

Abstract
The aim of the present study was to determine the effect of 60% normobaric oxygen (NBO) on neurological function, brain edema and the expression of hypoxia-inducible factor-1α (HIF-1α), aquaporin 4 (AQP4) and Na+/H+ exchanger 1 (NHE1) in a rat model of cerebral ischemia-reperfusion injury. Male Sprague-Dawley rats underwent transient focal cerebral ischemia via right middle cerebral artery occlusion (MCAO) for 120 min followed by 48 h of reperfusion. The rats were exposed to NBO at 60 and 100% or no treatment during reperfusion for 48 h. Neurological impairment score (NIS) was evaluated prior to the sacrifice of all rats. Hematoxylin-eosin staining was performed after 48 h of reperfusion with NBO treatment. The infarct volume and brain water content (BWC) were determined to assess brain ischemic injury at 24 and 48 h. The levels of HIF-1α, AQP4 and NHE1 expression in brain tissue samples were determined by western blotting and reverse transcription-quantitative polymerase chain reaction analysis. During reperfusion, the protein and mRNA expression of HIF-1α, AQP4 and NHE1 increased over time (up to 48 h). Exposure to 60 and 100% NBO during reperfusion following MCAO improved NIS, and alleviated BWC and infarct volume after 24 and 48 h, with further improvements in the 100% NBO group, compared with 60%. Additionally, the molecular mechanisms involved in the effects of NBO may be associated with reduced AQP4 and NHE1 expression and increased HIF-1α expression. However, 60% NBO therapy during reperfusion following an acute ischemic stroke did not achieve the same effects as 100% NBO. Further experimental studies should be performed to elucidate the mechanism and beneficial effects of 60% NBO, as it is more cost-effective to use, compared with 100% NBO.
Keywords: normobaric oxygen, cerebral ischemic/reperfusion, hypoxia-inducible factor-1α, aquaporin 4, Na+/H+exchanger 1
Introduction
Stroke is a major health concern globally (1). The insufficient blood supply and tissue hypoxia caused by a stroke is a leading cause of mortality and disability, which results in a high socioeconomic burden (2,3). Normobaric oxygen (NBO) therapy experimentally induces an increase in penumbral O2 pressure, which may be promising as a treatment during reperfusion to increase tissue protection (4,5). NBO is neuroprotective, increases the pressure of oxygen in brain tissue, improves aerobic metabolism, reduces damage to the blood-brain barrier (BBB), and reduces ischemic brain injury and infarct volume in animal models of acute ischemic stroke, and in patients in pilot studies (5-10). However, the optimal concentration and duration of NBO treatment have not been established, and the molecular mechanisms of the neuroprotective effect are unclear. In previous studies, an excessive oxygen concentration and duration of NBO may result in oxidative stress and free radical damage, whereas insufficient oxygen concentration and duration will have a minimal therapeutic effect (8-10). NBO at 60%, which is frequently used in an attempt to improve the clinical situation (11), may be an attractive treatment option during the acute phase of an ischemic stroke.
At the molecular level, hypoxia-inducible factor-1α (HIF-1α), as an intrinsic marker of cellular hypoxia that is sensitive to oxygen levels, is considered to be part of the potential downstream pathways involved in oxygen rehabilitation (12). Additionally, the membrane proteins Na+/H+ exchanger 1 (NHE1) and aquaporin 4 (AQP4) regulate the hydromineral balance across the BBB to maintain the appropriate internal environment in brain tissue (13). However, whether NBO alters NHE1 and AQP4 protein expression following an ischemic stroke has not yet been reported. Therefore, the aim of the present study was to elucidate the effect of 60% NBO therapy during reperfusion following brain ischemia on neurological damage, infarct volume and brain edema, and to determine whether NHE1 and AQP4 protein expression is associated with the effects of NBO.
Materials and methods
Animals
A total of 150 adult male Sprague-Dawley rats [obtained from the Experimental Animal Center of Zhengzhou University; (Zhengzhou, China); license no: SCXK (Henan) 2015-0004] weighing 250-300 g and aged 8-10 weeks were used in the present study. Animals were maintained in a 12 h light/dark cycle at room temperature (23±2°C) in 60% humidity, and the animals were allowed free access to food and water. All experimental procedures followed the instructions of and were approved by the Zhengzhou University Animal Care and Use Committee, with every effort made to minimize discomfort during the surgery and recovery period. All surgical procedures were performed under chloral hydrate anesthesia.
Middle cerebral artery occlusion and reperfusion (MCAO)
The rats were anesthetized with 10% chloral hydrate (350 mg/kg) via intraperitoneal injection. No peritonitis was observed in the rats following the administration of the chloral hydrate. During surgery, the rectal temperature was maintained at 37.0±0.5°C using a feedback-heating pad. MCAO was performed as described previously (14). Briefly, following a midline neck incision, the right common and external carotid arteries were isolated and ligated. The flow through the internal carotid artery was temporarily stopped. A 0.26-mm monofilament (cat. no. A4-263650; Beijing Cinontech Co., Ltd., Beijing, China) was inserted into the common carotid artery through a small incision. Reperfusion was initiated 2 h after the onset of ischemia by gentle removal of the monofilament.
Experimental protocol and NBO treatment
All rats (n=150) were assigned to one of four main groups: Sham-operated; control; 60% NBO; and 100% NBO groups. The sham-operated group rats (n=18) were subjected to surgery of the neck region without ligation or ischemia. The control group rats (n=36) were subjected to brain ischemia for 2 h via MCAO followed by 48 h of reperfusion. In the NBO treatment groups, male Sprague-Dawley rats were administered with NBO (60 or 100%; n=36 each) during 2 h MCAO, followed by 48 h reperfusion. A total of 6 subgroups (2, 4, 6, 12, 24 and 48 h during reperfusion) of animals were studied within each of the groups. Animals (n=14) that succumbed during reperfusion were excluded from the present study. Failed model rats (n=10) were euthanized by cervical dislocation prior to 10% chloral hydrate (700 mg/kg) via intraperitoneal injection at the end of experiment. NBO was delivered using a ventilator with the animal placed in a sealed chamber. An oxygen probe assured a constant O2 concentration in the chamber.
Neurological impairment score (NIS)
NIS was determined prior to and following NBO therapy for 2, 4, 6, 12, 24 and 48 h during reperfusion following MCAO. The NIS was based on the 5-point scale of Longa et al (15) and was assessed by a blinded observer. Neuroscores were graded as follows: 0, no neurological deficit; 1, mild focal neurological deficit (failure to fully extend left forepaw); 2, moderate focal neurological deficit (circling to the left); 3, severe focal deficit (falling to the left); and 4, did not walk spontaneously and had a depressed level of consciousness (15). All rats were graded as 0 prior to the experiment. The scores were analyzed using the Kruskal-Wallis H test.
