

Abstract
Copper is essential for normal growth and development of human organisms. The role of copper as a cofactor of important metabolic enzymes, such as cytochrome c oxidase, superoxide dismutase, lysyl oxidase, dopamine-β-hydroxylase, and many others, has been well established. In recent years, new regulatory roles of copper have emerged. Accumulating evidence points to the involvement of copper in lipid metabolism, antimicrobial defense, neuronal activity, resistance of tumor cells to platinum-based chemotherapeutic drugs, kinase-mediated signal transduction, and other essential cellular processes. For many of these processes, the precise mechanism of copper action remains to be established. Nevertheless, it is increasingly clear that many regulatory and signaling events are associated with changes in the intracellular localization and abundance of copper transporters, as well as distinct compartmentalization of copper itself. In this review, we discuss current data on regulation of the localization and abundance of copper transporters in response to metabolic and signaling events in human cells. Regulation by kinase-mediated phosphorylation will be addressed along with the emerging area of the redox-driven control of copper transport. We highlight mechanistic questions that await further testing.
1. COPPER AUTO REGULATES ITS CYTOPLASMIC LEVELS BY MODULATING THE AMOUNTS OF A HIGH-AFFINITY TRANSPORTER CTR1 AT THE PLASMA MEMBRANE
1.1. Acute Regulation of CTR1 through Copper-Dependent Trafficking
Copper enters human cells through at least two independent pathways. The high-affinity copper transporter CTR1 is responsible for the 60–70% of total copper influx, while the remaining uptake is mediated through a low-affinity transport mechanism (Lee, Petris, & Thiele, 2002). The identity of the low-affinity transporter(s) remains unknown. Several candidates have been proposed (Arredondo, Munoz, Mura, & Nunez, 2003; Zimnicka, Ivy, & Kaplan, 2011); the role of the divalent metal transporter DMT1 in copper acquisition in the brain has recently been questioned (Zheng, Chen, & Zheng, 2012). Human CTR1, hCTR1, is a homotrimer with a relatively long His/Met-rich N-terminal tail exposed at the extracellular side, three transmembrane (TM) segments, and a very short cytosolic C-terminus containing the His-Cys-His motif (Kaplan & Lutsenko, 2009; Puig et al., 2007). In the vast majority of cells, hCTR1 is responsible for acquisition of copper from the blood; in intestine, it plays an essential role in the dietary copper absorption (Nose, Kim, & Thiele, 2006). Copper uptake by hCTR1 is tightly controlled: the short-term regulation is mediated via copper-dependent changes in the abundance of hCTR1 at the plasma membrane (PM). Copper depletion increases the amount of hCTR1 at the PM through the recruitment from the intracellular pools (Kuo, Gybina, Pyatskowit, Gitschier, & Prohaska, 2006), whereas elevated copper induces rapid endocytosis of the transporter from the PM to vesicles (Guo, Smith, Lee, Thiele, & Petris, 2004) (Fig. 1).
Figure 1.
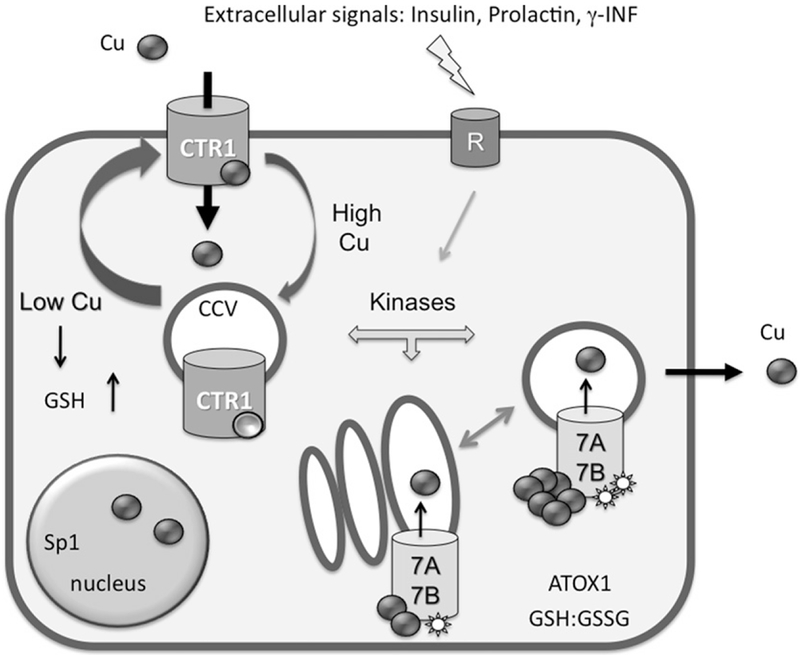
Main regulation modes of copper transporters in human cells. The high-affinity copper transporter hCTR1 is located at the PM, where it facilitates copper entry into the cell. It is currently unknown whether hCTR1 is functional in the intracellular compartments. The cycling between two compartments may be constitutive and involves the clathrin-dependent pathway (CCV, clathrin-coated vesicles). Copper binding to “sensor” sites favors the redistribution of hCTR1 toward intracellular vesicles, whereas copper depletion facilitates the delivery of hCTR1 to the PM. Copper-transporting ATPases 7A and 7B are located in the TGN under basal conditions where they have basal level of phosphorylation (indicated by a star). In response to copper elevation and hormonal signaling, the Cu-ATPases become hyperphosphorylated and traffic to vesicles, where they sequester excess copper. The return to the TGN is linked to the decrease in copper levels and return to a basal level of phosphorylation. Changes in glutathione (GSH) levels and/or alterations of the cellular redox balance GSH:GSSG influence the levels and activity of copper transporters.
The mechanisms through which copper triggers hCTR1 internalization and, reciprocally, the loss of copper initiates the return of hCTR1 to the PM are unclear. Biochemical and molecular modeling experiments revealed the presence of copper-binding sites at both the extracellular and cytosolic portions of hCTR1 (De Feo, Aller, Siluvai, Blackburn, & Unger, 2009; Haas, Putterman, White, Thiele, & Franz, 2011; Tsigelny et al., 2012). Mutations to Ala of residues at the entrance of the copper translocation pathway (Met150 and Met154 in the TM2) eliminate copper uptake. In contrast, mutations/deletions of Met, His, or Cys residues in the N-terminal or C-terminal tails of hCTR1, respectively, change the rate of transport (Du, Wang, Li, & Sun, 2012), but do not fully abolish the transport activity, pointing to the regulatory functions for the N-terminal and C-terminal copper-binding sites. The binding of copper to one or more of such regulatory sites and subsequent conformational changes in the protein (Eisses & Kaplan, 2002) are likely to be the first steps toward hCTR1 internalization. Further steps involve a clathrin-dependent pathway, as suggested by the co-localization of hCTR1 with the transferrin receptor and the inhibition of hCTR1 endocytosis by chlorpromazine and methyl-β-cyclodextrin (Petris, Smith, Lee, & Thiele, 2003).
