

Abstract
Pharmacological treatments to decrease low-density lipoprotein (LDL) cholesterol (LDL-C) have limited effects on patients with homozygous familial hypercholesterolemia (HoFH). Since LDL receptors are located mainly in the liver, liver transplantation is considered to be the only way to correct the hepatic cholesterol metabolism abnormalities in HoFH. Liver transplantations, including those combined with heart transplantation, for HoFH have been increasing since 1984, making this a globally established therapeutic option for HoFH. Plasma LDL-C is reported to be dramatically lowered, by 80%, after transplantation, with the rapid regression of cutaneous and tendinous xanthomas. However, long-term cardiovascular benefits remain unclear. The major concerns about liver transplantation include surgical complications, the need for lifelong immunosuppressive therapy, and rejection. In addition, organ transplantations from deceased donors are extremely rare in Japan. We experienced two pediatric siblings with HoFH who received living-donor liver transplantations from their heterozygous parents. Their plasma LDL-C levels decreased immediately and stabilized at approximately 200 mg/dL. Both developed normally with the administration of lipid-lowering medications and have been free of severe problems for more than 10 years, to date, since transplantation. In Japan, where the shortage of deceased donors is critical, the combination of living-donor liver transplant from a heterozygous donor, that is, usually a parent, and medication is regarded as a valid therapeutic option for HoFH. Further studies and clinical experience are required to establish liver transplantation as a safe and effective treatment for HoFH.
Keywords: Familial hypercholesterolemia, Liver transplantation, Cholesterol metabolism
Introduction
Familial hypercholesterolemia (FH) is an autosomal dominant genetic disorder of cholesterol metabolism, resulting in elevated serum low-density lipoprotein (LDL) cholesterol (LDL-C) and a high incidence of coronary heart disease1–3). With the use of statins over the last few decades, the management of LDL-C in FH heterozygotes has improved markedly. On the other hand, pharmacological treatment has a limited effect on both homozygous and compound heterozygous FH cases. In recent years, Lomitapide, which reduces the serum level of LDL-C by inhibiting the function of the microsomal triglyceride transport protein, has been an available therapeutic option for homozygous FH (HoFH) in Japan4, 5). However, it has several adverse effects, such as hepatic steatosis and gastrointestinal disorders. LDL apheresis, which intensively lowers both the LDL-C level and the incidence of cardiovascular events, has been established as a standard treatment for HoFH in Japan3). However, LDL apheresis is known to be associated with several difficulties, including high cost, long-term maintenance of blood access, and poor quality of life in small infants6). In addition, the treatment must be repeated every one to two weeks.
The main pathogenic causes of FH are gene mutations affecting the LDL receptor or the pathways related to LDL receptor metabolism. Since an estimated 75% of LDL receptors are located in the liver7), liver transplantation achieves the replacement of dysfunctional hepatic LDL receptors in patients with HoFH, resulting in near-normal lipoprotein metabolism. Although advanced approaches aimed at achieving adequate LDL metabolism, such as gene therapy or cell transplantation, have also been research goals aiming to treat HoFH, liver transplantation is currently regarded as the only way to correct hepatic abnormal cholesterol metabolism in FH.
Positioning of Liver Transplantation for HoFH Treatment
The first patient undergoing liver transplantation for HoFH was reported by Starzl in 19848). The patient suffered repeated severe attacks of angina, requiring coronary artery bypass graft surgery. At age 6 years, she received combined heart and liver transplantation, resulting in her serum total cholesterol level decreasing from 1,225 to 300 mg/dL, or even lower. Unfortunately, the patient died 3 years after transplantation.