Hematoxylin-eosin staining for evaluation of pathological changes in the rat brain
Animals were anesthetized by intra-peritoneal injection of 10% chloral hydrate (350 mg/kg) prior to sacrifice. Hematoxylin-eosin staining of brain was assessed in three rats from each group at 48 h. Brains were removed, fixed in 4% paraformaldehyde for 24 h and embedded in paraffin. The tissue was cut into longitudinal sections at a thickness of 4 µm using a microtome (Leica RM2255; Leica Microsystems GmbH, Wetzlar, Germany). Following dewaxing by dimethylbenzene and hydration in gradient ethanol (anhydrous ethanol I; anhydrous ethanol II; 95% ethanol; 90% ethanol; 80% ethanol; 70% ethanol; tap water; distilled water I; and distilled water II), the sections were stained with hematoxylin for 3 min, rinsed with tap water, rinsed in 1% alcohol hydrochloric acid for 1 sec to remove excess stain, rinsed with tap water again for 10 min, stained with eosin for 3 min, dehydrated with reversed gradient ethanol (70% ethanol, 80% ethanol, 90% ethanol, 95% ethanol, anhydrous ethanol II and anhydrous ethanol I), made transparent with xylene and mounted. Between each step, the sections were rinsed with PBS (pH 7.4) three times, for 5 min each time. All the aforementioned steps were performed at 25°C. The sections were observed under a light microscope (Nikon Eclipse TS 100; Nikon Corporation, Tokyo, Japan) at a magnification of ×200.
Cerebral infarct volume
The brain infarct volume was evaluated at 24 and 48 h after NBO administration using 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich; Merck KGaA Darmstadt, Germany) staining. Coronal brain slices (n=6) with a 2-mm thickness were cut for treatment with 2% TTC at 37°C for 30 min and then fixed in 4% phosphate-buffered paraformaldehyde. After 24 h, the sections were imaged with a camera and infarction volumes were determined using Image-Pro Plus software version 6.0 (Media Cybernetics, Inc., Rockville, MD, USA). The percentage of infarction (infarct ratio) was calculated by dividing the infarct volume by the total volume of the slices.
Brain water content (BWC) determination
BWC, as a direct index of brain edema, was determined using the wet/dry weight method, as previously described (16). The wet weight (WW) of each hemisphere was carefully weighed and recorded. The dry weight (DW) was recorded following drying the sample in an oven at 85°C for 72 h. Brain edema (%) was evaluated by measuring the water content using the following formula: (WW-DW)/WW ×100%.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis of HIF-1α, AQP4 and NHE1 mRNA
Total RNA was extracted from ischemic hemisphere brain tissue using TRIzol® reagent (Thermo Fisher Scientific, Inc., Waltham, MA, USA), according to the manufacturer's protocol. cDNA was synthesized using a cDNA synthesis kit (Takara Biotechnology Co., Ltd., Dalian, China) according to the manufacturer's protocols. RT-qPCR was performed at 50°C for 2 min, 94°C for 15 min, followed by 40 cycles of 94°C for 15 sec, 58°C for 30 sec and 72°C for 30 sec.
The expression of HIF-1α, NHE1, AQP4 and β-actin were evaluated by RT-qPCR using Platinum SYBR® Green qPCR Supermix (Takara Biotechnology Co., Ltd.). The specific primers used are presented in Table I. The gene-specific mRNA expression was determined by the cycle quantification (Cq) method and Cq values for β-actin were used as the internal control. The 2−ΔΔCq method (17) was used for relative quantitation.
Table I.
The gene primer sequence and product size used for AQP4, NHE1 and β-actin.
| Gene name | Oligonucleotides used for RT-qPCR primer sequence (5'-3') | Production size (bp) |
|---|---|---|
| HIF-1α | F: AACAGAATGGAACGGAGCAA | |
| R: TTCACAATCGTAACTGGTCAGC | 119 | |
| AQP4 | F: CTCATCTCCCTCTGCTTTGG | |
| R: CGATGCTGATCTTTCGTGTG | 124 | |
| NHE1 | F: GACAAGAAGCACTTCCCCATGT | |
| R: GGTCATGCCCTGCACAAAG | 87 | |
| β-actin | F: GCCAACACAGTGCTGTCTG | |
| R: TACTCCTGCTTGCTGATCCA | 104 |
RT-qPCR, reverse transcription-quantitative polymerase chain reaction; F, forward; R, reverse.
Western blot analysis of HIF-1α, AQP4 and NHE1
Rats were sacrificed at 2, 4, 6, 12, 24 and 48 h post-reperfusion. Ischemic cerebrum hemisphere samples were homogenized in high effect-radioimmunoprecipitation assay lysis buffer (cat. no. R0010; Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) containing a protease inhibitor (cat. no. 88669; Thermo Fisher Scientific, Inc.) and sonicated. The protein concentrations were determined by the bicinchoninic acid method and 50 µg of protein was separated on 10% SDS-PAGE [H2O (47%), 40% Acr/bis (25%), 1.5 M tris-Hcl (26%) (pH=8.8), 10% SDS (1%), 10% APS (1%), TEMED (0.04%)] and transferred onto 0.45 mm polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). Following blocking in 5% milk containing TBS and 0.05% Tween-20 at 25°C for 1 h, the membranes were incubated with the appropriate antibodies, including: Anti-HIF-1α (1:500; cat. no. bs-0737R; BIOSS, Beijing, China); AQP-4 (1:500; cat. no. sc-20812; Santa Cruz Biotechnology, Inc., Dallas, TX, USA); anti-NHE-1 antibody (1:1,000; cat. no. ab67314; Abcam, Cambridge, MA, USA); and β-tubulin (1:4,000; cat. no. TA-10; OriGene Technologies, Inc., Beijing, China) overnight at 4°C. Anti-rabbit IgG (1:5,000; cat. no. 7074; Cell Signaling Technology, Inc., Danvers, MA, USA) and goat anti-mouse IgG (1:6,000; cat. no. CW0102S; CoWin Bioscience, Beijing China) horseradish peroxidase-conjugated secondary antibodies were used for 2 h at room temperature. After four washes, enhanced chemiluminescence detection system (Beijing Solarbio Science & Technology, Co., Ltd.) was used to visualize protein bands. Blots were quantified using Image Quant TL 8.1 (GE Healthcare, Chicago, IL, USA) as a proportion of the signal of a housekeeping protein band (β-tubulin).