1.2. Does Low or High Copper Serve as a Physiologically Relevant Signal for CTR1 Trafficking?
It should be noted that most of the current evidence for hCTR1 internalization and recycling has been obtained using the recombinant overexpressed protein. There is little doubt that the recombinant hCTR1 traffics to the intracellular compartments upon exposure to copper, whereas treatment with the copper chelator (or cisplatin) stabilizes protein levels at the PM (Guo et al., 2004). Given these observations, it might be worth considering why the results of early experiments on the endogenous hCTR1 are notably different from those with the overexpressed protein. In cells of various origin, the location of the endogenous hCTR1 was found to be predominantly intracellular. In addition to vesicles, the PM location was observed in HEK293 cells (Eisses et al., 2005), Caco-2 cells (Klomp, Tops, Van Denberg, Berger, & Klomp, 2002) and in polarized Jeg-3 cells (Hardman et al., 2006). High copper did not affect the PM localization of endogenous CTR1 in any of these cells, and no obvious internalization was observed. More recently, endocytosis of recombinant and endogenous hCTR1 was examined using surface biotinylation (Molloy & Kaplan, 2009). In these experiments, partial endocytosis (20–60%) was detected for both the recombinant and endogenous hCTR1 in some, but not all cell types. Internalization of endogenous CTR1 was seen in polarized MDCK cells and in HepG2 cells; no significant internalization of hCTR1 in response to extracellular copper was detected in HeLa and T47D cells (Molloy & Kaplan, 2009).
In mouse tissues, mCTR1 is also predominantly intracellular with a notable exception of the liver where it was immuno-detected at the PM (Hardman et al., 2006; Kuo et al., 2006; Ralle et al., 2010)]. In copper-deficient tissues, there is a distinct upregulation and recruitment of mCTR1 to the PM, especially in the intestine (Kuo et al., 2006). High copper has an opposite effect on protein localization, but not under all circumstances. The decreased levels of mCTR1 at the PM were observed in the liver tissue with a prolonged (20 weeks) intracellular accumulation of copper (Ralle et al., 2010), whereas high dietary copper did not seem to have such an effect (Araya et al., 2012). Altogether, these observations raise an interesting possibility that under physiologically relevant conditions the distribution of CTR1 between the intracellular and PM pools is controlled by the metabolic demands of a cell (i.e. by the need for copper for biosynthetic purposes) rather than by extracellular copper. This hypothesis would be consisted with a repeatedly observed increase in the CTR1 levels (total and at the PM) in response to copper deprivation. Hardman et al. (2006) have also found decreased intracellular staining of CTR1 in the insulin-treated Jeg-3 cells and proposed that insulin influenced the recruitment of hCTR1 to the PM through the mechanism resembling that of the glucose transporter GLUT4.
1.3. Copper Sensor Sites in CTR1 are Probably Cytosolic
How copper levels are sensed by CTR1 and then transferred into a signal for redistribution between cellular compartments is an interesting unresolved problem. The fact that the endogenous hCTR1 in HEK293 cells does not traffic in response to extracellular additions of copper, whereas a stably transfected hCTR1 does, suggests that the rate of copper entry and accumulation in the cytosol (which is higher in cells overexpressing hCTR1) may play an important role in triggering the internalization of the transporter. Consistent with this hypothesis, the recombinant CTR1, which lost its transport activity due to mutations of critical Met150 and Met154 (see above), does not traffic from the PM to vesicles despite the presence of the extracellular copper-binding sites at the N-terminal tail (Guo et al., 2004). Finally, since CTR1 internalization is a reversible process (i.e. copper depletion triggers the return of CTR1 to the PM) it seems that a “copper sensing” site is present at the cytosolic face of the molecule, where copper can be easily exchanged. Consistent with this idea, exposure of cells to higher copper delays the return of internalized CTR1 to the PM (Molloy & Kaplan, 2009), although this phenomenon could also be due to time needed to clear extra copper by the efflux systems.
1.4. Regulation of Copper Uptake by Modulation of Total Levels of CTR1
The long-term regulation of CTR1 is achieved through transcriptional/translational changes. The basal levels of CTR1 are influenced by the HIF2α transcription factor (Pourvali et al., 2012); however, which proteins directly control expression of CTR1 in response to changing copper levels or other metabolic needs is not entirely clear. Furthermore, similar to studies on CTR1 trafficking, there is some discrepancy in the literature on whether an elevated copper influences the levels of CTR1 messenger RNA (mRNA). Recent studies demonstrating that regulation of the CTR1 abundance could be affected by various metabolic factors offer potential explanation for such inconsistencies between the reports. For example, incubation of HepG2 cells in high copper results in decreased amounts of CTR1 mRNA and protein, whereas treatment with estradiol alleviates this copper effect (Arredondo et al., 2010). It is also interesting that overexpression of the exogenous CTR1 is associated with downregulation of the endogenous CTR1 mRNA indicating a tight homeostatic control of the total hCTR1 levels (Liang, Tsai, Lee, Savaraj, & Kuo, 2012).
The role of glutathione in regulating copper transport machinery is another intriguing finding. Overexpression of recombinant gamma-glutamyl-cysteine synthase γ-GCSht0090, a rate-limiting enzyme in the biosynthesis of glutathione, in human lung cancer cell line is associated with a two- to three-fold increase in the cellular levels of glutathione (Yamane et al., 1998). This, in turn, coincides with an increase in the CTR1 mRNA and protein level as well as increased copper uptake (Chen et al., 2008). In another set of experiments, the expression of CTR1 was shown to be upregulated upon copper depletion and downregulated under replete conditions. These changes in CTR1 levels paralleled changes in the levels of the transcription factor specificity protein 1 (Sp1) leading to the suggestion that Sp1 oscillations in response to copper availability regulate hCTR1 expression (Liang et al., 2012; Song et al., 2008).
Increases in the CTR1 mRNA and protein have also been observed in a cell model of inflammation. Treatment of BV-2 microglial cells with γ-interferon resulted in a two-fold increase of the CTR1 mRNA levels, increased rates of copper uptake, and higher copper accumulation (Zheng et al., 2010). Very similar phenomena, i.e. an increase in the CTR1 mRNA along with higher copper accumulation was observed in RAW264.7 cells exposed to hypoxic conditions (4% oxygen) (White, C., Kambe, T., Fulcher, Y.G., Sachdev, S.W., Bush, A.I., Fritsche, K., et al., 2009a). The mechanisms behind these interesting and potentially important regulatory events remain largely unexplored; however, the role of a kinase-mediated signaling appears increasingly likely. Recent study revealed an unexpected requirement for the CTR1-mediated copper uptake in signaling via the Ras/mitogen-activated protein (MAP) kinase pathway (Turski et al., 2012). In these studies, genetic inactivation of CTR1 in flies and in mouse cells as well as copper deficiency induced by metal chelation were shown to reduce the ability of the MAP kinase kinase Mek1 to phosphorylate the MAP kinase Extracellular signal regular Kinases (ERK1/2), whereas Akt kinase remained unaffected (Turski et al., 2012). Surprisingly, silver (which would be expected to mimic the effects of copper) had effect similar to copper chelation, i.e. it decreased cells’ response to insulin, as evidenced by the diminished ERK1/2 phosphorylation. An important finding in this set of experiments was the observation that the addition of copper to the CTR1+/+ cells did not potentiate insulin signaling, whereas the same treatment of the copper-deficient CTR1−/− cells partially restores the ability of insulin to signal via the ERK1/2 phosphorylation pathway. These results seem to imply that the CTR1 function in insulin signaling is to maintain normal cell metabolism, rather than directly deliver copper to a copper-dependent kinase(s), although there is a convincing evidence for the direct Cu2+ binding to Mek1 kinase in vitro (Turski et al., 2012).
