

Abstract
MicroRNAs (miRNAs) post-transcriptionally regulate gene expression by targeting the 3′ untranslated region (UTR) of target genes, and serve diverse roles in cell proliferation, differentiation and apoptosis. However, the association between miR-16-2-3p and 3-phosphoinositide-dependent protein kinase-1 (PDPK1) in nonsyndromic cleft lip (NSCL) remains unclear. In the present study, a luciferase activity assay indicated that miR-16-2-3p negatively regulated PDPK1 in maxillary primordium mesenchymal cells (MPMCs). In addition, it was confirmed that the expression levels of miR-16-2-3p was markedly increased in cleft lip tissues compared with those in adjacent normal lip tissues. A negative correlation between miR-16-2-3p and PDPK1 in cleft lip tissues was observed. Furthermore, miR-16-2-3p inhibited cell proliferation and migration, and induced apoptosis of MPMCs via repressing PDPK1. Finally, miR-16-2-3p exerted its suppressive role in MPMCs by inhibiting the PDPK1/protein kinase B signaling pathway. These results indicate that miR-16-2-3p may inhibit cell proliferation and migration, and promote apoptosis in MPMCs through repression of PDPK1 and may be a potential target for future clinical prevention and treatment of NSCL.
Keywords: microRNA-16-2-3p, nonsyndromic cleft lip, 3-phospho-inositide-dependent protein kinase-1, proliferation, migration, apoptosis, maxillary primordium mesenchymal cells
Introduction
Nonsyndromic cleft lip (NSCL) is one of most common birth defects that negatively affects facial appearance and lip function (1-3). Multiple factors have been demonstrated to contribute to the etiology of NSCL, including geographical distribution, ethnic background, economic status, genetics and environmental factors (4,5). Surgery is the primary treatment for NSCL (6-10), but identifying potential regulators of the underlying mechanisms of NSCL may aid in the development of novel treatment strategies.
MicroRNAs (miRNAs) are small non-coding RNAs consisting of ~22 nucleotides that post-transcriptionally regulate gene expression by targeting the 3′ untranslated region (UTR) of target genes. miRNAs also serve diverse roles in cell proliferation, differentiation and apoptosis (11-13). Accumulating evidence demonstrates that tissue miRNAs are involved in functional regulation of orofacial cleft development (14-16). However, the molecular mechanisms underlying the effects of miRNAs on NSCL have not been fully investigated.
We previously demonstrated that miRNA-16-2-3p (miR-16-2-3p) was markedly elevated in plasma samples from patients with NSCL compared with samples from patients without NSCL. Therefore, miR-16-2-3p may be regarded as a potential biomarker for diagnosing NSCL (17). 3-phos-phoinositide-dependent protein kinase-1 (PDPK1), a member of the AGC serine/threonine kinase family, has been established as critical in cell proliferation, apoptosis and invasion (18,19). However, the association between miR-16-2-3p and PDPK1 in NSCL remains unclear. Any factors inhibiting fusion of paired maxillary prominences mesenchyme, including inhibiting cell proliferation and migration, and inducing apoptosis of maxillary primordium mesenchymal cells, (MPMCs), or apoptosis of medial edge epithelium (MEE) vs. epithelial-to-mesenchymal transition (EMT) may ultimately result in cleft lip (20,21). Interactions between the neural crest, facial mesoderm, surrounding endoderm and ectoderm result in 5 facial prominences by embryonic day (E)9.5: The singular frontonasal prominence, paired maxillary prominences and paired mandibular prominences (22). Several studies have suggested that the upper lip forms from fusion between the maxillary and the medial nasal processes (23,24). Compared with other mesenchymal cells, MPMCs are involved in formation of maxillary primordium mesenchymal structures (25). Therefore, the present study was designed to examine the effect of miR-16-2-3p on MPMCs. miR-16-2-3p expression levels in tissue samples from 25 patients with NSCL were measured and its role in regulating cell proliferation, apoptosis, and migration in embryonic MPMCs was evaluated in vitro. Additional analysis indicated that PDPK1 was a direct downstream target of miR-16-2-3p.
Materials and methods
Tissue samples
Cleft lip tissue samples and adjacent normal lip tissue samples were surgically collected from 25 patients with NSCL (median age, 4 months; age range, 1-7 months; 15 males and 10 females), who underwent cleft lip repair operation in the Department of Burns and Plastic Surgery at Children's Hospital of Nanjing Medical University (Nanjing, China) from July 2016 to December 2017. None of the patients in the present study received any other therapies prior to surgery. All patients with NSCL were definitively diagnosed to exclude any other craniofacial deformities. All tissue samples were immediately frozen in liquid nitrogen and stored in −80°C until protein or RNA isolation. All study protocols were conducted subsequent to receiving consent from the parents of the patients with NSCL to use the tissue specimens for academic research purposes. The present study was approved by the Ethics Committee of the Children's Hospital of Nanjing Medical University.
RNA isolation and reverse transcription quantitative polymerase chain reaction (RT-qPCR)
Total RNA from tissue samples and cultured cells was extracted and isolated using TRIzol® Reagent (Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the manufacturer's protocol. RNA quantity was measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc., Wilmington, DE, USA). Total RNA was reverse-transcribed into cDNA using a PrimeScript® RT reagent kit (Takara Biotechnology, Co., Ltd., Dalian, China). qPCR was performed using the SYBR Green PCR master mix (Takara Biotechnology, Co., Ltd.) in 20 μl reactions on the Cobas z480 analyzer (Roche Molecular Diagnostics, Pleasanton, CA, USA). mRNA levels of miR-16-2-3p and PDPK1 were normalized to β-actin. The thermocycling conditions used were as follows: Initial denaturation at 95°C for 30 sec; followed by 40 cycles of denaturation at 95°C for 5 sec and annealing/extension at 60°C for 30 sec. All RT-qPCR reactions were performed in triplicate. Relative mRNA levels were calculated using the 2−ΔΔCq method (26). The primer sequences were as follows: miR-16-2-3p forward 5′-GGG ACC AAT ATT ATT GTG CTG C-3′ and reverse 5′-GCT GTC AAC GAT ACG CTA CGT AAC G-3′; PDPK1 forward 5′-CTG TAT GAC GCT GTG CCC ATT and reverse 5′-AAG GGG TTG GTG CTT GGT C; and β-actin forward 5′-GAG ACC TTC AAC ACC CCA GC and reverse 5′-GGG CAC GAA GGC TCA TCA TT.
MPMC culture and transfection
MPMC culture was performed according to a previously established protocol described by Zhao and Wang (27). C57BL/6 J mice were purchased from Shanghai Laboratory Animal Resource Center (Shanghai, China). Maxillary primordium tissues were separated from E12.5 mouse embryos and dissociated in 0.25% trypsin at 37°C for 30 min. Dissociated cells were transferred to a 6-well plate and cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.) containing 15% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.). The isolated MPMCs were continuously cultured in DMEM medium containing 10% FBS for 3-4 days. All experimental protocols were approved by the Animal Care and Use Committee of Nanjing Medical University (Nanjing, China).