Immunofluorescence of AQP4 and NHE1 co-expression with glial fibrillary acidic protein (GFAP)
The indirect immunofluorescence method was employed as described previously (18) following rat brains being harvested at 48 h during reperfusion. The samples were sliced into 4-µm histological sections. Tissue sections were dewaxed and rehydrated through xylene and graded ethanol series (anhydrous ethanol I, anhydrous ethanol II, 95% ethanol, 90% ethanol, 80% ethanol, 70% ethanol, tap water, distilled water I and distilled water II), and the slides were then incubated for 10 min at 95°C in a citrate solution for antigen retrieval. The nonspecific binding sites on sections were blocked with 0.5% bovine serum albumin (Sigma-Aldrich, Merck KGaA) for 1 h at room temperature. Subsequently, the tissue sections were incubated with primary antibodies: GFAP (1:100; cat. no. ab10062; Abcam, Cambridge, MA, USA); AQP-4 (1:100; cat. no. sc-20812; Santa Cruz Biotechnology, Inc., Dallas, TX, USA); anti-NHE-1 antibody (1:100; cat. no. ab67314; Abcam) at 4°C overnight, followed by incubation with secondary antibodies: Alexa Fluor® 488 goat anti-mouse IgG (1:200; A11001; Thermo Fisher Scientific, Inc.) or Alexa Fluor® 568 goat anti-rabbit IgG (1:200; A11011; Thermo Fisher Scientific, Inc.) for 2 h at room temperature. DAPI (Invitrogen; Thermo Fisher Scientific, Inc.) was used for counterstaining for 10 min at room temperature, then washed three times with PBS. The immunostained specimens were observed with an Olympus fluorescence microscope (Olympus; IX73; Olympus Corporation, Tokyo, Japan). The double-labeled positive cells were measured under a magnification of ×400 using the cellSens Dimension 1.17 analysis software (Olympus Corporation).
Statistical analysis
NISs are expressed as the median ± interquartile range. Other values are presented as the mean ± standard deviation. The differences between groups were calculated using Kruskal Wallis analysis followed by Dunn's post-hoc method for non-parametric comparisons. Statistically significant differences were evaluated by two-way repeated measures analysis of variance, multiple comparisons followed by Bonferroni's post-hoc test for parametric comparisons. Statistical analyses were performed using SPSS version 22.0 software (IBM Corp., Armonk, NY, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
NBO improves NIS
During reperfusion the rats' nerve function were checked prior to and following NBO treatment via NIS assessment. The NIS was 0 in all sham-operated groups (date not show) and significantly increased in the ischemic control animals. Table II depicts that the median NIS was reduced in rats receiving 60 and 100% NBO ≥12 h, compared with the control group. Furthermore, the NIS was significantly reduced in all NBO treatment groups at 24 h (P=0.012, Kruskal Wallis test) and 48 h (P=0.004, Kruskal Wallis test), compared with the control group.
Table II.
Effect of NBO on NIS.
| Group | NIS at time points [median (IQR)]
|
|||||
|---|---|---|---|---|---|---|
| 2 h | 4 h | 6 h | 12 h | 24 h | 48 h | |
| 100% NBO | 2.0 (1.75-3.0) | 2.0 (1.0-2.5) | 2.0 (1.75-2.25) | 1.5 (1.0-3.0) | 1.5 (1.0-2.0)a | 1.0 (1.0-2.0)a |
| 60% NBO | 2.5 (1.75-3.0) | 2.0 (1.75-3.0) | 2.0 (1.75-3.0) | 2.0 (1.0-2.25) | 2.0 (1.0-2.0)a | 2.0 (1.0-2.0)a |
| Control | 2.0 (2.0-2.0) | 2.0 (2.0-3.0) | 2.0 (2.0-3.0) | 2.5 (2.0-3.0) | 3.0 (2.0-3.25) | 3.0 (2.75-4.0) |
| H-value | 1.195 | 1.123 | 0.821 | 2.698 | 8.833 | 11.152 |
| P-value | 0.550 | 0.570 | 0.663 | 0.260 | 0.012 | 0.004 |
The median (IQR) NIS for each group after NBO therapy, compared with the ischemic control group (n=6 per group). Data are expressed as median ± IQR.
P<0.05 vs. the control group. NIS, neurologic impairment score; IQR, interquartile range; NBO, normobaric oxygen.
Assessment of BWC and infarct volume
The sham-group animals had almost no infarctions (Fig. 1A). The infarction ratio increased to 48.67±3.51% at 24 h and to 50.33±1.53% at 48 h after MCAO of the right hemisphere in the control group; however, it was significantly decreased in both NBO treatment groups at 48 h, compared with the ischemic control group (60% NBO vs. control, P=0.039; 100% NBO vs. control, P=0.005). Compared with 60% NBO, 100% NBO decreased the infarction ratio at both time points (24 h, 60% NBO vs 100% NBO, P=0.028; 48 h, 60% NBO vs. 100% NBO, P=0.041; Fig. 1B).
Figure 1.

Quantification of infraction rate and brain water content at 24 and 48 h. (A) Brain sections stained with 2,3,5-triphenyltetrazolium chloride exhibiting ischemic infarctions. Red-colored regions indicate non-ischemia, and pale-colored regions indicate the ischemic portion of the brain. (B) Infraction rate at 24 and 48 h. (C) BWC at 24 and 48 h. *P<0.05 vs. control group; #P<0.05 vs. 60% NBO group. Values present the mean ± standard deviation, n=3 per group. BWC, brain water content; NBO, normobaric oxygen.
Following ischemia and reperfusion, the BWC of the infarcted hemisphere of control-ischemia rats significantly increased to 85.43±1.40% at 24 h, and to 86.24±1.23% at 48 h within the control group. Compared with the control-ischemia group, 60 and 100% NBO therapy significantly reduced the BWC at 48 h (60% NBO vs. control, P=0.028; 100% NBO vs. control, P=0.015). However, only 100% NBO supplementation reduced BWC at 24 h (P=0.018; Fig. 1C). These results indicate that the administration of NBO may decrease brain edema during reperfusion following ischemia.
Hematoxylin and eosin staining of the brain tissue
Rats were sacrificed following assessment of NIS. Hematoxylin and eosin staining was used to determine the histopathological alterations induced by MCAO and reperfusion in all groups at 48 h. The intermediate region between under-stained infarcted areas and normal brain tissue was the ischemic penumbral zone. The structure of brain tissue in the sham group was normal. There were numerous red-stained neurons (eosinophilic degeneration), indicating neuronal death, nuclear shrinkage and widespread loss of Nissl bodies in the infarcted area following cerebral ischemia-reperfusion injury in the ischemia control group. Brain edema, neutrophil infiltration and increased perivascular space were also observed. NBO treatment attenuated these pathological changes, with a decrease in the ischemic penumbral zone area and a reduction in the number of red-stained neurons. Brain edema, neutrophil infiltration and increased perivascular space persisted in the NBO-treated groups (Fig. 2).
