

Abstract
The aim of the present study was to characterize the roles of two microRNAs (miRNAs), miR-122 and miR-199, in oral lichen planus (OLP). miRNA microarray analysis was performed to detect potential miRNAs involved in OLP, while in-silicon analysis, reverse transcription-quantitative polymerase chain reaction (RT-qPCR), western blot and immunohistochemistry (IHC) analyses were utilized to explore the molecular mechanisms underlying the roles of miR-199 and miR-122 in OLP. The results from the microarray and RT-qPCR analyses demonstrated that the expression levels of miR-122 and miR-199 were significantly decreased in the peripheral blood mononuclear cells (PBMCs) collected from the OLP group compared with the control group. In addition, miR-122 and miR-199 directly targeted AKT serine/threonine kinase 1 (AKT1) and mammalian target of rapamycin (mTOR), respectively, by binding to their 3′ UTRs. AKT1 and mTOR were highly expressed in PBMCs derived from OLP patients. In fact, a negative regulatory relationship was observed between miR-122 and AKT1, and between miR-199 and mTOR, with negative correlation coefficients of −0.41 and −0.51, respectively. Furthermore, the protein levels of AKT1, mTOR and microtubule associated protein 1 light chain 3β (LC3B) were upregulated in the OLP group compared with the control group. Finally, overexpression of miR-122 inhibited the expression of AKT1 and LC3B, while overexpression of miR-199 reduced the levels of mTOR and LC3B. In conclusion, the present study demonstrated that miR-199 and miR-122 are implicated in the pathogenesis of OLP by regulating the expression of mTOR and AKT1.
Keywords: miR-122, miR-199, AKT serine/threonine kinase 1, mammalian target of rapamycin, autophagy
Introduction
As one of most common immune-mediated mucocutaneous diseases, oral lichen planus (OLP) is characterized by chronic inflammation (1). The prevalence of OLP in the general population is ~1–2%, but its incidence among 30-60-year-old females is the highest (2). It has been demonstrated in previous investigations that, in activated T cells, autophagy is associated with the pathogenesis of some autoimmune diseases, via mediating cell survival, apoptosis, differentiation and proliferation (3). In fact, five differentially expressed autophagy-associated genes, autophagy-related 9B (ATG9B), estrogen receptor 1 (ESR1), synuclein α (SNCA), hepatocyte growth factor-regulated tyrosine kinase substrate (HGS), and insulin-like growth factor 1 (IGF1), have been identified from the peripheral blood T cells of OLP patients using autophagy arrays (4).
As a threonine/serine kinase, mammalian target of rapamycin (mTOR) acts as a central regulator of autophagy, cell survival, proliferation, growth and protein synthesis (5). In addition, mTOR complex 1 (mTORC1) acts as an inhibitor of autophagy, a process of cell survival and programmed catabolic activity that is initialized under nutrient-rich conditions and enhanced under cellular stress (6). Numerous cellular processes, such as cell cycle, glucose transport, proliferation, vesicle trafficking, cytoskeletal organization, and apoptosis, are regulated by AKT serine/threonine kinase (Akt; also known as protein kinase B) (7). It has been suggested that the phosphoinositide 3-kinase (PI3K)/Akt/mTOR pathway significantly affects the process of autophagy in mammalian cells by mediating anti-apoptotic signaling (8).
A previous study has demonstrated that the activation of the Akt/mTOR/pS6 signaling is frequently observed in malignant and premalignant oral lesions, but is only occasionally found in a subset of OLP cases (9). The correlation between the frequency of mTOR/pS6/Akt dysregulation and the clinical manifestation of OLP has drawn great interest, while the specific subtypes of OLP, such as atrophic, plaque-like, and/or erosive OLP, have been attributed to a higher rate of malignant transformation (10). Additionally, the expression of Akt and PI3K enzymes can trigger a cascade of pro-survival and pro-proliferative signals, whose transduction may affect the basal cells in OLP patients and promote their proliferation. In fact, it has been suggested that the above process can be induced by the secretion of RANTES, a chemokine expressed by T-cells during OLP, and the activation of normal T-cells. As a result, the extended life of these cells may cause dysplastic variations and may increase the risk of malignant transformation during OLP (11).
As single-strand noncoding RNAs with 19-22 nucleotides in length, microRNAs (miRNAs) can bind with partial complementarity to the 3′ untranslated region (UTR) of their target mRNAs and induce post-transcriptional silencing (12). The expression of multiple proteins can be regulated by a single miRNA, while several miRNAs can also regulate the expression of a specific protein. It has been demonstrated that miRNAs are highly conserved in the genome and are involved in many fundamental biological processes, including growth control, differentiation, development, apoptosis, autophagy, and tumorigenesis (13).
In the present study, samples from both OLP patients and normal controls were collected. Using miRNA microarrays, the miRNA expression profiles of the case and control groups were compared, and miR-122 and miR-199 were demonstrated to be significantly downregulated in the OLP group. By searching an online miRNA database, mTOR was predicted as a target of miR-199, and AKT1 was predicted as a target of miR-122. Both mTOR and AKT have been implicated in the control of autophagy, a major process underlying the pathogenesis of OLP (4,14,15). The present findings confirmed the regulatory relationship between miR-199 and mTOR, and between miR-122 and AKT1, in OLP.
Materials and methods
Samples
The present study was conducted according to the Declaration of Helsinki and approval was obtained from the Ethics and Research Committee of Ningbo No. 2 Hospital (Nigbo, China). All patients signed an informed consent form for study participation, after the details of the study were carefully explained to them. A total of 22 patients clinically diagnosed with OLP and 19 control subjects (normal tissue adjacent to cancer tissue) were recruited for the study at the Ningbo No. 2 Hospital from May 2017 to September 2017. The clinicopathological characteristics of the patients, including age, sex, clinical classification (atrophic, erosive, reticular and/or plaque-like OLP), and lesion site (buccal mucosa, buccal mucosa and gingiva), are listed in Table I. Patients with any systemic disease or receiving any systemic or topical medications during the three months prior to the start of the present study were excluded. In addition, patients with gingival inflammations or suspected restoration-related reactions were also excluded.