We identified 44 patients who underwent liver transplantation for HoFH in the literature since 1985, as shown in Table 1. Ten of these patients had received combined liver and cardiac transplantation, because some had already developed severe coronary artery disease due to hypercholesterolemia9). Case reports describing transplantation are now rarely accepted for publication by international peer-reviewed journals. Thus, the current number of liver transplantation cases for HoFH is unclear. In 2015, a retrospective study involving 36 patients with FH undergoing liver transplantation from 2008 to 2014 was reported from Iran10). In that study, though several transplant cases were collected, 20% of recipients were FH heterozygotes. The patient characteristics in the Iranian study suggested that the indications for liver transplantation for FH are still controversial. More recently, 8 patients undergoing liver transplantation for HoFH were reported from Turkey11). These reports suggest that liver transplantation has become globally established as a therapeutic option for HoFH.
Table 1. Reported Cases of Liver Transplantation for Familial Hypercholesterolemia.
| Age of recipient | Status of donor | Combined transplantation | Complications | Survival time at report | Year | Reference |
|---|---|---|---|---|---|---|
| 6 | DD | heart | 10 weeks | 1984 | 8 | |
| 17 | DD | heart | LF, HF | death after transplantation | 1985 | 19 |
| 12 | DD | heart | LF, HF | 6 months | 1986 | 20 |
| 6 | DD | heart | hypertension | 2 years | 1987 | 21 |
| 12 | DD | 6 months | 1988 | 22 | ||
| 6 | DD | heart | 5 years | 190 | 23 | |
| 17 | DD | heart | 1 month | 1990 | 23 | |
| 33 | DD | heart | HF | 1992 | 24 | |
| 4 | DD | 1 year | 1993 | 25 | ||
| 33 | DD | heart | 3.7 years | 1995 | 26 | |
| 9 | DD | heart | 9 years | 1995 | 27 | |
| 15 | DD | 3.8 years | 1995 | 28 | ||
| 10 | DD | LF | 1 year | 1995 | 28 | |
| 11 | DD | 1995 | 28 | |||
| 18 | DD | 13 months | 2000 | 29 | ||
| 16 | DD | 7 months | 2000 | 29 | ||
| 39 | DD | heart | HF | 3 years | 2000 | 9 |
| 46 | DD | heart | LF | 4 years | 2001 | 30 |
| 2 | LD | 3 years | 2003 | 16 | ||
| 25 | DD domino | 7 years | 2003 | 31 | ||
| 2008 | ||||||
| 3 | DD | 17 months | 2004 | 32 | ||
| 4 | LD | 1 year | 2005 | 33 | ||
| 16 | DD | 9 years | 2007 | 34 | ||
| 2 | LD | biliary obstruction | 2 years | 2007 | 17 | |
| 17 | DD domino | 1 year | 2009 | 35 | ||
| 15 | DD | 1 month | 2009 | 36 | ||
| 11 | LD | 1 month | 2009 | 36 | ||
| 13 | LD | 3 months | 2011 | 37 | ||
| 14 | LD | 3 months | 2011 | 37 | ||
| 9 | LD | 3 months | 2011 | 37 | ||
| 7 | DD | 14 months | 2011 | 38 | ||
| 7 | DD | LF | 3 years | 2011 | 38 | |
| 11 | DD | 6 months | 2011 | 14 | ||
| 14 | DD | 10 years | 2011 | 39 | ||
| 27 | DD | 3 months | 2014 | 40 | ||
| 7 | DD | 3 years | 2016 | 15 | ||
| 3 | DD | 6 years | 2016 | 13 | ||
| 8 | DD | 5 years | 2016 | 13 | ||
| 17 | DD | 6 years | 2016 | 13 | ||
| 15 | DD | 4 years | 2016 | 13 | ||
| 11 | DD | 1 year | 2016 | 13 | ||
| 3 | DD | 1 year | 2016 | 13 | ||
| 4 | DD | 1 year | 2016 | 13 | ||
| 2 | DD | 1 year | 2016 | 13 |
DD; deceased donor, LD; living donor, LF; liver failure, HF; heart failure
Worldwide, organs for medical transplantation are obtained from deceased donors. As shown in Table 1, livers can be transplanted from deceased donors in most HoFH cases. In contrast, a total of 7,474 liver transplantations, mainly for patients with cholestatic diseases, hepatocellular disease and acute liver failure, were carried out in Japan during the period up to 2013. This total included 7,255 living-donor and 219 deceased-donor transplantations, meaning that 97% of transplanted livers were donated by living subjects, while cadaveric liver transplantation was infrequent12). This bias in organ donation is a significant issue in the field of medical transplantation in Japan.
