

Abstract
In this trans-ethnic multi-omic study we reinterpret the genetic architecture of blood pressure to identify genes, tissues, phenome, and medication contexts of blood pressure homeostasis. We discovered 208 novel common blood pressure SNPs and 53 rare variants in GWASs of systolic, diastolic and pulse pressure in up to 776,078 participants from the Million Veteran Program (MVP) and collaborating studies, with analysis of the blood pressure clinical phenome in MVP. Our transcriptome-wide association study detected 4,043 blood pressure associations with genetically-predicted gene expression of 840 genes in 45 tissues, and murine renal single-cell RNA sequencing identified upregulated blood pressure genes in kidney tubule cells.
Editorial summary:
Analysis of blood pressure data from the Million Veteran Program trans-ethnic cohort identifies common and rare variants, and genetically predicted gene expression across multiple tissues associated with systolic, diastolic and pulse pressure in over 775,000 individuals.
Decades of scientific evidence implicate elevated blood pressure in the etiology of cardiovascular disease, including coronary artery disease, peripheral arterial disease, and stroke, as well as renal and ocular damage. Elevated blood pressure accounts for at least 13% of annual deaths worldwide1,2. The risk of death from ischemic heart disease and stroke increases linearly with systolic blood pressure (SBP) and diastolic blood pressure (DBP) elevations greater than 115 mmHg and 75 mmHg, respectively3. Recent treatment guidelines emphasize the benefit of blood pressure-lowering strategies, including drug treatments, at lower thresholds of SBP or DBP4. These guidelines also identify a substantial patient population who are untreated or undertreated for elevated blood pressure, or do not have sufficient treatment response to anti-hypertensive drugs and highlight the need to identify new gene targets for therapies5.
Large-scale genome-wide association studies (GWAS) have reported over 250 loci associated with blood pressure traits, establishing that blood pressure traits are complex with many genetic determinants of modest effect6–27. Large blood pressure GWAS meta-analyses combine evidence from many cohorts and identify genetic determinants of SBP, DBP and pulse pressure levels. Regulatory effects may account for substantial heritability in GWAS studies, and GWAS sentinel SNPs are enriched for regulatory SNPs compared to the proportion of the genome containing regulatory elements28–30. Most blood pressure SNPs are noncoding, are not in strong linkage disequilibrium (LD) with trait-associated coding variants, and are often found in regulatory elements12. Several methods were recently developed to use multiple SNPs to perform gene-based tests of association between imputed gene expression levels and phenotypes, which are tissue-specific and provide interpretable direction and magnitude of effects31–34
In this trans-ethnic study, we meta-analyzed data for 459,777 participants, 318,891 from the Million Veteran Program (MVP) and 140,886 from the UK Biobank (UKB)12. We subsequently performed independent replication in 316,301 participants from the International Consortium for Blood Pressure (ICBP)25 and Vanderbilt University’s BioVU cohort to study common variant associations with minor allele frequency (MAF) greater than 1% (Figure 1). With 318,891 participants from the MVP as the discovery sample, we conducted two studies of rare variants, one focused on variants across the genome with independent replication in 445,360 participants from UKB and the other focused on exonic regions with replication in up to 420,704 participants from the Blood Pressure-International Consortium of Exome chip studies (BP-ICE) consortium. We report associations between blood pressure and common and rare SNPs, and blood pressure and genetically predicted gene expression (GPGE). We also evaluated gene-drug relationships and toxicities, conducted a phenome-wide association study (PheWAS) of blood pressure genetic risk scores, conducted pathway and tissue gene set enrichment analyses, and performed studies to identify murine kidney cell types where expression for implicated genes is increased.
Figure 1. Study design schematic.
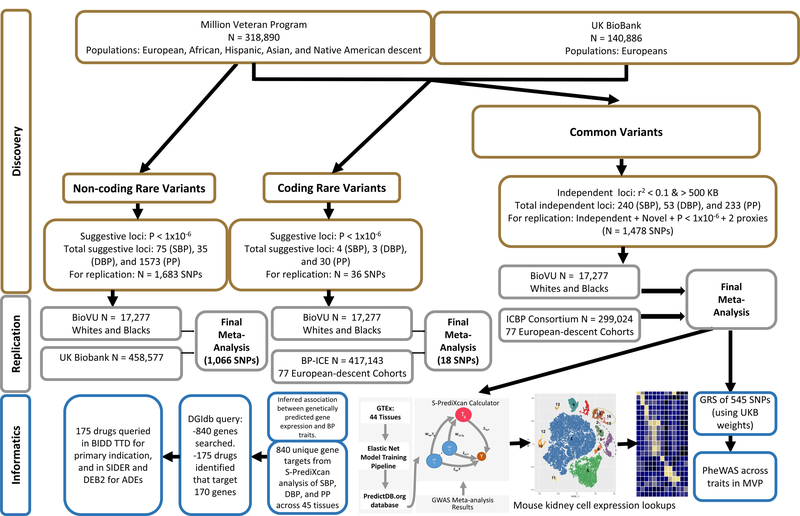
Flowchart depicting strategy for the three association analysis strategies (common, rare, and exonic variants), as well as replication selection criteria and numbers of samples and SNPs by stage. Subsequent TWAS and PheWAS analyses using common variant summary statistics are also presented. SBP, systolic blood pressure; DBP, diastolic blood pressure; PP, pulse pressure; SNP, single nucleotide polymorphism; ICBP, International Consortium for Blood Pressure; BP-ICE, Blood Pressure-International Consortium for Exomechip; BioVU, Vanderbilt University biorepository; ADE, adverse drug events
RESULTS
MVP participants (N = 318,891), representing the majority of the discovery sample size, were predominantly male (91.5%), and were administratively identified as non-Hispanic whites (69.1%), with non-Hispanic blacks, Hispanics, non-Hispanic Asians and non-Hispanic Native Americans representing 18.8%, 6.7%, 0.77% and 0.85% of the population respectively (Supplementary Table 1). Blacks were older on average [mean = 60.6 years, (standard deviation = 11.4)], followed by whites [58.9 (12.6)], Native Americans [58.9 (12.6)], Hispanics [52.7 (14.5)], and Asians [48.6 (16.1)]. Approximately half of the MVP participants were on antihypertensive medications and a quarter had diabetes. Participants from the UKB interim release, originating from UKB application number 236, (N = 140,886) were also included in the discovery analysis; their characteristics are reported elsewhere12.
Single Variant Analyses
Common Variants
We identified a total of 505 independent loci (201 novel loci, 304 previously reported) associated with one or more blood pressure traits: SBP, DBP and pulse pressure. Among the previously reported loci, 216 were associated with SBP, 76 with DBP and 208 with pulse pressure (Table 1; Figure 2a-c; Supplementary Tables 2a-c) and were not evaluated for replication. Sentinel variants from loci that were deemed to be novel by comparison with the GWAS catalog (accessed March 2017 or literature report; P-value < 1 × 10−6 in the discovery phase; greater than 500 kb from a known sentinel SNP; r2 ≤ 0.1 with known sentinel SNPs) and up to two proxies (N = 1,478) were carried forward for replication with the ICBP consortium. Replicated variants had consistent directions of effect in the discovery and replication phases, had P-values < 0.05 in the replication stage, and had meta-analysis (discovery + replication) P-values < 5 × 10−8 (Online methods for details). These replicated SNPs represented 201 novel loci and included 124 loci for SBP, 4 loci for DBP and 123 loci for pulse pressure (Supplementary Tables 3a-c). Comparison of mean effect estimates of blood pressure-trait increasing alleles showed that, on average, novel loci had smaller magnitudes of association (0.24, 0.14, and 0.18 mmHg per allele for SBP, DBP and pulse pressure, respectively) than known loci (0.32, 0.27, and 0.27 mmHg per allele, for SBP, DBP and pulse pressure, respectively; Table 1). Sentinel SNPs at all independent loci from meta-analysis of common variants explained 3.56%, 1.06% and 3.72% of the total variance for SBP, DBP and pulse pressure respectively. Novel variants contributed to 0.80%, 0.24% and 0.72% of the total variance explained by all independent loci for SBP, DBP, and pulse pressure, respectively.
Table 1.
Summary of known and novel loci achieving statistical significance from analysis of common variants.
| Known Loci |
Novel Loci |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| All novel | P-value** | Tier 1 | Tier 2 | Tier 3 | |||||||
| N Loci | Average Effect* (SD) | N Loci | Average Effect* (SD) | N Loci | Average Effect* (SD) | N Loci | Average Effect* (SD) | N Loci | Average Effect* (SD) | ||
| SBP | 216 | 0.32 (0.15) | 124 | 0.24 (0.12) | 1.03×10−7 | 26 | 0.29 (0.16) | 39 | 0.23 (0.12) | 59 | 0.22 (0.07) |
| DBP | 76 | 0.27 (0.15) | 4 | 0.14 (0.02) | 7.73×10−13 | 2 | 0.14 (0.02) | 0 | 0 | 2 | 0.14 (0.002) |
| PP | 208 | 0.27 (0.15) | 123 | 0.18 (0.09) | 9.57×10−12 | 21 | 0.19 (0.08) | 46 | 0.18 (0.09) | 56 | 0.17 (0.09) |
| Total | 304 | - | 201 | - | - | - | - | - | - | - | |
Known loci: known loci were only tested for significance in the discovery sample (N = 459,776). Novel loci: tested in discovery sample and ICBP (N-Discovery = 459,776; N-replication = 299,024; Total N = 758,800)
Mean beta = average and standard deviation of the absolute value of beta-estimates for each trait;
Represents P-value from two-sample t-test comparing mean beta for known loci and all novel loci. SBP = systolic blood pressure; DBP = diastolic blood pressure; PP = pulse pressure; Tier 1 = First tier significance criteria: GWAS significance at discovery + replication passing Bonferroni threshold + consistent directions of associations between discovery and replication sets + GWAS significant at final meta-analysis; Tier 2 = Second tier significance criteria: GWAS significance at discovery + replication P-value < 0.05 + consistent directions of associations between discovery and replication sets + GWAS significant at final meta-analysis; Tier 3 = Third tier significance criteria: Suggestive significance at discovery (P-value < 1 × 10−6 & > 5 × 10−8) + replication P-value < 0.05 + consistent directions of associations between discovery and replication sets + GWAS significant at final meta-analysis.
Figure 2. Manhattan plots summarizing discovery and replication meta-analysis for (a) SBP, (b) DBP, and (c) pulse pressure.