Table I.
Demographic and clinicopathological characteristics of subjects recruited in the study.
| Characteristic | OLP (n=22) | Control (n=19) | P-value |
|---|---|---|---|
| Age (years) | |||
| Mean ± SD | 45.5±15.8 | 43.7±10.7 | 0.814 |
| Range | 18-79 | 20-69 | |
| Sex | |||
| Female/male | 14/8 | 12/7 | 0.672 |
| Clinical classification | |||
| Erosive | 12 | - | |
| Reticular | 10 | - | |
| Location | |||
| Cheek | 13 | - | |
| Tongue | 8 | - | |
| Gingiva | 1 | - | |
Comparisons between the two groups were performed using the Chi-square test. No significant difference was observed with respect to patient characteristics, including age, sex, clinical classification and location.
miRNA microarray analysis
Microarray expression profiling was performed using 5 µg total RNA extracted from peripheral blood mononuclear cells (PBMCs) collected from 22 cases and 19 controls. MiRCURYTM Array Labeling kit (Exiqon, Vedbaek, Denmark) was utilized to label miRNAs fluorescently, and then miRNA microarray chips (Exiqon) were utilized to hybridize miRNAs. Hybridization data were collected using GenePix 4000B laser scanner (Axon Instruments, Foster City, CA, USA), and GenePix 4.0 software (Axon Instruments) was utilized to digitize and analyze the images.
RNA isolation and reverse transcription-quantitative poly- merase chain reaction (RT-qPCR)
TRIzol reagent (Thermo Fisher Scientific, Inc., Waltham, MA, USA) was utilized to isolate total RNA from THP-1 cells and U937 cells and tissue samples, following a standard protocol. Chloroform was then added into lysates and centrifuged at 4°C and 13,000×g for 15 min. Subsequently, isopropanol was used to treat the lysate and the pellet was washed with ethanol. Finally, the pellets were resus-pended in RNase-free water, and UV spectrophotometry was used to measure RNA concentration. A reverse transcription kit (Applied Biosystems; Thermo Fisher Scientific, Inc.) was used to synthesize cDNA from 2 µg of total RNA. A house-keeping gene, 18S RNA, was used as an internal control for normalizing the expression of miR-199 and miR-122, while GAPDH was used as an internal control for normalizing the expression of mTOR and AKT1. The sequences were as follows: 18S RNA forward, 5′-CAG CCA CCC GAG ATT GAG CA-3′ and reverse, 5′-TAG TAG CGA CGG GCG GTG TG-3′; miR-199 forward, 5′-ACA GTA GTC TGC ACA TTG GTTA-3′ and reverse, 5′-GCG AGC ACA GAA TTA ATA CGA C-3′; miR-122 forward, 5′-TGG AGT GTG ACA ATG GTG TTT G-3′ and reverse, 5′-GCG AGC ACA GAA TTA ATA CGA C-3′; mTOR forward, 5′-ACG CCT GCC ATA CTT GAG TC-3′ and reverse, 5′-TTG TGT CCA TCT TCT TG T CG-3′; AKT1 forward, 5′-CCT CCA CGA CAT CGC ACT G-3′ and reverse, 5′-TCA CAA AGA GCC CTC CAT TAT CA-3; and GAPDH forward, 5′-TGC ACC ACC AAC TGC TTA-3′ and reverse, 5′-GGA TGC AGG GAT GAT GTT-3′.
The thermocycling conditions were set as follows: 15 min at 95°C for initial activation, followed by 60 sec at 94°C for denaturation, 60 sec at 53°C for annealing, and 120 sec at 72°C for extension. A SYBR-Green PCR Master Mix (Applied Biosystems; Thermo Fisher Scientific, Inc.) was used to measure the expression levels of miR-199, miR-122, mTOR and AKT1. The relative expression levels of miR-199, miR-122, mTOR and AKT1 were determined by calculating the cycle threshold (Cq) values (16). Three independent reactions were performed.
Cell culture and transfection
THP-1 cells and U937 cells were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and cultured in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Inc.), 100 mg/ml streptomycin sulfate and 100 U/ml penicillin sodium. The cells were maintained in an incubator at 37°C and 5% CO2. Subsequently, lipopolysaccharide (LPS) was used to treat THP-1 and U937 cells. When the cells grew to 80% confluence, Lipofectamine 2000 (Thermo Fisher Scientific, Inc.) was used to transfect miR-199 mimics (5′-GCC AACC CAG UGU UCA GAC UAC CUG UUC AGG AGG CUC UCA AUG UGU ACA GUA GUC UGC ACA UUG GUU AGG C-3′) and miR-122 mimics (5′-CCU UAG CAG AGC UGU GGA GUG UGA CAA UGG UGU UUG UGU CUA AAC UAU CAA ACG CCA UUA UCA CAC UAA AUA GCU ACU GCU AGG C-3′) into LPS-treated THP-1 cells and U937 cells, following the protocol provided by the manufacturer. Each transfection was conducted using 30 nM of the target oligonucleotide and subsequent functional analyses were performed 48 h following transfection. Three independent experiments were performed.