MPMCs were transfected with precursor miR-16-2-3p (pre16-2-3p; Ambion; Thermo Fisher Scientific, Inc.), precursor negative control (NC) miRNA (preNC; Ambion; Thermo Fisher Scientific, Inc.), siRNA targeting miR-16-2-3p (si16-2-3p, Shanghai GenePharma Co., Ltd., Shanghai, China), or siRNA negative control (siNC; Shanghai GenePharma Co., Ltd.) at a concentration of 10 nmol/l, using Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. The expression plasmids of PDPK1 and NC for PDPK1 (Shanghai GenePharma Co., Ltd.) were transfected into MPMCs that had been transfected with pre16-2-3p, while siRNA targeting PDPK1 (siPDPK1; Shanghai GenePharma Co., Ltd.) and siNC (Shanghai GenePharma Co., Ltd.) were transfected into si16-2-3p-transfected-MPMCs using Lipofectamine® 2000. Following transfection for 48 h, MPMCs were harvested for subsequent assays. The siRNA sequences were as follows: 5′-AAA GCA GCA CAA UAA UAU UGG U-3′ for si16-2-3p; and 5′-GCA AGA CGA CGA CAG UUA UUA-3′ for siPDPK1; and 5′-CAG UAC UUU UGU GUA GUA CAA-3′ for siNC.
Luciferase activity assay
The binding sites between miR-16-2-3p and PDPK1 were predicted using TargetScan (http://www.targetscan.org). MPMCs were co-transfected with luciferase expressing pMIR-REPORT vector (Ambion; Thermo Fisher Scientific, Inc.) containing wild-type or mutated PDPK1 (Invitrogen; Thermo Fisher Scientific, Inc.) and miR-16-2-3p or preNC, respectively, using Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. Luciferase activity in different groups was measured 48 h post-transfection using the Dual Luciferase Reporter Assay System (Promega Corporation, Madison, WI, USA) according to the manufacturer's protocol, and the data were normalized to Renilla luciferase activity.
MTT assay
MPMCs proliferation was measured using an MTT assay. MPMCs were seeded into 96-well plates at 5,000 cells/well and cultured for 12, 24, 48, 72 and 96 h, respectively. Following the addition of 20 μl MTT solution (5 mg/ml), MPMCs were additionally cultured at 37°C for 4 h, then 200 μl dimethyl sulfoxide was added to dissolve the crystals. Absorbance at 450 nm was read on a microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
Flow cytometry assays
The FITC (fluorescein isothiocyanate) Annexin V Apoptosis Detection kit (BestBio Science, Shanghai, China) was used to detect apoptotic cells according to the manufacturer's protocol. Transfected MPMCs were seeded into 96-well plates at a density of 3x104 cells/well. Annexin V-FITC (5 μl) and propidium iodide (PI) (1 μg/ml) were added into cells, and incubated at room temperature for 15 min in the dark, then the cell apoptosis rate was processed using a flow cytometer. Flow cytometry results were analyzed using FlowJo software version 7.6.3 (TreeStar, Inc., Ashland, OR, USA).
Migration assays
A wound healing assay was used for the detection of the migration of cells in vitro. After 48 h of transfection of pre16-2-3p, si16-2-3p or their corresponding NCs, MPMCs were seeded into 6-well plates at 5×105 cells/well. Once the cells reached 90% confluence, the cell monolayer was scratched with a sterile yellow pipette tip (tip perpendicular to the surface). Following washing of the wells with PBS twice, the cells were additionally cultured at 37°C for 48 h. Images were captured at 0 and 48 h after the scratch wound using an inverted light microscope (magnification, ×100).
Cell cycle analysis
At 48 h after transfection, MPMCs were seeded into 96-well plates at a density of 3×104 cells/well. The cells were washed with pre-cooled PBS and fixed with 70% cold ethanol overnight at 4°C. Subsequently, MPMCs were treated with PI (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 20 min at room temperature and RNaseA (Sigma-Aldrich; Merck KGaA) for 30 min at room temperature. Following incubation, the percentage of cells in each cell cycle phase was analyzed using a flow cytometer and FlowJo software version 7.6.3 (Tree Star Inc.).
5-ethynyl-2'-deoxyuridine (EdU) assays
At 48 h after trans-fection, MPMCs were exposed to 10 mM EdU (Guangzhou RiboBio Co., Ltd., Guangzhou, China) for 24 h at 37°C. The cultured cells were then fixed with 4% paraformalde-hyde (PFA) for 30 min at room temperature and permeabilized with 0.5% Triton X-100. Next, the 1X Apollo reaction cocktail (Guangzhou RiboBio Co., Ltd.) was added to the cells and incubated for 30 min at room temperature. The cells were then stained using a Cell-Light™ EdU Apollo®488 in vitro imaging kit (Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. EdU-stained cells were observed using a fluorescence microscope (magnification, ×400; Nikon Corporation, Tokyo, Japan).
Western blot analysis
Frozen tissues or transfected cells were lysed in radioimmunoprecipitation assay lysis buffer (Beyotime Institute of Biotechnology, Haimen, China) and centrifuged at 12,700 × g for 20 min at 4°C. Protein concentration was measured using the Bradford assay (Bio-Rad Laboratories,Inc., Hercules, CA, USA). A total of 20 μg protein was separated by gel electrophoresis using 8% SDS-PAGE and then transferred to a 0.22 μm polyvinylidene difluoride membrane. Total protein was analyzed using immunoblot with the indicated antibodies following SDS-PAGE analysis. Following blocking of nonspecific binding with 5% non-fat milk in TBS containing 0.1% Tween-20 (TBS-T) at 25°C for 1 h, the membranes were incubated with the following primary antibodies: Anti-PDPK1 (cat. no. 3062; 1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA), anti-phosphorylated protein kinase B (p-Akt; cat. no. 2965S; 1:1000; Cell Signaling Technology, Inc.), anti-proliferating cell nuclear antigen (PCNA; cat. no. 2586S; 1:1,000; Cell Signaling Technology, MA, USA), anti-cleaved caspase-3 (cat. no. 9660; 1:1,000; Cell Signaling Technology, Inc.), anti-matrix metalloproteinase-9 (MMP-9; cat. no. 10375-2-AP; 1:2,000; Proteintech Group, Wuhan, China), anti-MMP-2 (cat. no. 10373-2-AP; 1:2,000; Proteintech Group), and anti-β-actin (cat. no. 10230-1-AP; 1:2,000; Proteintech Group). The membranes were incubated for 2 h at room temperature with a horseradish peroxidase-conjugated secondary antibody (cat. no. ab99697; 1:2,000; Abcam, Cambridge, MA, USA). Protein bands were visualized by enhanced chemiluminescence (Pierce; Thermo Fisher Scientific, Inc.). β-actin was used as an internal control.