Figure 2.

Histological were evaluated by haematoxylin and eosin staining of cerebral ischemic penumbra at 48 h. Morphological improvement in slice following NBO treatment. (A) The normal brain tissue was depicted. (B) The control group numerous shrunken and triangular-shaped neurons with nuclei shrinkage distributed in the damaged brain tissue brain edema, neutrophil infiltration and increased perivascular space were also observed. (C) Brain tissue damage exhibited less damage in the 60% NBO group, compared with the vehicle control. (D) The 100% NBO treatment ameliorated brain damage, compare with 60% NBO treatment. x200 magnification. Arrow head, neurons; asterisk, capillaries; arrow, neutrophils; NBO, normobaric oxygen.
Effect of NBO treatment on HIF-1α, NHE1 and AQP4 protein expression levels
The protein expression levels of HIF-1α, AQP4 and NHE1 were determined by western blot analysis (Figs. 3, 4). Semi-quantitative western blot analysis of ischemic hemisphere samples indicated that HIF-1α expression was markedly increased at 2 h and reached the maximum level at 24 h following reperfusion in the ischemic-control group, compared with the sham group (P<0.05). Compared with the control group, the expression of HIF-1α protein significantly increased at 6, 12 and 24 h in the 60% NBO group (P<0.05), while at 2-24 h in the 100% NBO group (P<0.05; Bonferroni test following repeated measures ANOVA); however, compared with the 60% NBO group, HIF-1α protein was significantly increased at 2-24 h in 100% NBO treatment group (P<0.05), with no significant differences at 48 h (Fig. 3A and B).
Figure 3.

Time course of HIF-1α and AQP4 protein expression for the control and NBO treatment groups. (A) Representative photographs of western blot analysis of HIF-1α and AQP4 expression in brain tissue during reperfusion at 2, 4, 6, 12, 24 and 48 h following the control and NBO treatment. β-tubulin was used as the loading control. (B) Quantification of HIF-1α expression. (C) Quantification of AQP4 expression. Values are presented as the mean ± standard deviation, n=3 per group. *P<0.05 vs. the sham group; #P<0.05 vs. the control group; &P<0.05 vs. the 60% NBO group. HIF-1α, hypoxia-inducible factor-1α; AQP4, aquaporin-4; NBO, normobaric oxygen therapy; MCAO, middle cerebral ischemic occlusion; I/R, ischemic/reperfusion.
Figure 4.
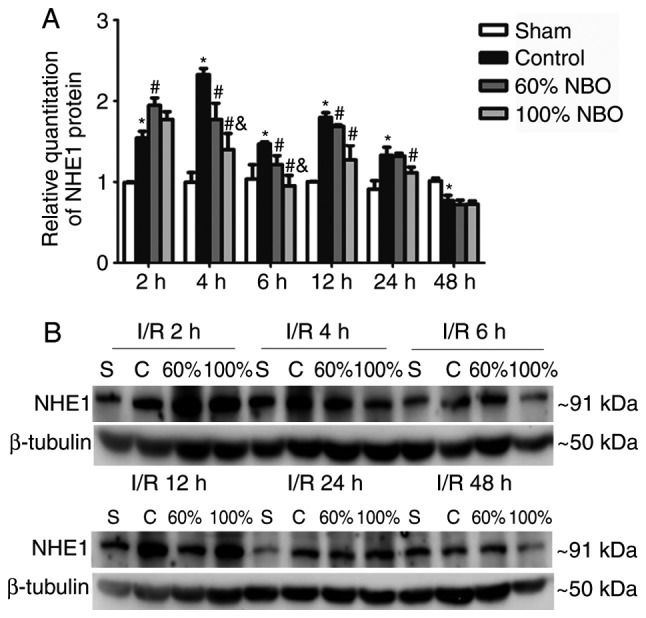
Time course of NHE1 protein expression for the control and NBO treatment groups. (A) Quantification of NHE1 expression. (B) Representative photographs of western blot analysis of NHE1 in the ischemic brain during reperfusion at 2, 4, 6, 12, 24 and 48 h following the control and NBO treatment. Values are presented as the mean ± standard deviation, n=3 per group. β-tubulin was used as the loading control. *P<0.05 vs. the sham group; #P<0.05 vs. the control group; &P<0.05 vs. the 60% NBO group. NHE1, Na+/H+ exchanger 1; NBO, normobaric oxygen; S, sham; C, control; I/R, ischemic/reperfusion.
Compared with the sham group, semi-quantitative western blot analysis of AQP4 protein expression was significantly increased after 2, 4, 12 and 24 h in the ischemic control group, except at 6 and 48 h, where the opposite trend was observed (Fig. 3A and C). The 60% NBO treatment significantly increased AQP4 expression at 2 h, but expression was decreased in the subsequent time points, compared with the control group, and the changes were statistically significant at the 4-48 h time points except at 6 h (P<0.05; Bonferroni test following repeated measures ANOVA). Compared with the 60% NBO group, the expression of AQP4 in the 100% NBO group was significantly reduced when NBO treatment was performed for 2 h and ≥12 h (P<0.05; Fig. 3A and C).
The results demonstrate that the NHE1 protein expression of the ischemic-control group was significantly increased at 2-24 h, with the highest results at 4 and 12 h, and significantly decreased at 48 h, compared with the sham group (Fig. 4A and B). Compared with the ischemic control group, the expression of NHE1 protein was significantly increased at 2 h, and then decreased at ≥4 h in the 60% NBO treatment group, with the significant decrease at 4, 6 and 12 h (P=0.015, P=0.036 and P=0.047, respectively). The 100% NBO treatment significantly decreased the NHE1 expression at 4 and 6 h, compared with the 60% NBO group. However, the results for 2 and ≥12 h are not significantly different.
Effect of NBO treatment on HIF-1α, NHE1 and AQP4 mRNA expression
The mRNA expression levels of HIF-1α, NHE1 and AQP4 were analyzed by RT-qPCR (Fig. 5). The graph depicts that the levels of HIF-1α mRNA were increased by ~25% at 2 h in the ischemia-control rats, compared with the sham group, and the results were significantly increased at all time points (P<0.05). Following NBO treatment, a significant increase in the expression of HIF-1α mRNA was observed in the 60 and 100% NBO groups from 2-48 h following ischemia and reperfusion, except the 60% NBO at 48 h. A significant increase in the expression of HIF-1α mRNA was demonstrated in the 100% NBO treatment group at 4-24 h, compared with the 60% NBO group (P<0.05; Fig. 5A).
Figure 5.