Vector construction, mutagenesis and luciferase assay
A High-Capacity cDNA reverse transcription kit (Applied Biosystems; Thermo Fisher Scientific, Inc.) was used to synthesize mTOR and AKT1 cDNA containing miR-199 and miR-122 binding sites, respectively. Subsequently, the 3′-UTR of the target genes (mTOR or AKT1), and mutant (Mut) 3′-UTR fragments for both genes (as a negative control), were subcloned into pmiR-REPORT vectors (RiboBio Co., Ltd., Guangzhou, China) to generate luciferase reporter constructs. Following overnight culture, Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) was used to co-transfect THP-1 cells and U937 cells with mimic control (Ribobio Co., Ltd.) or miR-199/miR-122 mimics in conjunction with vectors carrying wild-type (WT)-mTOR/AKT1 3′-UTRs or Mut-mTOR/AKT1 3′-UTRs. A Dual-Luciferase Reporter Assay System (Promega Corporation, Madison, WI, USA) was used to measure the luciferase activity in the cells at 48 h post-transfection following the manufacturer's protocol. Renilla luciferase was used as the endogenous control. Each assay was repeated three times.
Western blot analysis
To analyze the expression of mTOR, AKT1 and microtubule associated protein 1 light chain 3β (LC3B) proteins, cells were lyzed with ice-cold lysis buffer (1% NP-40, 0.1% sodium dodecyl sulfate, 50 mM Tris-HCl pH 7.4, and 150 mM NaCl) supplemented with protease inhibitors (Roche Diagnostics). Total protein (35 µg as determined by a Bradford's assay) was separated on 8–12% SDS-PAGE, and electro-transferred to a nitrocellulose membrane (Bio-Rad Laboratories, Inc., Hercules, CA, USA) for 2 h at 90 V. PBS containing 5% nonfat dry milk was used to block the membrane for 60 min in order to eliminate non-specific binding. Subsequently, the membrane was incubated at 4°C overnight with polyclonal primary antibodies against mTOR (cat. no. sc-1550-R), AKT1 (cat. no. sc-81434), LC3B (cat. no. sc-271625, 1:5,000 dilution; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and β-actin (cat. no. sc-571582, 1:10,000 dilution; Santa Cruz Biotechnology, Inc.). After the membrane was washed twice with PBS, it was incubated at room temperature for 2 h with horseradish peroxidase (HRP)-conjugated secondary antibodies (cat. no. sc-51625, 1:12,000 dilution; Santa Cruz Biotechnology, Inc.). A chemiluminescent kit (Pierce; Thermo Fisher Scientific, Inc.) was used to visualize antigen-antibody complexes. All experiments were performed in triplicate.
Immunohistochemistry
Tongue tissue samples were cut into 4 µm sections and blocked with a blocking reagent (Protein Block Serum-Free; Dako; Agilent Technologies, Inc., Santa Clara, CA, USA) in an autoclave at 100°C for 30 min; 4% paraformaldehyde was used to perform fixation on the tissues collected (4°C for 2 h), which were later dehydrated and embedded in paraffin and sliced into 4 µm sections. Subsequently, the sections were incubated with rabbit anti-mTOR (cat. no. ab2732), anti-AKT1 (cat. no. ab81283), anti-p-AKT1 (cat. no. ab18206) and anti-LC3B monoclonal antibodies (cat. no. ab192890, 1:800 dilution; Abcam, Cambridge, MA, USA) at 4°C for 12 h, followed by 30 min of room temperature incubation with HRP-conjugated secondary antibodies (1:1,000 dilution; Histofine, Simple stain MAX-PO; Nichirei, Tokyo, Japan). A Dako REAL EnVision Detection System (Dako; Agilent Technologies, Inc.) was used to visualize the immune-complexes following the protocol from the manufacturer. Hematoxylin was used to counter-stain the cytoplasm. Samples were analyzed using an Olympus microscope (magnification, ×100; Olympus Corporation, Tokyo, Japan). ImageJ software (version 1.48; National Institutes of Health, Bethesday, MD, USA) to calculate the immunoreactivity score. All tests were run in triplicate.
Statistical analysis
All data were presented as mean ± standard deviation. SAS software version 6.1 (SAS institute, Inc., Cary, NC, USA) was used to perform all statistical analyses. Wilcoxon rank test or χ2 test were used to analyze the differences between treated and untreated groups. Continuous dependent variables between two groups were compared using an independent t-test. Correlation between groups was analyzed using Spearman correlation analysis. P<0.05 was considered to indicate a statistically significant difference.
Results
Characteristics of the participants
A total of 41 subjects, 22 OLP patients and 19 controls, were recruited for the present study. The demographic and clinicopathological characteristics of the participants, including age, sex, clinical classification (erosive, atrophic), and lesion location (cheek, tongue and gingiva), are summarized in Table I. A χ2 test was used to perform the statistical analysis between the two groups, and no difference was observed with respect to age and sex.
Identification of differentially expressed miRNAs
PBMC samples were collected from 22 OLP patients and 19 controls, and total RNA was extracted. To identify miRNAs that are potentially involved in the development of OLP, a microarray study was performed to compare the expression of different miRNAs between the two groups. The results identified 16 miRNAs (miR-223-3p, miR-186, miR-423, miR-181a, miR-155, miR-375, miR-133b, miR-497, miR-92, miR-199, miR-564, miR-1304, miR-296, miR-346, miR-19a and miR-122) as differentially expressed, and therefore these were selected for subsequent analyses. RT-qPCR was performed to confirm the microarray results. As presented in Fig. 1, the expression levels of miR-122 and miR-199 were decreased the most in the OLP group compared with the control group.
Figure 1.