2. REGULATION OF COPPER EXPORT VIA PROTEIN TRAFFICKING AND KINASE-MEDIATED PHOSPHORYLATION
2.1. ATP7A and ATP7B are Copper-Transporting ATPases Important for Maintaining Cellular Copper Homeostasis
Human cells express two homologous copper-exporting ATPases, ATP7A and ATP7B, that transport copper across a membrane bilayer by using the energy derived from ATP hydrolysis. Both ATP7A and ATP7B belong to the P-type ATPases family, i.e. during their catalytic cycle they form a transient phosphorylated intermediate (catalytic phosphorylation) that is stimulated by binding of the exported ion, copper. ATP7A/B contains eight TM domains and several cytosolic domains (Fig. 2). The cytosolic N-terminal domain is composed of six metal-binding domains (MBDs) that are connected by flexible loops. Each MBD binds one copper (Lutsenko, Petrukhin, Cooper, Gilliam, & Kaplan, 1997); the binding induces conformational changes within the domain (DiDonato, Hsu, Narindrasorasak, Que, & Sarkar, 2000) that are accompanied by the rearrangements of the loops connecting MBDs (Bartee, Ralle, & Lutsenko, 2009). The nucleotide-binding domain (NBD), the phosphorylation (P) domain and the actuator (A) domain are central for the P-type ATPase function. The NBD and the P-domain are involved in ATP binding and hydrolysis, whereas the A-domain is required for conformational changes during ATP hydrolysis. Both the N-terminal domain and C-terminal tails of ATP7A/B are important for proper targeting of the protein. The 1–63 region of the N-terminal domain of ATP7B contains a region (F37AFDNVGYE45), which is involved in trans-Golgi network (TGN) retention/apical targeting (Braiterman et al., 2009), whereas the C-terminal tail contains residues important for endocytosis (Cater, La Fontaine, Shield, Deal, & Mercer, 2006; Francis et al., 1999; Stephenson, Dubach, Lim, Mercer, & La Fontaine, 2005) and copper-responsive trafficking to the apical membrane (Braiterman, Nyasae, Leves, & Hubbard, 2011).
Figure 2.
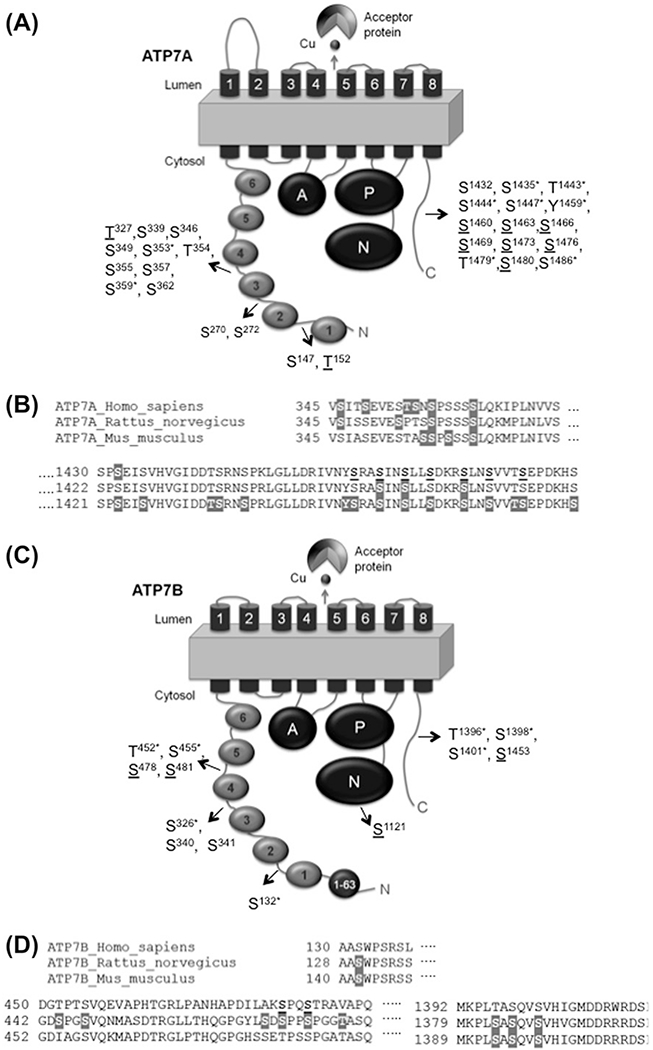
The location of phosphorylated sites in ATP7A and ATP7B. (A, C) Schematic presentation of the domain organization of ATP7A (A) and ATP7B (C). Filled circles represent the N-terminal MBDs and are numbered 1–6. TM domains are numbered 1–8. The other domains are as follows: A: actuator domain; P: phosphorylation domain; N: NBD. The 1–63 region of ATP7B contains a region (F37AFDNVGYE45) important for TGN retention/apical targeting (Braiterman et al., 2009). The positions of identified phosphosites are shown. Underlined residues are putative copper-responsive phosphosites; the star (*) labels the putative phosphosites identified in other species and aligned with human ATP7A or ATP7B. (B,D) Sequence alignment of ATP7A (B) and ATP7B (D) orthologs in the regions with identified phosphosites. Sites for basal/constitutive phosphorylation identified by MS are in gray background. Copper-responsive phosphosites are in bold and underlined.
2.2. Localization of Human Copper Efflux Transporters is Regulated
ATP7A and ATP7B have two main localizations within cells. One compartment is the TGN where these copper-ATPases supply the copper cofactor to cuproenzymes; the other compartment is vesicles-PM, to which copper-ATPases traffic in order to remove excess copper out of the cells (Gupta & Lutsenko, 2009). Copper regulates the distribution of ATP7A and ATP7B between these two locations. In basal or low copper, the TGN is the primary site for the localization and function of copper-ATPases; in high copper, the transporters exit the TGN and relocate to vesicles, which eventually fuse with the PM. This copper-regulated trafficking of ATP7A/7B is important for cellular copper homeostasis. Despite its central role, the exact mechanism of ATP7A7B trafficking between cellular compartments remains only partially characterized. It has been established that both ATP7A/7B undergo kinase-mediated phosphorylation and the level of phosphorylation is copper dependent and reversible. Therefore, the kinase-mediated phosphorylation of ATP7A/7B is likely to be an important regulatory step for trafficking of ATP7A/7B and/or for modulation of their copper transport function.
2.3. Both Copper-ATPases are Phosphorylated on Multiple Sites
Biochemical and mass spectrometry (MS) experiments suggest that both ATP7A and ATP7B are phosphorylated by kinases on serine residues (Vanderwerf, Cooper, Stetsenko, & Lutsenko, 2001; Vanderwerf & Lutsenko, 2002; Voskoboinik et al., 2003). The level of kinase-mediated phosphorylation is linked to the intracellular localization of copper-ATPases. Under basal conditions, while in TGN, ATP7A/B are basally phosphorylated. Upon copper elevation, the kinase-mediated phosphorylation increases, and this coincides with the trafficking of ATP7A/B to vesicles. Return to the basal media or copper depletion with chelators triggers trafficking back to the TGN. In parallel, the regulatory phosphorylation of ATP7A/7B reverts to its basal levels; suggesting that copper-dependent phosphorylation might be required for either exit from the TGN or retention in vesicles (Vanderwerf et al., 2001; Voskoboinik et al., 2003).
The identification of phosphorylation sites is necessary for determining the precise physiological role of ATP7A/7B phosphorylation. The in-cell and in vitro studies indicate that more than one residue is phosphorylated in either ATP7A or ATP7B (Pilankatta, Lewis, Adams, & Inesi, 2009; Vanderwerf et al., 2001; Voskoboinik et al., 2003). For ATP7B, it has been shown that the copper-responsive phosphorylation occurs at different site(s) than the basal phosphorylation site(s) and different regions of the protein are involved in these events (Vanderwerf et al., 2001). The C-terminal tail and the N-terminal domain of ATP7B are not required for basal phosphorylation, indicating that at least some of the basal phosphorylation site(s) are located in the Ser 796-Tyr 1384 region of ATP7B. On the other hand, the copper-induced phosphorylation requires the presence of the N-terminal domain (Vanderwerf et al., 2001), which is a target of phosphorylation both in vitro and in cells (Bartee et al., 2009). MS analysis of the in vitro phosphorylated N-terminal domain of ATP7B pointed to the loop connecting MBD3-MBD4 as a site for the kinase-mediated modification (Bartee et al., 2009). Other putative copper-responsive phosphorylation sites were identified by MS analysis of the full-length ATP7B expressed in mammalian cells (Table 1): these sites are located in the N-terminal domain (Ser 478 and Ser 481), the NBD (Ser 1121) and the C-terminal tail (Ser 1453) (Pilankatta et al., 2009; Pilankatta, Lewis, & Inesi, 2011).
