoms from the crystals, insert missing atoms in incomplete residues, protonate the structure of EGFR kinase domain using the Chemistry at HARvard Macromolecular Mechanics (CHARMM) force field (11), and optimise the side-chain conformation of residues with inserted atoms. The binding site of each EGFR kinase domain was defined as the volume of co-crystallised inhibitor present in the initial crystal structure.
2D-QSAR modeling
Multiple linear regression (MLR) and a support vector machine (SVM) were employed to construct the 2D-QSAR models using MATLAB R2010b (https://www.mathworks.com/products/matlab.html) and LibSVM v.2.91 (https://www.csie.ntu.edu.tw/~cjlin/libsvm/) (12), respectively. All 38 compounds were divided randomly into training and test sets (28 and 10 compounds, respectively), and their IC50 values were determined as pIC50 values (−logIC50). The genetic function approximation (13) protocol in DS2.5 was employed to identify the suitable molecular descriptors for the prediction models.
Docking simulation
The docking poses for each compound were calculated using the LigandFit protocol (14) in DS2.5 using a shape filter and Monte-Carlo ligand conformation generation, followed by optional minimisation with the CHARMM force field (11). Similar docking poses of each compound were filtered using the leader algorithm (15). The optimal docking pose of each compound was determined according to its interactions with the EGFR kinase domains and three scoring functions, -PLP1, -PLP2 and -PMF (16–18), which were evaluated by summing two types of pairwise interaction, hydrogen bonding (H-bonding) and steric interactions.
Comparative force field analysis (CoMFA) and comparative similarity indices analysis (CoMSIA)
CoMFA (19) and CoMSIA (20) were used on the 28 compounds in the training set to construct 3D-QSAR models using SYBYL-X v1.1 (http://www.tripos.com). CoMFA, with the distance-dependent dielectric method, was performed to evaluate the steric and electrostatic field descriptors using Lennard-Jones and Coulombic potential energies, respectively. CoMSIA may produce more stable models than CoMFA, as it was performed with a Gaussian function based on distance in order to evaluate five physicochemical parameters (steric, electrostatic and hydrophobic properties, and H-bond donor and acceptor). The partial least-squares regression method was used to analyse the 3D-QSAR models.
Results
2D-QSAR modelling
The chemical scaffolds of the anthraquinone derivatives are shown in Fig. 1, and their pIC50 values are listed in Table I. The seven representative descriptors for building 2D-QSAR models, identified using genetic function approximation, were 'ES_Count_ssO', 'Num_H_Donors', 'Jurs_FPSA_3′, 'Jurs_RNCS', 'Jurs_RPCS', 'Shadow_XYfrac' and 'Shadow_XZ'.
The 'ES_Count_ssO' descriptor is an electrotopological state key (21,22) and represents the count of oxygen atoms with two single bonds. For the 'Num_H_Donors' descriptor, H-bond donors are defined as heteroatoms (O, N, S and P) with ≥1 attached H atom. The 'Jurs' descriptors (23) combine shape and electronic information to characterise molecules, by mapping atomic partial charges on the solvent-accessible surface areas of individual atoms. The 'Jurs_FPSA_3′ descriptor covers the fractional charged partial surface areas, which are obtained by dividing the total charge-weighted positive surface area by the total molecular solvent-accessible surface area. The 'Jurs_RNCS' and 'Jurs_RPCS' descriptors report the relative negatively and positively charged surface areas, respectively. The 'Shadow_XYfrac' and 'Shadow_XZ' descriptors are shadow indices (24), which calculate the area of the molecular shadow in the xy- and xz-plane, respectively.
An MLR model was established with the training set of 28 compounds, using the aforementioned seven descriptors selected by genetic function approximation, and the test set of 10 compounds was used to evaluate the final MLR model. The equation of the MLR model was as follows: pIC50 =−5.513 − 0.235 × 'ES_Count_ssO' − 0.895 × 'Num_H_ Donors' + 100.724 × 'Jurs_FPSA_3′ − 0.078 × 'Jurs_ RNCS' − 0.124 × 'Jurs_RPCS' + 6.406 × 'Shadow_ XYfrac' + 0.058 × 'Shadow_XZ'.
A non-linear SVM model was established with the same training set and descriptors as the MLR model. The predicted pIC50 values obtained by the MLR and SVM models are listed in Table II, and the correlation between the predicted and experimental pIC50 values for the two models are shown in Fig. 2A and B.
Table II.