Microarray and RT-qPCR analyses were used to identify potential miRNAs differentially expressed in OLP. The results revealed that miR-122 and miR-199 were significantly downregulated in OLP patients compared with healthy controls. RT-qPCR, reverse transcription-quantitative polymerase chain reaction; miRNA, microRNA; OLP, oral lichen planus (experiment was repeated three times).
miR-122 and miR-199 respectively target AKT1 and mTOR
The levels of miR-199 and miR-122 were compared between the PBMC samples from the OLP and the control groups, using RT-qPCR. As illustrated in Fig. 2A and B, the levels of miR-199 (Fig. 2A) and miR-122 (Fig. 2B) in the OLP group were significantly lower compared with the control group. In addition, in silicon analyses and luciferase assays were performed to explore the molecular mechanisms underlying the roles of miR-199 or miR-122 in the development of OLP. As illustrated in Fig. 2C, AKT1 was identified as a candidate target gene of miR-122, with a miR-122 binding site located in the AKT1 3′UTR. Results from the luciferase activity assay in cells co-transfected with miR-122 mimics and wild-type AKT1 3′UTR demonstrated that luciferase activity was significantly decreased (Fig. 2D), while the luciferase activity of cells co-transfected with miR-122 mimics and mutant AKT1 3′UTR had no obvious difference (Fig. 2D). Furthermore, the results of bioinformatics analyses predicted that miR-199 may directly bind to the mTOR 3′UTR (Fig. 2E). Results from the luciferase activity assay in cells co-transfected with miR-199 mimics and wild-type mTOR 3′UTR demonstrated that the luciferase activity was significantly decreased (Fig. 2D), while the luciferase activity of cells co-transfected with miR-199 mimics and mutant mTOR 3′UTR had no obvious difference. These results collectively suggested that miR-122 and miR-199 directly and negatively regulated the expression of AKT1 and mTOR, respectively.
Figure 2.

miR-122 and miR-199 directly target AKT1 and mTOR, respectively. (A) miR-122 levels (*P<0.05, vs. control group) and (B) miR-199 levels were lower in the OLP group compared with the control group (*P<0.05, vs. control group). (C) The AKT1 3′-UTR was predicted to contain a binding site for miR-122. (D) miR-122 inhibited the luciferase activity of wild-type AKT1 3′UTR, but did not affect the luciferase activity of mutant AKT1 3′UTR (*P<0.05, vs. miR cont group). (E) The mTOR 3′-UTR was predicted to contain a binding site for miR-199. (F) miR-199 inhibited the luciferase activity of wild-type mTOR 3′UTR, but did not affect the luciferase activity of mutant mTOR3′-UTR (*P <0.05, vs. miR cont group). AKT1, AKT serine/threonine kinase 1; mTOR, mammalian target of rapamycin; miR cont, control microRNA; OLP, oral lichen planus; UTR, untranslated region; WT, wild-type; MUT, mutant; cont, control; luc, luciferase.
Negative relationships of miR-122/AKT1 and miR-199/mTOR
RT-qPCR was performed to compare the mRNA levels of AKT1 and mTOR between OLP and control groups. As presented in Figs. 3A and B, the mRNA expression levels of AKT1 (Fig. 3A) and mTOR (Fig. 3B) were significantly increased in PBMCs derived from OLP patients compared with the control group. Subsequently, the miRNA/mRNA regulatory relationship between miR-122 and AKT1, as well as between miR-199 and mTOR, was evaluated by correlation analysis. According to the results, miR-122 and AKT1 (Fig. 3C) were negatively correlated. Similarly, miR-199 and mTOR (Fig. 3D) were also negatively correlated. Their negative correlation coefficients were -0.41 and -0.51, respectively.
Figure 3.

Negative relationships between miR-122/AKT1 and miR-199/mTOR. (A) mRNA levels of AKT1 (*P<0.05, vs. control group) and (B) mTOR in the OLP and control groups (*P<0.05, vs. control group). (C) Spearman correlation analysis for miR-122 and AKT1 and for (D) miR-199 and mTOR relative expression levels. AKT1, AKT serine/threonine kinase 1; mTOR, mammalian target of rapamycin; OLP, oral lichen planus.
Differential protein expression of AKT1, mTOR and LC3B between OLP and control groups
Tongue tissue samples were collected from 22 OLP patients and 19 controls and analyzed by immunohistochemistry to compare their protein levels of AKT1, mTOR and LC3B. As illustrated in Fig. 4, the protein levels of AKT1 in the OLP group were markedly increased compared with the control group. Similarly, the protein levels of mTOR (Fig. 5) and of LC3B (Fig. 6) were significantly increased in the OLP group compared with the control group.
Figure 4.

Immunohistochemistry analysis of AKT1 protein expression levels in tongue tissue samples from OLP and control subjects. (A) Representative images with positive staining indicated in brown. (B) Quantification of immunoreactivity score. AKT1, AKT serine/threonine kinase 1; OLP, oral lichen planus; (*P<0.05, vs. control group).
Figure 5.

Immunohistochemistry analysis of mTOR protein expression levels in tongue tissue samples from OLP and control subjects. (A) Representative images with positive staining indicated in brown. (B) Quantification of immunoreactivity score. mTOR, mammalian target of rapamycin; OLP, oral lichen planus; (*P<0.05, vs. control group).
Figure 6.

Immunohistochemistry analysis of LC3B protein expression levels in tongue tissue samples from OLP and control subjects. (A) Representative images with positive staining indicated in brown. (B) Quantification of immunoreactivity score. LC3B, microtubule associated protein 1 light chain 3β; OLP, oral lichen planus; (*P<0.05, vs. control group).
Confirmation of miR-122/AKT1 and miR-199/mTOR regulation
THP-1 cells (Fig. 7) and U937 cells (Fig. 8) were transfected with miR-122 mimics, miR-199 mimics, miR-122 mimics + miR-199 mimics, or miR-122 mimics + miR-199 mimics, respectively, and the levels of AKT1, mTOR and LC3B were detected using RT-qPCR and western blot analysis. As illustrated in Fig. 7 (THP-1 cells) and 8 (U937 cells), miR-122 mimics and miR-122 mimics + miR-199 mimics both significantly inhibited the mRNA and protein expression of AKT1. By contrast, miR-199 mimics alone had no effect on AKT1 expression. Additionally, miR-199 mimics and miR-122 mimics + miR-199 mimics both significantly decreased the mRNA and protein expression of mTOR (Figs. 7 and 8). By contrast, miR-122 mimics alone had no effect on mTOR expression. As illustrated in Fig. 7C (THP-1 cells) and 8C (U937 cells), LC3B expression was lower in the groups of miR-122 mimics and miR-199 mimics compared with the scramble control group. Of note, the LC3B levels in the group of miR-122 mimics + miR-199 mimics was the lowest compared with the other transfection groups.