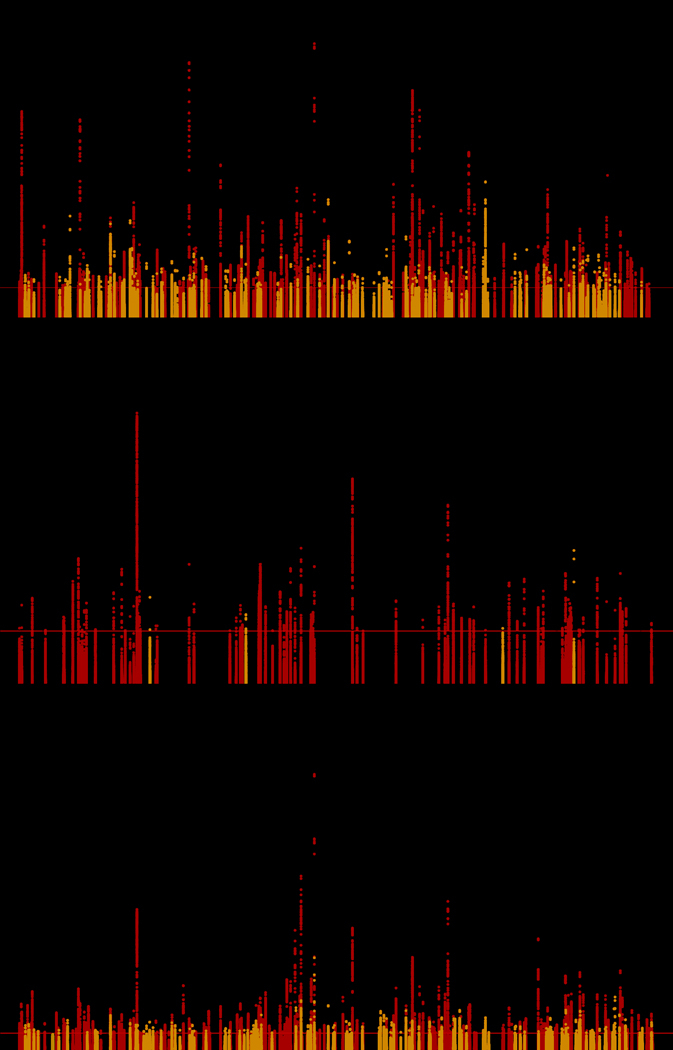
Manhattan plot of the discovery + replication meta-analysis. The y axis shows the –log10 P-values and the x axis shows the chromosomal positions. The horizontal red line represents the thresholds of P-value = 5 × 10−8 for genome-wide significance. SNPs in red are in previously identified loci (includes discovery only; Neff-max = 459,670 for SBP, 459,093 for DBP, and 459,305 for pulse pressure) whereas SNPs in orange are in novel loci (includes discovery + replication; Neff-max = 760,226 for SBP, 767,920 for DBP, and 759,768 for pulse pressure). All P-values are computed for associations between genotyped/imputed SNPs and blood pressure traits as dependent variables in multivariable adjusted logistic regression models. SBP, systolic blood pressure; DBP, diastolic blood pressure
Identification of conditionally independent signals using discovery meta-analysis results identified an additional 37 SNPs from 29 loci (7 novel, 3 within the boundaries of both known and novel loci) as significant in one or more blood pressure traits (Supplementary Table 4).
Trans-ancestry Comparisons
To update the observations reported by Franceschini et al. in 20139, where 29 SNP effects on blood pressure reported by Ehret et al. in 201135 were consistent across European, African, East Asian, and South Asian ancestries, we compared known and novel loci across ancestral groups in MVP. We examined the correlation between effect sizes in white, black, and Hispanic samples. Observed correlations of effect sizes between race/ethnic groups in MVP were weaker than those previously reported, though the directions of effects were largely consistent (Supplementary Figure 1; Supplementary Table 5).
Rare Exonic Variants
Rare exonic variants (MAF < 1%) with suggestive evidence of association (P-value < 1 × 10−6) from the discovery sample were queried for replication in populations from BioVU (N = 17,277) and the BP-ICE (Nmax = 420,704) consortium. Eighteen variants were on the exome chip and available for final meta-analysis. Ten missense variants from seven genes were associated with blood pressure traits (P < 5 × 10−8; Table 2). Five variants were associated with SBP and/or DBP (rs141328069 [PDE3A; Arg→Gln], rs139491786, [SLC9A3R2; Arg→Trp], rs61760904 [RRAS; Asp→Asn], rs73181210 [PHC3; Lys→Glu], rs3085380 [DBH; Gly → Ala]) with consistent directions of effect for SBP and DBP. Three rare variants from COL21A1 (rs118079907 [Cys→Arg], rs200999181 [Gly→Val], and rs2764043 [Leu→Pro]) and one variant from NOX4 (rs139341533; Leu→Phe) were significantly associated with pulse pressure but not with SBP or DBP and in fact had opposite directions effect for SBP and DBP. SNPs in RRAS, DBH, and one of the three SNPs in COL21A1 (rs200999181) have been previously reported22–24. Average absolute values of effect estimates for SBP, DBP and pulse pressure in these variants were 1.52, 0.63, and 1.50 mmHg per allele, respectively.
Table 2.
Associations between missense variants identified in collaboration with consortia evaluating exonic variants and rare-variants.
| SNP | Chr:BP | Gene | Amino Acid Change | EA/RA | EAF | SBP | DBP | Pulse Pressure | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Effect | P-value | Neff | Info | Effect | P-value | Neff | info | Effect | P-value | Neff | info | ||||||
| rs141325069 | 12:20769270 | PDE3A | R→Q | A/G | 0.0030 | 1.42 | 8.7×10−9 | 700,771 | 0.99 | 0.72 | 2.7 ×10−5 | 700,575 | 0.99 | 0.76 | 5.2×10−5 | 700,391 | 0.99 |
| rs139491786#† | 16:2086421 | SLC9A3R2 | R→W | T/C | 0.0068 | −1.92 | 4.6×10−21 | 651,069 | 0.97 | −1.32 | 1.9×10−22 | 651,707 | 0.97 | −0.64 | 1.9×10−5 | 649,540 | 0.96 |
| rs61760904* | 19:50139932 | RRAS | D→N | T/C | 0.0073 | 1.16 | 1.1×10−12 | 844,155 | 0.99 | 0.52 | 8.8×10−7 | 843,327 | 0.98 | 0.67 | 2.0×10−8 | 843,773 | 0.99 |
| rs73181210 | 3:169831268 | PHC3 | K→E | T/C | 0.0107 | 0.86 | 3.7×10−8 | 845,000 | 0.93 | 0.61 | 1.8×10−9 | 844,834 | 0.93 | 0.23 | 4.2×10−2 | 842,255 | 0.93 |
| rs3025380* | 9:136501756 | DBH | G→A | C/G | 0.0046 | −1.14 | 1.9×10−8 | 864,699 | 0.98 | −0.83 | 7.2×10−10 | 863,755 | 0.98 | −0.32 | 3.0×10−2 | 864,042 | 0.98 |
| rs139341533# | 11:89182666 | NOX4 | L→F | A/C | 0.0037 | −0.81 | 2.1×10−4 | 851,884 | 0.99 | 0.22 | 1.2×10−1 | 850,481 | 0.99 | −0.93 | 8.7×10−9 | 852,260 | 0.99 |
| rs115079907 | 6:55924005 | COL21A1 | C→R | T/C | 0.0023 | 1.26 | 2.1×10−3 | 837,965 | 0.99 | −0.58 | 3.2×10−2 | 831,917 | 0.97 | 1.70 | 1.4×10−8 | 830,745 | 0.97 |
| rs200999181*# | 6:55935568 | COL21A1 | G→V | A/C | 0.0014 | 1.90 | 9.2×10−6 | 724,111 | 0.80 | −1.04 | 4.3×10−4 | 718,225 | 0.79 | 2.96 | 3.3×10−21 | 724,615 | 0.80 |
| rs2764043# | 6:56035643 | COL21A1 | L→P | A/G | 0.0016 | −1.53 | 1.9×10−5 | 812,384 | 0.93 | 0.35 | 1.4×10−1 | 814,667 | 0.93 | −1.93 | 2.4×10−13 | 810,623 | 0.93 |
| rs138582164 | 8:95264265 | GEM | R→. | A/G | 0.0011 | 3.25 | 2.1×10−7 | 633,292 | 1.00 | 0.13 | 7.6×10−1 | 632,884 | 1.00 | 3.16 | 6.0×10−12 | 632,845 | 1.00 |
| rs202102042^ | 1:11907171 | NPPA | R→Q | T/C | 0.0004 | 3.86 | 3.1×10−7 | 765,853 | 0.78 | 1.06 | 3.1×10−2 | 765,853 | 0.78 | 3.20 | 2.4×10−8 | 765,853 | 0.78 |
SNPs have been previously reported in the literature.
SNPs were identified in two different replication strategies: exonic set and rare-variant set.
SNPs were replicated in the rare variants replication set.
SNP was no longer significant after conditioning on GWAS sentinel SNP at this locus. GWAS significance set at P < 5 × 10−8) For SNPs that were available in both rare-variant and exonic analyses, table reports results with the largest sample size. Neff represents the effective N available for each SNP, defined by the sum of N* imputation info score across each analysis strata. EA, effect allele; RA, reference allele, EAF, effected allele frequency.
We conditioned these SNPs on the sentinel common variant in each respective locus, where available, in the MVP whites discovery sample and compared effect estimates before and after conditioning. SNP rs139491786 in SLC9A3R2 showed a >50% reduction in effect size after conditioning on common variant rs140869992 (r2 = 0.35). Effect sizes for all other rare exonic variants were considered independent as no substantial differences in effect estimates were observed after conditioning (Supplementary Table 6).
All Rare Variants
Discovery analysis in the MVP samples only (excluding UKB) identified 1,684 rare variants with suggestive evidence for association across the three blood pressure traits; 1,066 of these variants were available for meta-analysis in UKB. We observed statistically significant associations (P-value < 5 × 10−8) between 48 rare variants and one or more blood pressure traits. We identified 40 SNPs for pulse pressure, eight SNPs for SBP, and two SNPs for DBP (Supplementary Table 7). Average absolute values of effect estimates for SBP, DBP and pulse pressure were 9.67, 2.33 and 13.89 mmHg per allele, respectively. The missense variants from NOX4 (rs139341533), SLC9A3R2 (rs139491786), and COL21A1 (rs200999181, rs2764043) were evaluated in the both the exonic and rare-variant analyses separately (Table 2; Supplementary Table 7).
Transcriptome-Wide Association Analyses
Common variants from the final meta-analysis were used to evaluate the associations between blood pressure traits and genetically predicted gene expression (GPGE) levels across 44 Genotype-Tissue Expression Project (GTEx)36 tissues and the human kidney reference described by Ko et al.37, using S-PrediXcan31. We identified statistically significant GPGE associations for 1,552 gene-tissue pairs with SBP, 521 with DBP, and 1,970 for pulse pressure (Supplementary Tables 8a-c; Supplementary Figures 2–4). We identified 409 genes with this analysis that would not be identified if SNPs were annotated using the nearest gene. MTHFR was the top result from SBP and showed decreasing SBP with increasing GPGE in skeletal muscle, aorta, and several other tissues.