Experimental and predicted pIC50 values obtained by the MLR and SVM models.
| Compound | Experimental pIC50 | MLR
|
SVM
|
||
|---|---|---|---|---|---|
| pIC50 | Error | pIC50 | Error | ||
| Emodin | 4.33 | 4.33 | 0.00 | 4.21 | −0.12 |
| Physicona | 3.46 | 4.45 | 0.99 | 4.00 | 0.54 |
| 3 | 3.93 | 4.19 | 0.26 | 4.05 | 0.12 |
| 4 | 3.96 | 3.88 | −0.08 | 3.84 | −0.12 |
| 5 | 4.48 | 4.27 | −0.21 | 4.35 | −0.13 |
| 6 | 4.45 | 3.99 | −0.46 | 4.33 | −0.12 |
| 7a | 4.34 | 4.29 | −0.05 | 4.41 | 0.07 |
| 8 | 4.37 | 4.53 | 0.16 | 4.46 | 0.09 |
| 9 | 4.41 | 4.57 | 0.16 | 4.54 | 0.13 |
| 10 | 3.63 | 4.25 | 0.62 | 3.93 | 0.30 |
| 11 | 3.79 | 3.49 | −0.30 | 3.66 | −0.13 |
| 12a | 4.06 | 3.76 | −0.30 | 3.97 | −0.09 |
| 13a | 3.51 | 3.18 | −0.33 | 2.84 | −0.67 |
| 14 | 2.64 | 2.70 | 0.06 | 2.77 | 0.13 |
| 15 | 4.83 | 4.68 | −0.15 | 4.71 | −0.12 |
| 16a | 4.39 | 4.20 | −0.19 | 4.09 | −0.30 |
| 17a | 5.14 | 4.86 | −0.28 | 4.63 | −0.51 |
| 18 | 3.04 | 3.14 | 0.10 | 3.16 | 0.12 |
| 19 | 3.74 | 3.77 | 0.03 | 3.87 | 0.13 |
| 20a | 4.83 | 4.81 | −0.02 | 4.81 | −0.02 |
| 21 | 4.42 | 4.67 | 0.25 | 4.66 | 0.24 |
| 22 | 3.91 | 3.90 | −0.01 | 3.87 | −0.04 |
| 23 | 3.77 | 3.87 | 0.10 | 3.90 | 0.13 |
| 24a | 4.38 | 4.43 | 0.05 | 3.91 | −0.47 |
| 25 | 4.49 | 4.12 | −0.37 | 4.16 | −0.33 |
| 26 | 4.44 | 4.39 | −0.05 | 4.31 | −0.13 |
| 27 | 4.01 | 3.96 | −0.05 | 3.93 | −0.08 |
| 28a | 3.81 | 4.23 | 0.42 | 4.07 | 0.26 |
| 29 | 4.95 | 4.36 | −0.59 | 4.15 | −0.80 |
| 30 | 4.23 | 4.31 | 0.08 | 4.27 | 0.04 |
| 31 | 4.25 | 4.25 | 0.00 | 4.12 | −0.13 |
| 32 | 4.40 | 4.42 | 0.02 | 4.46 | 0.06 |
| 33 | 4.40 | 4.25 | −0.15 | 4.28 | −0.12 |
| 34 | 3.16 | 3.62 | 0.46 | 3.55 | 0.39 |
| 35 | 4.65 | 4.72 | 0.07 | 4.56 | −0.09 |
| 36 | 4.17 | 4.17 | 0.00 | 4.30 | 0.13 |
| 37a | 4.48 | 4.29 | −0.19 | 4.31 | −0.17 |
| 38 | 4.40 | 4.47 | 0.07 | 4.28 | −0.12 |
Test set. MLR, multiple linear regression; SVM, support vector machine; IC50, half maximal inhibitory concentration; pIC50, −log(IC50).
Figure 2.
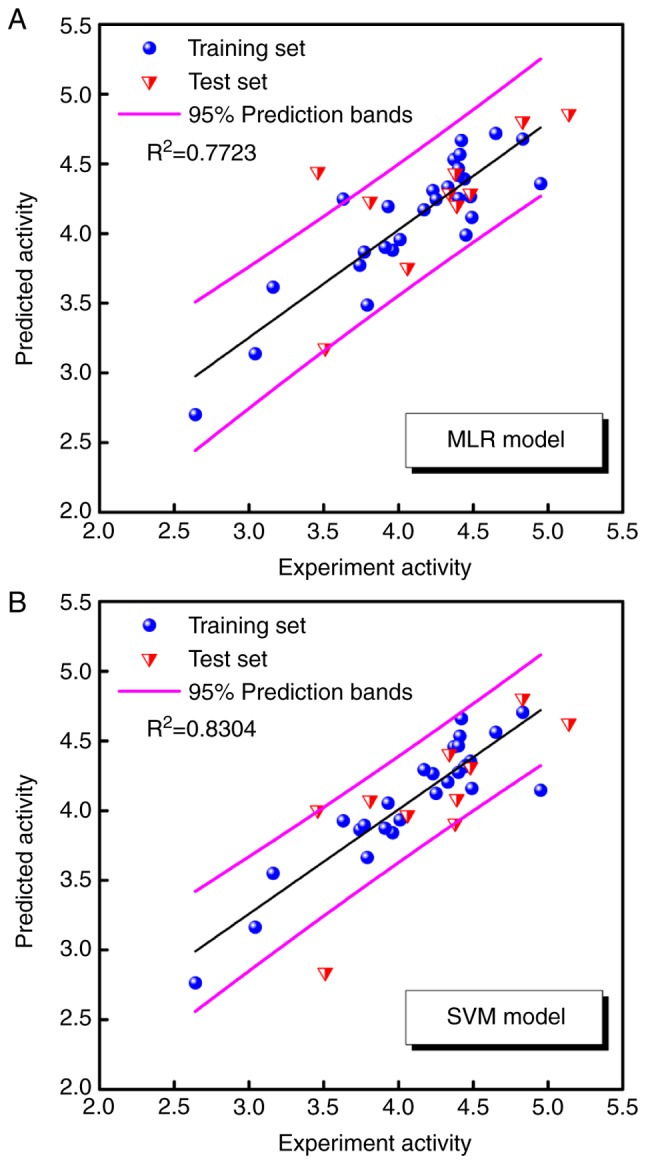
Correlation between the predicted and experimental pIC50 values of the MLR and SVM models. (A) MLR model and (B) SVM model. MLR, multiple linear regression; SVM, support vector machine; IC50, half maximal inhibitory concentration; pIC50, -log(IC50).