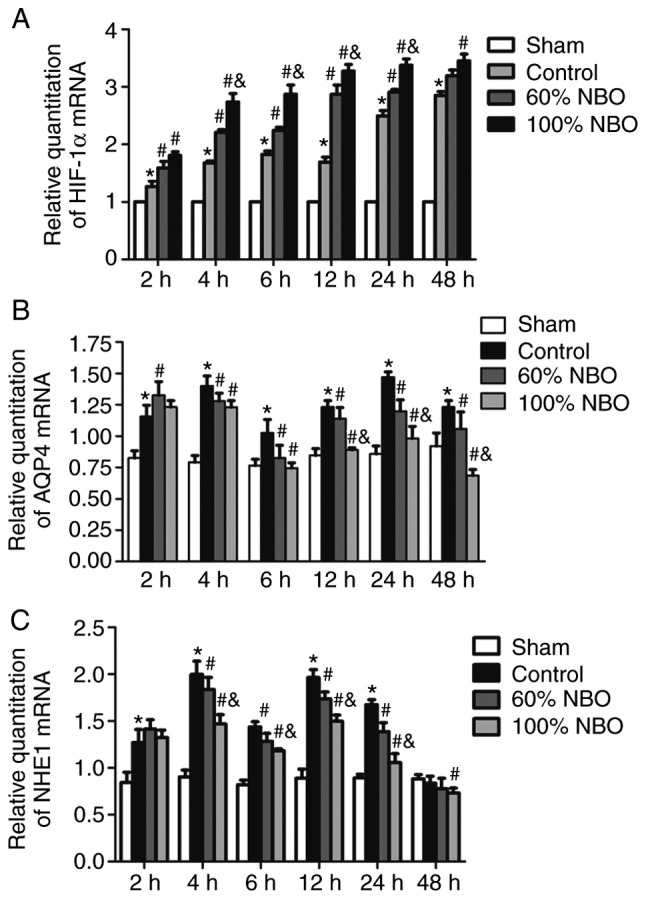
The expression of (A) HIF-1α, (B) AQP4 and (C) NHE1 mRNA in the control and NBO groups at: 2, 4, 6, 12, 24 and 48 h. Bonferroni test following repeated measures analysis of variance. Values are presented as the mean ± standard deviation, n=3 per group. *P<0.05 vs. the sham group; #P<0.05 vs. the control group; &P<0.05 vs. the 60% NBO group. NBO, nor-mobaric oxygen; HIF-1α, hypoxia-inducible factor-1α; AQP4, aquaporin-4; NHE1, Na+/H+ exchanger 1.
The AQP4 mRNA expression levels were also determined. The expression levels of AQP4 mRNA were significantly increased in the ischemic-control group, reaching a ~1.5-fold increase at the 2 h time point, with a peak at 4 and 24 h during reperfusion, compared with the sham group. The 60 and 100% NBO treatment significantly reduced AQP4 mRNA expression at ≥4 h of reperfusion following ischemia (P<0.05). Following 100% NBO treatment, the decrease of AQP4 mRNA expression was significantly enhanced at 12, 24 and 48 h, compared with the 60% NBO treatment group (P<0.05; Fig. 5B).
NHE1 mRNA expression levels were evaluated using RT-qPCR. The expression levels of NHE1 mRNA were significantly increased by ~40% at the 2 h in the ischemia-control group, compared with sham rats (P<0.05), and reached a ~2-fold increase at 4 and 12 h during the reperfusion. However, NHE1 mRNA levels demonstrated an opposite trend in analysis at 48 h. In the 60% NBO treatment groups, NHE1 mRNA exhibited a transient increase at 2 h and was then decreased by ~20% throughout the remaining recording period, compared with the control group. Following 100% NBO treatment, the decrease in NHE1 was reduced by a further 5-35% at ≥4 h, but was not statistically significant at 48 h, compared with the 60% NBO group (P<0.05; Fig. 5C).
Furthermore, immunofluorescence was performed to detect AQP4 and NHE1 co-expression with GFAP in astro-cytes from the experimental rats after 48 h of reperfusion. Expression of AQP4 and NHE1 with GFAP was observed in perivascular endothelial cells, ependymal cells and the end-feet of astrocytes in sham rats; whereas, expression was rarely detected in the cortical or subcortical regions of these rats (data not shown). The number of AQP4(+)/GFAP(+) and NHE1(+)/GFAP(+) perivascular endothelial cells was increased in the ischemic penumbra following ischemia and reperfusion. NBO treatment (60%) decreased the AQP4(+)/GFAP(+) and NHE1(+)/GFAP(+) expression in the demarcation zone (perile-sion and ischemic core), and the markers were decreased further in the 100% NBO treatment group, compared with the ischemia control rats (P<0.05; Figs. 6 and 7).
Figure 6.

Characterization of AQP4 expression in the astrocytic compartment of the control and NBO treatment rat brains at 48 h. Triple immunofluorescent staining of GFAP (green), AQP4 (40) and DAPI (blue). (A) The immunofluorescence of AQP4(+)/GFAP(+) expression in the control and NBO treatment groups. (B) The quantification of AQP4(+)/GFAP(+) immunofluorescence in the control and NBO treatment rats group (3-5 fields was observed). *P<0.05 vs. the control group; #P<0.001 vs. the 60% NBO group. Scale bars, 50 µm. Arrow head, co-expression of AQP4(+)/GFAP(+) astrocytes; NBO, normobaric oxygen; AQP4, aquaporin-4; GFAP, glial fibrillary acidic protein.
Figure 7.

Characterization of NHE1 in the astrocytic compartment and effect of NBO on NHE1 expression astrocytes activation at 48 h. Triple immunofluores-cent staining of GFAP (green), NHE1 (40) and DAPI (blue). (A) The immunofluorescence of NHE1(+)/GFAP(+) expression in the control and NBO treatment groups. (B) The quantification of NHE1(+)/GFAP(+) immunofluorescence in control and NBO treatment rat groups (3-5 fields was observed). *P<0.05 vs. the control group; #P<0.05 vs. the 60% NBO group; Scale bars, 50 µm. Arrow head, co-expression of NHE1(+) and GFAP(+) astrocytes. NBO, normobaric oxygen; NHE1, Na+/H+ exchanger 1; GFAP, glial fibrillary acidic protein.
Discussion
The primary observations of the present study included that exposure to 60 and 100% NBO reduced neurological impairment, BWC and infarct volume in rats following an acute ischemic stroke, and 100% NBO produced greater effects at the 4-24 h time points, compared with 60% NBO. Additionally, NBO treatment during reperfusion following ischemia upregulated HIF-1α expression and downregulated AQP4 and NHE1 expression, of which the latter are transporters predominantly localized to astrocytic end-feet at the brain vasculature (13). During reperfusion following ischemia, the expression of HIF-1α, AQP4 and NHE1 were increased significantly, compared with the expression in sham animals. Therefore, it may be possible to use these proteins as markers of the development of ischemic brain edema.