Figure 7.
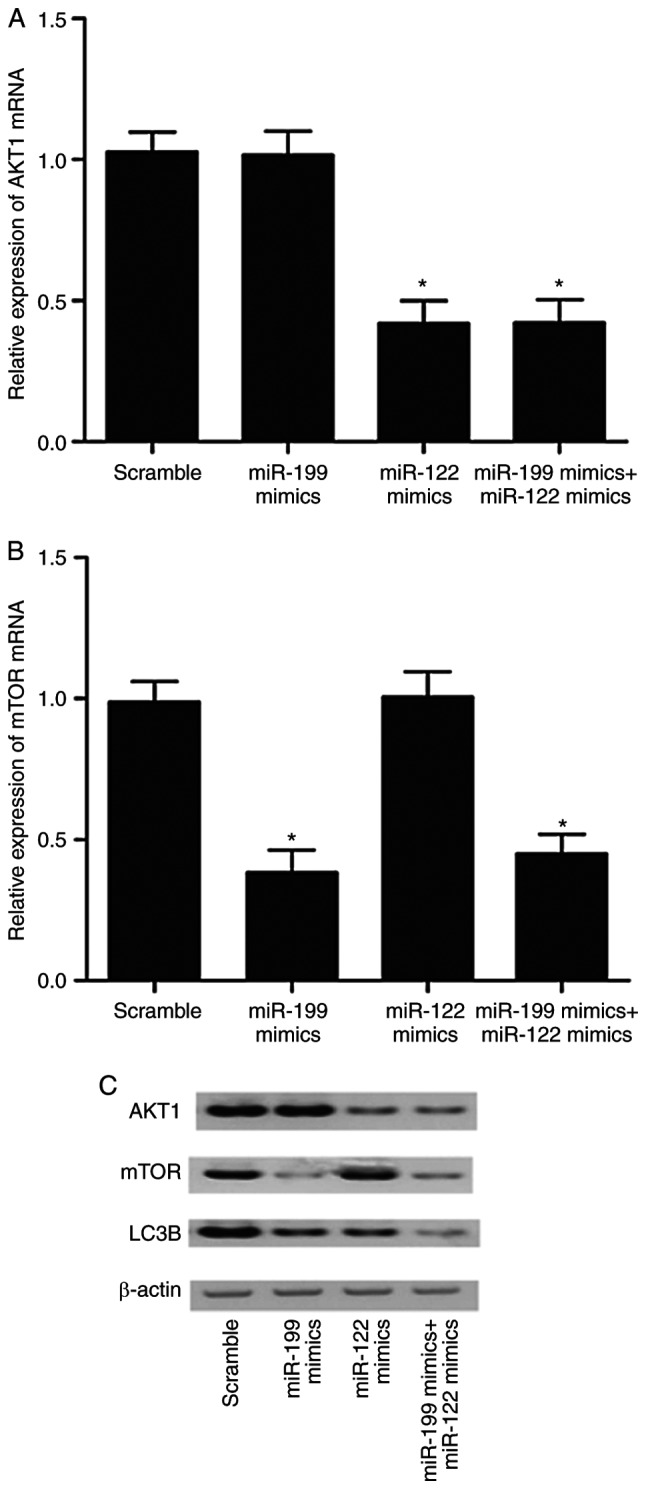
Effect of miR-122 and miR-199 overexpression on the levels of AKT1, mTOR and LC3B in THP-1 cells. THP-1 cells were transfected with either scramble control, miR-199 mimics alone, miR-122 mimics alone, or a miR-199 and miR-122 mimics combination. (A) mRNA levels of AKT1 (*P<0.05, vs. scramble control group). (B) mRNA levels of mTOR (*P<0.05, vs. scramble control group). (C) Protein levels of AKT1, mTOR and LC3B, as detected by western blotting. β-actin was used as an internal loading control. AKT1, AKT serine/threonine kinase 1; mTOR, mammalian target of rapamycin; LC3B, microtubule associated protein 1 light chain 3β.
Figure 8.