Table 1.
Predicted phosphorylation sites in ATP7B
| Phosphosites/phosphopeptides | Type of phosphorylation | Effect of mutation | Ref. |
|---|---|---|---|
| [Ser 478 (N-terminal), Ser 481(N-terminal), Ser 1121 (NBD), Ser 1453 (C-terminal)]h | Copper responsive | Quadruple S > A mutant is not trafficking in response to copper. | (Pilankatta et al., 2009, 2011) |
| (340 SSSSHSPGSPPR)N,h | Basal/constitutive | S340/341 mutants are localized in vesicles. | (Bartee et al., 2009) |
| (270 SCVLNIEENIGQLLGVQSIQVSLENK)N,h (352 NQVQGTCSTTL)N,h (407 VISPEELR)N,h (415 AIEDMGFEASVVSESCSTNPLGNHSAGN)N,h (439 HSAGNSMVQTTDGTPTSVQEVAPHTGR)N,h (444 SMVQTTDGTPTSVQEVAPHTGR)N,h (496 GMTCASCVSNIER)N,h (512 EAGVLSVLVALMAGK)N,h (589 TNGITYASVALATSK)N,h (595 ASVALATSK)N,h |
Basal/constitutive | ND | (Bartee et al., 2009) |
| (140 AAS142WPSR)N,m | Basal/constitutive | ND | (Huttlin et al., 2010) |
| (1389 MKPLS1393AS1395QVS1398VHIGMDDR)*,C,m | Basal/constitutive | ND | (Huttlin et al., 2010) |
| (1389 MKPLSAS1395QVSVHI)C,m | Insulin dependent | ND (putative kinase motif: NEK6) | (Monetti et al., 2011) |
| (458 GLLTHQGPGYLSDSPPS474PGGTASQ)N,r | Insulin dependent | ND | (Fedjaev et al., 2012) |
| (128 AAS130WPSR)N,r | Basal/constitutive | ND | (Lundby et al., 2012) |
| (323 VS324LPDGLEK)N,r | Basal/constitutive | ND | (Lundby et al., 2012) |
| (431 VSSGNSVPQAVGDS444PGS447VQNMASDTR)*, N,r | Basal/constitutive | ND | (Lundby et al., 2012) |
| (457 GLLTHQGPGYLS468DS470PPS473PGGT477ASQK)*, N,r | Basal/constitutive | ND | (Lundby et al., 2012) |
| (1381 PLS1383AS1385QVS1388VHVGMDDR)*, C,r | Basal/constitutive | ND | (Lundby et al., 2012) |
| (1439 WSLLLS1444DR)*, C,r | Basal/constitutive | ND | (Lundby et al., 2012) |
Species: m-mouse; r-rat; h-human; N-N-terminal; C-C-terminal; ND: not determined.
The identified phosphopeptide has different phosphorylated residue combinations identified by MS, and different phospho-occupancy.
Similar to ATP7B, ATP7A has multiple phosphorylation sites. Residues associated with the basal and copper-induced phosphorylation have been mapped to the N-terminal domain and the C-terminal tail of ATP7A (Veldhuis et al., 2009). Several other phosphosites/phosphopeptides have been identified by the MS analysis and the results are summarized in Table 2 (Fedjaev et al., 2012; Huttlin et al., 2010; Lundby et al., 2012; Monetti, Nagaraj, Sharma, & Mann, 2011). Despite these important advances, it remains unclear which, if any, of these sites represent primary targets of phosphorylation and how stoichiometry of phosphorylation is regulated in response to changing copper levels. Mutation of two putative phosphorylation sites revealed that the corresponding ATP7A variants, ATP7AS1469A and ATP7AS1432A, have different intracellular localization, indicating that phosphorylation may have different consequences even when residues are located in the same domain (Veldhuis et al., 2009). At the same time, different signaling events may yield phosphorylation of the same region/sites triggering trafficking response similar to that induced by copper. For example, treatment of placental Jeg-3 cells with insulin stimulates ATP7A exit from the TGN and, interestingly, downregulates the ATP7B protein levels resulting in lower copper levels in cells (Hardman et al., 2006). In those earlier studies, the mechanism of insulin action was not explored. However, more recent studies using the large-scale MS identified numerous liver proteins phosphorylated following insulin signaling, including ATP7A and ATP7B. The residues phosphorylated in response to insulin were found in regions that were also known to be phosphorylated in response to changing copper level (Tables 1 and 2). How different signaling pathways intersect and interact with each other is a complex and fascinating question for further studies.
Table 2.
Predicted phosphorylation sites in ATP7A
| Phosphosites/phosphopeptides | Type of phosphorylation | Effects of mutation | Ref. |
|---|---|---|---|
| (141 ELVPELS147LDTGT152LEKK)*, N,h | Ser 147-Basal/constitutive Thr 152 copper responsive | ND | (Veldhuis et al., 2009) |
| (270 S270PS272YTNDSTATFIID)*,N,h | Basal/constitutive | ND | (Veldhuis et al., 2009) |
| (321 YNASSVT327PESLR)N,h | Copper responsive | ND | (Veldhuis et al., 2009) |
| (334 AIEAVS339PGLYR)N,h | Basal/constitutive | ND | (Veldhuis et al., 2009) |
| (345 VS346ITS349EVEST354S355NS357PSSSS362LQK)*,N,h | Basal/constitutive | ND | (Veldhuis et al., 2009) |
| (1430 SPS1432EISVHVGIDDTSR)C,h | Basal/constitutive | S1432A is mislocalized (vesicles in high Cu, proximal to basolateral PM) | (Veldhuis et al., 2009) |
| (1456 IVNYS1460RAS1463INS1466LLS1469DK)*,C,h | Copper responsive | S1469A is mislocalized (vesicles in high Cu, but no PM localization) | (Veldhuis et al., 2009) |
| (1473 S1473LNS1476VVTS1480EPDK)*,C,h | Copper responsive | S1473A; basal localization and Cu-induced trafficking similar to WT. | (Veldhuis et al., 2009) |
| (345 VSIASEVESTAS356S357PS359SSS362LQK)*,N,m | Basal/constitutive | ND | (Huttlin et al., 2010) |
| (1421 SPS1423EIS1426VHVGIDDT1434S1435RNS1438PR)*,C,m | Basal/constitutive | ND | (Huttlin et al., 2010) |
| (1447 IVNY1450S1451RAS1454INS1457LLS1460DKR)*,C,m | Basal/constitutive | ND | (Huttlin et al., 2010) |
| (1464 S1464LNS1467VVT1470S1471EPDKHS1477 LLVGDFEDDDTTL)*,C,m | Basal/constitutive | ND | (Huttlin et al., 2010) |
| (1446 DRIVNYS1452RAS1455INS1458LLS1461DKRSLN)*,C,m | Insulin dependent | ND; (putative kinase motifs: AURORA, AURORA-1, CK1, PKA, GSK3) | (Monetti et al., 2011) |
| (345 VS346ISSEVES353PTSS357PSSSSLQK)*,C,r | Basal/constitutive | ND | (Lundby et al., 2012) |
| (1454 AS1455INS1458LLSDK)*,C,r | Basal/constitutive | ND | (Lundby et al., 2012) |
| (1465 S1465LNSVVTSEPDK)C,r | Basal/constitutive | ND | (Lundby et al., 2012) |
Species: m-mouse; r-rat; h-human; N-N-terminal; C-C-terminal; ND: not determined.