NBO treatment has been reported to reduce lesion volume, and improve functional outcomes in the acute and chronic stages of cerebral ischemia (2,4,5,19,20). In terms of ischemic cerebrovascular disease, blood circulation should be restored quickly to minimize the degree of damage (11,21). Neurocytic lesions induced by blood reperfusion injury do further harm to the ischemic brain (22). From the perspective of treatment, NBO treatment during reperfusion is worthy of further investigation, and numerous studies have used this model (23-26). Whether 60% is the optimal 'dose' of NBO remains unclear. Li et al (16) reported that 60% NBO decreases brain edema in MCAO rats at 24 h after occlusion, but there was no decrease in infarct size (the hemispheric volumes were 22±17, 27±18 and 31±22 mm3 in the 60, 40 and 100% NBO groups). Our previous study demonstrated that 60% NBO may inhibit the apoptotic pathway by further reducing caspase-3 and -9 expression compared with 33 and 45% NBO concentrations, following 72 h of reperfusion after 2 h of ischemia in a rat model (14). In the present study, 60% NBO did not improve NIS, or decrease infarct volume and brain edema compared with the higher concentration of NBO (100%). This finding is similar to a previous Indian trial that used 61% NBO (10 l/min for 12 h) for anterior circulation ischemic stroke patients within 12 h after stroke, which failed to improve clinical outcomes (27). This may be due to the differential effects of NBO duration and stroke severity on treatment outcomes.
The optimum duration of NBO treatment for stroke remains disputable. Early NBO (95% O2, 5% CO2 and 3 l/min) supplementation initiated at 15 min after MCAO and continued for 90 min does not protect the brain from vaso-genic edema following an acute ischemic stroke in adult male rats (28). NBO treatment for 120 min initiated at 30 min post-MCAO onset failed to significantly decrease infarct volume in rats with 150-min ischemia plus 7-day reperfu-sion (25). In the present study, NBO treatment for <2 h did not improve NIS score, or reduce BWC and infarct volume, and it may cause ischemic brain damage if initiated at <2 h post-ischemia in rats. In a previous study, NBO therapy (1 l/min pure oxygen) completely prevented sensorimotor deficits (P<0.02) and almost completely reversed selective neuronal loss following brief focal ischemia (5). In a stroke oxygen pilot study (20), neurological recovery at 1 week was more notably improved in the oxygen group (2 or 3 l/min for 72 h), compared with the controls, and following correction for differences in baseline stroke severity and prognostic factors, there was a trend for improved outcomes following oxygen treatment at 6 months. In the present study, it was determined that the longer the NBO treatment (within 48 h), the more that neurological impairment, BWC and infarct volume were decreased. Therefore, the effects of long NBO durations require further investigation.
The membrane protein AQP4 is expressed by perivascular astrocyte end-feet, ependymal glial cells and the glia limitary of the brain (29), and AQP4 is associated with BBB function (30). An increase in AQP4 may damage BBB integrity, leading to water entering brain tissues and resulting in brain edema; furthermore, AQP4 knockout has been reported to aggravate ischemia/reperfusion injury in mice (31). Cerebral edema is a major, potentially fatal complication of an acute ischemic stroke (32). Therefore, downregulation of AQP4 following cerebral ischemia may provide a therapeutic effect on brain edema. Previous studies reported that knockdown of AQP4 gene expression, or the use of an AQP4 inhibitor (TGN-020), ameliorates brain swelling and improves outcomes following a brain ischemia stroke (29,33). Hyperbaric oxygen preconditioning for 8 h per day for 3 or 7 days was demonstrated to increase AQP4 and vascular endothelial growth factor expression in cultured astrocytes to induce ischemic tolerance (34). However, in the present study, NBO reduced AQP4 expression in the ischemic brain, and the number of GFAP(+)/AQP4(+) cells was signifi-cantly reduced at the cortical border zone in the NBO treatment groups, compared with ischemia controls (Figs. 3 and 6). This indicates that the neuroprotective mechanism of NBO may be associated with AQP4 (+) astrocytes, and AQP4 may be a key marker and target for the development of edema.
NHE1, the major NHE protein isoform present in the brain, is crucial for H+ extrusion in exchange for Na+ in astrocytes, and is required for BBB function (35). NHE1 activity is also required for astrocytic homeostasis and correct brain functioning (13). Excessive NHE1 activation during hypoxia-ischemia results in intracellular Na+ overload, which subsequently promotes Ca2+ entry via reversal of the Na+/Ca2+ exchanger (36). The increased cytosolic Ca2+ then triggers a neurotoxic cascade (35,37). Genetic ablation of NHE1 or pharmacological inhibition with its potent inhibitor (HOE 642) protects against astrocyte damage and improves neurological function following an ischemic stroke (13,38). In the present study, NHE1 immunoreactivity was distributed throughout the brain tissue (data not shown), and strong signals were observed in the demarcation zone, as reported in previous studies (35,39-41). Additionally, NHE1 expression was signifi-cantly increased in ischemic brain tissue, compared with tissue from sham rats (Fig. 3). Treatment with NBO abolished the increase of NHE1 expression in astrocytes at 48 h (P<0.05; Fig. 7). Furthermore, the AQP4 overexpression was accompanied by an increase in NHE1 expression in the ischemic brain tissue. It can be hypothesized that NHE1 and AQP4 may cooperatively contribute to the pathogenesis of brain edema.
Collectively, the observations of the present study demonstrated that 60 and 100% NBO reduced neurological impairment, alleviated BWC and decreased the infarct volume in rats at 2-48 h following an acute ischemic stroke; whereas, 100% NBO induced further improvements in these parameters. NHE1, AQP4 and HIF-1α may cooperatively contribute to the pathogenesis of brain edema. NBO treatment suppressed NHE1 expression, which may decrease Na+ influx, resulting in the movement of water through AQP4 to reduce brain edema. The present study described a potential novel mechanism of NBO implementation during clinical practice in patients who experienced an ischemic stroke, and the appropriate concentration and duration requires further investigation.
A number of limitations of the present study should be noted. The short-term (<48 h) benefits of NBO were demonstrated; however, long-term effects, such as at 14 and 28 days post-injury, were not observed. Additionally, the side effects of high-dose NBO on edema formation and BBB disruption following ischemia-reperfusion were not investigated. Although the observations indicate that a molecular pathway is potentially involved in the protective effect of oxygen therapy, a causal association between oxygen therapy and modulation of NHE1, AQP4 and HIF-1α activation was not established. Furthermore, as the major channel proteins expressed in astrocytes, the interactions between NHE1 and AQP4 were not investigated.