Effects of Liver Transplantation for HoFH
The plasma LDL-C levels were dramatically lowered, by 80%, post transplantation in patients with HoFH. At best, the lipid profile would be expected to improve beyond the LDL-C level of the donor, because the deficits of extrahepatic LDL receptors in recipients persist. The marked LDL-C reduction induced the rapid regression of cutaneous and tendinous xanthomas. Although long-term cardiovascular benefits are still unclear, coronary artery stenosis, as evaluated by angiography or intravascular ultrasound, has shown gradual improvement in parallel with LDL-C reductions in many cases13). On the other hand, several cases required coronary reperfusion procedures and there were a few mortalities due to progressive coronary artery disease after transplantation14). In addition, aortic valve stenosis may develop, despite liver transplantation with normalization of lipid profiles and no history of graft rejection15). The clinical course of the cases reported previously suggested that, even with a dramatic LDL-C reduction in response to liver transplantation, it may not be possible to slow the rates of aortic valve disease progression in HoFH once vascular disease has been established. Taken together, these observations indicate that taking the opportunity for liver transplantation is critical for patients with HoFH. The aggressive early lowering of LDL-C by liver transplantation, aimed at preventing the development of severe atherosclerosis and aortic valve stenosis, should be considered before irreversible vascular disease has occurred.
Critical Issues in Liver Transplantation for HoFH
Although liver transplantation for HoFH is presently viewed as the most effective strategy in preventing cardiovascular complications and achieving a definitive cure of this disease, critical concerns remain to be addressed. First, liver transplantation is a very high-risk procedure, necessitating extensive invasion and involving severe potential complications, although its outcomes are generally favorable. The 10-year survival rate exceeds 70% in Japan. Liver transplantation for HoFH cannot be viewed in the same way as that for other fatal diseases, which have no treatment options other than transplantation.
Second, long-term immunosuppressant therapy carries risks of numerous adverse effects. For instance, glucocorticoid administration may induce glucose intolerance, osteoporosis, and psychosomatic disorders. Tacrolimus, cyclosporine, and mycophenolate mofetil, which are widely used immunosuppressant agents at present, can cause renal damage, heart failure, and myelosuppression. Moreover, insufficient immunosuppression may lead to rejection, which is one of the most serious complications encountered in medical transplantation. Finally, a critical problem in Japan is the lack of organs from deceased donors. At the same time, candidate living donors, usually close relatives of the patient, are restricted in the case of living-donor transplantation.
Sibling Cases of Living-Donor Liver Transplantation for HoFH
We experienced two pediatric siblings with HoFH who received living-donor liver transplantations from their parents who were heterozygous for FH. The elder sibling, a boy, was the subject of the first case report of living-donor liver transplantation for FH worldwide16). Both patients developed normally with the administration of lipid-lowering medications and have remained free of severe problems for more than 10 years, to date, since undergoing transplantation.