The identified phosphopeptide has different phosphorylated residue combinations identified by MS, and different phospho-occupancy
Several studies have been done to identify the kinases involved in the phosphorylation of ATP7A and ATP7B. Protein kinase D (PKD) (Pilankatta et al., 2011) and casein kinase II (Vanderwerf et al., 2001) were suggested to be involved in kinase-mediated phosphorylation of ATP7B. In contrast, PKD does not seem to affect ATP7A, whereas protein kinase A and Rho GTPase Cdc 42 were implicated in ATP7A anterograde trafficking (Cobbold, Ponnambalam, Francis, & Monaco, 2002).
2.4. Copper-Induced Trafficking of ATP7B Depends on the Conformational State of the Transporter
The cytosolic copper chaperone Atox1 was shown to transfer copper to the regulatory MBDs of the Cu-ATPases and regulate their occupancy (Walker et al., 2004). Current data indicate that copper binding to N-terminal domain of both ATP7A and ATP7B is necessary for their copper-responsive phosphorylation (Vanderwerf et al., 2001; Voskoboinik et al., 2003). Upon binding, copper induces rearrangements of the loops connecting MBDs in the N-terminal domain of ATP7B and also facilitates phosphorylation by a kinase, suggesting that conformational changes due to copper binding might be important in the exposure of additional phosphorylation sites (Bartee et al., 2009). An ATP7A mutant which cannot bind copper (due to mutation of CxxC in MBDs to SxxS) (Strausak et al., 1999) has a basal level of phosphorylation, but shows no increase in phosphorylation upon copper treatment and no trafficking (Voskoboinik et al., 2003).
The Wilson disease-causing mutant ATP7BG591D is mislocalized to the endoplasmic reticulum (ER) under basal conditions (de Bie et al., 2007) and has a basal level of phosphorylation but no copper-induced phosphorylation (Vanderwerf et al., 2001). Similarly, the ATP7BR875 variant is also localized in ER under basal/copper-depleted conditions and is basally phosphorylated (Hasan et al., 2012). These findings indicate that the basal phosphorylation of ATP7B occurs early in the secretory pathway (presumably in the ER). The difference between ATP7BG591D and ATP7BR875 is that ATP7BR875 binds copper and under elevated copper conditions traffics to vesicles, which is also coupled with the increase in the kinase-mediated phosphorylation (Hasan et al., 2012).
2.5. The Sequence of Events Associated with the TGN Exit/Trafficking of Copper-ATPases
It is not yet clear how hyperphosphorylation and trafficking are linked, or which occurs first. Copper binding to the regulatory N-terminal domain of ATP7A/7B might expose additional phosphorylation sites and the addition of an extra negative charge (hyperphosphorylation) may induce further structural changes necessary for the exit of copper-ATPases from the TGN. Alternatively, copper binding to the ATPase and the subsequent structural changes may alone enable protein sorting within the TGN/interaction with the trafficking machinery, whereas hyperphosphorylation may be a result of the relocalization of ATP7A/7B to the correct vesicular compartment in which kinase(s) might be present.
Several sets of data support the important role of copper-induced structural changes in the initiation of ATP7B exit from the TGN. Copper binding not only influences the rearrangement of the N-terminal domain of ATP7B but also alters the domain–domain interactions within the entire ATP7B molecule. Relative to the copper-free form, copper-bound N-terminal domain interacts poorly with the NBD (Tsivkovskii, MacArthur, & Lutsenko, 2001), but strongly with the A-domain (Gupta et al., 2011). Since copper binding to the N-terminus of ATP7B alters the inter-domain contacts, the increased phosphorylation might be a result of the exposure of additional phosphorylation sites in more than one domain.
An alternative model is that the stabilization of ATP7B in a distinct conformational state is sufficient to cause its trafficking from TGN to vesicles. This model is supported by the observation that mutating residues that are not directly involved in copper binding may significantly alter the targeting and trafficking of ATP7B. The mutant 858TGE860 > AAA, which is catalytically inactive, is found in vesicles under basal copper conditions (Cater, La Fontaine, & Mercer, 2007). This targeting is associated with the stabilization of this mutant protein in the so-called E2 conformational state, as was demonstrated by the high level of catalytic phosphorylation (whether this hyperphosphorylation caused by the loss of phosphatase activity by the mutant is also coupled to the increase in regulatory kinase-mediated phosphorylation has not been explored). Mutating the six N-terminal MBDs and/or the TM Cys-Proc-Cys(CPC) motif individually results in a non-trafficking ATP7B; however, simultaneous mutation of all sites produces protein targeted to vesicles even in basal conditions (Cater et al., 2007). The fact that the conformation acquired by the ATP7BTGE>AAA variant is sufficient to trigger its trafficking to vesicles suggests that trafficking is driven by structural changes in ATP7B, rather than interactions with other copper-binding proteins or phosphorylation by copper-dependent kinases.
Similar results were obtained by mutating S340/341 residues of the N-terminal domain of ATP7B. The ATP7B mutants with S340/341 mutations are found in vesicles under basal conditions. Metabolic labeling in cells with inorganic phosphate directly demonstrated that mutation of S340/341 residues significantly decreased the level of ATP7B phosphorylation, indicating that these residues were important for ATP7B modification by a kinase (Hasan et al., 2012). Therefore, vesicular localization of the ATP7BSer340/341Gly protein was not due to mutation-induced hyperphosphorylation of some other residues, but rather due to conformational changes in ATP7B. Further studies revealed weakened interactions between the N-terminal and NBD domains in ATP7BS340/341A compared to control ATP7B (Hasan et al., 2012). Thus, the role of kinase-mediated phosphorylation might be to keep the N-terminal and NBD domains apart and stabilize the protein in a particular confirmation that can be recognized by the trafficking machinery, rather than acting as a specific recognition sequence for such machinery (Hasan et al., 2012). Although 858TGE860 and S340/341 residues are located in different domains of ATP7B, the mutations of these sites may have similar effects on the overall conformation of ATP7B and thus produce a similar trafficking response.
2.6. Copper Binding to the Regulatory N-terminal Sites and Catalytically Important TM Sites Has Different Effect on the Kinase-Mediated Phosphorylation of ATP7B
Mutating the N-terminal copper-binding site in MBD6 (ATP7BC575/578A) interferes with a kinase-mediated phosphorylation, whereas mutating the intramembrane TM6 copper-binding site (ATP7BC983/985A) does not have such an effect. Thus, copper binding to the MBD6 of N-terminal domain might be important for inducing the conformational changes within the domain so the phosphorylation sites can be accessed by the kinase. Neither mutant traffics in response to copper, i.e. the kinase-mediated phosphorylation of ATP7BC983/985A is not sufficient for triggering its relocalization. At the same time, the ATP7BC983/985A mutant has a lower phosphorylation level (or possibly a slower rate of phosphorylation) compared to the wild-type (WT) ATP7B, suggesting that the extent of phosphorylation might be important for ATP7B exit from the TGN or that this mutation indirectly altered other sites (Pilankatta et al., 2011).