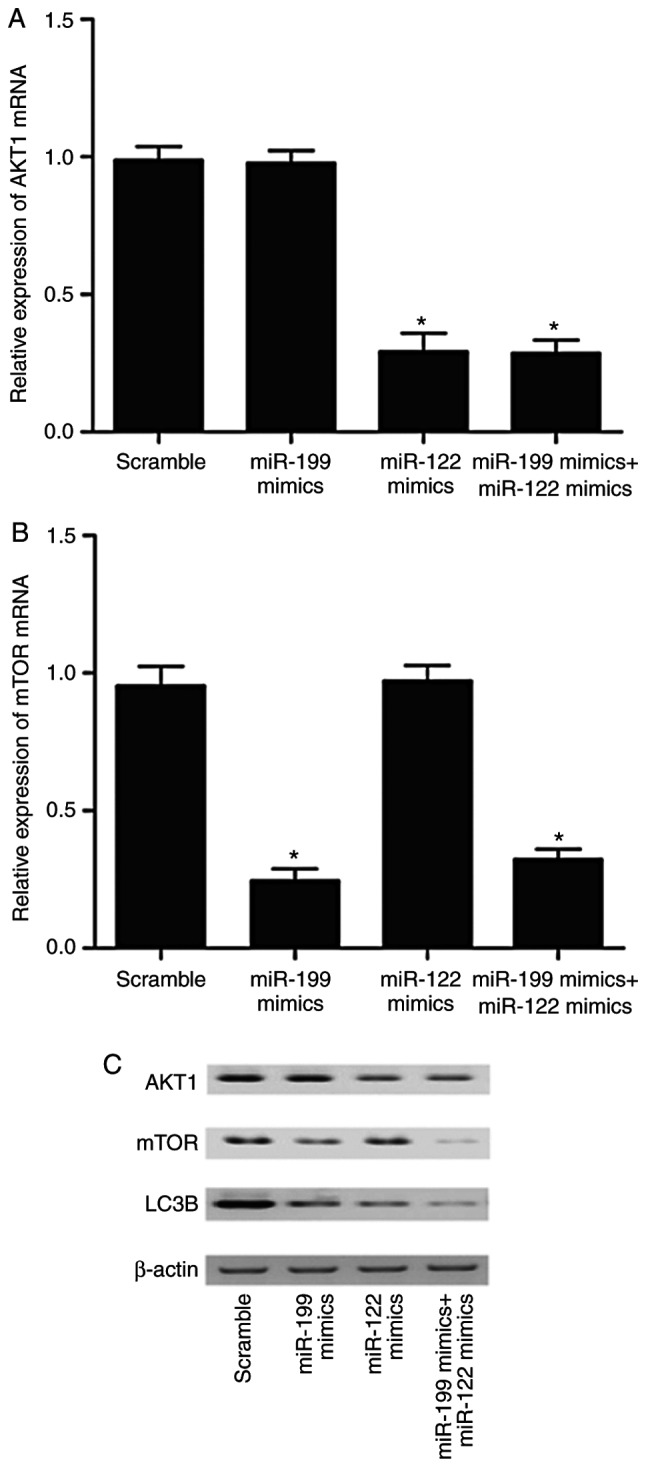
Effect of miR-122 and miR-199 overexpression on the levels of AKT1, mTOR and LC3B in U937 cells. U937 cells were transfected with either scramble control, miR-199 mimics alone, miR-122 mimics alone, or a miR-199 and miR-122 mimics combination. (A) mRNA levels of AKT1 (*P<0.05, vs. scramble control group). (B) mRNA levels of mTOR (*P<0.05, vs. scramble control group). (C) Protein levels of AKT1, mTOR and LC3B, as detected by western blotting. β-actin was used as an internal loading control. AKT1, AKT serine/threonine kinase 1; mTOR, mammalian target of rapamycin; LC3B, microtubule associated protein 1 light chain 3β.
Discussion
During OLP, T cells are triggered by antigens presented via major histocompatibility complex (MHC) class I and class II molecules (11). Autophagy is associated with many types of adaptive and innate immune processes, such as antigen processing for MHC presentation, lymphocyte function and development, inflammatory regulation, and pathogen recognition and destruction (17). Upon antigen recognition, T cells can induce autophagy to promote their own survival, apoptosis, differentiation or proliferation (18). Therefore, autophagy has become a fundamental event in regulating T cell-mediated immunity (18). In the present study, it was hypothesized that the autophagy of T cells is implicated in the immunopathogenesis of OLP.
The current study enrolled 41 subjects, consisting of 22 OLP patients and 19 controls, and performed miRNA microarrays and RT-qPCR to identify candidate miRNAs involved in OLP. The results demonstrated that miR-122 and miR-199 were significantly inhibited in the OLP group compared with the control group. Using a miRBase database, thirteen nucleotides were found to have a certain degree of complementarity to the sequences of mTOR and miR-199a-3p. In addition, it has been reported previously via luciferase reporter gene assays that miR-199a-3p can modulate cell proliferation by inhibiting mTOR expression (19). miR-122 also serves as a tumor suppressor and has a critical role in inhibiting the tumorigenesis of breast cancer by regulating the mTOR/p70S6K/PI3K/AKT pathway and by targeting IGF1 receptor (20). It has been demonstrated from in vivo studies that the angiogenesis and growth of xenograft tumors are suppressed by miR-122 (21). In addition, miR-122 suppresses angiogenesis by reducing the levels of AKT, mTOR and vascular endothelial growth factor C in tumor tissues (21). In the present study, using luciferase assays, it was confirmed that miR-122 and miR-199 directly targeted the expression of AKT1 and mTOR, respectively.
As a 289-kD threonine/serine multi-domain protein, containing a FKBP12 binding domain and a kinase domain, mTOR can regulate various physiological processes. For example, mTOR interacts with multiple upstream signal components, including PI3K/Akt, growth factors, insulin, glycogen synthase kinase 3 (GSK-3), and AMP-activated protein kinase (22). It has also been demonstrated in recent investigations that the dysregulation of mTOR is involved in many diseases, including cardiovascular disease, diabetes, cancer, obesity, aging and neurodegenerative diseases (23-28). In addition, the level of autophagy can be significantly enhanced by suppressing mTORC1, while the nutrient-insensitive mTOR can render cells unresponsive to starvation-induced autophagy (29). Furthermore, active mTOR can suppress autophagy by inhibiting the formation of ULK1/ATG13/FIP200 complexes, especially via the inhibitory phosphorylation of ULK1 (30). In the present study, we compared the protein levels of AKT1, mTOR and LC3B between OLP and control groups, and found that the protein levels of AKT1, mTOR and LC3B in the OLP group were significantly higher. Furthermore, RT-qPCR and western blot analyses were used to measure the expression of AKT1, mTOR and LC3B in cells transfected with miR-122 mimics, miR-199 mimics, and miR-122 mimics + miR-199 mimics. The results demonstrated that miR-122 negatively regulated the expression of AKT1 and LC3B, while miR-199 negatively regulated the expression of mTOR and LC3B.