Immunohistochemistry
PFA-fixed paraffin sections (4 μm) were mounted on slides, dewaxed with conventional xylene at 60°C and rehydrated with gradient alcohol for 5 min respectively. Slides were washed with PBS and boiled in 10 mM sodium citrate buffer (pH 6.0) for 2 min for antigen retrieval, cooled on the bench top for 30 min, treated with 3% H2O2 at 37°C for 15 min to block endogenous peroxidase activity, and then blocked with 20% goat serum (cat. no. ab138478; Abcam) in PBS-T for 1 h at room temperature. Samples were incubated with primary antibodies (anti-PDPK1; cat. no. ab52893; 1:50; Abcam) overnight at 4°C. Following washing with PBS-T, sections were incubated with an horseradish peroxidase-conjugated secondary anti-rabbit antibody for 20 min at room temperature, rinsed again with PBS-T, and incubated with Lab Vision™ polyvalent detection kit (Thermo Fisher Scientific, Inc.) and DAB [Tiangen Biotech (Beijing) Co., Ltd., Shanghai, China] for 10 min at room temperature. Slides were then observed under an inverted light microscope (magnification, ×400).
Statistical analysis
SPSS 19.0 (IBM Corp., Armonk, NY, USA) and GraphPad Prism 6.0 (GraphPad Software, Inc., La Jolla, CA, USA) were used for statistical analyses. The data are expressed as the mean ± standard deviation from three independent experiments. Unpaired Student's t-tests were used to analyze differences between two groups. Correlation between miR-16-2-3p and PDPK1 mRNA level in cleft lip tissues was determined using Pearson correlation analysis. P<0.05 was considered to indicate a statistically significant significance.
Results
miR-16-2-3p directly regulates the 3′-UTR of PDPK1
Using TargetScan, it was predicted that tmiR-16-2-3p targets the 3′-UTR of PDPK1 (Fig. 1A). To confirm this prediction, a dual-luciferase reporter assay was performed to determine if miR-16-2-3p interacts with the 3′-UTR of PDPK1. The position of the mutation in the PDPK1 3′-UTR is demonstrated in Fig. 1B. The miR-16-2-3p transfection efficiency in MPMCs was measured using RT-PCR. The relative luciferase activity in MPMCs that were co-transfected with pre16-2-3p and wild-type 3′-UTR PDPK1 was significantly suppressed compared with the activity in MPMCs co-transfected with pre16-2-3p and mutant-type3′-UTR PDPK1 (Fig. 1C). Taken together, these data indicate that miR-16-2-3p directly regulates PDPK1 expression via binding to its 3′-UTR.
Figure 1.

miR-16-2-3p directly regulates the 3′-UTR of PDPK1. (A) The miR-16-2-3p predicted binding sequence in the 3′-UTR of PDPK1. (B) The mutation introduced in the 3′-UTR of PDPK1. (C) miR-16-2-3p transfection efficiency was measured by reverse transcription quantitative polymerase chain reaction in MPMCs. The luciferase reporter gene assay was used to determine PDPK1 activity in MPMCs that were co-transfected with wild-type or mut 3′-UTR PDPK1 and pre16-2-3p. Independent experiments were repeated at least three times, and the values are expressed as the mean ± standard deviation. **P<0.01 vs. NC group. miR, microRNA; UTR, untranslated region; PDPK1, 3-phosphoinositide-dependent protein kinase-1; hsa, Homo sapiens; wild, wild-type; mut, mutant; NC, negative control.
miR-16-2-3p and PDPK1 expression in cleft lip tissues
miR-16-2-3p and PDPK1 expressions were measured in cleft lip and adjacent normal lip tissue samples. mRNA levels of miR-16-2-3p were significantly increased in cleft lip tissues compared with normal lip tissues, while PDPK1 mRNA levels were decreased in cleft lip tissues compared with adjacent normal lip tissues (Fig. 2A). Subsequent analysis indicated that miR-16-2-3p and PDPK1 expression were negatively correlated (n=25; r=−0.6436, P=0.0005; Fig. 2B). Consistent with the mRNA results, decreased PDPK1 protein expression levels were observed in cleft lip tissues, as determined by western blot analysis (Fig. 2C). Furthermore, the immuno-histochemical analysis of PDPK1 showed that cleft lip tissues exhibited lower membranous PDPK1 expression compared with adjacent normal lip tissues (Fig. 2D).
Figure 2.

miR-16-2-3p is upregulated in human cleft lip tissue samples. (A) Reverse transcription quantitative polymerase chain reaction was used to measure miR-16-2-3p and PDPK1 mRNA levels in cleft lip tissue samples and adjacent normal lip tissue samples. (B) Pearson analysis for the correlation of miR-16-2-3p and PDPK1 mRNA levels in cleft lip tissue samples and adjacent normal lip tissue samples (n=25; r=-0.6436; P=0.0005). (C) Western blot analysis of PDPK1 protein expression in cleft lip tissue samples and adjacent normal lip tissue samples. (D) Immunostaining of PDPK1 (magnification, x400). Independent experiments were repeated at least 3 times, and the values are expressed as the mean ± standard deviation. **P<0.01 and ***P<0.001 vs. normal samples. miR, microRNA; PDPK1, 3-phosphoinositide-dependent protein kinase-1.
Overexpression of miR-16-2-3p suppresses MPMC proliferation and induces cell cycle arrest at G1 phase
To determine the role of miR-16-2-3p in MPMCs, cells were transfected with pre16-2-3p or preNC (Fig. 3A). PDPK1 mRNA levels were significantly decreased in MPMCs transfected with pre16-2-3p compared with the preNC group (Fig. 3B). The changes in MPMC proliferation were measured using an MTT assay. Cell proliferation was significantly decreased in MPMCs transfected with pre16-2-3p compared with the NC (Fig. 3C). In addition, EdU staining demonstrated that proliferation was markedly decreased in pre16-2-3p-transfected cells compared with the NC (Fig. 3D). With regards to cell cycle distribution, a significant increase in the number of cells in G0/G1-phase and a significant decrease in the number of cells in S-phase were observed in the pre16-2-3p group compared with the preNC group (Fig. 3E).
Figure 3.