Murine Kidney Single Cell Sequencing Analysis
Homologs of human genes identified as significant in the S-PrediXcan analysis of kidney tissue were investigated for kidney cell type-specific RNA expression using single cell sequencing in murine kidney cells. Cells were clustered into 11 groups representing structural features and other cell types found in the kidney. Sixteen of the 28 genes were most expressed in any of the five tubule-related cell types: proximal tubules, loop of Henle, distal convoluted tubules, collecting duct principal cells and collecting duct intercalated cells (Figure 3; Supplementary Table 9a–c). Cross-referencing protein expression levels in the Human Protein Atlas38 confirmed findings from murine kidney, including higher expression of CDC16, SRR, SFXN2 and CLCN6 proteins in tubules compared to glomeruli (Supplementary Table 10).
Figure 3. Mapping blood pressure-associated genes to murine kidney cell type clusters for (a) SBP, (b) DBP, and (c) pulse pressure.
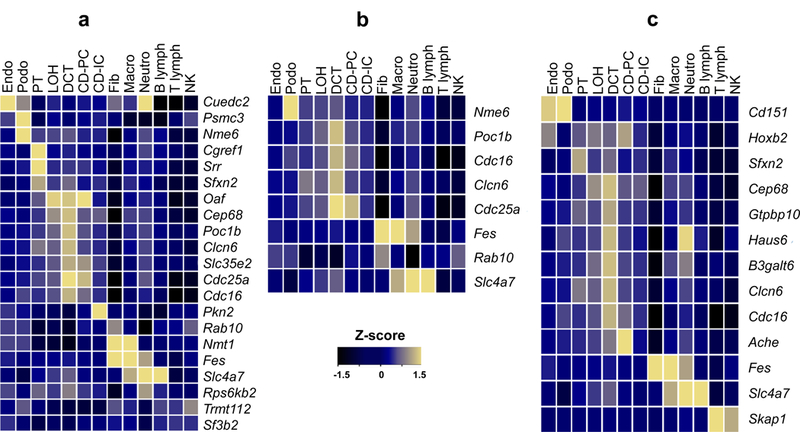
Average expression level of GWAS/eQTL defined genes in murine kidney cell types. Expression levels were determined in 43,745 kidney cells derived from seven mice. Mean expression values of the genes were calculated in each cluster. Color scheme is based on Z-score distribution obtained from two-sided Wald test. Z-scores are not corrected for multiple comparisons. Each row represents one gene and each column is single cell type cluster (as defined by Park et al.) on the heat map. Endo: endothelial, vascular, descending loop of Henle, Podo: podocyte, PT: proximal tubule, LOH: ascending loop of Henle, DCT: distal convoluted tubule, CD-PC: collecting duct principal cell, CD-IC: CD intercalated cell, Fib: fibroblast, Macro: macrophage, Neutro: neutrophil, NK: natural killer cell.
Assessment of Gene-Drug Relationships
To better understand how genes identified in the study relate to medications, associations identified from the GPGE analyses were investigated for overlap with gene targets of known antihypertensive medications, non-antihypertensive medications, and medications with adverse drug events (ADEs) for hypertension and hypotension (Supplementary Tables 11–14). A total of 2,177 tissue- and blood pressure trait-specific drug-gene interactions were identified in this analysis. Of these, there were 617 unique drug-gene interactions, with 175 (28.36%) with antagonistic effects.
The genes PDE3A, PSMB9, and SH2B3 targeted by the non-antihypertensive drugs theophylline, carfilzomib, and pazopanib, respectively, have adverse drug events of either hypo- or hypertension and increase blood pressure with increasing GPGE. The most significant gene in S-PrediXcan targeted by an antihypertensive medication was CLCN6 (SBP β = −2.76, P-value = 8.14 × 10−45) in tibial artery tissue (Supplementary Table 11). The gene most significant in S-PrediXcan with a positive effect size and targeted by a non-antihypertensive medication was PRKAR2B (β = 1.38, P-value = 1.39 × 10−81) in aortic artery tissue identified from the pulse pressure analysis (Supplementary Table 12). The gene most significant in S-PrediXcan with a positive effect size and targeted by a drug with an ADE for hypertension was PSMB9 (pulse pressure β = 0.42, P-value = 7.57 × 10−9) in tibial artery tissue (Supplementary Table 13).
PheWAS with blood pressure Genetic Risk Scores
To systematically evaluate pleiotropy between genetic predictors of blood pressure-traits and diseases throughout the phenome, we performed PheWAS using blood pressure-trait weighted genetic risk scores (w-GRS) separately in self-reported/administratively identified non-Hispanic white, non-Hispanic black, and Hispanic individuals in the MVP. We used all known and novel common sentinel SNPs from the final meta-analysis and trait-specific weights from the UKB discovery sample to generate w-GRSs for each blood pressure trait and regressed PheWAS outcomes from MVP onto those scores, adjusted for the top 10 genetic principal components. Eighty-eight of 1,813 phenotypes were significantly associated with any w-GRSs at a Bonferroni-corrected P-value threshold < 2.76 × 10−5 (Supplementary Table 15). Hypertension (smallest P-value < 1 × 10−305), essential hypertension (smallest P-value < 1 × 10−305) and hypertensive heart and/or renal disease (smallest P-value = 3.3 × 10−173) were the top associations for each of the nine w-GRSs. PheWAS associations were consistent across race/ethnic groups (Supplementary Figure 5). Associations with phenotypes in the circulatory system (N = 52) accounted for more than 50% of the significant results in whites. The phenotype groups with the next most associations were endocrine/metabolic (N = 28), genitourinary (N = 10) and hematopoietic (N = 6).
Among significant associations, 45 were significant for all three w-GRSs, 10 were significant for both SBP and DBP, and 15 were significant for both SBP and pulse pressure, demonstrating substantial overlap between signals captured by genetically predicted blood pressure traits (Supplementary Figure 6; Supplementary Table 15). Thirteen associations were significant only for the pulse pressure w-GRS, of which five were the diabetes sequelae ophthalmic manifestations, neurological manifestations, diabetic retinopathy, other abnormal glucose, and polyneuropathy. Aortic and other aneurysms were only associated with the DBP w-GRS, but not with other w-GRSs.
Enrichment and pathway analyses
We evaluated whether statistically significant genes from S-PrediXcan analyses were enriched in one or more tissues. Compared to other tissues, aorta showed the greatest evidence for enrichment of significant genes across all three traits (SBP P-value = 3.7 × 10−3; DBP P-value = 5.7 × 10−3; and pulse pressure P-value = 1.2 × 10−9; Supplementary Table 16). We provided sentinel SNPs from known and novel loci for each blood pressure trait (Supplementary Tables 17a–c and 18a-c) to DEPICT39 and detected enrichment of 36 tissues across seven systems for pulse pressure (FDR < 5%). The greatest enrichment was seen in arteries (P-value = 3.43 × 10−10) and 11 out of the 36 tissues are grouped in the cardiovascular system (Supplementary Table 17c). Gene-set enrichment of the pulse pressure GWAS results identified 574 enriched gene-sets (FDR < 5%); abnormal vascular smooth muscle morphology (MP:0005592; P-value = 1.47 × 10−8) was the top gene set (Supplementary Table 18c).
We prioritized statistically significant results from the S-PrediXcan aorta tissue analyses for all three traits and investigated pathways using the Ingenuity Pathway Analysis (IPA) software (Supplementary Figures 7–9). Cardiovascular Disease (SBP P-value = 7.2 × 10−6; pulse pressure P-value = 9.53 × 10−4), and Cardiovascular System Development and Function (SBP P-value = 7.7 × 10−5; pulse pressure P-value = 9.53 × 10−4) networks were among the top enriched networks. Notable features in the SBP IPA results included the TGF-β and NOTCH signaling pathways (Supplementary Figure 7), while pulse pressure IPA results featured atherosclerosis genes including CDH13, TCF7L2, PHACTR1 and MTHFR (Supplementary Figure 9).
Convergence of evidence
We collated evidence for genes that were associated in two or more types of investigations that inform relevant gene targets (rare coding variants, predicted gene expression, single-cell sequencing expression enrichment, and drug query) and highlight noteworthy genes (Table 3). We identified 46 known and 7 novel genes satisfying this criterion, including three Mendelian hypo- or hypertension genes and 15 genes targeted by antihypertensive medications. Nine genes were expressed in murine kidney tubule cell types and 19 genes were identified in at least one aorta IPA network.
Table 3.
Converging evidence across analyses.