The elder sibling had presented with an LDL-C level of 898 mg/dL and orange cutaneous xanthomas on the ankles and wrists bilaterally, leading to a diagnosis of HoFH at the age of 1 year. It took several years for him to become accustomed to LDL apheresis, and therapeutic liver transplantation was thus considered. Since the likelihood of receiving a transplant from a deceased donor is very low in Japan, we searched for a normolipidemic subject who might serve as a living donor for liver transplantation, but no suitable candidates were identified. His father's LDL-C level was approximately 170 mg/dL with the administration of simvastatin 10 mg, and that of his mother was approximately 200 mg/dL without medication, suggesting that both his parents were undiagnosed FH heterozygotes. The patient underwent living-donor liver transplantation at age 2 years and 5 months, with his father serving as the donor. His LDL-C concentration immediately dropped below 100 mg/dL postoperatively, and then rose gradually to approximately 200 mg/dL. The administration of pravastatin resulted in an LDL-C decrease to the range expected based on the LDL-C level of the donor. Fig. 1A shows the long-term LDL-C level course and the medications used after transplantation. The pravastatin dose was increased gradually, up to 20 mg orally per day. In addition, low-dose ezetimibe was administered, starting 9 years after liver transplantation, and was also increased gradually up to 10 mg daily. As shown in Fig. 1, his lipid profile remained well controlled for over 15 years. Moreover, the orange cutaneous xanthomas regressed slowly, and had disappeared 6 years after transplantation. No abnormalities were detected by occasional ultrasound examinations of the carotid artery and heart. Overall, he was growing and developing appropriately for his age, with no episodes of rejection or liver dysfunction.
Fig. 1.
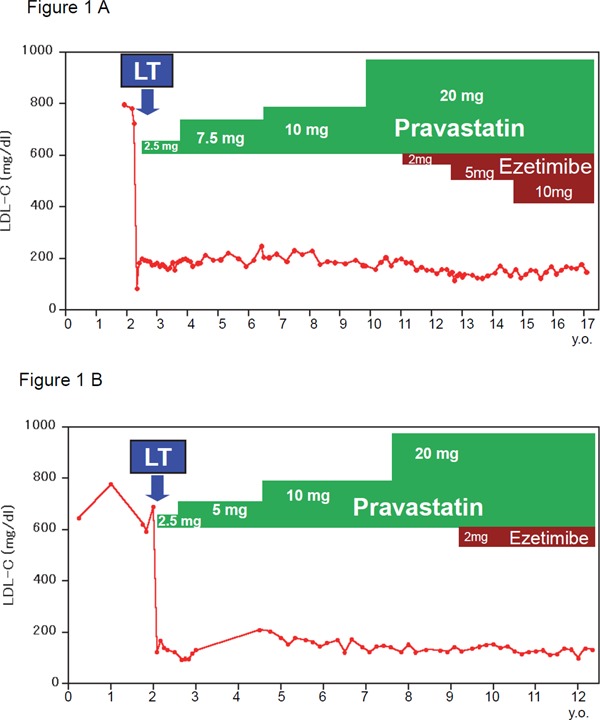
Time course of lipid profiles and lipid-lowering medication.
(A) Elder sibling, male; (B) younger sibling, female.
Line plot showing plasma LDL-C levels.
LT; liver transplantation.
Two years after the first patient underwent transplantation, his younger sister was born17). Her LDL-C was 776 mg/dL just after birth, similar to that of her brother. As the progress of her brother had been good after transplantation, liver transplantation was also considered as a therapeutic strategy for the baby. She was initially listed on the brain-dead donor series of Japan, and waited for 1 year, but no suitable donors became available. She underwent ABO-incompatible living-donor liver transplantation, at age 2 years, with her mother serving as the donor. Her LDL-C concentration decreased immediately after the operation. Pravastatin, as in her brother's case, was administered. Soon after transplantation, the younger sister suffered druginduced liver dysfunction, though not rejection, as demonstrated by a liver biopsy. Subsequently, 7 months after surgery, she developed common bile duct obstruction, but recovered with stent placement. As shown in Fig. 1B, the pravastatin dose was increased gradually to 20 mg per day, and the LDL-C concentration stabilized subsequently within the range of 130–170 mg/dL in response to combined statin and ezetimibe therapy. Despite health problems associated with liver transplantation, such as liver dysfunction and biliary congestion, she developed appropriately for her age.