2.7. The Catalytic and Kinase-Mediated Phosphorylation Occur through Different Mechanisms and are Independent Events
Both catalytic and kinase-mediated phosphorylation are stimulated by copper, but through different mechanisms (Pilankatta et al., 2011). Supporting the model that both phosphorylations are independent events, inhibition of the kinase-mediated phosphorylation using a kinase inhibitor does not affect the formation of catalytic phosphorylation by ATP7B (Pilankatta et al., 2011). Reciprocally, the catalytically inactive ATP7BD1027A and ATP7BD1027N mutants are phosphorylated indicating that the catalytic transfer of phosphate from ATP to D1027 is not necessary for their kinase-mediated phosphorylation (Bartee et al., 2009; Pilankatta et al., 2011). The same phenomenon was observed for the ATP7BC983/985A mutant, which had no catalytic phosphorylation, but showed kinase-mediated phosphorylation (Pilankatta et al., 2011). In the cases of ATP7BD1027N and ATP7BC983/985A mutants, the kinase-mediated phosphorylation level was lower (or the phosphorylation rate was slower compared to the WT), suggesting that the overall phosphorylation level of ATP7B might be altered as a result of catalytic impairment (Pilankatta et al., 2011).
In summary, both ATP7A and ATP7B undergo kinase-mediated phosphorylation, which is a regulatory posttranslational modification coupled to the intracellular localization of the protein. Copper binding to the N-terminal domain and/or trafficking to the post-Golgi compartments (vesicular/PM) is associated with hyperphosphorylation, but in some cases (ATP7BS340/341G or ATP7BTGE>AAA) the conformational state attained by the protein determines its localization irrespective of copper binding. Several putative phosphorylation sites have been identified; however, it remains unclear whether the stoichiometry of phosphorylation or phosphorylation of distinct residues are important for the copper-responsive trafficking of ATP7A/B.
3. REDOX CONTROL OF COPPER UPTAKE AND EFFLUX FROM HUMAN CELLS
As described in Section 1, copper deficiency is associated with the increase of cellular glutathione levels, which in turn stimulate production of hCTR1 (Tennant, Stansfield, Yamaji, Srai, & Sharp, 2002). Recent studies indicate that the cellular glutathione status and the intracellular redox potential may also have a significant effect on the copper export activity of ATP7A and ATP7B. It has been suggested that under basal conditions, both copper-ATPases can be glutathionylated, presumably at their metal-binding sites and that the glutathionylation of ATP7A/7B and their interaction with glutaredoxin 1 are affected by changes in copper levels (Singleton et al., 2010). The downregulation of glutaredoxins by Small interfering RNA(siRNA) resulted in increased intracellular copper accumulation (Singleton et al., 2010), which was interpreted as evidence for direct regulation of copper export by glutaredoxin 1. While the data are compelling, it remains to be established more firmly whether the diminished copper export is due to a direct oxidation of copper-ATPases or due to the effect of experimental manipulation on a cellular redox environment and interference with the copper delivery system. Recent data suggest that copper export can be controlled at the stage of copper transfer from the cytosolic chaperone to the copper-ATPases.
The copper-ATPases receive copper from a copper chaperone Atox1. This small cytosolic protein has a characteristic copper-binding site CxxC, which is maintained in the reduced state by glutathione (Hatori, Clasen, Hasan, Barry, & Lutsenko, 2012). Decrease in the cellular ratio of reduced to oxidized glutathione (GSH:GSSG) results in Atox1 oxidation and a diminished delivery of copper to the secretory pathway, as evidenced by the decreased levels of secreted holo-ceruloplasmin (Hatori et al., 2012). The redox potential of Atox1 is estimated at approximately −220 mV and, in resting cells, Atox1 is expected to be fully reduced. However, upon cell proliferation and differentiation, the intracellular environment changes significantly (Dong-Yun, Yu-Ru, Shan-Lin, Ya-Dong, & Lian, 2003; Markovic et al., 2009; Ranjan et al., 2006). In Caco-2 cells, where both ATP7A and ATP7B are expressed, the redox state of cellular GSH/GSSG pair changes to be 40 mV more oxidizing, when cells progress from proliferation to contact inhibition and differentiation (Nkabyo, Ziegler, Gu, Watson, & Jones, 2002). This shift would be sufficient to produce a partial oxidation of Atox1 and presumably influence copper excretion. Direct testing of these predictions would greatly expand our understanding between copper transport and cellular redox balance.
Overall, the current results suggest that copper transport in and out of cells is likely to fluctuate depending on cellular metabolic status, the stage of cell division, or differentiation. An example of significant changes in the routes of copper distribution is provided by the effects of hypoxia in macrophages (White, C., Kambe, T., Fulcher, Y.G., Sachdev, S.W., Bush, A.I., Fritsche, K., et al., 2009a). Oxygen limitation in RAW264.7 macrophage cells stimulates copper uptake likely due to increased expression of hCTR1. Under these conditions, copper transfer to the secretory pathway is also enhanced, as inferred from the trafficking of ATP7A to vesicles. At the same time, the activities of cytosolic superoxide dismutase and mitochondria cytochrome c oxidase are reduced in response to hypoxia (White, C., Kambe, T., Fulcher, Y.G., Sachdev, S.W., Bush, A.I., Fritsche, K., et al., 2009a). Thus, under hypoxic conditions copper delivery to the secretory pathway becomes the primary pathway of copper distribution, presumably to facilitate copper incorporation into ceruloplasmin.
In conclusion, human copper transporters are subject of a complex multilayer regulation by various metabolic signals. Recent success in understanding the structure and biochemistry of these transporters (see other chapters in this volume) provides strong foundation for trying to link the molecular characteristics of the transporters to their behavior in cells.
ACKNOWLEDGMENTS
We thank Dr Arnab Gupta and Dr Jack H. Kaplan for stimulating discussions. This work was supported by the grants from the National Institute of Health P01GM01067166 and R01DK071865 to S.L.