Docking simulation
The binding sites of the wild-type (PDB ID, 5CAV) and TMLR mutant (PDB ID, 5CAS) EGFR kinase domains were defined as the volume of co-crystallised inhibitors present in the initial crystal structures. A docking simulation was performed to validate the accuracy of the LigandFit protocol by redocking the co-crystallised EGFR inhibitor into the binding site of each EGFR kinase domain. The values of the root-mean-square deviation between the crystallised structure and the docking pose of the inhibitor were 0.4706 and 0.4322 for the wild-type and TMLR mutant, respectively.
The results of the docking simulations for all 38 anthraquinone derivatives in the binding site of each EGFR kinase domain are listed in Table III. The docking poses of three high-affinity compounds, 15, 20 and 35, in each EGFR kinase domain, are shown in Fig. 3A–F.
Table III.
Scoring functions of each complex obtained by docking simulation.
| Compound | pIC50 | 5CAV
|
5CAS
|
||||
|---|---|---|---|---|---|---|---|
| −PLP1 | −PLP2 | −PMF | −PLP1 | −PLP2 | −PMF | ||
| Emodin | 4.33 | 64.62 | 71.20 | 91.21 | 73.07 | 76.99 | 93.72 |
| Physicona | 3.46 | 44.51 | 43.67 | 96.99 | 43.12 | 41.73 | 62.42 |
| 3 | 3.93 | 65.20 | 59.92 | 111.74 | 83.79 | 76.72 | 81.25 |
| 4 | 3.96 | 79.81 | 86.22 | 88.59 | 87.77 | 89.76 | 88.21 |
| 5 | 4.48 | 62.67 | 55.15 | 122.56 | 79.83 | 71.08 | 126.61 |
| 6 | 4.45 | 72.30 | 61.25 | 99.36 | 61.80 | 53.20 | 127.27 |
| 7a | 4.34 | 102.78 | 101.73 | 86.88 | 75.07 | 70.05 | 150.17 |
| 8 | 4.37 | 103.66 | 94.66 | 101.74 | 95.34 | 90.63 | 109.19 |
| 9 | 4.41 | 56.21 | 51.77 | 104.36 | 55.03 | 54.33 | 105.17 |
| 10 | 3.63 | 53.83 | 52.39 | 76.19 | 60.32 | 63.13 | 76.37 |
| 11 | 3.79 | 64.02 | 65.84 | 51.38 | 61.56 | 54.57 | 79.38 |
| 12a | 4.06 | 53.95 | 54.22 | 57.21 | 57.18 | 57.87 | 80.13 |
| 13a | 3.51 | 67.65 | 72.88 | 42.90 | 54.8 | 49.32 | 76.73 |
| 14 | 2.64 | 59.11 | 56.33 | 68.86 | 58.39 | 60.44 | 83.99 |
| 15 | 4.83 | 47.60 | 48.20 | 89.25 | 50.42 | 53.99 | 101.97 |
| 16a | 4.39 | 49.73 | 53.42 | 103.26 | 62.46 | 69.88 | 102.27 |
| 17a | 5.14 | 45.57 | 48.18 | 87.04 | 55.28 | 58.20 | 95.57 |