Both donors have been doing well, without major problems, for more than 10 years, to date, since hepatic transplantation. Both have continued to take lipid-lowering medications, resulting in reasonably good LDL-C concentrations given their heterozygous FH status.
Gene mutation analysis of the LDL receptor gene revealed that the father had a gene mutation at intron 12 (c.1845 + 2T > C) and the mother had a mutation at exon 15 (c.2193dupC). Thus, these siblings, who underwent liver transplantations from their parents, have compound heterozygous mutations of the LDL receptor gene.
Parents of homozygous FH patients are nearly always heterozygous for FH, and thus benefit from statin therapy. Given that, in Japan, the shortage of deceased donors is critical, a therapeutic strategy combining liver transplantation from a donor heterozygous for FH with cholesterol-lowering drugs is thought to be a promising option for patients who are homozygous or compound heterozygous for FH.
Conclusion
The consensus panel on HoFH of the European Atherosclerosis Society noted that liver transplantation corrects the molecular defects of LDL clearance, resulting in a marked improvement in LDL-C levels; however, there are obvious disadvantages, including surgical complications and the necessity for lifelong immunosuppressive therapy18). Therefore, they positioned liver transplantation as a limited management option applicable only to selected patients.
Based on both the fundamental therapeutic effects and the critical problems of liver transplantation for HoFH, treatment decisions in these cases require careful deliberation. Especially, further studies and clinical experience are needed to establish living-donor liver transplantation, from heterozygous FH donors to HoFH recipients, as a safe and effective therapeutic option.
Acknowledgments
We greatly appreciate the generous assistance provided by Hiroshi Mabuchi, Atsushi Nohara, and Hayato Tada from Kanazawa University in performing the genetic analyses.
This work was supported by Research on rare and intractable disease, Health, Labour and Welfare Sciences Research Grants.
Conflicts of Interest Statement
The authors reported the following disclosures: Yasushi Ishigaki has received honoraria from Astellas Pharmaceutical Co., Novartis Pharma K.K., Tanabe- Mitsubishi Pharma Co., Ltd., MSD Co., Ltd., Takeda Pharmaceutical Co., and clinical research funding from Ono Pharmaceutical Co., Ltd.; Hideki Katagiri has received honoraria from Astellas Pharmaceutical Co., Daiichi Sankyo Co. Ltd., Taisho Toyama Pharmaceutical Co., Nippon Boehringer Ingelheim Co. Ltd., and clinical research funding from Ono Pharmaceutical Co., Ltd., Novo Nordisk., Astellas Pharmaceutical Co., Daiichi Sankyo Co. Ltd., Tanabe-Mitsubishi Pharma Co., Ltd., Taisho Toyama Pharmaceutical Co., and Sanofi K.K.