| Gene | Locus Novelty | Common/Rare Coding SNPs | Nearest Gene | CVD Tissues (S-PrediXcan) | Mouse Kidney Single Cell Clusters | Supplementary Drug Table Reference | Count/Drug | Aorta IPA |
|---|---|---|---|---|---|---|---|---|
| ADK | novel | rs34868542 | ADK | --↑x-xx | ST12 | 12 | PP;SBP | |
| FDFT1e | novel | NA | NA | x-↑↑↑xx | ST11, ST12 | LOVASTATIN, LAPAQUISTAT ACETATE | - | |
| PLAUe | novel | rs34868542 | ADK | ↑-xxxx- | ST11, ST12 | 55 | - | |
| PSMB7 | novel | NA | NA | xx-x↑xx | ST12 | 5 | - | |
| PSMB9 | novel | NA | NA | x--x↑x- | ST12, ST13 | 5 | - | |
| RAB10 | novel | NA | NA | xx-xxx↑ | Immune | - | ||
| RXFP2 | novel | rs9603376 | RXFP2 | xxxx-↑x | ST12 | 4 | - | |
| ACHEb | known | rs374292503 | Intergenic | −x↓↓↓↓↓ | Tubules | ST12 | 56 | DBP |
| ACP2e | known | rs11039216 | SLC39A13 | ↑↑xx-xx | ST11 | ATENOLOL, VERAPAMIL | - | |
| AGERa | known | rs1061808 | AGPAT1 | x↑↑x↑-x | ST12 | VITAMIN B12 | DBP | |
| ARL3 | known | rs140473396 | CNNM2 | ↑↑↑↑↑xx | ST12 | CHEMBL384759 | - | |
| ARVCF | known | rs2240716 | ARVCF | xx↓x↓xx | ST12, ST13 | 3 | PP | |
| AS3MT | known | rs140473396 | CNNM2 | --↑↑↑x- | ST12 | LK-204545 | - | |
| ATP1B1f | known | rs4656180 | NME7 | xx↑x-x- | ST12 | 4 | - | |
| BACE1e | known | rs573455 | CEP164 | xxxx-↑x | ST11, ST12 | 28 | - | |
| BAG6 | known | rs1800629 | TNF | --↑↑↑xx | ST13 | CARBAMAZEPINE | - | |
| BCAR1e | known | rs12449170 | CFDP1 | −x↑x↑x- | ST11 | CELECOXIB | SBP | |
| C4Ab | known | rs1061808 | AGPAT1 | ↓↓↓↓↓↓x | ST12 | IMMUNE GLOBULIN | DBP | |
| CDC16 | known | rs11617448 | CDC16 | ↑↑↑↑↑↑↑ | Tubules | - | ||
| CDC25A | known | rs35979968 | MAP4 | ↓↓↓↓↓-↓ | Tubules | SBP | ||
| CEP68 | known | rs74181299 | CEP68 | ↑↑↑-↑-↑ | Tubules | PP | ||
| CLCN6e | known | rs6669371 | CLCN6 | xx-x↓-↓ | Tubules | ST11, ST13, ST14 | 9 | - |
| COL21A1c | known | rs12203179/ rs115079907, rs200999181, rs2764043 | COL21A1 | xxx-↓x↓ | - | |||
| CYP21A2a | known | rs1061808 | AGPAT1 | xx↑x↑xx | ST13 | KETOCONAZOLE | - | |
| FES | known | rs2071382 | FES | −x↓↓↓-↓ | Immune | ST13 | NAPROXEN | SBP |
| FURIN | known | rs2071382 | FES | xx↑xxxx | ST12 | 3 | SBP | |
| GFAPe | known | rs6503413 | NMT1 | xx↓xxx- | ST11, ST13, ST14 | 28 | - | |
| GUCY1A3e | known | rs3796592 | GUCY1A3 | xx↓xxx↓ | ST11, ST14 | 6 | SBP | |
| GUCY1B3e | known | rs3796592 | GUCY1A3 | xx↓xx-x | ST11, ST14 | 6 | SBP | |
| HLA-Bg | known | rs1800629 | TNF | ---↑--x | ST12, ST13 | 6 | - | |
| HLA-DRB5b | known | rs1061808 | AGPAT1 | --↓↓↓-x | ST12 | 1D09C3 | - | |
| IGFBP3e | known | rs10260816 | Intergenic | x↑-xxxx | ST11, ST14 | FLUOROURACIL, CELECOXIB | - | |
| MADDe | known | rs11039216 | SLC39A13 | ↑↑-x--x | ST11 | VERAPAMIL | - | |
| NME6 | known | rs35979968 | MAP4 | x--x↑↑↑ | Glomerulus | - | ||
| NMT1 | known | rs6503413 | NMT1 | --↑↑↑-↑ | Immune | ST12 | CHEMBL355497 | SBP |
| NOTCH4b | known | rs1061808/ rs139341533 | AGPAT1 | x↓↓x↓↓x | ST12 | NIROGACESTAT, REGN-421 | DBP | |
| NOV | known | rs11783703 | NOV | x↓↑x↑xx | ST12 | INSULIN | DBP | |
| NPPBef | known | rs6669371 | CLCN6 | x↓xxxxx | ST11, ST14 | OXYMETHOLONE, CARVEDILOL | - | |
| NPR3e | known | rs12656497 | NPR3 | xxxxx↑x | ST11, ST12, ST13 | 4 | - | |
| NSF | known | rs80335285 | GOSR2 | ↑↑-↑-xx | ST12 | BITOSCANATE | - | |
| NT5C2 | known | rs140473396 | CNNM2 | xx↑x↑↓x | ST12 | 4 | SBP | |
| OPRL1 | known | rs6090040 | TCEA2 | ↓x↓x↓↓x | ST12 | 13 | - | |
| PDE3A | known | rs60691990/ rs141325069 | Intergenic | xxx-xx- | ST12, ST13 | 19 | - | |
| PKN2 | known | rs12035750 | GTF2B | x-xxxx↑ | Tubules | ST12 | 3 | - |
| POC1B | known | rs11105354 | ATP2B1 | x--x-x↑ | Tubules | - | ||
| PRKAR2Bd | known | rs12705390 | AF086203 | x-↑x--x | ST12 | 5 | ALL | |
| SBF2e | known | rs58068637 | SWAP70 | xx↓xxxx | ST11 | PAROXETINE | - | |
| SFXN2 | known | rs140473396 | CNNM2 | −↑x-xx↑ | Tubules | - | ||
| SLC12A2e | known | rs6595838 | FBN2 | x↑xx-xx | ST11, ST12 | 6 | - | |
| SLC4A7 | known | rs2643826 | SLC4A7 | xx↓xxx↓ | Immune | SBP | ||
| SRR | known | rs12952051 | SMG6 | ↑↑-↑↑↑↑ | Tubules | ST12 | PYRIDOXAL PHOSPHATE, SERINE | - |
| THBS2a | known | rs533414974 | Intergenic | xx↑-↑xx | ST12 | BEVACIZUMAB, CORTICOTROPIN | DBP | |
| TUBB1 | known | rs6026739 | ZNF831 | xxxxxx↑ | ST12, ST13 | 10 | - |
Selection criteria: evidence from at least 2 categories (coding variant gene, S-PrediXcan gene, gene implicated in mouse kidney single-cell expression, or drug target) and from at least 4 analyses. Gene = Gene showed enriched expression in one or more cell type in murine kidney single-cell RNA sequencing experiment, or was significant in genetically predicted gene expression analysis for any of the 45 tissues; Novel = Indicator variable denotes whether variants in a given gene region have previously been reported in genome-wide association studies of blood pressure traits. Sentinel SNPs = Sentinel SNP from common or rare variant analyses from each independent locus. Nearest Gene = Column reports genes that would have been identified if the nearest gene annotation strategy was used to link GWAS significant variants. CVD Tissues = Column identifies genes that were significantly associated with genetically predicted gene expression and blood pressure traits (using S-PrediXcan) in the following cardiovascular related tissues: heart atrial appendage, heart left ventricle, aorta, coronary artery, tibial artery, adrenal gland and kidney. Up (down) arrow indicates a positive (negative) association between GPGE and at least one blood pressure trait. Dash indicates data was not significant and x indicates data were unavailable. For a full-set of tissue specific results, see Supplementary Tables 8a–8c. Mouse kidney single-cell clusters = Column highlights the cell-type in which specific genes were enriched in murine single-cell RNA sequencing expression analyses. Supplementary Drug Table Reference = References the supplementary tables containing the gene-drug relationship. Count/Drug = Column summarizes drugs targeting genes when the number of drugs is ≤2 or the total number of drugs targeting the gene when the number of drugs is >2 which were identified to interrelate with the gene from the following databases: Guide to Pharmacology Interactions, Drugbank, PharmGKB, TDG Clinical Trials, TEND, and/or ChEMBL Interactions. Aorta IPA = Trait(s) for which the gene was identified in the aorta IPA network, SBP = systolic blood pressure; DBP = diastolic blood pressure; PP = pulse pressure;
Genetically predicted expression of these genes was positively associated with pulse pressure, inversely associated with DBP, and non-significant for SBP
Genetically predicted expression of these genes was positively associated with DBP, inversely associated with pulse pressure, and non-significant for SBP
Genetically predicted expression of these genes was positively associated with DBP and inversely associated with SBP and pulse pressure
Genetically predicted expression of these genes was positively associated with SBP and pulse pressure and inversely associated with DBP
Targeted by an antihypertensive medication
Mendelian gene for hypertension
Mendelian gene for hypotension
DISCUSSION
We present results from multi-omic analyses of a trans-ethnic GWAS consortium for blood pressure traits. By incorporating large sample sizes, bioinformatics, and measures of gene expression, we re-interpret the genetic architecture of blood pressure and identify tissues and anatomical features where blood pressure genes exert effects. Interrogation of gene-drug relationships and toxicities for GPGE associations provides additional evidence for known and novel blood pressure genes, and suggests genes as potential leads for drug development and repurposing potential for existing drugs. We emphasize the utility of large-scale blood pressure GWAS as a requisite starting point for analyses providing insight into clinical factors, genetic etiology, pathophysiology, and pharmacology of blood pressure homeostasis.
The MVP comprises US military veterans, and has an overrepresentation of male and black participants compared to the US population. We had a larger collection of Hispanic participants than the largest previous study of blood pressure traits in that population13, and almost twice the number of black participants as the largest previous study of African ancestry populations10. Although consistent, our comparison of SNP effects on blood pressure traits demonstrated a lower correlation between race/ethnic groups than Franceschini et al.9; however, the effects we compared were much more subtle than the first 29 blood pressure SNPs detected by Ehret et al.6. We compared the effects on clinical outcomes of genetically-imputed blood pressure traits in a PheWAS, and observed consistent effects between racial/ethnic groups in MVP. These results support the previous observation9 that genetic effects on blood pressure of common SNPs are consistent between populations, and suggest that an increasing burden of blood pressure-increasing alleles has a similar effect on health across white, black, and Hispanic populations.
Evidence from S-PrediXcan and DEPICT identified arteries as the tissue type with greatest evidence for gene enrichment. These findings agree with results from Warren et al. 201712, which reported arteries as the top tissue from a DEPICT analysis of blood pressure traits, and also with results from Gamazon et al. 201840, which reported aorta as the top PrediXcan tissue with highly enriched gene signals for SBP. The lack of enrichment of genes in other relevant tissues such as the kidney maybe due to the smaller sample of kidney tissues available in prediction training sets or that tissue-specific gene expression is differentially enriched.
The SBP IPA analysis highlights genes linked to TGF-β and notch signaling pathways including FURIN, GUCY1A3 (Syn: GUCY1A1), and GUCY1B3. (Table 3; Supplementary Figure 7). GPGE of FURIN in the aorta was positively associated with SBP. This effect is likely mediated by furin-induced activation of pro-TGF-β1 to TGF-β1, which works along with the RAS pathway to increase blood pressure41–44. Predicted expression of FES, a gene <1 kb upstream of FURIN, is inversely associated with SBP in aorta, coronary artery, tibial artery, and kidney (Supplementary Table 8a), suggesting the presence of two proximal blood pressure loci with different regulatory mechanisms. Naproxen, a non-steroidal anti-inflammatory drug, has an inhibitory effect on FES (Supplementary Tables 12 and 13) and hypertension is one of its known side-effects45. Findings highlight the importance of the role of soluble guanylyl cyclase (sGC) expression via the Notch signaling pathway in the mouse aorta on hypertension46. GUCY1A3 and GUCY1A1 encode subunits of sGC which is a major nitric oxide (NO) receptor in the vascular wall46–48. As mediators of the vasodilatory effects of NO, increased expression of these genes predicted a decrease in SBP in aorta (Table 3 and Supplementary Table 8a).