| 18 | 3.04 | 64.69 | 64.20 | 58.02 | 65.39 | 63.78 | 86.34 |
| 19 | 3.74 | 54.02 | 55.81 | 91.70 | 60.02 | 63.80 | 84.75 |
| 20a | 4.83 | 49.31 | 49.87 | 83.80 | 61.24 | 62.95 | 90.19 |
| 21 | 4.42 | 47.57 | 49.08 | 89.96 | 59.00 | 60.92 | 87.61 |
| 22 | 3.91 | 55.02 | 57.83 | 94.92 | 56.47 | 56.86 | 84.56 |
| 23 | 3.77 | 50.46 | 54.35 | 99.94 | 60.46 | 67.85 | 97.32 |
| 24a | 4.38 | 56.09 | 54.18 | 89.29 | 55.40 | 47.37 | 75.34 |
| 25 | 4.49 | 58.32 | 56.39 | 68.49 | 58.22 | 62.31 | 92.49 |
| 26 | 4.44 | 54.69 | 54.25 | 46.94 | 56.09 | 55.54 | 76.29 |
| 27 | 4.01 | 58.49 | 58.55 | 62.80 | 61.69 | 58.24 | 50.50 |
| 28a | 3.81 | 53.69 | 53.67 | 81.66 | 62.51 | 64.48 | 79.19 |
| 29 | 4.95 | 55.19 | 55.90 | 82.85 | 62.27 | 63.53 | 85.81 |
| 30 | 4.23 | 55.05 | 55.05 | 62.42 | 56.17 | 56.77 | 79.84 |
| 31 | 4.25 | 55.41 | 55.12 | 56.78 | 60.56 | 57.02 | 44.52 |
| 32 | 4.40 | 51.96 | 52.18 | 83.14 | 59.91 | 61.83 | 77.87 |
| 33 | 4.40 | 47.67 | 46.35 | 69.79 | 53.19 | 54.67 | 79.28 |
| 34 | 3.16 | 48.39 | 50.31 | 95.00 | 55.33 | 58.62 | 82.15 |
| 35 | 4.65 | 58.96 | 56.96 | 110.18 | 65.63 | 66.48 | 101.84 |
| 36 | 4.17 | 65.34 | 62.50 | 118.53 | 74.46 | 72.09 | 108.60 |
| 37a | 4.48 | 63.10 | 60.99 | 130.19 | 80.10 | 77.80 | 122.05 |
| 38 | 4.40 | 68.72 | 68.51 | 129.06 | 81.43 | 79.45 | 127.00 |
Test set. IC50, half maximal inhibitory concentration; pIC50, −log(IC50).
Figure 3.
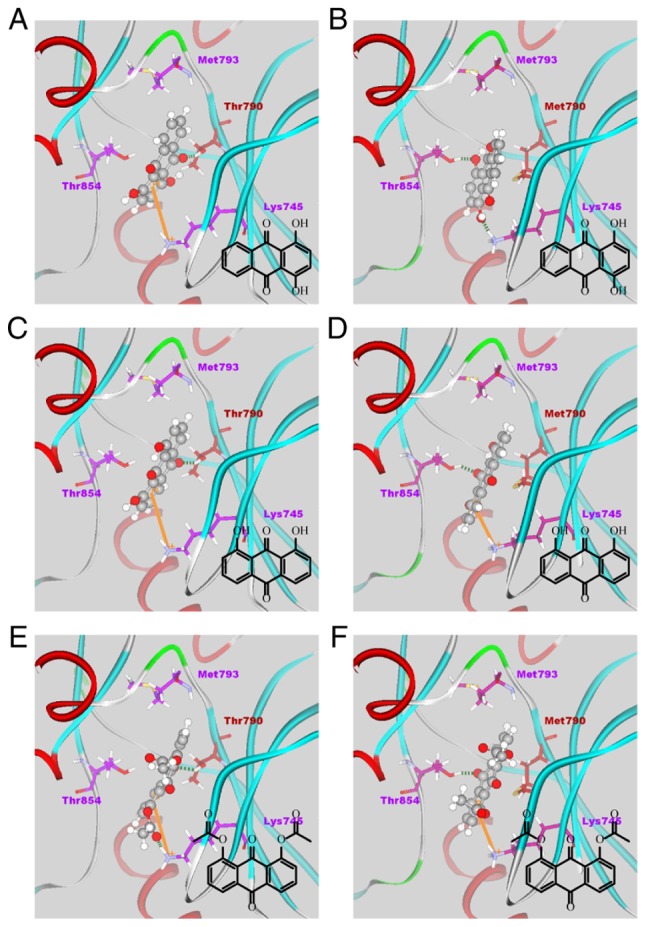
Docking poses of compound 15 in the binding site of the (A) wild-type (PDB ID, 5CAV) and (B) mutant (PDB ID, 5CAS) EGFR domain; compound 20 in (C) the wild-type and (D) mutant EGFR domain and compound 35 in (E) the wild-type and (F) mutant EGFR domain. EFGR, epidermal growth factor receptor.
CoMFA and CoMSIA
In order to identify the association between the functional groups of the compounds and their bioactivities, the CoMFA and CoMSIA models were established using the wild-type EGFR kinase domain (PDB ID, 5CAV). Therefore, the associations between the bioactivities and five physicochemical parameters (steric, electrostatic and hydrophobic properties, and H-bond donor and acceptor) were analysed.