References
- 1). Mabuchi H: Half a Century Tales of Familial Hypercholesterolemia (FH) in Japan. J Atheroscler Thromb, 2017; 24: 189-207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2). Harada-Shiba M, Arai H, Ishigaki Y, Ishibashi S, Okamura T, Ogura M, Dobashi K, Nohara A, Bujo H, Miyauchi K, Yamashita S, Yokote K: Guidelines for Diagnosis and Treatment of Familial Hypercholesterolemia 2017. J Atheroscler Thromb, 2018; 25: 751-770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3). Harada-Shiba M, Ohta T, Ohtake A, Ogura M, Dobashi K, Nohara A, Yamashita S, Yokote K: Guidance for Pediatric Familial Hypercholesterolemia 2017. J Atheroscler Thromb, 2018; 25: 539-553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4). Harada-Shiba M, Ikewaki K, Nohara A, Otsubo Y, Yanagi K, Yoshida M, Chang Q, Foulds P: Efficacy and Safety of Lomitapide in Japanese Patients with Homozygous Familial Hypercholesterolemia. J Atheroscler Thromb, 2017; 24: 402-411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5). Nohara A, Otsubo Y, Yanagi K, Yoshida M, Ikewaki K, Harada-Shiba M, Jurecka A: Safety and Efficacy of Lomitapide in Japanese Patients with Homozygous Familial Hypercholesterolemia (HoFH): Results from the AEGR-733-301 Long-Term Extension Study. J Atheroscler Thromb, 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6). Makino H, Tamanaha T, Harada-Shiba M: LDL apheresis in Japan. Transfus Apher Sci, 2017; 56: 677-681 [DOI] [PubMed] [Google Scholar]
- 7). Bilheimer DW, Goldstein JL, Grundy SM, Starzl TE, Brown MS: Liver transplantation to provide low-density-lipoprotein receptors and lower plasma cholesterol in a child with homozygous familial hypercholesterolemia. N Engl J Med, 1984; 311: 1658-1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8). Starzl TE, Bilheimer DW, Bahnson HT, Shaw BW, Jr., Hardesty RL, Griffith BP, Iwatsuki S, Zitelli BJ, Gartner JC, Jr., Malatack JJ, Urbach AH: Heart-liver transplantation in a patient with familial hypercholesterolaemia. Lancet, 1984; 1: 1382-1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9). Alkofer BJ, Chiche L, Khayat A, Deshayes JP, Lepage A, Saloux E, Reznik Y: Liver transplant combined with heart transplant in severe heterozygous hypercholesterolemia: report of the first case and review of the literature. Transplant Proc, 2005; 37: 2250-2252 [DOI] [PubMed] [Google Scholar]
- 10). Mansoorian M, Kazemi K, Nikeghbalian S, Shamsaeefar A, Mokhtari M, Dehghani SM, Bahador A, Salahi H, Amoozgar H, Malek Hosseini SA: Liver transplantation as a definitive treatment for familial hypercholesterolemia: A series of 36 cases. Pediatr Transplant, 2015; 19: 605-611 [DOI] [PubMed] [Google Scholar]
- 11). Alim A, Tokat Y, Erdogan Y, Gokkaya Z, Dayangac M, Yuzer Y, Oezcelik A: Liver transplantation for homozygote familial hypercholesterolemia: the only curative treatment. Pediatr Transplant, 2016; 20: 1060-1064 [DOI] [PubMed] [Google Scholar]
- 12). Umeshita K, Inomata Y, Furukawa H, Kasahara M, Kawasaki S, Kobayashi E, Kokudo N, Sakisaka S, Shimada M, Tanaka E, Uemoto S: Liver transplantation in Japan: Registry by the Japanese Liver Transplantation Society. Hepatol Res, 2016; 46: 1171-1186 [DOI] [PubMed] [Google Scholar]
- 13). Martinez M, Brodlie S, Griesemer A, Kato T, Harren P, Gordon B, Parker T, Levine D, Tyberg T, Starc T, Cho I, Min J, Elmore K, Lobritto S, Hudgins LC: Effects of Liver Transplantation on Lipids and Cardiovascular Disease in Children With Homozygous Familial Hypercholesterolemia. Am J Cardiol, 2016; 118: 504-510 [DOI] [PubMed] [Google Scholar]