Overexpression of miR-16-2-3p suppresses MPMC proliferation and induces cell cycle arrest at G1 phase. Induction of miR-16-2-3p by pre16-2-3p transfection and relative expression levels of (A) miR-16-2-3p and (B) PDPK1 were determined by reverse transcription quantitative polymerase chain reaction. (C) MTT assay was performed to measure cell proliferation. (D) The number of Edu-stained cells was significantly decreased following pre16-2-3p transfection (magnification, x400). (E) Effect of miR-16-2-3p overexpression on cell cycle in MPMCs. Independent experiments were repeated at least 3 times, and the values are expressed as the mean ± standard deviation. *P<0.05 and **P<0.01 vs. preNC. miRNA, microRNA; MPMCs, maxillary primordium mesenchymal cells; preNC, precursor negative control; pre16-2-3p, precursor miR-16-2-3p; PDPK1, 3-phosphoinositide-dependent protein kinase-1; Edu, 5-ethynyl-2′-deoxyuridine; NC, negative control; OD, optical density.
miR-16-2-3p increases MPMC apoptosis
Flow cytometry analysis was performed to determine cell apoptosis among the different MPMC groups. Apoptosis was significantly increased in the pre16-2-3p group compared with the preNC group (Fig. 4A and B). Using western blot analysis, it was identified that miR-16-2-3p overexpression upregulated cleaved caspase-3 expression, additionally supporting the data suggesting that miR-16-2-3p regulates apoptosis (Fig. 4C).
Figure 4.

Effect of miR-16-2-3p on MPMC apoptosis. (A) Apoptosis in preNC and pre16-2-3p MPMC groups was assessed by flow cytometry analysis. (B) Quantification of the level of apoptosis in preNC and pre16-2-3p groups. (C) Western blot analysis of cleaved caspase-3 protein expression. Independent experiments were repeated at least 3 times, and the values are expressed as the mean ± standard deviation. **P<0.01 vs. preNC. miR, microRNA; MPMC, maxillary primordium mesenchymal cells; preNC, precursor negative control; pre16-2-3p, precursor miR-16-2-3p; NC, negative control.
miR-16-2-3p overexpression inhibits MPMC migration
To additionally investigate the role of miR-16-2-3p in cell migration, wound healing assays were performed on MPMCs transfected with pre16-2-3p or preNC for 48 h. The results demonstrated that MPMC migration was significantly decreased in the pre16-2-3p-transfected groups compared with the preNC group (Fig. 5A and B). Consistently, it was identified that MMP-9 and MMP-2 protein expression levels were decreased in MPMCs transfected with pre16-2-3p compared with the preNC group (Fig. 5C).
Figure 5.

Overexpression of miR-16-2-3p inhibits MPMC migration. (A) Migration capabilities of MPMCs that were transfected with pre16-2-3p or preNC were determined using a wound healing assay at 0 and 48 h (magnification, ×100). (B) Quantification of the wound healing assay data. (C) MMP-9 and MMP-2 protein expression was measured in MPMCs transfected with pre16-2-3p or preNC. Independent experiments were repeated at least 3 times, and the values are expressed as the mean ± standard deviation. **P<0.01 vs. preNC. miR, microRNA; MPMC, maxillary primordium mesenchymal cells; NC, negative control; MMP, matrix metalloproteinase.
miR-16-2-3p inhibition promotes MPMC growth
To investigate if miR-16-2-3p knockdown promoted MPMC growth, cells were transfected with either si16-2-3p or siNC, and the knockdown efficiency was measured using RT-qPCR (Fig. 6A). PDPK1 mRNA expression was significantly increased following knockdown of miR-16-2-3p (Fig. 6B). Cell proliferation, confirmed by MTT assay, was significantly increased in the si16-2-3p-transfected MPMCs compared with the siNC (Fig. 6C). Consistently, EdU staining demonstrated that miR-16-2-3p knockdown markedly increased MPMC growth (Fig. 6D). Flow cytometry analysis demonstrated that miR-16-2-3p inhibition significantly promoted the number of MPMCs in S phase (Fig. 6E).
Figure 6.

miR-16-2-3p inhibition promotes MPMC growth. (A) miR-16-2-3p and (B) PDPK1 mRNA levels were measured in MPMCs transfected with si16-2-3-p or siNC. (C) The MTT assay was used to measure cell proliferation. (D) The EdU assay was performed to determine cell proliferation (magnification, ×400). (E) Effects of miR-16-2-3p knockdown on the cell cycle in MPMCs as determined by flow cytometry analysis. Independent experiments were repeated at least 3 times, and the values are expressed as the mean ± standard deviation. *P<0.05 and **P<0.01 vs. siNC. miR, microRNA; MPMC, maxillary primordium mesenchymal cells; PDPK1, 3-phosphoinositide-dependent protein kinase-1; si, small interfering; Edu, 5-ethynyl-2'-deoxyuridine; NC, negative control; OD, optical density.
miR-16-2-3p knockdown decreases MPMC apoptosis
Flow cytometry analysis was performed to measure cell apop-tosis among the different MPMC groups. As indicated in Fig. 7A and B, miR-16-2-3p knockdown significantly inhibited MPMC apoptosis. Furthermore, western blot analysis revealed that miR-16-2-3p knockdown markedly decreased cleaved caspase-3 protein expression (Fig. 7C).
Figure 7.

miR-16-2-3p inhibition decreases MPMC apoptosis. (A) Flow cytometry analysis was performed to measure cell apoptosis in siNC and si16-2-3-p MPMC groups. (B) Quantification of the flow cytometry data. (C) Western blot analysis of cleaved caspase-3 protein expression. Independent experiments were repeated at least 3 times, and the values are expressed as the mean ± standard deviation. *P<0.05 vs. siNC. miR, microRNA; MPMC, maxillary primordium mesenchymal cells; si, small interfering; NC, negative control.
miR-16-2-3p knockdown promotes MPMC migration
Wound healing assays were performed to determine the effects of miR-16-2-3p knockdown on MPMC migration. si16-2-3p significantly increased cell migration compared with the siNC at 48 h (Fig. 8A and B). Furthermore, MMP-9 and MMP-2 protein expression levels were increased following miR-16-2-3p knockdown in MPMCs (Fig. 8C).
Figure 8.

miR-16-2-3p inhibition promotes MPMC migration. (A) The wound healing assay was used to measure cell migration in MPMCs transfected with si16-2-3p or siNC (magnification, x100). (B) Quantification of the wound healing assay data (C) MMP-9 and MMP-2 protein expression levels were determined in MPMCs transfected with si16-2-3p or siNC by western blot analysis. Independent experiments were repeated at least 3 times, and the values are expressed as the mean ± standard deviation. *P<0.05 vs. siNC. miR, microRNA; MPMC, maxillary primordium mesenchymal cells; si, small interfering; NC, negative control.