To understand how genetically-predicted blood pressure is associated with the clinical phenome, we calculated w-GRSs for each blood pressure trait and evaluated them with a PheWAS (Supplementary Table 15). The pulse pressure w-GRS was associated with diabetic complications, while w-GRS for SBP or DBP had fewer diabetes-related associations. Pulse pressure is an independent predictor of cardiovascular disease and incident diabetes49,50. Elevated pulse pressure is a marker for arterial stiffness51, which is positively associated with diabetic retinopathy and neuropathy52. These findings are supported by the pulse pressure IPA results where the top cardiovascular gene network includes at least four genes which may directly or indirectly mediate arterial stiffness or atherosclerosis, including TCF7L2, CDH13, PHACTR1, and MTHFR (Supplementary Figure 9)53–59. Our finding of a positive association between the DBP w-GRS and aortic and other aneurysms supports evidence from a previous study of 1.25 million individuals where an association between DBP and aortic aneurysm was reported60. Our study is the first to provide evidence for a genetic etiology for this observation.
Convergent evidence from multiple analyses identified several blood pressure genes with strong biologic importance, including PDE3A and novel genes RXFP2 and ADK. RXFP2 is a receptor for the hormone relaxin61, which causes vasodilation, increases cardiac output and renal perfusion, and has been evaluated in clinical trials as a treatment for acute heart failure62–64. RXFP2 is expressed in multiple tissues, which likely underlies the multiple physiological effects of the relaxin hormone throughout the circulatory system65.
The product of ADK, adenosine kinase, catalyzes the transfer of gamma-phosphate from adenosine triphosphate to adenosine to form adenosine monophosphate and has widespread effects on multiple systems including cardiovascular, nervous and respiratory systems66. Adenosine terminates supraventricular tachycardia (SVT) involving the atrioventricular (AV) node and has been attributed to cardiac brady-arrhythmias67,68. Intravenous adenosine injection in humans induces vasodilation and systemic hypotension69, and is the primary drug used in the treatment of stable narrow-complex SVT70. It is known to reduce blood pressure and blood pressure variability in rats, and its actions are mediated through adenosine receptors71. We show a positive association between GPGE of ADK and SBP and pulse pressure in aortic tissue (Supplementary Tables 8a and 8c), consistent with the known directional effects of adenosine on blood pressure.
PDE3A is targeted by a wide variety of inhibitors for indications including congestive heart failure, hypertension, and heart disease. The PDE3A inhibitor theophylline used to treat chronic obstructive pulmonary disease has a hypotension ADE, which is consistent with the effects of increased gene expression in our analysis (Supplementary Tables 12 and 13). The autosomal dominant Mendelian condition Hypertension and Brachydactyly Syndrome (HTNB; OMIM: 112410) is caused by at least six distinct rare PDE3A mutations72,73. HTNB features include brachydactyly type E, severe salt-independent but age-dependent hypertension, increased fibroblast growth rate, neurovascular contact at the rostral-ventrolateral medulla, altered baroreflex blood pressure regulation, and death from stroke before age 50 years when untreated74,75. Associations between common variants in the PDE3A locus and blood pressure have been reported previously25,26.
A novel aspect of this study is the incorporation of single-cell gene expression data from cells derived from murine kidneys. We show that a majority of blood pressure genes identified by S-PrediXcan analyses in human kidneys are enriched in tubule cell types derived from murine kidneys. This observation suggests that genes detected through GPGE associations and expressed in tubules, including SRR, ACHE, SFXN2 and CLCN6, may play a role in blood pressure regulation. Several lines of evidence implicate the SRR gene, while the nearest gene annotation strategy identifies SMG6 as associated with blood pressure at this locus. A SNP in SMG6, rs216172, has been associated with coronary artery disease76; however, this SNP is an eQTL for SRR and not for SMG6 (GTEx portal). SNPs near SRR have been associated with type 2 diabetes (T2D) and T2D mediated arterial stiffness53. ACHE terminates signal transduction at the neuromuscular junction by rapid hydrolysis of acetylcholine released into the synaptic cleft77. Inhibition of acetylcholinesterase is an effective treatment for orthostatic hypotension, especially in patients with supine hypertension78. Dimetacrine, a tricyclic antidepressant, and decamethonium, a muscle relaxant, have inhibitory effects on ACHE and are not currently prescribed as anti-hypertension medications.
This work helps to clarify the complex MTHFR locus by providing unique tissue-specific evidence for several genes in the region in relation to blood pressure79,80. In addition to MTHFR, our study provides evidence for the role of NPPA (novel missense variant; Table 2), NPPB (GPGE association with SBP and pulse pressure in the left ventricle; Supplementary Tables 8a and 8c), and CLCN6 (inverse association in kidney S-PrediXcan, and enrichment in murine kidney; Supplementary Tables 8a-c and 9a-c) in blood pressure. NPPA and NPPB are exclusively expressed in the heart and have biological functions that include natriuresis, diuresis, vasorelaxation, inhibition of renin and aldosterone secretion, and play a key role in cardiovascular homeostasis81. GPGE of CLCN6, a putative chloride antiporter82, was consistently associated across blood pressure traits. CLCN6 is targeted by the antihypertensive medication chlorthalidone, and NPPB is targeted by the antihypertensive medication carvedilol (Supplementary Table 11). Findings for this locus highlight that effects of multiple associated genes from the same locus may vary by tissue type, and several nearby genes with very different biological functions may jointly contribute to the trait of interest.
In conclusion, we applied multiple post-GWAS analyses to identify genes with effects on blood pressure regulation. We report hundreds of novel SNPs and genes, several with strong biological plausibility, and tissue-specific gene associations with directions of effect. We provide insight into the natural experiments of gene regulation and direct perturbation of proteins by mutation with regard to effects on blood pressure, which enhances the biological understanding of blood pressure traits. Our study identifies a refined set of genes that often have coordinated expression across multiple tissues that may have relevance to blood pressure traits and these gene-tissue pairs are prime candidates for causal investigation.
ONLINE METHODS
We conducted a multi-stage GWAS of common and rare variants in over 750,000 participants. We then performed additional bioinformatics analyses of GPGE for blood pressure traits, evaluated cell types where associated genes are expressed, performed a phenome-wide association study of genetic risk scores for blood pressure traits from the electronic health records of MVP participants, and screened known drugs to evaluate potential for repurposing and validate observed associations. A flow chart for analyses is presented in Figure 1.
Discovery Cohorts
The Million Veteran Program
The Million Veteran’s Program (MVP) is a large cohort of fully consented participants who were recruited from the patient populations of 63 Department of Veterans Affairs (VA) medical facilities. The MVP is recruited at VA hospitals from men and women who are veterans of the US armed forces. It is enriched with African American and Hispanic participants compared with the general US population, and males are overrepresented. Across race groups, the average age ranged between 49 for Asian and 61 for black participants (Supplementary Table 1). Average BMI ranged between 27.8 for Asian and 30.9 for Native Americans. The proportion of males ranged between 87% for Native Americans and 93% for whites. Average SBP ranged between 132 mmHg for Asians and 140 mmHg for blacks, average DBP ranged between 81 mmHg for Asians and 85 mmHg for Blacks, and average pulse pressure ranged between 51 mmHg for Asians and 57 mmHg for whites. The proportion of participants on an anti-hypertensive drug at the time of blood pressure measure ranged between 31% for Asians and 53% for blacks.
Recruitment began in 2011 and is conducted in-person, initiated by an invitation letter and completed by answering baseline and lifestyle questionnaires, providing a blood sample, providing access to medical records, and giving permission for re-contact. Consent to participate is provided after counseling by research staff and mailing of informational materials. All documents and protocols are approved by the VA Central Institutional Review Board. Blood samples are collected by phlebotomists and banked at the VA Central Biorepository in Boston, MA. Genotyping was conducted using a customized Affymetrix Axiom Biobank Array chip with content added to provide coverage of African and Hispanic haplotypes, as well as markers for common diseases in the VA population. Researchers are provided with de-identified data, and do not have the ability or authorization to link these details with a participants’ identity.
MVP Genotype Quality Control
Blood samples drawn from consenting MVP participants were shipped to the Central Biorepository in Boston, MA, where DNA was extracted and shipped to two external centers for genotyping on an Affymetrix Axiom Biobank array designed specifically for the MVP. The MVP genomics working group applied standard quality control and genotype calling algorithms to the data in batches using the Affymetrix Power Tools Suite (v1.18). Standard quality control pipelines were used to exclude duplicate samples, samples with more heterozygosity than expected, samples with an excess (>2.5%) of missing genotype calls, and samples with discordance of genetically inferred sex versus self-report. We excluded related individuals (halfway between 2nd and 3rd degree relatives or closer) as measured by the KING software83. Prior to imputation, variants that were poorly called or that deviated from their expected allele frequency based on reference data from the 1000 Genomes Project84 were excluded. After pre-phasing using EAGLE v285, genotypes from the 1000 Genomes Project84 phase 3, version 5 reference panel were imputed into Million Veteran Program (MVP) participants via Minimac3 software86. Principal component analysis was performed using the FlashPCA87, to generate the top 10 genetic principal components explaining the greatest variability.
Race/ethnicity
Information on race (whites, blacks, Asians, and Native Americans) and ethnicity (Hispanic: Yes or No) were obtained based on self-report through centralized VA data collection methods using standardized survey forms, or through the use of information from corporate data warehouse (CDW), or Observational Medical Outcomes Partnership (OMOP) data, when information from self-report survey was missing. Race and ethnicity categories were then merged to form the following race/ethnicity variables: non-Hispanic whites (whites), non-Hispanic blacks (blacks), non-Hispanic Asians (Asians), non-Hispanic Native Americans (Native Americans) and Hispanics. Individuals for whom race and ethnicity could not be assigned due to conflicting records and missing data, were categorized as unknown. Prior to analysis QC, there were 15,710 Veterans with unknown status for race/ethnicity. For these individuals, we used a K-means clustering approach in R (McQueen algorithm) with the top 10 genetic principal components as input. To obtain the most reliable cluster designations for the missing data, the K-means approach was applied to the maximum available samples: the 1000 Genomes reference populations and all individuals for whom PCs were available regardless of whether race/ethnicity designations were unknown. K-clusters were optimized by testing values K=2 through K=10. K = 4 was ultimately chosen as the most optimal value, as visual examination of these most closely corresponded to whites (N=5,265), blacks (N=4,671), Asians (N= 3,936) and Hispanics (N= 1,838).