- 14). El-Rassi I, Chehab G, Saliba Z, Alawe A, Jebara V: Fatal cardiac atherosclerosis in a child 10 years after liver transplantation: a case report and a review. J Clin Lipidol, 2011; 5: 329-332 [DOI] [PubMed] [Google Scholar]
- 15). Greco M, Robinson JD, Eltayeb O, Benuck I: Progressive Aortic Stenosis in Homozygous Familial Hypercholesterolemia After Liver Transplant. Pediatrics, 2016; 138: [DOI] [PubMed] [Google Scholar]
- 16). Shirahata Y, Ohkohchi N, Kawagishi N, Syouji M, Tsukamoto S, Sekiguchi S, Koyamada N, Oikawa S, Satomi S: Living-donor liver transplantation for homozygous familial hypercholesterolemia from a donor with heterozygous hypercholesterolemia. Transpl Int, 2003; 16: 276-279 [DOI] [PubMed] [Google Scholar]
- 17). Kawagishi N, Satoh K, Akamatsu Y, Sekiguchi S, Ishigaki Y, Oikawa S, Satomi S: Long-term outcome after living donor liver transplantation for two cases of homozygous familial hypercholesterolemia from a heterozygous donor. J Atheroscler Thromb, 2007; 14: 94-98 [DOI] [PubMed] [Google Scholar]
- 18). Cuchel M, Bruckert E, Ginsberg HN, Raal FJ, Santos RD, Hegele RA, Kuivenhoven JA, Nordestgaard BG, Descamps OS, Steinhagen-Thiessen E, Tybjaerg-Hansen A, Watts GF, Averna M, Boileau C, Boren J, Catapano AL, Defesche JC, Hovingh GK, Humphries SE, Kovanen PT, Masana L, Pajukanta P, Parhofer KG, Ray KK, Stalenhoef AF, Stroes E, Taskinen MR, Wiegman A, Wiklund O, Chapman MJ: Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J, 2014; 35: 2146-2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19). Shaw BW, Jr., Bahnson HT, Hardesty RL, Griffith BP, Starzl TE: Combined transplantation of the heart and liver. Ann Surg, 1985; 202: 667-672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20). Figuera D, Ardaiz J, Martin-Judez V, Pulpon LA, Pradas G, Cuervas-Mons V, Burgos R, Arcas M, Pardo F, Cienfuegos JA: Combined transplantation of heart and liver from two different donors in a patient with familial type IIa hypercholesterolemia. J Heart Transplant, 1986; 5: 327-329 [PubMed] [Google Scholar]
- 21). Fricker FJ, Griffith BP, Hardesty RL, Trento A, Gold LM, Schmeltz K, Beerman LB, Fischer DR, Mathews RA, Neches WH, Park SC, Zuberbuhler JR, Lenox CC, Bahnson HT: Experience with heart transplantation in children. Pediatrics, 1987; 79: 138-146 [PubMed] [Google Scholar]
- 22). Valdivielso P, Escolar JL, Cuervas-Mons V, Pulpon LA, Chaparro MA, Gonzalez-Santos P: Lipids and lipoprotein changes after heart and liver transplantation in a patient with homozygous familial hypercholesterolemia. Ann Intern Med, 1988; 108: 204-206 [DOI] [PubMed] [Google Scholar]
- 23). Bahnson HT, Gordon RD: Transplantation of other organs with the heart. Cardiovasc Clin, 1990; 20: 237-248 [PubMed] [Google Scholar]
- 24). Barbir M, Khaghani A, Kehely A, Tan KC, Mitchell A, Thompson GR, Yacoub M: Normal levels of lipoproteins including lipoprotein(a) after liver-heart transplantation in a patient with homozygous familial hypercholesterolaemia. Q J Med, 1992; 85: 807-812 [PubMed] [Google Scholar]
- 25). Sokal EM, Ulla L, Harvengt C, Otte JB: Liver transplantation for familial hypercholesterolemia before the onset of cardiovascular complications. Transplantation, 1993; 55: 432-433 [DOI] [PubMed] [Google Scholar]