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
DY designed the experiments and drafted the manuscript. LM contributed to designing the experiment and critical revised the manuscript. PW performed the histological examination of the ischemic rat brain. DY conducted the western blot analysis experiment. YZ created the MCAO model and protected the rats throughout the experiment. XZ conducted the immu-nofluorescence experiment. JL conducted the RT-qPCR experiment. JZ contributed to data acquisition and analysis. ZZ contributed to the conception of the experiments. FG assisted with performing the analysis with constructive discussions. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All procedures were approved by Zhengzhou University Animal Care and Use Committee (Zhengzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Jayakumar T, Hsu WH, Yen TL, Luo JY, Kuo YC, Fong TH, Sheu JR. Hinokitiol, a natural tropolone derivative, offers neuro-protection from thromboembolic stroke in vivo. Evid Based Complement Alternat Med. 2013;2013:840487. doi: 10.1155/2013/840487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Michalski D, Härtig W, Schneider D, Hobohm C. Use of normobaric and hyperbaric oxygen in acute focal cerebral ischemia-a preclinical and clinical review. Acta Neurol Scand. 2011;123:85–97. doi: 10.1111/j.1600-0404.2010.01363.x. [DOI] [PubMed] [Google Scholar]
- 3.Brainin M, Teuschl Y, Kalra L. Acute treatment and long-term management of stroke in developing countries. Lancet Neurol. 2007;6:553–561. doi: 10.1016/S1474-4422(07)70005-4. [DOI] [PubMed] [Google Scholar]
- 4.Singhal AB, Benner T, Roccatagliata L, Koroshetz WJ, Schaefer PW, Lo EH, Buonanno FS, Gonzalez RG, Sorensen AG. A pilot study of normobaric oxygen therapy in acute ischemic stroke. Stroke. 2005;36:797–802. doi: 10.1161/01.STR.0000158914.66827.2e. [DOI] [PubMed] [Google Scholar]
- 5.Ejaz S, Emmrich JV, Sitnikov SL, Hong YT, Sawiak SJ, Fryer TD, Aigbirhio FI, Williamson DJ, Baron JC. Normobaric hyperoxia markedly reduces brain damage and sensorimotor deficits following brief focal ischaemia. Brain. 2016;139:751–764. doi: 10.1093/brain/awv391. [DOI] [PubMed] [Google Scholar]
- 6.Singhal AB, Dijkhuizen RM, Rosen BR, Lo EH. Normobaric hyperoxia reduces MRI diffusion abnormalities and infarct size in experimental stroke. Neurology. 2002;58:945–952. doi: 10.1212/WNL.58.6.945. [DOI] [PubMed] [Google Scholar]
- 7.David HN, Haelewyn B, Degoulet M, Colomb DG, Jr, Risso JJ, Abraini JH. Prothrombolytic action of normobaric oxygen given alone or in combination with recombinant tissue-plasminogen activator in a rat model of thromboembolic stroke. J Appl Physiol. 2012;1985(112):2068–2076. doi: 10.1152/japplphysiol.00092.2012. [DOI] [PubMed] [Google Scholar]
- 8.Singhal AB, Wang X, Sumii T, Mori T, Lo EH. Effects of normobaric hyperoxia in a rat model of focal cerebral ischemia reperfusion. J Cereb Blood Flow Metab. 2002;22:861–868. doi: 10.1097/00004647-200207000-00011. [DOI] [PubMed] [Google Scholar]
- 9.Shi SH, Qi ZF, Luo YM, Ji XM, Liu KJ. Normobaric oxygen treatment in acute ischemic stroke: A clinical perspective. Med Gas Res. 2016;6:147–153. doi: 10.4103/2045-9912.191360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazdeh M, Taher A, Torabian S, Seifirad S. Effects of normobaric hyperoxia in severe acute stroke: A randomized controlled clinical trial study. Acta Med Iran. 2015;53:676–680. [PubMed] [Google Scholar]
- 11.Ji Z, Liu K, Cai L, Peng C, Xin R, Gao Z, Zhao E, Rastogi R, Han W, Rafols JA, et al. Therapeutic effect of tPA in ischemic stroke is enhanced by its combination with normobaric oxygen and hypothermia or ethanol. Brain Res. 2015;1627:31–40. doi: 10.1016/j.brainres.2015.08.019. [DOI] [PubMed] [Google Scholar]
- 12.Correia SC, Moreira PI. Hypoxia-inducible factor 1: A new hope to counteract neurodegeneration? J Neurochem. 2010;112:1–12. doi: 10.1111/j.1471-4159.2009.06443.x. [DOI] [PubMed] [Google Scholar]
- 13.Begum G, Song S, Wang S, Zhao H, Bhuiyan MIH, Li E, Nepomuceno R, Ye Q, Sun M, Calderon MJ, et al. Selective knockout of astrocytic Na+/H+ exchanger isoform 1 reduces astrogliosis, BBB damage, infarction, and improves neurological function after ischemic stroke. Glia. 2018;66:126–144. doi: 10.1002/glia.23232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen S, Peng H, Rowat A, Gao F, Zhang Z, Wang P, Zhang W, Wang X, Qu L. The effect of concentration and duration of normobaric oxygen in reducing caspase-3 and -9 expression in a rat-model of focal cerebral ischaemia. Brain Res. 2015;1618:205–211. doi: 10.1016/j.brainres.2015.05.027. [DOI] [PubMed] [Google Scholar]
- 15.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.STR.20.1.84. [DOI] [PubMed] [Google Scholar]
- 16.Li JS, Zhang W, Kang ZM, Ding SJ, Liu WW, Zhang JH, Guan YT, Sun XJ. Hyperbaric oxygen preconditioning reduces ischemia-reperfusion injury by inhibition of apoptosis via mitochondrial pathway in rat brain. Neuroscience. 2009;159:1309–1315. doi: 10.1016/j.neuroscience.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Sugimoto K, Nishioka R, Ikeda A, Mise A, Takahashi H, Yano H, Kumon Y, Ohnishi T, Tanaka J. Activated microglia in a rat stroke model express NG2 proteoglycan in peri-infarct tissue through the involvement of TGF-β1. Glia. 2014;62:185–198. doi: 10.1002/glia.22598. [DOI] [PubMed] [Google Scholar]
- 19.Esposito E, Mandeville ET, Hayakawa K, Singhal AB, Lo EH. Effects of normobaric oxygen on the progression of focal cerebral ischemia in rats. Exp Neurol. 2013;249:33–38. doi: 10.1016/j.expneurol.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singhal AB. A review of oxygen therapy in ischemic stroke. Neurol Res. 2007;29:173–183. doi: 10.1179/016164107X181815. [DOI] [PubMed] [Google Scholar]