PDPK1 is the functional downstream target of miR-16-2-3p that regulates MPMC proliferation, cell cycle, apoptosis and migration
To investigate whether PDPK1 is a direct target of miR-16-2-3p involved in proliferation, apoptosis and migration, PDPK1 was overexpressed in pre16-2-3p-transfected MPMCs (Fig. 9A). It was identified that cell proliferation, the percentage of cells in S phase and cell migration increased significantly in the pre16-2-3p group compared with the preNC group (Fig. 9B-D). However, PDPK1 overexpression significantly inhibited cell apoptosis in MPMCs transfected with pre16-2-3p (Fig. 9E). Accordingly, it was identified that PDPK1 knockdown markedly inhibited cell proliferation, the percentage of cells in S phase and cell migration (Fig. 9A-D), but induced cell apoptosis in si16-2-3p-transfected MPMCs (Fig. 9E).
Figure 9.

miR-16-2-3p targets PDPK1 to regulate MPMC proliferation, cell cycle, apoptosis and migration. (A) PDPK1 protein expression measured by western blot analysis in pre16-2-3p-transfected-MPMCs followed by transfection of PDPK1 expression plasmid or NC, and in si16-2-3p-transfected-MPMCs followed by transfection with siPDPK1 or siNC. Altering PDPK1 expression partly abolished the functional effect of miR-16-2-3p on (B) proliferation, (C) cell cycle, (D) migration and (E) apoptosis in MPMCs. Independent experiments were repeated at least 3 times, and the values are expressed as the mean ± standard deviation. *P<0.05 and **P<0.01 vs. siNC. miR, microRNA; PDPK1, 3-phosphoinositide-dependent protein kinase-1; MPMC, maxillary primordium mesenchymal cells; pre16-2-3p, precursor miR-16-2-3p; si, small interfering; NC, negative control; OD, optical density.
miR-16-2-3p regulates the PDPK1/Akt signaling pathway in MPMCs
Previous studies demonstrated that the PDPK1/Akt pathway regulates cell proliferation, apoptosis and migration in various types of cancer (28,29). Therefore, the regulatory effect of miR-16-2-3p on this pathway was additionally investigated by measuring p-AKT, AKT, PDPK1 and PCNA expression. Overexpression of miR-16-2-3p markedly inhibited, and knockdown of miR-16-2-3p promoted, PDPK1, p-Akt and PCNA expression, whereas the Akt level was unaffected (Fig. 10A and B). These results suggest that miR-16-2-3p exerts it suppressive role in MPMCs by inhibiting the PDPK1/Akt signaling pathway.
Figure 10.
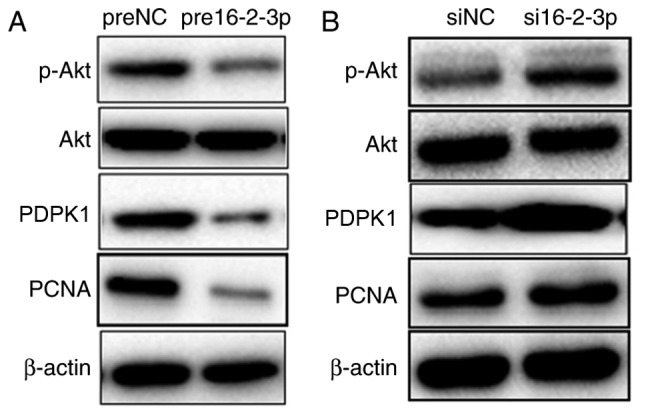
miR-16-2-3p regulates the PDPK1/Akt signaling pathway in MPMCs. At 72 h after transfection with (A) pre16-2-3p/preNC or (B) si16-2-3p/siNC, MPMCs were subjected to western blot analysis to detect PDPK1, p-Akt, total Akt and PCNA expression. Independent experiments were repeated at least 3 times. miR, microRNA; PDPK1, 3-phosphoinositide-dependent protein kinase-1; Akt, protein kinase B; MPMC, maxillary primordium mesenchymal cells; pre16-2-3p, precursor miR-16-2-3p; preNC, precursor negative control; si, small interfering; NC, negative control; p, phosphorylated; PCNA, proliferating cell nuclear antigen.
Discussion
NSCL is a common type of oral cleft defect that results in cleft palate or other developmental syndromes. Tissue fusion during lip development is an extremely complicated progress (22,30). Any factors inhibiting fusion of the paired maxillary prominences mesenchyme or apoptosis of MEE and EMT may cause NSCL (21,31,32).
Several miRNAs have been associated with NSCL (14,16). At present, a number of studies have identified that miR-16-2-3p is significantly upregulated in hepatocellular carcinoma (33) and multiple sclerosis (34). A decline in miR-16-2-3p expression was also confirmed in mandibular prognathism (35), suggesting that miR-16-2-3p suppresses cell proliferation and growth. Our previous study also showed that miR-16-2-3p maybe a potential biomarker for early detection of NSCL (17). However, the molecular mechanism by which miR-16-2-3p affects cell processes underlying NSCL development is not entirely clear.
Therefore, the aim of the present study was to explore the regulatory role of miR-16-2-3p in NSCL development. It was confirmed that miR-16-2-3p expression is markedly increased in cleft lip tissues compared with adjacent normal lip tissues in patients with NSCL. In addition, pre16-2-3p inhibited cell proliferation and migration and induced apoptosis of MPMCs via repressing PDPK1. By contrast, si16-2-3p promoted cell proliferation and migration and inhibited apoptosis of MPMCs. MMP-2 and MMP-9 are highly associated with cell migration and invasiveness, and numerous studies have demonstrated a positive correlation between cell migration and the expression of MMP-2 and MMP-9 (36,37). The protein levels of MMP-2 and MMP-9 were also detected, and the results were consistent with wound healing assay. Finally, it was demonstrated that miR-16-2-3p exerts its suppressive role by inhibiting the PDPK1/Akt signaling pathway in MPMCs.
Previously, PDPK1 has received considerable attention as a critical factor regulating cell proliferation, apoptosis and metastasis of various cancer cells (38-41). Furthermore, PDPK1 has been demonstrated as the functional downstream target of numerous miRNAs (42-45). As evidenced by the luciferase assay results in the present study, miR-16-2-3p significantly repressed PDPK1 expression through directly regulating the 3′-UTR of PDPK1. These data indicate that miR-16-2-3p negatively regulates PDPK1 in MPMCs. The mRNA and protein levels of PDPK1 were significantly decreased in cleft lip tissues compared with the adjacent normal lip tissues. Furthermore, a negative correlation between miR-16-2-3p and PDPK1 expression in cleft lip tissue samples was observed. The data also indicated that miR-16-2-3p inhibited MPMC proliferation and apoptosis. In addition, overexpression of PDPK1 reversed miR-16-2-3p-mediated repression of cell proliferation, migration and the miR-16-2-3p-mediated increase in MPMC apoptosis.