Akt is a threonine/serine kinase member of the AGC family, that has important roles in cell survival, proliferation, growth and protein translation (31). Akt is recruited to the plasma membrane by phosphatidylinositol (3,4,5)-triphosphate (PIP3) upon the activation of PI3K. On the cell membrane, Akt is phosphorylated at sites Ser473 and Thr308 by mTORC2 and 3-phosphoinositide-dependent protein kinase-1 (PDK1), respectively (32). Once activated, Akt phosphorylates multiple targets located on the surface of the endoplasmic reticulum (ER) and in the nucleus, mitochondria and cytoplasm (33). The deregulation of the Akt pathway has been implicated in both human cancers and the development of cancer in mouse models (34). It has also been demonstrated that AKT activation reduces the excessive level of autophagy in cells by inducing autosis, the autophagy-dependent cell death (35). The present results confirmed previous findings, where it was demonstrated the basal autophagy in prostate cancer and glioma cells is suppressed by AKT (36). As a threonine/serine protein kinase, AKT acts as a critical mTOR regulator and is activated by growth factors and nutrients in a PI3K-dependent manner (5). Aberrations in the Akt/mTOR signaling pathway have been considered as one of the most important reasons leading to the pathogenesis of various types of human cancer, such as ovine squamous cell carcinoma (37). Therefore, it is reasonable to hypothesize that the activation of Akt/mTOR signaling in OLP patients may increase the premalignant potential of individuals (9). Previous studies have reported that the expression of matrix metallopeptidase (MMP)-2 is directly targeted by miR-125b in HaCaT cells. In addition, in an LPS-induced OLP model, the proliferation of keratinocytes is suppressed, while the apoptosis of keratinocytes is promoted by the mTOR/PI3K/Akt signaling pathway (38). Furthermore, Akt/mTOR signaling is a nutrient-sensitive pathway that can partially suppress autophagy by phosphorylating Unc-51 Like Autophagy Activating Kinase 1 (ULK1), a kinase involved in the initiation of autophagy, upon the activation by growth factors or under conditions with excessive nutrients (39). The expression of ULK1, LC3B, phosphorylated (p-) mTOR, and p-Akt is increased in lesion sites of OLP, indicating that the level of Akt/mTOR-induced autophagy is increased in OLP patients (15).
In conclusion, the present results demonstrated that mTOR and AKT were targets of miR-199 and miR-122, respectively. In addition, the AKT/mTOR signaling pathway was implicated in the regulation of autophagy, a major process underlying the pathogenesis of OLP. The present findings suggest that miR-122 and miR-199 may serve as potential therapeutic targets for OLP treatment.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Nature and Science Foundation of Ningbo (grant no. 2016A610147) and the Chinese Traditional Medicine Foundation of Zhejiang province (grant no. 2016ZA175).
Availability of data and materials
The analyzed datasets generated during the study are available from the corresponding author on reasonable request.
Authors' contributions
YC agreed to be accountable for all aspects of the study in ensuring that questions related to the accuracy or integrity of any part of the study are appropriately investigated and resolved. LW and YC made substantial contributions to the design of the present study. LW and WW, JC conducted the experimental study, acquired and analyzed the data. LW also prepared the manuscript with YL and MX, which was edited by YC. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The present study was approved by the Ethics and Research Committee of Ningbo No. 2 Hospital. All patients signed an informed consent form for study participation.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Farhi D, Dupin N. Pathophysiology, etiologic factors, and clinical management of oral lichen planus, part I: Facts and controversies. Clin Dermatol. 2010;28:100–108. doi: 10.1016/j.clindermatol.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Silverman S, Jr, Gorsky M, Lozada-Nur F. A prospective follow-up study of 570 patients with oral lichen planus: Persistence, remission, and malignant association. Oral Surg Oral Med Oral Pathol. 1985;60:30–34. doi: 10.1016/0030-4220(85)90210-5. [DOI] [PubMed] [Google Scholar]
- 3.Kovacs JR, Li C, Yang Q, Li G, Garcia IG, Ju S, Roodman DG, Windle JJ, Zhang X, Lu B. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ. 2012;19:144–152. doi: 10.1038/cdd.2011.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan YQ, Zhang J, Du GF, Lu R, Chen GY, Zhou G. Altered autophagy-associated genes expression in T cells of oral lichen planus correlated with clinical features. Mediators Inflamm. 2016;2016:4867368. doi: 10.1155/2016/4867368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 6.Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13:805–811. doi: 10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manning BD, Cantley LC. AKT/PKB signaling: Navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: Cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 9.Prodromidis G, Nikitakis NG, Sklavounou A. Immunohistochemical analysis of the activation status of the Akt/mTOR/pS6 signaling pathway in oral lichen planus. Int J Dent. 2013;2013:743456. doi: 10.1155/2013/743456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez-Moles MA, Scully C, Gil-Montoya JA. Oral lichen planus: Controversies surrounding malignant transformation. Oral Dis. 2008;14:229–243. doi: 10.1111/j.1601-0825.2008.01441.x. [DOI] [PubMed] [Google Scholar]
- 11.Roopashree MR, Gondhalekar RV, Shashikanth MC, George J, Thippeswamy SH, Shukla A. Pathogenesis of oral lichen planus-a review. J Oral Pathol Med. 2010;39:729–734. doi: 10.1111/j.1600-0714.2010.00946.x. [DOI] [PubMed] [Google Scholar]