For alignment of the 38 compounds, all were superimposed according to their docking poses. The predicted pIC50 values obtained by the CoMFA model using three components and the CoMSIA model using two components are listed in Table IV, and the correlations between the predicted and experimental pIC50 values of the CoMFA and CoMSIA models are shown in Fig. 4A and B. The squared correlation coefficients (R2) values of the training set were 0.7078 and 0.7069 for the CoMFA and CoMSIA models, respectively. The results of the CoMFA and CoMSIA models with high-affinity compound 20 are graphically represented by field contribution maps in Fig. 5.
Table IV.
Experimental and predicted pIC50 values obtained by the CoMFA and CoMSIA models for 28 compounds in the training set and 10 compounds in the test set.
| Compound | Experimental pIC50 | CoMFA
|
CoMSIA
|
||
|---|---|---|---|---|---|
| pIC50 | Error | pIC50 | Error | ||
| Emodin | 4.33 | 3.83 | −0.50 | 3.83 | −0.21 |
| Physicona | 3.46 | 4.22 | 0.76 | 4.22 | 0.57 |
| 3 | 3.93 | 3.91 | −0.02 | 3.91 | 0.19 |
| 4 | 3.96 | 3.81 | −0.15 | 3.81 | 0.17 |
| 5 | 4.48 | 4.49 | 0.01 | 4.49 | 0.23 |
| 6 | 4.45 | 4.70 | 0.25 | 4.70 | 0.03 |
| 7a | 4.34 | 3.33 | −1.01 | 3.33 | −0.11 |
| 8 | 4.37 | 4.31 | −0.06 | 4.31 | 0.11 |
| 9 | 4.41 | 4.29 | −0.12 | 4.29 | −0.30 |
| 10 | 3.63 | 4.20 | 0.57 | 4.20 | −0.12 |
| 11 | 3.79 | 3.72 | −0.07 | 3.72 | 0.07 |
| 12a | 4.06 | 4.08 | 0.02 | 4.08 | 0.11 |
| 13a | 3.51 | 4.41 | 0.90 | 4.41 | 0.35 |
| 14 | 2.64 | 3.37 | 0.73 | 3.37 | 0.58 |
| 15 | 4.83 | 4.36 | −0.47 | 4.36 | −0.74 |
| 16a | 4.39 | 3.84 | −0.55 | 3.84 | −0.46 |
| 17a | 5.14 | 3.57 | −1.57 | 3.57 | −0.84 |
| 18 | 3.04 | 2.90 | −0.14 | 2.90 | 0.13 |
| 19 | 3.74 | 3.90 | 0.16 | 3.90 | −0.03 |
| 20a | 4.83 | 4.52 | −0.31 | 4.52 | −0.62 |
| 21 | 4.42 | 4.36 | −0.06 | 4.36 | 0.01 |
| 22 | 3.91 | 3.67 | −0.24 | 3.67 | 0.13 |
| 23 | 3.77 | 3.69 | −0.08 | 3.69 | 0.03 |
| 24a | 4.38 | 4.12 | −0.26 | 4.12 | −0.77 |
| 25 | 4.49 | 4.40 | −0.09 | 4.40 | −0.62 |
| 26 | 4.44 | 4.25 | −0.19 | 4.25 | −0.05 |
| 27 | 4.01 | 3.74 | −0.27 | 3.74 | 0.17 |
| 28a | 3.81 | 4.34 | 0.53 | 4.34 | 0.65 |
| 29 | 4.95 | 4.94 | −0.01 | 4.94 | 0.21 |
| 30 | 4.23 | 4.25 | 0.02 | 4.25 | −0.01 |
| 31 | 4.25 | 4.23 | −0.02 | 4.23 | 0.13 |
| 32 | 4.40 | 4.62 | 0.22 | 4.62 | 0.11 |
| 33 | 4.40 | 4.15 | −0.25 | 4.15 | −0.56 |
| 34 | 3.16 | 3.72 | 0.56 | 3.72 | 0.35 |
| 35 | 4.65 | 4.52 | −0.13 | 4.52 | −0.23 |
| 36 | 4.17 | 4.36 | 0.19 | 4.36 | 0.17 |
| 37a | 4.48 | 4.25 | −0.23 | 4.25 | −0.21 |
| 38 | 4.40 | 4.60 | 0.20 | 4.60 | 0.06 |
Test set. CoMFA, comparative force field analysis; CoMSIA, comparative similarity indices analysis; IC50, half maximal inhibitory concentration; pIC50, −log(IC50).
Figure 4.