REFERENCES
- Araya M, Nunez H, Pavez L, Arredondo M, Mendez M, Cisternas F, et al. (2012). Administration of high doses of copper to capuchin monkeys does not cause liver damage but induces transcriptional activation of hepatic proliferative responses. Journal of Nutrition, 142(2), 233–237. [DOI] [PubMed] [Google Scholar]
- Arredondo M, Nunez H, Lopez G, Pizarro F, Ayala M, & Araya M (2010). Influence of estrogens on copper indicators: in vivo and in vitro studies. Biological Trace Element Research, 134(3), 252–264. [DOI] [PubMed] [Google Scholar]
- Arredondo M, Munoz P, Mura CV, & Nunez MT (2003). DMT1, a physiologically relevant apical Cu1+ transporter of intestinal cells. American Journal of Physiology Cell Physiology, 284(6), C1525–C1530. [DOI] [PubMed] [Google Scholar]
- Bartee MY, Ralle M, & Lutsenko S (2009). The loop connecting metal-binding domains 3 and 4 of ATP7B is a target of a kinase-mediated phosphorylation. Biochemistry, 48(24), 5573–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braiterman L, Nyasae L, Guo Y, Bustos R, Lutsenko S, & Hubbard A (2009). Apical targeting and Golgi retention signals reside within a 9-amino acid sequence in the copper-ATPase, ATP7B. American Journal of Physiology Gastrointestinal Liver Physiology, 296(2), G433–G444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braiterman L, Nyasae L, Leves F, & Hubbard AL (2011). Critical roles for the COOH terminus of the Cu-ATPase ATP7B in protein stability, trans-Golgi network retention, copper sensing, and retrograde trafficking. American Journal of Physiology Gastrointestinal Liver Physiology, 301(1), G69–G81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cater MA, La Fontaine S, & Mercer JF (2007). Copper binding to the N-terminal metal-binding sites or the CPC motif is not essential for copper-induced trafficking of the human Wilson protein (ATP7B). Biochemical Journal, 401(1), 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cater MA, La Fontaine S, Shield K, Deal Y, & Mercer JF (2006). ATP7B mediates vesicular sequestration of copper: insight into biliary copper excretion. Gastroenterology, 130(2), 493–506. [DOI] [PubMed] [Google Scholar]
- Chen HH, Song IS, Hossain A, Choi MK, Yamane Y, Liang ZD, et al. (2008). Elevated glutathione levels confer cellular sensitization to cisplatin toxicity by upregulation of copper transporter hCtr1. Molecular Pharmacology, 74(3), 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobbold C, Ponnambalam S, Francis MJ, & Monaco AP (2002). Novel membrane traffic steps regulate the exocytosis of the Menkes disease ATPase. Human Molecular Genetics, 11(23), 2855–2866. [DOI] [PubMed] [Google Scholar]
- de Bie P, van de Sluis B, Burstein E, van de Berghe PV, Muller P, Berger R, et al. (2007). Distinct Wilson’s disease mutations in ATP7B are associated with enhanced binding to COMMD1 and reduced stability of ATP7B. Gastroenterology, 133(4), 1316–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Feo CJ, Aller SG, Siluvai GS, Blackburn NJ, & Unger VM (2009). Three-dimensional structure of the human copper transporter hCTR1. Proceedings of the National Academy of Sciences of the United States of Amercia, 106(11), 4237–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato M, Hsu HF, Narindrasorasak S, Que L Jr., & Sarkar B (2000). Copper-induced conformational changes in the N-terminal domain of the Wilson disease copper-transporting ATPase. Biochemistry, 39(7), 1890–1896. [DOI] [PubMed] [Google Scholar]
- Dong-Yun S, Yu-Ru D, Shan-Lin L, Ya-Dong Z, & Lian W (2003). Redox stress regulates cell proliferation and apoptosis of human hepatoma through Akt protein phosphorylation. FEBS Letters, 542(1–3), 60–64. [DOI] [PubMed] [Google Scholar]
- Du X, Wang X, Li H, & Sun H (2012). Comparison between copper and cisplatin transport mediated by human copper transporter 1 (hCTR1). Metallomics. 4(7), 679–685. [DOI] [PubMed] [Google Scholar]
- Eisses JF, Chi Y, & Kaplan JH (2005). Stable plasma membrane levels of hCTR1 mediate cellular copper uptake. Journal of Biological Chemistry, 280(10), 9635–9639. [DOI] [PubMed] [Google Scholar]
- Eisses JF, & Kaplan JH (2002). Molecular characterization of hCTR1, the human copper uptake protein. Journal of Biological Chemistry, 277(32), 29162–29171. [DOI] [PubMed] [Google Scholar]
- Fedjaev M, Parmar A, Xu Y, Vyetrogon K, Difalco MR, Ashmarina M, et al. (2012). Global analysis of protein phosphorylation networks in insulin signaling by sequential enrichment of phosphoproteins and phosphopeptides. Molecular BioSystems, 8(5), 1461–1471. [DOI] [PubMed] [Google Scholar]
- Francis MJ, Jones EE, Levy ER, Martin RL, Ponnambalam S, & Monaco AP (1999). Identification of a di-leucine motif within the C terminus domain of the Menkes disease protein that mediates endocytosis from the plasma membrane. Journal of Cell Science, 112(Pt 11), 1721–1732. [DOI] [PubMed] [Google Scholar]
- Guo Y, Smith K, Lee J, Thiele DJ, & Petris MJ (2004). Identification of methionine-rich clusters that regulate copper-stimulated endocytosis of the human Ctr1 copper transporter. Journal of Biological Chemistry, 279(17), 17428–17433. [DOI] [PubMed] [Google Scholar]
- Gupta A, Bhattacharjee A, Dmitriev OY, Nokhrin S, Braiterman L, Hubbard AL, et al. (2011). Cellular copper levels determine the phenotype of the Arg875 variant of ATP7B/Wilson disease protein. Proceedings of the National Academy of Sciences of the United States of America, 108(13), 5390–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, & Lutsenko S (2009). Human copper transporters: mechanism, role in human diseases and therapeutic potential. Future Medicinal Chem, 1(6), 1125–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas KL, Putterman AB, White DR, Thiele DJ, & Franz KJ (2011). Model peptides provide new insights into the role of histidine residues as potential ligands in human cellular copper acquisition via Ctr1. Journal of American Chemical Society, 133(12), 4427–4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardman B, Manuelpillai U, Wallace EM, Monty JF, Kramer DR, Kuo YM, et al. (2006). Expression, localisation and hormone regulation of the human copper transporter hCTR1 in placenta and choriocarcinoma Jeg-3 cells. Placenta, 27(9–10), 968–977. [DOI] [PubMed] [Google Scholar]
- Hasan NM, Gupta A, Polishchuk E, Yu CH, Polishchuk R, Dmitriev OY, et al. (2012). Molecular events initiating the exit of a copper-transporting ATPase ATP7B from the trans-Golgi network. Journal of Biological Chemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatori Y, Clasen S, Hasan NM, Barry AN, & Lutsenko S (2012). Functional partnership of the copper export machinery and glutathione balance in human cells. Journal of Biological Chemistry, 287(32), 26678–26687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, et al. (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell, 143(7), 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan JH, & Lutsenko S (2009). Copper transport in mammalian cells: special care for a metal with special needs. Journal of Biological Chemistry, 284(38), 25461–25465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klomp AE, Tops BB, Van Denberg IE, Berger R, & Klomp LW (2002). Biochemical characterization and subcellular localization of human copper transporter 1 (hCTR1). Biochemical Journal, 364(Pt 2), 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YM, Gybina AA, Pyatskowit JW, Gitschier J, & Prohaska JR (2006). Copper transport protein (Ctr1) levels in mice are tissue specific and dependent on copper status. Journal of Nutrition, 136(1), 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Petris MJ, & Thiele DJ (2002). Characterization of mouse embryonic cells deficient in the ctr1 high affinity copper transporter. Identification of a Ctr1-independent copper transport system. Journal of Biological Chemistry, 277(43), 40253–40259. [DOI] [PubMed] [Google Scholar]