- 12.Ambros V, Lee RC, Lavanway A, Williams PT, Jewell D. MicroRNAs and other tiny endogenous RNAs in C. elegans. Curr Biol. 2003;13:807–818. doi: 10.1016/S0960-9822(03)00287-2. [DOI] [PubMed] [Google Scholar]
- 13.Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 14.Rana S, Gupta R, Singh S, Mohanty S, Gupta K, Kudesia M. Localization of T-cell subsets in cutaneous lichen planus: An insight into pathogenetic mechanism. Indian J Dermatol Venereol Leprol. 2010;76:707–709. doi: 10.4103/0378-6323.72452. [DOI] [PubMed] [Google Scholar]
- 15.Zhang N, Zhang J, Tan YQ, Du GF, Lu R, Zhou G. Activated Akt/mTOR-autophagy in local T cells of oral lichen planus. Int Immunopharmacol. 2017;48:84–90. doi: 10.1016/j.intimp.2017.04.016. [DOI] [PubMed] [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 17.Münz C. Enhancing immunity through autophagy. Annu Rev Immunol. 2009;27:423–449. doi: 10.1146/annurev.immunol.021908.132537. [DOI] [PubMed] [Google Scholar]
- 18.Li C, Capan E, Zhao Y, Zhao J, Stolz D, Watkins SC, Jin S, Lu B. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol. 2006;177:5163–5168. doi: 10.4049/jimmunol.177.8.5163. [DOI] [PubMed] [Google Scholar]
- 19.Wu D, Huang HJ, He CN, Wang KY. MicroRNA-199a-3p regulates endometrial cancer cell proliferation by targeting mammalian target of rapamycin (mTOR) Int J Gynecol Cancer. 2013;23:1191–1197. doi: 10.1097/IGC.0b013e31829ea779. [DOI] [PubMed] [Google Scholar]
- 20.Wang B, Wang H, Yang Z. MiR-122 inhibits cell proliferation and tumorigenesis of breast cancer by targeting IGF1R. PLoS One. 2012;7:e47053. doi: 10.1371/journal.pone.0047053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Xing QF, Liu XQ, Guo ZJ, Li CY, Sun G. MiR-122 targets VEGFC in bladder cancer to inhibit tumor growth and angiogenesis. Am J Transl Res. 2016;8:3056–3066. [PMC free article] [PubMed] [Google Scholar]
- 22.O' Neill C. PI3-kinase/Akt/mTOR signaling: Impaired on/off switches in aging, cognitive decline and Alzheimer's disease. Exp Gerontol. 2013;48:647–653. doi: 10.1016/j.exger.2013.02.025. [DOI] [PubMed] [Google Scholar]
- 23.Chong ZZ, Shang YC, Maiese K. Cardiovascular disease and mTOR signaling. Trends Cardiovasc Med. 2011;21:151–155. doi: 10.1016/j.tcm.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Habib SL, Liang S. Hyperactivation of Akt/mTOR and deficiency in tuberin increased the oxidative DNA damage in kidney cancer patients with diabetes. Oncotarget. 2014;5:2542–2550. doi: 10.18632/oncotarget.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edlind MP, Hsieh AC. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J Androl. 2014;16:378–386. doi: 10.4103/1008-682X.122876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martínez-Martínez E, Jurado-López R, Valero-Muñoz M, Bartolomé MV, Ballesteros S, Luaces M, Briones AM, López-Andrés N, Miana M, Cachofeiro V. Leptin induces cardiac fibrosis through galectin-3, mTOR and oxidative stress: Potential role in obesity. J Hypertens. 2014;32:1104–1114. doi: 10.1097/HJH.0000000000000149. discussion 1114. [DOI] [PubMed] [Google Scholar]
- 27.Gouras GK. mTOR: at the crossroads of aging, chaperones, and Alzheimer's disease. J Neurochem. 2013;124:747–748. doi: 10.1111/jnc.12098. [DOI] [PubMed] [Google Scholar]
- 28.Sarkar S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: Autophagy dysfunction in neuro-degenerative diseases and therapeutic application of autophagy enhancers. Biochem Soc Trans. 2013;41:1103–1130. doi: 10.1042/BST20130134. [DOI] [PubMed] [Google Scholar]
- 29.Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. 2016;16:20–33. doi: 10.1038/nrc.2015.2. [DOI] [PubMed] [Google Scholar]
- 30.Chen J, Ghorai MK, Kenney G, Stubbe J. Mechanistic studies on bleomycin-mediated DNA damage: Multiple binding modes can result in double-stranded DNA cleavage. Nucleic Acids Res. 2008;36:3781–3790. doi: 10.1093/nar/gkn302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: Implications for therapeutic targeting. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- 32.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 33.Hosoi T, Hyoda K, Okuma Y, Nomura Y, Ozawa K. Akt up- and down-regulation in response to endoplasmic reticulum stress. Brain Res. 2007;1152:27–31. doi: 10.1016/j.brainres.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 34.Fayard E, Tintignac LA, Baudry A, Hemmings BA. Protein kinase B/Akt at a glance. J Cell Sci. 2005;118:5675–5678. doi: 10.1242/jcs.02724. [DOI] [PubMed] [Google Scholar]
- 35.Liu Y, Levine B. Autosis and autophagic cell death: The dark side of autophagy. Cell. Death Differ. 2015;22:367–376. doi: 10.1038/cdd.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Degtyarev M, De Mazière A, Orr C, Lin J, Lee BB, Tien JY, Prior WW, van Dijk S, Wu H, Gray DC, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysoso-motropic agents. J Cell Biol. 2008;183:101–116. doi: 10.1083/jcb.200801099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simpson DR, Mell LK, Cohen EE. Targeting the PI3K/AKT/mTOR pathway in squamous cell carcinoma of the head and neck. Oral Oncol. 2015;51:291–298. doi: 10.1016/j.oraloncology.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 38.Wang J, Luo H, Xiao Y, Wang L. miR-125b inhibits keratinocyte proliferation and promotes keratinocyte apoptosis in oral lichen planus by targeting MMP-2 expression through PI3K/Akt/mTOR pathway. Biomed Pharmacother. 2016;80:373–380. doi: 10.1016/j.biopha.2016.02.043. [DOI] [PubMed] [Google Scholar]
- 39.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The analyzed datasets generated during the study are available from the corresponding author on reasonable request.