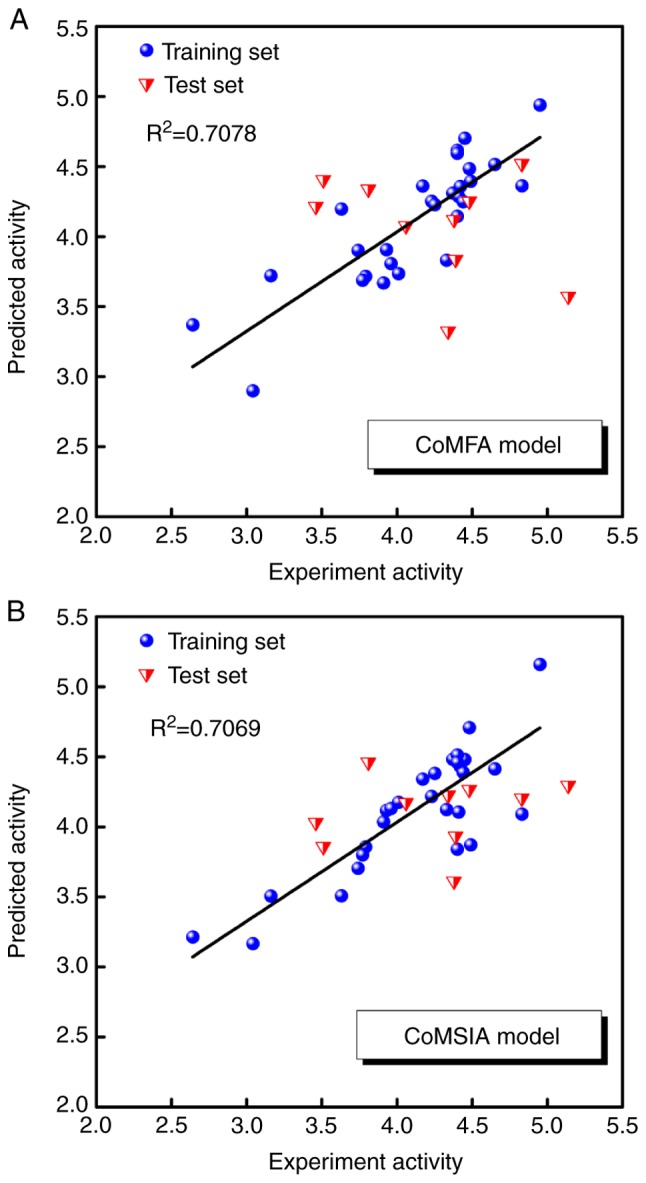
Correlations between the predicted and experimental pIC50 values of the CoMFA and CoMSIA models. (A) CoMFA model and (B) CoMSIA model. CoMFA, comparative force field analysis; CoMSIA, comparative similarity indices analysis; IC50, half maximal inhibitory concentration; pIC50, −log(IC50).
Figure 5.
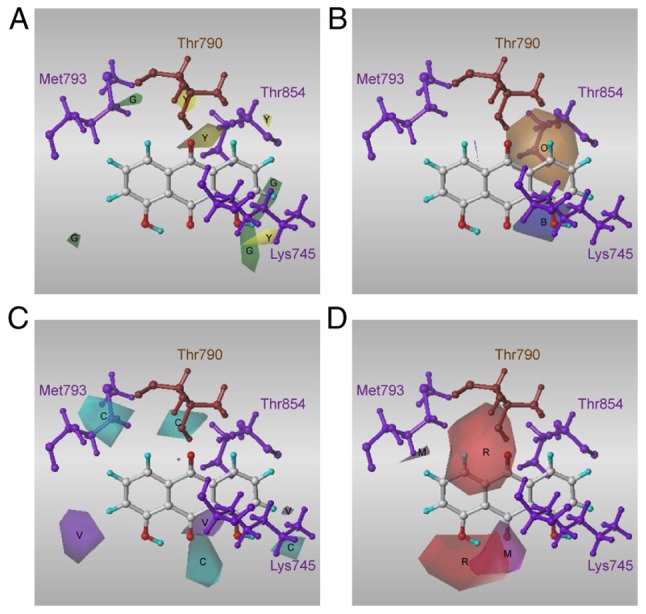
Contour plots of properties. (A) Steric properties revealed by the comparative force field analysis model and (B) hydrophobic, (C) hydrogen bond donor and (D) H-bond acceptor properties revealed by the comparative similarity indices analysis model. The template is high affinity compound 20. B, blue; C, cyan; G, green; M, magenta; O, orange; R, red; V, violet; Y, yellow.
In the CoMFA model (Fig. 5A), the proportions of steric and electrostatic fields were 100 and 0%, respectively. The favoured and disfavoured cut-off energies were set at 80 and 20%, respectively, for the steric field contributions.
In the CoMSIA model (Fig. 5B), the fractions of the hydrophobicity, H-bond donor and H-bond acceptor fields were 36.0, 43.5 and 20.5%, respectively. The favoured and disfavoured cut-off energies were set at 80 and 20%, respectively, for the hydrophobic contributions. For the field contributions of the H-bond donor properties (Fig. 5C), the favoured and disfavoured cut-off energies were set at 80 and 20%, respectively. For the field contributions of the H-bond acceptor properties (Fig. 5D), the favoured and disfavoured cut-off energies were set at 85 and 15%, respectively.