MVP Blood Pressure Phenotypes
We selected adults (age ≥ 18) and used the median eligible non-Emergency Department outpatient measured SBP in the entire available EHR, and used the corresponding DBP from this measure. In individuals where the median value was observed at multiple clinical encounters on distinct dates, we used the earliest of those measures to identify the DBP, age, BMI, and anti-hypertensive treatment status of the individual at that time. Measures were ineligible if they occurred at or after an ICD-9 code from the groups 585 (chronic kidney disease), 405 (secondary hypertension), or 428 (heart failure). If pain scores were available, blood pressure measures taken during encounters when a pain score ≥ 5 was recorded were also ineligible, because severe pain can elevate blood pressure88,89. For measures taken while a patient was on an antihypertensive medication we added 15 mmHg to SBP and 10 mmHg to DBP8,90.
MVP Analysis
For the MVP GWAS we performed linear regression association tests with additive models for untransformed blood pressure traits, after adjusting for medication use. We adjusted linear regression models analyzing SNP associations for age at blood pressure measure, age2, sex, BMI measured within 1 year of blood pressure measure, and the top 10 genetic principal components in analyses. All primary analyses for the MVP were conducted by either strata of administratively assigned race/ethnicity or by their empirically designated clusters. All regression based analyses were conducted in SNPTEST-v2.5.4-beta91. Inference was limited to genotyped and imputed variants with SNPTEST Info scores of 0.4 or higher, with Hardy Weinberg equilibrium P-value > 5 × 10−8 for common variant analysis (minor allele frequency > 0.1). Inference on rare variants, SNPs with MAF ≤ 1%, was restricted to variants with an effective minor allele count (SNPTEST Info score multiplied by minor allele count) of ≥10 in each analysis sub-cohort.
The UK Biobank
Summary statistics from the analysis of the interim data from the UK Biobank (UKB) were utilized in our meta-analysis. These results have been previously reported by Warren et al.12. Briefly, following central and study-specific quality control protocols, 140,886 empirically classified white individuals were analyzed for SBP, DBP, and pulse pressure traits. Blood pressure measures were averaged over two measures, and adjusted for medication use by adding 15 and 10 mmHg to SBP and DBP, respectively. Linear models were adjusted for the top 10 principal components of ancestry, age, age2, sex, an indicator for genotyping platform, and BMI.
Meta-Analysis of Discovery Datasets
Inverse-variance weighted fixed-effects meta-analysis of common variants across MVP subsets and summary statistics from UKB was performed using the METAL software. Genomic inflation factor was calculated, and λGC for the discovery from MVP were 1.195, 1.149, and 1.171 for SBP, DBP and pulse pressure, respectively, 1.303, 1.315, and 1.270 respectively, from UKB, and 1.275, 1.140, and 1.244, respectively, in the overall discovery analysis (Supplementary Figure 10). Subsequently, we utilized the LD Score Regression approach92 to ascertain whether inflation was due to residual population stratification or polygenicity. Calculation of the intercept in the MVP Whites discovery analysis dataset were 1.05 (standard error = 0.01), 1.03 (standard error = 0.01), and 1.04 (standard error = 0.01), for SBP, DBP, and pulse pressure respectively, suggesting that little of the observed inflation in the lambda is due to population stratification.
Selection of SNPs for Replication
Common Variants
For common variants, we considered for follow-up SNPs in loci non-overlapping with previously reported loci according to both an LD threshold of r2 ≤ 0.1 and a 1Mb interval. We obtained a list of these SNPs with P-value < 1 × 10−6 for any of the three blood pressure traits, a minor allele frequency (MAF) ≥ 1%, and concordant directions of effect between UKB and MVP.
In silico replication summary statistics were provided for 942 SNPs by the International Consortium for Blood Pressure Genetics (ICBP)25 after meta-analysis of 77 individual participating cohorts for a total maximum of 299K individuals, who were genotyped and analyzed according to study-specific protocols. Additional replication results were provided from Vanderbilt University’s BioVU EMR-linked biorepository, among which genotypes from the MEGA array and phenotype data were available from 17,277 participants. Discovery and replication data were combined using fixed-effects inverse-variance weighted meta-analysis implemented in METAL93.
Rare Variants
We conducted an in silico replication analysis of 18 rare exonic SNPs from our discovery analysis in 417,143 participants from the BP-ICE consortium. SNPs were chosen for replication if they had a discovery P-value < 1 × 10−6, and a MAF < 1%.
BP-ICE used the exome array and did not have genome wide coverage of rare variants. Therefore, we also pursued replication utilizing the full release of the UKB data to capture non-exonic rare variation. Due to the inclusion of UKB data in the discovery set, for the second analysis we sought replication from variants suggestive only in MVP cohorts following meta-analysis as described above. 1,066 rare variants with P-value < 1 × 10−6 for any of the three phenotypes were selected for replication in 458,577 participants from UKB. Additional replication was provided from BioVU MEGA, and all data were meta-analyzed using fixed-effects meta-analysis in METAL93.
Classifying Results by Evidence for Association
For results that reached statistical significance of P-value ≤ 5 × 10−8 after final meta-analysis, and that had consistent direction of effect between discovery and replication stages, we established three tiers of evidence that are annotated in results tables (Supplementary Tables 3a-c):
-
1)
Genome-wide significance in the discovery stage, and Bonferroni-corrected significance in replication and consistent trait-specific direction of effect across stages.
-
2)
Genome-wide significance in the discovery stage, and P-value ≤ 0.05 in the replication stage and consistent trait-specific direction of effect across stages
-
3)
Variants had P-value less than 1 × 10−6 and > 5 × 10−8 in the discovery stage, and had P-value < 0.05 in the replication stage and had consistent trait-specific direction of effect across stages and was genome-wide significant after final analysis.
Conditional Analysis
For conditional analysis of common variants we used two parallel approaches implemented in the Genome-wide Complex Traits Analysis (GCTA) software94. Details are described in the Supplementary Note.
Proportion of Variance Explained
We approximated the proportion of blood pressure-trait specific variance explained in the trans-ethnic meta-analysis by all independent sentinel SNPs (novel and known) and novel SNPs, separately. Variance explained by each SNP was first estimated by the following equation:
The sum of the variances of the independent sentinel SNPs for common variants provided estimates for the proportion of variance explained for all SNPs, and novel SNPs for each of the blood pressure traits. The transformation of the relationship between t-statistic and r2 to statistic to r2 is described in Supplementary Note.
Genetic Risk Score Construction
We constructed a genetic risk score (GRS) for each blood pressure trait by calculating a linear combination of weights derived from the 140,886 participants from the UKB common variant analysis and sentinel SNPs at each statistically significant locus observed in the MVP. Weighted GRS (w-GRS) were constructed for self-reported/administratively assigned non-Hispanic whites, blacks and Hispanics in the MVP.
Phenome Wide Association Study Analysis
We performed a phenome-wide association study (PheWAS)95 of GRS for each blood pressure trait in MVP whites (Nmax = 188,088), blacks (Nmax = 52,530), and Hispanics (Nmax = 16,735), leveraging the diverse nature of MVP as well as the full catalog of ICD-9 diagnosis codes. We used logistic regression to separately model up to 1,813 PheWAS traits as a function of the three GRSs, adjusted for age, age2, sex, BMI, and 10 PCs. We report the results from these analyses as odds ratios where the estimate is the average change in odds of the PheWAS trait per weighted blood pressure-increasing allele. Interpretation of results were limited to phenotypes with 25 or more cases. Multiple testing thresholds for statistical significance were set to P-value ≤ 2.75 × 10−5 (0.05/1,813). All PheWAS analyses were conducted using the R PheWAS package96. Effect-estimates from significant PheWAS results from any one or more of the analysis were then compared between whites, blacks and Hispanics to report Pearson’s correlations (R2) for each pair per trait (Supplementary Figure 5).
S-PrediXcan Analysis
Genetically predicted gene expression was evaluated for the common variant subset with S-PrediXcan31, a gene-level approach which estimates the genetically determined component of gene expression in a given tissue and tests it for association with SNP-level summary statistics. We utilized all three blood pressure meta-analysis results for common variants and 44 tissues from GTEx36 for this analysis, as well as the collection of kidney reference data that was recently described by Ko et al.37, incorporating covariance matrices developed for European populations (1000 Genomes) as the majority of samples were European in origin.
Evaluation of Kidney Loci in Murine Kidneys
For the genes implicated by the S-PrediXcan analysis as having associated expression in kidney, we evaluated homologous genes in the single cell atlas of the mouse kidney, where expression levels are measured by single-cell RNAseq across 57,979 total mouse kidney cells from 7 healthy mice97. This enables us to observe what cell types in the mammalian kidney express the genes where there is evidence for association between expression and blood pressure traits. After quality filtering steps, a single cell-gene matrix containing the UMI (unique molecular identifier) counts for 43,745 cells and 16,273 transcripts were generated from 7 normal mouse kidneys using 10x Chromium™ Single cell solution97. Mouse homologs of the target human genes identified by the S-PrediXcan analysis were found using Ensembl BioMart. Genes that expressed less than 5% of the cell clusters were excluded from analysis. To calculate the average expression level for each cluster, a z-score of UMI count was first obtained for every single cell. Then, we calculated the mean z-scores for individual cells in the same cluster, resulting in z-score for each gene and each cell cluster.
Evaluation of Drug Classes for Genes with Associations with Gene Expression
To better understand the performance of the S-PrediXcan method, identify genes with potential to be leads for drug development, and identify drug-gene pairs that may be leads for repurposing, three comparisons were made among significant associations from S-PrediXcan analyses. 1) We identified all S-PrediXcan genes that are targeted by an antihypertensive drug to validate associations and identify the most credible genes in regions with many associations. 2) We provide a list of candidate genes that are potential leads for novel inhibitory antihypertensive therapies by considering genes with positive effects for GPGE on blood pressure that are also targeted by a non-antihypertensive drug. 3) To identify genes that may be leads for developing novel treatments and drugs with repurposing potential, we report gene-drug pairs for significant S-PrediXcan genes that are not targeted by an antihypertensive drug, but are targeted by a drug with a toxicity that involves hyper- or hypotension.
A list of medications with a primary indication for hypertension and a list of medications with adverse drug events (ADEs) of hypertension or hypotension were created using SIDER98 and the DEB2 database99. Gene targets for antihypertension medications, medications targeting genes significant in S-PrediXcan analyses with positive effect sizes, and medications targeting genes mapped from significant GWAS signals were queried using DGIdb100. Primary indications for medications targeting genes significant in S-PrediXcan analyses with positive effect sizes were compiled using the BIDD TTD database101.