Several studies have demonstrated that activation of the PDPK1/Akt signaling pathway is essential for cell proliferation, migration, angiogenesis and metastasis in various types of cancer (46-48). Akt maybe phosphorylated at Thr308, which is primarily regulated by PDPK1 (49,50). In the present study, it was identified that overexpressing miR-16-2-3p inhibited the PDPK1 protein expression, resulting in downregulation of p-Akt in MPMCs. PCNA is essential for DNA replication, cell proliferation and cell cycle control, and serves as an index of cell proliferation (51,52). It was also identified that miR-16-2-3p overexpression decreased PNCA expression. By contrast, knockdown of miR-16-2-3p promoted PDPK1, p-Akt and PCNA expression. Based on these results, we hypothesized that miR-16-2-3p exerts it suppressive role in MPMCs by inhibiting the PDPK1/Akt signaling pathway.
Disruptions in the Akt-regulated pathways have been demonstrated to be associated with cancer, diabetes, cardiovascular and neurological diseases (53-55). Therefore, miR-16-2-3p has potential to be used in multiple therapeutic areas as an Akt inhibitor. Mammalian target of rapamycin (mTOR), which is implicated in different pathologies including cancer, autoimmune diseases and Human immunodeficiency virus (HIV) infection (56-59), is downstream of the Akt signaling pathway. It is necessary to investigate the regulation of the mTOR pathway by miR-16-2-3p in future studies, which may provide an underlying intervention for pathologies charac-terized from mTOR hyperactivation including cancer, multiple sclerosis and HIV infection. In addition, the pure inhibitors of Akt including afuresertib (60), or compounds blocking the PI3k-Akt/mTOR pathway including NVP-BEZ235 (61), may be considered for the pharmacological treatment of certain cases of NSCL. Notably, NSCL is developed in embryonic phase, but our previous and present studies also indicated that miR-16-2-3p from plasma samples and surgical specimens was associated with cleft lip formation. It indicates that miR-16-2-3p may serve a persistent biological role in different phase of patients with NSCL. Taken together, the present study identified that miR-16-2-3p upregulation may partially mitigate NSCL development. miR-16-2-3p inhibits cell proliferation and migration, and promotes apoptosis via directly targeting PDPK1 in MPMCs. Therefore, the results of the present study demonstrate that miR-16-2-3p may be a potential target for future clinical prevention and treatment of NSCL.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Science and Technique Development Foundation of Nanjing Medical University (grant. no., 2016NJMUZD058).
Availability of data and materials
The analyzed data sets generated during this study are available from the corresponding author on reasonable request.
Authors' contributions
JZ and WS planned the experiments and conducted the data analyses. TH and NW performed clinical sample preparation and expression analysis of miR-16-2-3p and PDPK1 in NSCL specimens. YW performed the in vitro assays and bioinfor-matics analyses. NW performed statistical analyses. TH wrote and edited the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study obtained consent from all parents of the patients from whom samples were collected. The study protocol was approved by the Ethics Committee of the Children's Hospital of Nanjing Medical University, and all experimental protocols were approved by the Animal Care and Use Committee of Nanjing Medical University.
Patient consent for publication
Written informed consent for the use of tissues for research and publication purposes was provided by each patient prior to the surgery.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Rahimov F, Jugessur A, Murray JC. Genetics of nonsyndromic orofacial clefts. Cleft Palate Craniofac J. 2012;49:73–91. doi: 10.1597/10-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vieira AR. Unraveling human cleft lip and palate research. J Dent Res. 2008;87:119–125. doi: 10.1177/154405910808700202. [DOI] [PubMed] [Google Scholar]
- 3.Sitzman TJ, Marcus JR. Cleft lip and palate: Current surgical management. Clin Plast Surg. 2014;41:xi–xii. doi: 10.1016/j.cps.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Noorollahian M, Nematy M, Dolatian A, Ghesmati H, Akhlaghi S, Khademi GR. Cleft lip and palate and related factors: A 10 years study in university hospitalised patients at Mashhad-Iran. Afr J Paediatr Surg. 2015;12:286–290. doi: 10.4103/0189-6725.172576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuchler EC, Silva LAD, Nelson-Filho P, Sabóia TM, Rentschler AM, Granjeiro JM, Oliveira D, Tannure PN, Silva RAD, Antunes LS, et al. Assessing the association between hypoxia during craniofacial development and oral clefts. J Appl Oral Sci. 2018;26:e20170234. doi: 10.1590/1678-7757-2017-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mulliken JB, Kim DC. Repair of bilateral incomplete cleft lip: Techniques and outcomes. Plast Reconstr Surg. 2013;132:923–932. doi: 10.1097/PRS.0b013e31829f4b81. [DOI] [PubMed] [Google Scholar]
- 7.Chen J, Shen W, Cui J. Modification of the rotation-advancement principle in cleft lip repair. J Craniofac Surg. 2009;20:2215–2216. doi: 10.1097/SCS.0b013e3181bf84c2. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Shen W, Cui J. A marking for moderate bilateral cleft lip. J Craniofac Surg. 2015;26:2022–2023. doi: 10.1097/SCS.0000000000001900. [DOI] [PubMed] [Google Scholar]
- 9.Chen J, Shen W, Jie C. Mild incomplete cleft lip repair. J Craniofac Surg. 2012;23:1131–1132. doi: 10.1097/SCS.0b013e31824e6477. [DOI] [PubMed] [Google Scholar]
- 10.Jianbing C, Shen W, Cui J. A marking for repair of complete cleft lip. J Craniofac Surg. 2014;25:1122–1124. doi: 10.1097/01.scs.0000436742.98159.68. [DOI] [PubMed] [Google Scholar]
- 11.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 12.Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 13.Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM. Bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113:25–36. doi: 10.1016/S0092-8674(03)00231-9. [DOI] [PubMed] [Google Scholar]