Discussion
2D-QSAR modelling
The majority of the predicted pIC50 values are within the 95% prediction bands, and the R2 values for the training set were >0.7, at 0.7723 and 0.8304 in the MLR and SVM models, respectively. The two models predicted reasonable bioactivities for the anthraquinone derivatives. The results indicate that the seven representative descriptors of anthraquinone derivatives are associated with their antitumour function.
Docking simulation
As shown in Fig. 3, the compounds formed π-cation interactions with the Lys745 residue and an H-bond with Thr790. As the mutation of T790M reduces the activity of EFGR inhibitors through inhibition of part of their interaction, in the resistant T790M mutant EGFR, π-cation interactions or H-bonds with residue Lys745 were present, but the H-bond between the compounds and Met790 was not. Instead, all three of the high-affinity compounds formed an H-bond with Thr854.
CoMFA and CoMSIA
As shown in Fig. 5A, part of the favoured steric field was observed close to the R3 and R4 moieties and some disfavoured steric field for the R6 moiety was observed towards Thr790 and Thr854. As shown in Fig. 5B, a favoured hydrophobic field for the R6 moiety was observed towards Thr854, with some disfavoured hydrophobic field close to the R3 moieties. As shown in Fig. 5C and 5D, the introduction of an H-bond donor and acceptor in the region may improve the activity.
In the present study, the MLR and SVM models indicated seven representative molecular descriptors of anthraquinone derivatives associated with their antitumour activities. The docking simulation indicated the possible docking poses of anthraquinone derivatives binding in the wild-type and resistant T790M mutant EGFR kinase domain. These compounds formed π-cation interactions with Lys745 and H-bonds with Thr790, and in the mutant EGFR kinase domain, H-bonds were formed with Thr854 instead of Met790. The CoMFA and CoMSIA models indicated the favoured and disfavoured fields for four physicochemical properties (steric, hydrophobic, H-bond donor, and H-bond acceptor), which may further improve the antitumour functions. The favoured steric fields and disfavoured hydrophobic fields were revealed close to the R3 moieties. The disfavoured steric fields and favoured hydrophobic fields for the R6 moiety were observed towards Thr854. As the compounds of the training set used in QSAR models have similar main scaffolds, those compounds with different main scaffolds may not fit these models. In addition, the results of a docking simulation can indicate that these compounds have suitable docking poses in the EGFR kinase domain, but they cannot be used to evaluate the antiproliferative activity of compounds. However, the results suggested the benefit of further investigations being performed to develop lead compounds with improved anticancer bioactivity. In future experiments, a series of analogues may be synthesised by adding larger hydrophilic substitutes in the R3 or R4 moieties and small hydrophobic substitutes in the R6 moiety in order to determine suitable anthraquinone derivatives with superior anticancer bioactivity against EGFR-overexpressing cell lines.
Acknowledgments
Not applicable.
Abbreviations
- EFGR
epidermal growth factor receptor
- QSAR
quantitative structure-activity relationship
- DS2.5
Discovery Studio 2.5
- TMLR
T790M/L858R
- CHARMM
Chemistry at HARvard Macromolecular Mechanics
- MLR
multiple linear regression
- SVM
support vector machine
- H-bond
hydrogen bond
- CoMFA
comparative force field analysis
- CoMSIA
comparative similarity indices analysis
Funding
The present study was supported by the Ministry of Science and Technology of Taiwan (grant nos. MOST 105-2320-B-039-032, MOST 106-2320-B-039-011 and MOST 107-2320-B-039-058), China Medical University (grant no. CMU107-S-33) and the China Medical University Hospital (grant nos. DMR-107-135, DMR-107-136, DMR-108-108 and DMR-108-142).
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Authors' contributions
KCC and JCL conceived of and designed the experiments, performed the experiments, analysed the data and wrote the manuscript. SHJ performed the MTT assay to determine the efficacy of the antitumour activity and calculated the IC50 values. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interest
The authors declare that they have no competing interests.