- 21.Cai L, Stevenson J, Peng C, Xin R, Rastogi R, Liu K, Geng X, Gao Z, Ji X, Rafols JA, et al. Adjuvant therapies using normobaric oxygen with hypothermia or ethanol for reducing hyperglycolysis in thromboembolic cerebral ischemia. Neuroscience. 2016;318:45–57. doi: 10.1016/j.neuroscience.2016.01.010. [DOI] [PubMed] [Google Scholar]
- 22.He Q, Li Z, Wang Y, Hou Y, Li L, Zhao J. Resveratrol alleviates cerebral ischemia/reperfusion injury in rats by inhibiting NLRP3 inflammasome activation through Sirt1-dependent autophagy induction. Int Immunopharmacol. 2017;50:208–215. doi: 10.1016/j.intimp.2017.06.029. [DOI] [PubMed] [Google Scholar]
- 23.Zhao Q, Wang X, Chen A, Cheng X, Zhang G, Sun J, Zhao Y, Huang Y, Zhu Y. Rhein protects against cerebral ischemic/reperfusion-induced oxidative stress and apoptosis in rats. Int J Mol Med. 2018;41:2802–2812. doi: 10.3892/ijmm.2018.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, Zhou Y, Li H, Wang R, Yang D, Li B, Fu J. Intravenous administration of DPSCs and BDNF improves neurological performance in rats with focal cerebral ischemia. Int J Mol Med. 2018;41:3185–3194. doi: 10.3892/ijmm.2018.3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beynon C, Sun L, Marti HH, Heiland S, Veltkamp R. Delayed hyperbaric oxygenation is more effective than early prolonged normobaric hyperoxia in experimental focal cerebral ischemia. Neurosci Lett. 2007;425:141–145. doi: 10.1016/j.neulet.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 26.Tang X, Liu KJ, Ramu J, Chen Q, Li T, Liu W. Inhibition of gp91(phox) contributes towards normobaric hyperoxia afforded neuroprotection in focal cerebral ischemia. Brain Res. 2010;1348:174–180. doi: 10.1016/j.brainres.2010.05.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Padma MV, Bhasin A, Bhatia R, Garg A, Singh MB, Tripathi M, Prasad K. Normobaric oxygen therapy in acute ischemic stroke: A pilot study in indian patients. Ann Indian Acad Neurol. 2010;13:284–288. doi: 10.4103/0972-2327.74203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pasban E, Panahpour H, Vahdati A. Early oxygen therapy does not protect the brain from vasogenic edema following acute ischemic stroke in adult male rats. Sci Rep. 2017;7:3221. doi: 10.1038/s41598-017-02748-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Assentoft M, Larsen BR, MacAulay N. Regulation and function of AQP4 in the central nervous system. Neurochem Res. 2015;40:2615–2627. doi: 10.1007/s11064-015-1519-z. [DOI] [PubMed] [Google Scholar]
- 30.Sadana P, Coughlin L, Burke J, Woods R, Mdzinarishvili A. Anti-edema action of thyroid hormone in MCAO model of ischemic brain stroke: Possible association with AQP4 modulation. J Neurol Sci. 2015;354:37–45. doi: 10.1016/j.jns.2015.04.042. [DOI] [PubMed] [Google Scholar]
- 31.Tang G, Yang GY. Aquaporin-4: A potential therapeutic target for cerebral edema. Int J Mol Sci. 2016;17:E1413. doi: 10.3390/ijms17101413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kahle KT, Simard JM, Staley KJ, Nahed BV, Jones PS, Sun D. Molecular mechanisms of ischemic cerebral edema: Role of electroneutral ion transport. Physiology (Bethesda) 2009;24:257–265. doi: 10.1152/physiol.00015.2009. [DOI] [PubMed] [Google Scholar]
- 33.Katada R, Akdemir G, Asavapanumas N, Ratelade J, Zhang H, Verkman AS. Greatly improved survival and neuroprotection in aquaporin-4-knockout mice following global cerebral ischemia. FASEB J. 2014;28:705–714. doi: 10.1096/fj.13-231274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Kang K, Wang S, Yao J, Zhang X. Focal cerebral ischemic tolerance and change in blood-brain barrier permeability after repetitive pure oxygen exposure preconditioning in a rodent model. J Neurosurg. 2016;125:943–952. doi: 10.3171/2015.7.JNS142220. [DOI] [PubMed] [Google Scholar]
- 35.Uria-Avellanal C, Robertson NJ. Na(+)/H(+) exchangers and intracellular pH in perinatal brain injury. Transl Stroke Res. 2014;5:79–98. doi: 10.1007/s12975-013-0322-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leng T, Shi Y, Xiong ZG, Sun D. Proton-sensitive cation channels and ion exchangers in ischemic brain injury: New therapeutic targets for stroke? Prog Neurobiol. 2014;115:189–209. doi: 10.1016/j.pneurobio.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu H, Cala PM, Anderson SE. Na/H exchange inhibition protects newborn heart from ischemia/reperfusion injury by limiting Na+-dependent Ca2+ overload. J Cardiovasc Pharmacol. 2010;55:227–233. doi: 10.1097/FJC.0b013e3181cb599f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O'Donnell ME, Chen YJ, Lam TI, Taylor KC, Walton JH, Anderson SE. Intravenous HOE-642 reduces brain edema and Na uptake in the rat permanent middle cerebral artery occlusion model of stroke: Evidence for participation of the blood-brain barrier Na/H exchanger. J Cereb Blood Flow Metab. 2013;33:225–234. doi: 10.1038/jcbfm.2012.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishioka R, Sugimoto K, Aono H, Mise A, Choudhury ME, Miyanishi K, Islam A, Fujita T, Takeda H, Takahashi H, et al. Treadmill exercise ameliorates ischemia-induced brain edema while suppressing Na(+)/H(+) exchanger 1 expression. Exp Neurol. 2016;277:150–161. doi: 10.1016/j.expneurol.2015.12.016. [DOI] [PubMed] [Google Scholar]
- 40.Yuen N, Lam TI, Wallace BK, Klug NR, Anderson SE, O'Donnell ME. Ischemic factor-induced increases in cerebral microvascular endothelial cell Na/H exchange activity and abundance: Evidence for involvement of ERK1/2 MAP kinase. Am J Physiol Cell Physiol. 2014;306:C931–C942. doi: 10.1152/ajpcell.00021.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parmar S, Moore-Langston S, Fredrickson V, Kim JM, Rastogi R, Elmadoun O, Ding Y. Neuroprotective mechanisms of oxygen and ethanol: A potential combination therapy in stroke. Curr Med Chem. 2015;22:1194–1204. doi: 10.2174/0929867322666150114152945. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.