- 14.Kumari P, Singh SK, Raman R. A novel non-coding RNA within an intron of CDH2 and association of its SNP with non-syndromic cleft lip and palate. Gene. 2018;658:123–128. doi: 10.1016/j.gene.2018.03.017. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Zou J, Li Q, Chen L, Gao Y, Yan H, Zhou B, Li J. Assessment of differentially expressed plasma microRNAs in nonsyndromic cleft palate and nonsyndromic cleft lip with cleft palate. Oncotarget. 2016;7:86266–86279. doi: 10.18632/oncotarget.13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehrotra D. Genomic expression in non syndromic cleft lip and palate patients: A review. J Oral Biol Craniofac Res. 2015;5:86–91. doi: 10.1016/j.jobcr.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zou J, Li J, Li J, Ji C, Li Q, Guo X. Expression profile of plasma microRNAs in nonsyndromic cleft lip and their clinical significance as biomarkers. Biomed Pharmacother. 2016;82:459–466. doi: 10.1016/j.biopha.2016.05.033. [DOI] [PubMed] [Google Scholar]
- 18.Zheng N, Ding X, Sun A, Jahan R. PDK1 activity regulates proliferation, invasion and growth of hemangiomas. Cell Physiol Biochem. 2015;36:1903–1910. doi: 10.1159/000430159. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Yang C, Zhang L, Yang P. MicroRNA-210 induces endothelial cell apoptosis by directly targeting PDK1 in the setting of atherosclerosis. Cell Mol Biol Lett. 2017;22:3. doi: 10.1186/s11658-017-0033-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang R, Bush JO, Lidral AC. Development of the upper lip: Morphogenetic and molecular mechanisms. Dev Dyn. 2006;235:1152–1166. doi: 10.1002/dvdy.20646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnston MC, Bronsky PT. Prenatal craniofacial development: New insights on normal and abnormal mechanisms. Crit Rev Oral Biol Med. 1995;6:25–79. doi: 10.1177/10454411950060010301. [DOI] [PubMed] [Google Scholar]
- 22.Ray HJ, Niswander L. Mechanisms of tissue fusion during development. Development. 2012;139:1701–1711. doi: 10.1242/dev.068338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashique AM, Fu K, Richman JM. Endogenous bone morphogenetic proteins regulate outgrowth and epithelial survival during avian lip fusion. Development. 2002;129:4647–4660. doi: 10.1242/dev.129.19.4647. [DOI] [PubMed] [Google Scholar]
- 24.Cox TC. Taking it to the max: The genetic and developmental mechanisms coordinating midfacial morphogenesis and dysmorphology. Clin Genet. 2004;65:163–176. doi: 10.1111/j.0009-9163.2004.00225.x. [DOI] [PubMed] [Google Scholar]
- 25.Sun D, Baur S, Hay ED. Epithelial-mesenchymal transformation is the mechanism for fusion of the craniofacial primordia involved in morphogenesis of the chicken lip. Dev Biol. 2000;228:337–349. doi: 10.1006/dbio.2000.9946. [DOI] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 27.Zhao J, Wang R. Biologic characteristics and osteogenic differentiation of maxillary primordium mesenchymal cells. J Craniofac Surg. 2015;26:340–342. doi: 10.1097/SCS.0000000000001403. [DOI] [PubMed] [Google Scholar]
- 28.Yu HG, Ai YW, Yu LL, Zhou XD, Liu J, Li JH, Xu XM, Liu S, Chen J, Liu F, et al. Phosphoinositide 3-kinase/Akt pathway plays an important role in chemoresistance of gastric cancer cells against etoposide and doxorubicin induced cell death. Int J Cancer. 2008;122:433–443. doi: 10.1002/ijc.23049. [DOI] [PubMed] [Google Scholar]
- 29.Tsukamoto Y, Nakada C, Noguchi T, Tanigawa M, Nguyen LT, Uchida T, Hijiya N, Matsuura K, Fujioka T, Seto M, Moriyama M. MicroRNA-375 is downregulated in gastric carcinomas and regulates cell survival by targeting PDK1 and 14-3-3zeta. Cancer Res. 2010;70:2339–2349. doi: 10.1158/0008-5472.CAN-09-2777. [DOI] [PubMed] [Google Scholar]
- 30.Bush JO, Jiang R. Palatogenesis: Morphogenetic and molecular mechanisms of secondary palate development. Development. 2012;139:231–243. doi: 10.1242/dev.067082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu H, Ye X, Guo N, Nathans J. Frizzled 2 and frizzled 7 function redundantly in convergent extension and closure of the ventricular septum and palate: Evidence for a network of interacting genes. Development. 2012;139:4383–4394. doi: 10.1242/dev.083352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lidral AC, Murray JC, Buetow KH, Basart AM, Schearer H, Shiang R, Naval A, Layda E, Magee K, Magee W. Studies of the candidate genes TGFB2, MSX1, TGFA, and TGFB3 in the etiology of cleft lip and palate in the Philippines. Cleft Palate Craniofac J. 1997;34:1–6. doi: 10.1597/1545-1569_1997_034_0001_sotcgt_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Li T, Qiu Y, Zhang T, Guo P, Ma X, Wei Q, Han L. Serum microRNA panel for early diagnosis of the onset of hepa-tocellular carcinoma. Medicine (Baltimore) 2017;96:e5642. doi: 10.1097/MD.0000000000005642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keller A, Leidinger P, Steinmeyer F, Stähler C, Franke A, Hemmrich-Stanisak G, Kappel A, Wright I, Dörr J, Paul F, et al. Comprehensive analysis of microRNA profiles in multiple sclerosis including next-generation sequencing. Mult Scler. 2014;20:295–303. doi: 10.1177/1352458513496343. [DOI] [PubMed] [Google Scholar]
- 35.Gu Y, Zhang Y, Zhao C, Pan Y, Smales RJ, Wang H, Ni Y, Zhang H, Ni J, Ma J, Wang L. Serum microRNAs as potential biomarkers of mandibular prognathism. Oral Dis. 2014;20:55–61. doi: 10.1111/odi.12073. [DOI] [PubMed] [Google Scholar]
- 36.Foda HD, Zucker S. Matrix metalloproteinases in cancer invasion, metastasis and angiogenesis. Drug Discov Today. 2001;6:478–482. doi: 10.1016/S1359-6446(01)01752-4. [DOI] [PubMed] [Google Scholar]
- 37.Webb AH, Gao BT, Goldsmith ZK, Irvine AS, Saleh N, Lee RP, Lendermon JB, Bheemreddy R, Zhang Q, Brennan RC, et al. Inhibition of MMP-2 and MMP-9 decreases cellular migration, and angiogenesis in in vitro models of retinoblastoma. BMC Cancer. 2017;17:434. doi: 10.1186/s12885-017-3418-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Westmoreland JJ, Wang Q, Bouzaffour M, Baker SJ, Sosa-Pineda B. Pdk1 activity controls proliferation, survival, and growth of developing pancreatic cells. Dev Biol. 2009;334:285–298. doi: 10.1016/j.ydbio.2009.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wada M, Horinaka M, Yasuda S, Masuzawa M, Sakai T, Katoh N. PDK1 is a potential therapeutic target against angiosarcoma cells. J Dermatol Sci. 2015;78:44–50. doi: 10.1016/j.jdermsci.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 40.Gagliardi PA, di Blasio L, Primo L. PDK1: A signaling hub for cell migration and tumor invasion. Biochim Biophys Acta. 2015;1856:178–188. doi: 10.1016/j.bbcan.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 41.Lawlor MA, Mora A, Ashby PR, Williams MR, Murray-Tait V, Malone L, Prescott AR, Lucocq JM, Alessi DR. Essential role of PDK1 in regulating cell size and development in mice. EMBO J. 2002;21:3728–3738. doi: 10.1093/emboj/cdf387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie F, Huang Q, Liu CH, Lin XS, Liu Z, Liu LL, Huang DW, Zhou HC. miR-1271 negatively regulates AKT/MTOR signaling and promotes apoptosis via targeting PDK1 in pancreatic cancer. Eur Rev Med Pharmacol Sci. 2018;22:678–686. doi: 10.26355/eurrev_201802_14293. [DOI] [PubMed] [Google Scholar]