To identify genes that are attractive leads for novel inhibitory drugs, we report significant S-PrediXcan genes with a positive effect size (i.e. increasing gene expression is associated with increasing one or more blood pressure traits) that are targeted by a non-antihypertensive medication without an indication of ADEs for hypertension or hypotension. This list thereby represents a set of genes that are both likely to be involved in blood pressure regulation in one or more tissues, and can be targeted by drugs. Known targets for anti-hypertensive drugs significant by S-PrediXcan and a summary of the most significant S-PrediXcan result across tissues and blood pressure traits is presented in Supplementary Table 11. Significant S-PrediXcan genes that have positive effect sizes in any tissue and are targeted by a non-hypertension drug with no ADE for hypertension or hypotension are presented in Supplementary Table 12.
We report another group of genes that may be attractive for treatment development, on the basis of being both associated with blood pressure traits and targeted by non-antihypertensive drugs that feature an ADE of either hypo- or hypertension. These gene-drug pairs may be leads for either modification of drug molecules, modification of dosing or delivery strategies, or potential gene targeting by novel treatments. Genes that are significant by S-PrediXcan, are targeted by a drug, and that have an ADE involving hypertension or hypotension are presented in Supplementary Table 13. Gene-drug relationships for all genes mapped from significant association signals are presented in Supplementary Table 14.
Enrichment and Pathway Analyses
We investigated whether one or more of the 45 tissues evaluated with S-PrediXcan were enriched. We also performed enrichment analyses in DEPICT39 by using trait-specific GWAS significant sentinel SNPs as input. We evaluated significant genes from the top enriched S-PrediXcan tissue (aorta) for each trait with the Ingenuity Pathway Analysis (IPA) software (IPA®,QIAGEN Redwood City) (Supplementary Figures 7–9 and Supplementary Note).
Ethics statement
The central Veterans Affairs Institutional Review Board (IRB) and site-specific IRBs approved the Million Veteran Program study. The Vanderbilt University Medical Center IRB approved the use of BioVU data for this study. Each cohort within the ICBP and BP-ICE consortiums have ethical approval from their local institution. All relevant ethical regulations were followed.
Reporting Summary
Further information on research design is available in the Life Sciences Reporting Summary linked to this article.
Supplementary Material
ACKNOWLEDGEMENTS
This work is a product of the effort, initiative and funds made available to several individuals by multiple funding organizations. Detailed acknowledgements and funding details are provided in the Supplementary Note. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; the U. S. Department of Health and Human Services; the National Health Service (UK); the EC (UK); the National Institute for Health Research (UK); or the Department of Health and Social Care (UK). This publication does not represent the views of the Department of Veterans Affairs or the United States Government.
Peter Sever received support from Pfizer Inc.
Neil Poulter has received financial support from several pharmaceutical companies which manufacture blood pressure-lowering agents, for consultancy fees (Servier), research projects and staff (Servier, Pfizer) and for arranging and speaking at educational meetings (AstraZeneca, Lri Therapharma, Napi, Servier and Pfizer). He holds no stocks and shares in any such companies.
Mark J. Caulfield is Chief Scientist for Genomics England, a UK Government company.
Bruce M. Psaty serves on the DSMB of a clinical trial funded by Zoll LifeCor and on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson.
Dennis Mook-Kanamori works as a part-time clinical research consultant for Metabolon, Inc.
Robert A. Scott is an employee and shareholder in GlaxoSmithKline plc.
The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute (USA); the National Institutes of Health (USA); National Health Service (U.K.); National Institute for Health Research (U.K.); The Department of Health and Social Care (U.K.); the EC; or the U. S. Department of Health and Human Services. This publication does not represent the views of the Department of Veterans Affairs or the United States Government.
URLs
Affymetrix Power Tools 2.10.0: https://www.thermofisher.com/us/en/home/life-science/microarray-analysis/affymetrix.html
BIDD TTD database: https://db.idrblab.org/ttd/
Corporate Data Warehouse: https://www.hsrd.research.va.gov/for_researchers/vinci/cdw.cfm
DEPICT: https://data.broadinstitute.org/mpg/depict/
DGIdb: http://www.dgidb.org/
EAGLE v2: https://data.broadinstitute.org/alkesgroup/Eagle/
Ensembl BioMart: http://www.ensembl.org/biomart/martview
FlashPCA2: https://github.com/gabraham/flashpca
GCTA v1.91.4beta: http://cnsgenomics.com/software/gcta/#Overview
GTEx portal: https://www.gtexportal.org/home/
GWAS catalog: https://www.ebi.ac.uk/gwas/
Human Protein Atlas: https://www.proteinatlas.org/
Ingenuity Pathway Analysis: https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/
KING software: http://people.virginia.edu/~wc9c/KING/
Kmeans package: http://stat.ethz.ch/R-manual/R-devel/library/stats/html/kmeans.html
LDSC v1.0.0: https://github.com/bulik/ldsc
METAL software: http://csg.sph.umich.edu/abecasis/metal/
Minimac3: https://genome.sph.umich.edu/wiki/Minimac3
Observational Medical Outcomes Partnership: https://fnih.org/what-we-do/major-completed-programs/omop
PheWAS package: https://github.com/PheWAS/PheWAS
R statistical software: https://www.r-project.org/
SIDER: http://sideeffects.embl.de/
SNPDOC: https://wakegen.phs.wakehealth.edu/public/snpdoc3/index.cfm
SNPTEST-v2.5.4-beta: https://mathgen.stats.ox.ac.uk/genetics_software/snptest/old/snptest_v2.3.0.html
S-PrediXcan: https://github.com/hakyimlab/MetaXcan
Footnotes
COMPETING INTERESTS
DATA AVAILABILITY STATEMENT
Full summary statistics relating to the Million Veteran Program (MVP) are publically available and may be accessed here with the accession code phs001672.v1.p1: https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001672.v1.p1. The UK BioBank data are available upon application to the UKBiobank (https://www.ukbiobank.ac.uk). Combined summary statistics for common and rare variant analysis (discovery + replication) for sentinel SNPs for each blood pressure-trait, are available in supplementary tables. Statistically significant reports for S-PrediXcan results for all 45 tissues and PheWAS analyses for all blood pressure-traits evaluated are also made available in the Supplementary Tables. Murine single-cell sequencing data can be found under GEO (GSE107585).
REFERENCES
- 1.Lawes CMM, Vander Hoorn S, Rodgers A & International Society of Hypertension. Global burden of blood-pressure-related disease, 2001. Lancet Lond. Engl 371, 1513–1518 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Forouzanfar MH et al. Global Burden of Hypertension and Systolic Blood Pressure of at Least 110 to 115 mm Hg, 1990–2015. JAMA 317, 165–182 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Lewington S et al. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet Lond. Engl 360, 1903–1913 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Whelton PK et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol (2017). 10.1016/j.jacc.2017.11.006 [DOI] [PubMed]
- 5.Muntner P et al. Potential U.S. Population Impact of the 2017 ACC/AHA High Blood Pressure Guideline. J. Am. Coll. Cardiol 71, 109–118 (2018).29146532 [Google Scholar]
- 6.International Consortium for Blood Pressure Genome-Wide Association Studies et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 478, 103–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levy D et al. Genome-wide association study of blood pressure and hypertension. Nat. Genet 41, 677–687 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newton-Cheh C et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat. Genet 41, 666–676 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franceschini N et al. Genome-wide association analysis of blood-pressure traits in African-ancestry individuals reveals common associated genes in African and non-African populations. Am. J. Hum. Genet 93, 545–554 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang J et al. Single-trait and multi-trait genome-wide association analyses identify novel loci for blood pressure in African-ancestry populations. PLoS Genet 13, e1006728 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li C et al. Genome-Wide Association Study Meta-Analysis of Long-Term Average Blood Pressure in East Asians. Circ. Cardiovasc. Genet 10, e001527 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warren HR et al. Genome-wide association analysis identifies novel blood pressure loci and offers biological insights into cardiovascular risk. Nat. Genet 49, 403–415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sofer T et al. Genome-Wide Association Study of Blood Pressure Traits by Hispanic/Latino Background: the Hispanic Community Health Study/Study of Latinos. Sci. Rep 7, 10348 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adeyemo A et al. A genome-wide association study of hypertension and blood pressure in African Americans. PLoS Genet 5, e1000564 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parmar PG et al. International Genome-Wide Association Study Consortium Identifies Novel Loci Associated With Blood Pressure in Children and Adolescents. Circ. Cardiovasc. Genet 9, 266–278 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu X et al. Genome-wide association study in Chinese identifies novel loci for blood pressure and hypertension. Hum. Mol. Genet 24, 865–874 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He J et al. Genome-wide association study identifies 8 novel loci associated with blood pressure responses to interventions in Han Chinese. Circ. Cardiovasc. Genet 6, 598–607 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato N et al. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat. Genet 43, 531–538 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kato N et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat. Genet 47, 1282–1293 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly TN et al. Genome-wide association study meta-analysis reveals transethnic replication of mean arterial and pulse pressure loci. Hypertens. Dallas Tex 1979 62, 853–859 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wain LV et al. Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nat. Genet 43, 1005–1011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kraja AT et al. New Blood Pressure-Associated Loci Identified in Meta-Analyses of 475 000 Individuals. Circ. Cardiovasc. Genet 10, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Surendran P et al. Trans-ancestry meta-analyses identify rare and common variants associated with blood pressure and hypertension. Nat. Genet 48, 1151–1161 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu C et al. Meta-analysis identifies common and rare variants influencing blood pressure and overlapping with metabolic trait loci. Nat. Genet 48, 1162–1170 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wain LV et al. Novel Blood Pressure Locus and Gene Discovery Using Genome-Wide Association Study and Expression Data Sets From Blood and the Kidney. Hypertens. Dallas Tex 1979 (2017). 10.1161/HYPERTENSIONAHA.117.09438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ehret GB et al. The genetics of blood pressure regulation and its target organs from association studies in 342,415 individuals. Nat. Genet 48, 1171–1184 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffmann TJ et al. Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation. Nat. Genet 49, 54–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee D et al. A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet 47, 955–961 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finucane HK et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gusev A et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am. J. Hum. Genet 95, 535–552 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barbeira AN et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun 9, 1825 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gamazon ER et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet 47, 1091–1098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gusev A et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet 48, 245–252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Z et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet 48, 481–487 (2016). [DOI] [PubMed] [Google Scholar]