- Liang ZD, Tsai WB, Lee MY, Savaraj N, & Kuo MT (2012). Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Molecular Pharmacology, 81(3), 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundby A, Secher A, Lage K, Nordsborg NB, Dmytriyev A, Lundby C, et al. (2012). Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nature Communications, 3, 876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutsenko S, Petrukhin K, Cooper MJ, Gilliam CT, & Kaplan JH (1997). N-terminal domains of human copper-transporting adenosine triphosphatases (the Wilson’s and Menkes disease proteins) bind copper selectively in vivo and in vitro with stoichiometry of one copper per metal-binding repeat. Journal of Biological Chemistry, 272(30), 18939–18944. [DOI] [PubMed] [Google Scholar]
- Markovic J, Mora NJ, Broseta AM, Gimeno A, de-la-Concepcion N, Vina J, et al. (2009). The depletion of nuclear glutathione impairs cell proliferation in 3t3 fibroblasts. PLoS One, 4(7), e6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy SA, & Kaplan JH (2009). Copper-dependent recycling of hCTR1, the human high affinity copper transporter. Journal of Biological Chemistry, 284(43), 29704–29713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monetti M, Nagaraj N, Sharma K, & Mann M (2011). Large-scale phosphosite quantification in tissues by a spike-in SILAC method. Nature Methods, 8(8), 655–658. [DOI] [PubMed] [Google Scholar]
- Nkabyo YS, Ziegler TR, Gu LH, Watson WH, & Jones DP (2002). Glutathione and thioredoxin redox during differentiation in human colon epithelial (Caco-2) cells. American Journal of Physiology Gastrointestinal Liver Physiology, 283(6), G1352–G1359. [DOI] [PubMed] [Google Scholar]
- Nose Y, Kim BE, & Thiele DJ (2006). Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metabolism, 4(3), 235–244. [DOI] [PubMed] [Google Scholar]
- Petris MJ, Smith K, Lee J, & Thiele DJ (2003). Copper-stimulated endocytosis and degradation of the human copper transporter, hCtr1. Journal of Biological Chemistry, 278(11), 9639–9646. [DOI] [PubMed] [Google Scholar]
- Pilankatta R, Lewis D, Adams CM, & Inesi G (2009). High yield heterologous expression of wild-type and mutant Cuþ-ATPase (ATP7B, Wilson disease protein) for functional characterization of catalytic activity and serine residues undergoing copper-dependent phosphorylation. Journal of Biological Chemistry, 284(32), 21307–21316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilankatta R, Lewis D, & Inesi G (2011). Involvement of protein kinase D in expression and trafficking of ATP7B (copper ATPase). Journal of Biological Chemistry, 286(9), 7389–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourvali K, Matak P, Latunde-Dada GO, Solomou S, Mastrogiannaki M, Peyssonnaux C, et al. (2012). Basal expression of copper transporter 1 in intestinal epithelial cells is regulated by hypoxia-inducible factor 2alpha. FEBS Letters, 586(16): 2423–2427. [DOI] [PubMed] [Google Scholar]
- Puig S, Mira H, Dorcey E, Sancenon V, Andres-Colas N, Garcia-Molina A, et al. (2007). Higher plants possess two different types of ATX1-like copper chaperones. Biochemical and Biophysical Research Communications, 354(2), 385–390. [DOI] [PubMed] [Google Scholar]
- Ralle M, Huster D, Vogt S, Schirrmeister W, Burkhead JL, Capps TR, et al. (2010). Wilson disease at a single cell level: intracellular copper trafficking activates compartment-specific responses in hepatocytes. Journal of Biological Chemistry, 285(40), 30875–30883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjan P, Anathy V, Burch PM, Weirather K, Lambeth JD, & Heintz NH (2006). Redox-dependent expression of cyclin D1 and cell proliferation by Nox1 in mouse lung epithelial cells. Antioxidants & Redox Signaling, 8(9–10), 1447–1459. [DOI] [PubMed] [Google Scholar]
- Singleton WC, McInnes KT, Cater MA, Winnall WR, McKirdy R, Yu Y, et al. (2010). Role of glutaredoxin1 and glutathione in regulating the activity of the copper-transporting P-type ATPases, ATP7A and ATP7B. Journal of Biological Chemistry, 285(35), 27111–27121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song IS, Chen HH, Aiba I, Hossain A, Liang ZD, Klomp LW, et al. (2008). Transcription factor Sp1 plays an important role in the regulation of copper homeostasis in mammalian cells. Molecular Pharmacology, 74(3), 705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson SE, Dubach D, Lim CM, Mercer JF, & La Fontaine S (2005). A single PDZ domain protein interacts with the Menkes copper ATPase, ATP7A. A new protein implicated in copper homeostasis. Journal of Biological Chemistry, 280(39), 33270–33279. [DOI] [PubMed] [Google Scholar]
- Strausak D, La Fontaine S, Hill J, Firth SD, Lockhart PJ, & Mercer JF (1999). The role of GMXCXXC metal binding sites in the copper-induced redistribution of the Menkes protein. Journal of Biological Chemistry, 274(16), 11170–11177. [DOI] [PubMed] [Google Scholar]
- Tennant J, Stansfield M, Yamaji S, Srai SK, & Sharp P (2002). Effects of copper on the expression of metal transporters in human intestinal Caco-2 cells. FEBS Letters, 527(1–3), 239–244. [DOI] [PubMed] [Google Scholar]
- Tsigelny IF, Sharikov Y, Greenberg JP, Miller MA, Kouznetsova VL, Larson CA, et al. (2012). An all-atom model of the structure of human copper transporter 1. Cell Biochemistry and Biophysics, 63(3), 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsivkovskii R, MacArthur BC, & Lutsenko S (2001). The Lys1010-Lys1325 fragment of the Wilson’s disease protein binds nucleotides and interacts with the N-terminal domain of this protein in a copper-dependent manner. Journal of Biological Chemistry, 276(3), 2234–2242. [DOI] [PubMed] [Google Scholar]
- Turski ML, Brady DC, Kim HJ, Kim BE, Nose Y, Counter CM, et al. (2012). A novel role for copper in Ras/mitogen-activated protein kinase signaling. Molecular Cell Biology, 32(7), 1284–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderwerf SM, Cooper MJ, Stetsenko IV, & Lutsenko S (2001). Copper specifically regulates intracellular phosphorylation of the Wilson’s disease protein, a human copper-transporting ATPase. Journal of Biological Chemistry, 276(39), 36289–36294. [DOI] [PubMed] [Google Scholar]
- Vanderwerf SM, & Lutsenko S (2002). The Wilson’s disease protein expressed in Sf9 cells is phosphorylated. Biochemical Society Transactions, 30(4), 739–741. [DOI] [PubMed] [Google Scholar]
- Veldhuis NA, et al. (2009). Phosphorylation regulates copper-responsive trafficking of the Menkes copper transporting P-type ATPase. International Journal of Biochemistry & Cell Biology, 41(12), 2403–2412. [DOI] [PubMed] [Google Scholar]
- Voskoboinik I, Fernando R, Veldhuis N, Hannan KM, Marmy-Conus N, Pearson RB, et al. (2003). Protein kinase-dependent phosphorylation of the Menkes copper P-type ATPase. Biochemical and Biophysics Research Communications, 303(1), 337–342. [DOI] [PubMed] [Google Scholar]
- Walker JM, Huster D, Ralle M, Morgan CT, Blackburn NJ, & Lutsenko S (2004). The N-terminal metal-binding site 2 of the Wilson’s Disease Protein plays a key role in the transfer of copper from Atox1. Journal of Biological Chemistry, 279(15), 15376–15384. [DOI] [PubMed] [Google Scholar]
- White C, Kambe T, Fulcher YG, Sachdev SW, Bush AI, Fritsche K, et al. (2009a). Copper transport into the secretory pathway is regulated by oxygen in macrophages. Journal of Cell Science, 122(Pt 9), 1315–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane Y, Furuichi M, Song R, Van NT, Mulcahy RT, Ishikawa T, et al. (1998). Expression of multidrug resistance protein/GS-X pump and gamma-glutamylcysteine synthetase genes is regulated by oxidative stress. Journal of Biological Chemistry, 273(47), 31075–31085. [DOI] [PubMed] [Google Scholar]
- Zheng G, Chen J, & Zheng W (2012). Relative contribution of CTR1 and DMT1 in copper transport by the blood-CSF barrier: implication in manganese-induced neurotoxicity. Toxicology and Applied Pharmacology, 260(3), 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, White C, Lee J, Peterson TS, Bush AI, Sun GY, et al. (2010). Altered microglial copper homeostasis in a mouse model of Alzheimer’s disease. Journal of Neurochemistry, 114(6), 1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimnicka AM, Ivy K, & Kaplan JH (2011). Acquisition of dietary copper: a role for anion transporters in intestinal apical copper uptake. American Journal of Physiology Cell Physiology, 300(3), C588–C599 [DOI] [PMC free article] [PubMed] [Google Scholar]