- 43.Wang Z, Yao YJ, Zheng F, Guan Z, Zhang L, Dong N, Qin WJ. Mir-138-5p acts as a tumor suppressor by targeting pyruvate dehydrogenase kinase 1 in human retinoblastoma. Eur Rev Med Pharmacol Sci. 2017;21:5624–5629. doi: 10.26355/eurrev_201712_14005. [DOI] [PubMed] [Google Scholar]
- 44.Wang J, Yang S, Ge W, Wang Y, Han C, Li M. miR-613 suppressed the laryngeal squamous cell carcinoma progression through regulating PDK1. J Cell Biochem. 2018;119:5118–5125. doi: 10.1002/jcb.26468. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Sun X. MicroRNA-375 inhibits the proliferation, migration and invasion of kidney cancer cells by triggering apoptosis and modulation of PDK1 expression. Environ Toxicol Pharmacol. 2018;62:227–233. doi: 10.1016/j.etap.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Fu L, Cui M, Wang Y, Xu Y, Li M, Mi J. Amino acid transporter SLC38A3 promotes metastasis of non-small cell lung cancer cells by activating PDK1. Cancer Lett. 2017;393:8–15. doi: 10.1016/j.canlet.2017.01.036. [DOI] [PubMed] [Google Scholar]
- 47.Sargeant AM, Klein RD, Rengel RC, Clinton SK, Kulp SK, Kashida Y, Yamaguchi M, Wang X, Chen CS. Chemopreventive and bioenergetic signaling effects of PDK1/Akt pathway inhibition in a transgenic mouse model of prostate cancer. Toxicol Pathol. 2007;35:549–561. doi: 10.1080/01926230701338966. [DOI] [PubMed] [Google Scholar]
- 48.Mo Y, Lu Y, Wang P, Huang S, He L, Li D, Li F, Huang J, Lin X, Li X, et al. Long non-coding RNA XIST promotes cell growth by regulating miR-139-5p /PDK1/AKT axis in hepatocellular carcinoma. Tumour Biol. 2017;39:1010428317690999. doi: 10.1177/1010428317690999. [DOI] [PubMed] [Google Scholar]
- 49.Manning BD, Cantley LC. AKT/PKB signaling: Navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jia S, Agarwal M, Yang J, Horowitz JC, White ES, Kim KK. Discoidin domain receptor 2 signaling regulates fibroblast apop-tosis through PDK1/Akt. Am J Respir Cell Mol Biol. 2018;59:295–305. doi: 10.1165/rcmb.2017-0419OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maga G, Hubscher U. Proliferating cell nuclear antigen (PCNA): A dancer with many partners. J Cell Sci. 2003;116:3051–3060. doi: 10.1242/jcs.00653. [DOI] [PubMed] [Google Scholar]
- 52.Wang LF, Chai CY, Kuo WR, Tai CF, Lee KW, Ho KY. The prognostic value of proliferating cell nuclear antigen (PCNA) and p53 protein expression in patients with advanced nasopharyngeal carcinoma. Acta Otolaryngol. 2006;126:769–774. doi: 10.1080/00016480500469545. [DOI] [PubMed] [Google Scholar]
- 53.Dai B, Wu Q, Zeng C, Zhang J, Cao L, Xiao Z, Yang M. The effect of Liuwei Dihuang decoction on PI3K/Akt signaling pathway in liver of type 2 diabetes mellitus (T2DM) rats with insulin resistance. J Ethnopharmacol. 2016;192:382–389. doi: 10.1016/j.jep.2016.07.024. [DOI] [PubMed] [Google Scholar]
- 54.Nitulescu GM, Van De Venter M, Nitulescu G, Ungurianu A, Juzenas P, Peng Q, Olaru OT, Grădinaru D, Tsatsakis A, Tsoukalas D, et al. The Akt pathway in oncology therapy and beyond (Review) Int J Oncol. 2018;53:2319–2331. doi: 10.3892/ijo.2018.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xue M, Chen X, Guo Z, Liu X, Bi Y, Yin J, Hu H, Zhu P, Zhuang J, Cates C, et al. L-Carnitine attenuates cardiac dysfunction by ischemic insults through Akt signaling pathway. Toxicol Sci. 2017;160:429. doi: 10.1093/toxsci/kfx211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Steelman LS, Martelli AM, Cocco L, Libra M, Nicoletti F, Abrams SL, McCubrey JA. The therapeutic potential of mTOR inhibitors in breast cancer. Br J Clin Pharmacol. 2016;82:1189–1212. doi: 10.1111/bcp.12958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Donia M, Mangano K, Amoroso A, Mazzarino MC, Imbesi R, Castrogiovanni P, Coco M, Meroni P, Nicoletti F. Treatment with rapamycin ameliorates clinical and histological signs of protracted relapsing experimental allergic encephalomyelitis in Dark Agouti rats and induces expansion of peripheral CD4+CD25+Foxp3+ regulatory T cells. J Autoimmun. 2009;33:135–140. doi: 10.1016/j.jaut.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 58.Nicoletti F, Lapenta C, Donati S, Spada M, Ranazzi A, Cacopardo B, Mangano K, Belardelli F, Perno C, Aquaro S. Inhibition of human immunodeficiency virus (HIV-1) infection in human peripheral blood leucocytes-SCID reconstituted mice by rapamycin. Clin Exp Immunol. 2009;155:28–34. doi: 10.1111/j.1365-2249.2008.03780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nicoletti F, Fagone P, Meroni P, McCubrey J, Bendtzen K. mTOR as a multifunctional therapeutic target in HIV infection. Drug Discov Today. 2011;16:715–721. doi: 10.1016/j.drudis.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 60.Kinoshita S, Ri M, Kanamori T, Aoki S, Yoshida T, Narita T, Totani H, Ito A, Kusumoto S, Ishida T, et al. Potent antitumor effect of combination therapy with sub-optimal doses of Akt inhibitors and pomalidomide plus dexamethasone in multiple myeloma. Oncol Lett. 2018;15:9450–9456. doi: 10.3892/ol.2018.8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chang Z, Shi G, Jin J, Guo H, Guo X, Luo F, Song Y, Jia X. Dual PI3K/mTOR inhibitor NVP-BEZ235-induced apoptosis of hepatocellular carcinoma cell lines is enhanced by inhibitors of autophagy. Int J Mol Med. 2013;31:1449–1456. doi: 10.3892/ijmm.2013.1351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The analyzed data sets generated during this study are available from the corresponding author on reasonable request.