- 35.International Consortium for Blood Pressure Genome-Wide Association Studies et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 478, 103–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Consortium GTEx. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ko Y-A et al. Genetic-Variation-Driven Gene-Expression Changes Highlight Genes with Important Functions for Kidney Disease. Am. J. Hum. Genet 100, 940–953 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uhlén M et al. Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Pers TH et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat. Commun 6, 5890 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gamazon ER et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat. Genet 50, 956–967 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsuki K, Hathaway CK, Lawrence MG, Smithies O & Kakoki M The Role of Transforming Growth Factor β1 in the Regulation of Blood Pressure. Curr. Hypertens. Rev 10, 223–238 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lavoie P et al. Neutralization of transforming growth factor-beta attenuates hypertension and prevents renal injury in uremic rats. J. Hypertens 23, 1895–1903 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Dubois CM, Laprise MH, Blanchette F, Gentry LE & Leduc R Processing of transforming growth factor beta 1 precursor by human furin convertase. J. Biol. Chem 270, 10618–10624 (1995). [DOI] [PubMed] [Google Scholar]
- 44.Li N et al. Associations between genetic variations in the FURIN gene and hypertension. BMC Med. Genet 11, 124 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruschitzka F et al. Differential blood pressure effects of ibuprofen, naproxen, and celecoxib in patients with arthritis: the PRECISION-ABPM (Prospective Randomized Evaluation of Celecoxib Integrated Safety Versus Ibuprofen or Naproxen Ambulatory Blood Pressure Measurement) Trial. Eur. Heart J 38, 3282–3292 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rippe C et al. Hypertension reduces soluble guanylyl cyclase expression in the mouse aorta via the Notch signaling pathway. Sci. Rep 7, 1334 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bauersachs J et al. Vasodilator dysfunction in aged spontaneously hypertensive rats: changes in NO synthase III and soluble guanylyl cyclase expression, and in superoxide anion production. Cardiovasc. Res 37, 772–779 (1998). [DOI] [PubMed] [Google Scholar]
- 48.Ruetten H, Zabel U, Linz W & Schmidt HH Downregulation of soluble guanylyl cyclase in young and aging spontaneously hypertensive rats. Circ. Res 85, 534–541 (1999). [DOI] [PubMed] [Google Scholar]
- 49.Protogerou AD et al. Longitudinal Changes in Mean and Pulse Pressure, and All-Cause Mortality: Data From 71,629 Untreated Normotensive Individuals. Am. J. Hypertens 30, 1093–1099 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Yasuno S et al. Is Pulse Pressure a Predictor of New-Onset Diabetes in High-Risk Hypertensive Patients? Diabetes Care 33, 1122–1127 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Said MA, Eppinga RN, Lipsic E, Verweij N & Harst P van der. Relationship of Arterial Stiffness Index and Pulse Pressure With Cardiovascular Disease and Mortality. J. Am. Heart Assoc 7, e007621 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prenner SB & Chirinos JA Arterial stiffness in diabetes mellitus. Atherosclerosis 238, 370–379 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Xu M et al. Diabetes and Risk of Arterial Stiffness: A Mendelian Randomization Analysis. Diabetes 65, 1731–1740 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Bhosale SD et al. Serum Proteomic Profiling to Identify Biomarkers of Premature Carotid Atherosclerosis. Sci. Rep 8, 9209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee JH et al. Association between CDH13 Variants and Cardiometabolic and Vascular Phenotypes in a Korean Population. Yonsei Med. J 54, 1305–1312 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reschen ME, Lin D, Chalisey A, Soilleux EJ & O’Callaghan CA Genetic and environmental risk factors for atherosclerosis regulate transcription of phosphatase and actin regulating gene PHACTR1. Atherosclerosis 250, 95–105 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jarray R et al. Disruption of phactr-1 pathway triggers pro-inflammatory and pro-atherogenic factors: New insights in atherosclerosis development. Biochimie 118, 151–161 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Williams C, Kingwell BA, Burke K, McPherson J & Dart AM Folic acid supplementation for 3 wk reduces pulse pressure and large artery stiffness independent of MTHFR genotype. Am. J. Clin. Nutr 82, 26–31 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Girelli D et al. The Interaction between MTHFR 677 C→T Genotype and Folate Status Is a Determinant of Coronary Atherosclerosis Risk. J. Nutr 133, 1281–1285 (2003). [DOI] [PubMed] [Google Scholar]
- 60.Rapsomaniki E et al. Blood pressure and incidence of twelve cardiovascular diseases: lifetime risks, healthy life-years lost, and age-specific associations in 1·25 million people. Lancet Lond. Engl 383, 1899–1911 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bani D Relaxin: a pleiotropic hormone. Gen. Pharmacol 28, 13–22 (1997). [DOI] [PubMed] [Google Scholar]
- 62.Grossman J & Frishman WH Relaxin: a new approach for the treatment of acute congestive heart failure. Cardiol. Rev 18, 305–312 (2010). [DOI] [PubMed] [Google Scholar]
- 63.Teichman SL et al. Relaxin, a pleiotropic vasodilator for the treatment of heart failure. Heart Fail. Rev 14, 321–329 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Teerlink JR et al. Relaxin for the treatment of patients with acute heart failure (Pre-RELAX-AHF): a multicentre, randomised, placebo-controlled, parallel-group, dose-finding phase IIb study. Lancet Lond. Engl 373, 1429–1439 (2009). [DOI] [PubMed] [Google Scholar]
- 65.Bathgate R a. D. et al. Relaxin family peptides and their receptors. Physiol. Rev 93, 405–480 (2013). [DOI] [PubMed] [Google Scholar]
- 66.McNally T et al. Cloning and expression of the adenosine kinase gene from rat and human tissues. Biochem. Biophys. Res. Commun 231, 645–650 (1997). [DOI] [PubMed] [Google Scholar]
- 67.Shryock JC & Belardinelli L Adenosine and adenosine receptors in the cardiovascular system: biochemistry, physiology, and pharmacology. Am. J. Cardiol 79, 2–10 (1997). [DOI] [PubMed] [Google Scholar]
- 68.Böhm M [Cardiac effects of adenosine. Mechanism of action, pathophysiologic and clinical significance]. Klin. Wochenschr 65, 487–499 (1987). [DOI] [PubMed] [Google Scholar]
- 69.Echavarría-Pinto M et al. Low coronary microcirculatory resistance associated with profound hypotension during intravenous adenosine infusion: implications for the functional assessment of coronary stenoses. Circ. Cardiovasc. Interv 7, 35–42 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Camm AJ & Garratt CJ Adenosine and supraventricular tachycardia. N. Engl. J. Med 325, 1621–1629 (1991). [DOI] [PubMed] [Google Scholar]
- 71.Shen FM & Su DF The effect of adenosine on blood pressure variability in sinoaortic denervated rats is mediated by adenosine A2a-Receptor. J. Cardiovasc. Pharmacol 36, 681–686 (2000). [DOI] [PubMed] [Google Scholar]
- 72.Maass PG et al. PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat. Genet 47, 647–653 (2015). [DOI] [PubMed] [Google Scholar]
- 73.Houslay M Hypertension linked to PDE3A activation. Nat. Genet 47, 562–563 (2015). [DOI] [PubMed] [Google Scholar]
- 74.Schuster H et al. A cross-over medication trial for patients with autosomal-dominant hypertension with brachydactyly. Kidney Int 53, 167–172 (1998). [DOI] [PubMed] [Google Scholar]
- 75.Naraghi R et al. Neurovascular compression at the ventrolateral medulla in autosomal dominant hypertension and brachydactyly. Stroke 28, 1749–1754 (1997). [DOI] [PubMed] [Google Scholar]
- 76.Schunkert H et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet 43, 333–338 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Getman DK, Eubanks JH, Camp S, Evans GA & Taylor P The human gene encoding acetylcholinesterase is located on the long arm of chromosome 7. Am. J. Hum. Genet 51, 170–177 (1992). [PMC free article] [PubMed] [Google Scholar]
- 78.Singer W et al. Acetylcholinesterase inhibition: a novel approach in the treatment of neurogenic orthostatic hypotension. J. Neurol. Neurosurg. Psychiatry 74, 1294–1298 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Del Greco M,F et al. Genome-wide association analysis and fine mapping of NT-proBNP level provide novel insight into the role of the MTHFR-CLCN6-NPPA-NPPB gene cluster. Hum. Mol. Genet 20, 1660–1671 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Flister MJ et al. Identifying multiple causative genes at a single GWAS locus. Genome Res 23, 1996–2002 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Potter LR, Yoder AR, Flora DR, Antos LK & Dickey DM Natriuretic Peptides: Their Structures, Receptors, Physiologic Functions and Therapeutic Applications. Handb. Exp. Pharmacol 341–366 (2009). 10.1007/978-3-540-68964-5_15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brandt S & Jentsch TJ ClC-6 and ClC-7 are two novel broadly expressed members of the CLC chloride channel family. FEBS Lett 377, 15–20 (1995). [DOI] [PubMed] [Google Scholar]
- 83.Manichaikul A et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Loh P-R et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet 48, 1443–1448 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Das S et al. Next-generation genotype imputation service and methods. Nat. Genet 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Abraham G & Inouye M Fast Principal Component Analysis of Large-Scale Genome-Wide Data. PLOS ONE 9, e93766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chawla PS & Kochar MS Effect of pain and nonsteroidal analgesics on blood pressure. WMJ Off. Publ. State Med. Soc. Wis 98, 22–25, 29 (1999). [PubMed] [Google Scholar]
- 89.Maixner W, Gracely RH, Zuniga JR, Humphrey CB & Bloodworth GR Cardiovascular and sensory responses to forearm ischemia and dynamic hand exercise. Am. J. Physiol 259, R1156–1163 (1990). [DOI] [PubMed] [Google Scholar]
- 90.Taylor JY et al. A Genome-wide study of blood pressure in African Americans accounting for gene-smoking interaction. Sci. Rep 6, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marchini J, Howie B, Myers S, McVean G & Donnelly P A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet 39, 906–913 (2007). [DOI] [PubMed] [Google Scholar]
- 92.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinforma. Oxf. Engl 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang J, Lee SH, Goddard ME & Visscher PM GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet 88, 76–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Denny JC et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene–disease associations. Bioinformatics 26, 1205–1210 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carroll RJ, Bastarache L & Denny JC R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinforma. Oxf. Engl 30, 2375–2376 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Park J et al. Comprehensive single cell RNAseq analysis of the kidney reveals novel cell types and unexpected cell plasticity. bioRxiv 203125 (2017). 10.1101/203125 [DOI]
- 98.Kuhn M, Letunic I, Jensen LJ & Bork P The SIDER database of drugs and side effects. Nucleic Acids Res 44, D1075–1079 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Smith JC et al. Lessons Learned from Developing a Drug Evidence Base to Support Pharmacovigilance. Appl. Clin. Inform 4, 596–617 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cotto KC et al. DGIdb 3.0: a redesign and expansion of the drug-gene interaction database. Nucleic Acids Res (2017). 10.1093/nar/gkx1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li YH et al. Therapeutic target database update 2018: enriched resource for facilitating bench-to-clinic research of targeted therapeutics. Nucleic Acids Res 46, D1121–D1127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.