References
- 1.Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Bio Phys. 2004;59:21–26. doi: 10.1016/j.ijrobp.2003.11.041. [DOI] [PubMed] [Google Scholar]
- 2.Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, Greene MI. ErbB receptors: From oncogenes to targeted cancer therapies. J Clin Invest. 2007;117:2051–2058. doi: 10.1172/JCI32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walker F, Abramowitz L, Benabderrahmane D, Duval X, Descatoire V, Henin D, Lehy T, Aparicio T. Growth factor receptor expression in anal squamous lesions: Modifications associated with oncogenic human papillomavirus and human immunodeficiency virus. Hum Pathol. 2009;40:1517–1527. doi: 10.1016/j.humpath.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 4.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 5.Kuan CT, Wikstrand CJ, Bigner DD. EGF mutant receptor vIII as a molecular target in cancer therapy. Endocr-Relat Cancer. 2001;8:83–96. doi: 10.1677/erc.0.0080083. [DOI] [PubMed] [Google Scholar]
- 6.Liang FP, Lien JC, Wu YH, Chen CS, Juang SH. Em08red, a dual functional antiproliferative emodin analogue, is a downregulator of ErbB2 expression and inducer of intracellular oxidative stress. Drug Des Devel Ther. 2015;9:1499–1510. doi: 10.2147/DDDT.S66647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shia CS, Suresh G, Hou YC, Lin YC, Chao PD, Juang SH. Suppression on metastasis by rhubarb through modulation on MMP-2 and uPA in human A549 lung adenocarcinoma: An ex vivo approach. J Ethnopharmacol. 2011;133:426–433. doi: 10.1016/j.jep.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 8.Huang Q, Lu G, Shen HM, Chung MC, Ong CN. Anti-cancer properties of anthraquinones from rhubarb. Med Res Rev. 2007;27:609–630. doi: 10.1002/med.20094. [DOI] [PubMed] [Google Scholar]
- 9.Lee MS, Cha EY, Sul JY, Song IS, Kim JY. Chrysophanic acid blocks proliferation of colon cancer cells by inhibiting EGFR/mTOR pathway. Phytother Res. 2011;25:833–837. doi: 10.1002/ptr.3323. [DOI] [PubMed] [Google Scholar]
- 10.Heald R, Bowman KK, Bryan MC, Burdick D, Chan B, Chan E, Chen Y, Clausen S, Dominguez-Fernandez B, Eigenbrot C, et al. Noncovalent mutant selective epidermal growth factor receptor inhibitors: A lead optimization case study. J Med Chem. 2015;58:8877–8895. doi: 10.1021/acs.jmedchem.5b01412. [DOI] [PubMed] [Google Scholar]
- 11.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy minimization and dynamics calculations. J Comput Chem. 1983;4:187–217. doi: 10.1002/jcc.540040211. [DOI] [Google Scholar]
- 12.Fan R-E, Chen P-H, Lin C-J. Working set selection using second order information for training support vector machines. J Mach Learn Res. 2005;6:1889–1918. [Google Scholar]
- 13.Rogers D, Hopfinger AJ. Application of genetic function approximation to quantitative structure-activity relationships and quantitative structure-property relationships. J Chem Inf Comput Sci. 1994;34:854–866. doi: 10.1021/ci00020a020. [DOI] [Google Scholar]
- 14.Venkatachalam CM, Jiang X, Oldfield T, Waldman M. LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites. J Mol Graphics Modelling. 2003;21:289–307. doi: 10.1016/S1093-3263(02)00164-X. [DOI] [PubMed] [Google Scholar]
- 15.Hartigan JA. Clustering Algorithms. John Wiley & Sons Inc; [Google Scholar]
- 16.Gehlhaar DK, Verkhivker GM, Rejto PA, Sherman CJ, Fogel DB, Fogel LJ, Freer ST. Molecular recognition of the inhibitor Ag-1343 by Hiv-1 protease-conformationally flexible docking by evolutionary programmin g. Chem Biol. 1995;2:317–324. doi: 10.1016/1074-5521(95)90050-0. [DOI] [PubMed] [Google Scholar]
- 17.Gehlhaar Daniel K, Bouzida D, Rejto Paul A. Reduced dimensionality in ligand - Protein structure prediction: Covalent inhibitors of serine proteases and design of site-directed combinatorial libraries. Ration Drug Des Am Chem Soc. 1999;719:292–311. doi: 10.1021/bk-1999-0719.ch019. [DOI] [Google Scholar]
- 18.Muegge I, Martin YC. A general and fast scoring function for protein-ligand interactions: A simplified potential approach. J Med Chem. 1999;42:791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 19.Cramer RD, Patterson DE, Bunce JD. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc. 1988;110:5959–5967. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- 20.Klebe G, Abraham U, Mietzner T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J Med Chem. 1994;37:4130–4146. doi: 10.1021/jm00050a010. [DOI] [PubMed] [Google Scholar]
- 21.Hall LH, Mohney B, Kier LB. The electrotopological state: Structure information at the atomic level for molecular graphs. J Chem Inf Comput Sci. 1991;31:76–82. doi: 10.1021/ci00001a012. [DOI] [Google Scholar]
- 22.Hall LH, Kier LB. The E-state as the basis for molecular structure space definition and structure similarity. J Chem Inf Comput Sci. 2000;40:784–791. doi: 10.1021/ci990140w. [DOI] [PubMed] [Google Scholar]
- 23.Stanton DT, Jurs PC. Development and use of charged partial surface area structural descriptors in computer-assisted quantitative structure-property relationship studies. Anal Chem. 1990;62:2323–2329. doi: 10.1021/ac00220a013. [DOI] [PubMed] [Google Scholar]
- 24.Rohrbaugh RH, Jurs PC. Descriptions of molecular shape applied in studies of structure/activity and structure/property relationships. Analytica Chimica Acta. 1987;199:99–109. doi: 10.1016/S0003-2670(00)82801-9. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analysed during this study are included in this published article.