- 26). Rela M, Muiesan P, Heaton ND, Corbally M, Hajj H, Mowat AP, Williams R, Tan KC: Orthotopic liver transplantation for hepatic-based metabolic disorders. Transpl Int, 1995; 8: 41-44 [DOI] [PubMed] [Google Scholar]
- 27). Tellez de Peralta G, Burgos Lazaro R: [Multiple organ transplants]. Rev Esp Cardiol, 1995; 48 Suppl 7: 46-50 [PubMed] [Google Scholar]
- 28). Revell SP, Noble-Jamieson G, Johnston P, Rasmussen A, Jamieson N, Barnes ND: Liver transplantation for homozygous familial hypercholesterolaemia. Arch Dis Child, 1995; 73: 456-458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29). Lopez-Santamaria M, Migliazza L, Gamez M, Murcia J, Diaz-Gonzalez M, Camarena C, Hierro L, De la Vega A, Frauca E, Diaz M, Jara P, Tovar J: Liver transplantation in patients with homozygotic familial hypercholesterolemia previously treated by end-to-side portocaval shunt and ileal bypass. J Pediatr Surg, 2000; 35: 630-633 [DOI] [PubMed] [Google Scholar]
- 30). Offstad J, Schrumpf E, Geiran O, Soreide O, Simonsen S: Plasma exchange and heart-liver transplantation in a patient with homozygous familial hypercholesterolemia. Clin Transplant, 2001; 15: 432-436 [DOI] [PubMed] [Google Scholar]
- 31). Popescu I, Simionescu M, Tulbure D, Sima A, Catana C, Niculescu L, Hancu N, Gheorghe L, Mihaila M, Ciurea S, Vidu V: Homozygous familial hypercholesterolemia: specific indication for domino liver transplantation. Transplantation, 2003; 76: 1345-1350 [DOI] [PubMed] [Google Scholar]
- 32). Moyle M, Tate B: Homozygous familial hypercholesterolaemia presenting with cutaneous xanthomas: response to liver transplantation. Australas J Dermatol, 2004; 45: 226-228 [DOI] [PubMed] [Google Scholar]
- 33). Khalifeh M, Faraj W, Heaton N, Rela M, Sharara AI: Successful living-related liver transplantation for familial hypercholesterolemia in the Middle East. Transpl Int, 2005; 17: 735-739 [DOI] [PubMed] [Google Scholar]
- 34). Schmidt HH, Tietge UJ, Buettner J, Barg-Hock H, Offner G, Schweitzer S, Dedoussis GV, Rodeck B, Kallfelz HC, Schlitt HJ, Oldhafer K, Klempnauer J: Liver transplantation in a subject with familial hypercholesterolemia carrying the homozygous p.W577R LDL-receptor gene mutation. Clin Transplant, 2008; 22: 180-184 [DOI] [PubMed] [Google Scholar]
- 35). Liu C, Niu DM, Loong CC, Hsia CY, Tsou MY, Tsai HL, Wei C: Domino liver graft from a patient with homozygous familial hypercholesterolemia. Pediatr Transplant, 2010; 14: E30-33 [DOI] [PubMed] [Google Scholar]
- 36). Kakaei F, Nikeghbalian S, Kazemi K, Salahi H, Bahador A, Dehghani SM, Dehghani M, Nejatollahi SM, Shamsaeefar A, Khosravi MB, Malek-Hosseini SA: Liver transplantation for homozygous familial hypercholesterolemia: two case reports. Transplant Proc, 2009; 41: 2939-2941 [DOI] [PubMed] [Google Scholar]
- 37). Kucukkartallar T, Yankol Y, Kanmaz T, Topaloglu S, Acarli K, Kalayoglu M: Liver transplantation as a treatment option for three siblings with homozygous familial hypercholesterolemia. Pediatr Transplant, 2011; 15: 281-284 [DOI] [PubMed] [Google Scholar]
- 38). Maiorana A, Nobili V, Calandra S, Francalanci P, Bernabei S, El Hachem M, Monti L, Gennari F, Torre G, de Ville de Goyet J, Bartuli A: Preemptive liver transplantation in a child with familial hypercholesterolemia. Pediatr Transplant, 2011; 15: E25-29 [DOI] [PubMed] [Google Scholar]
- 39). Palacio CH, Harring TR, Nguyen NT, Goss JA, O'Mahony CA: Homozygous familial hypercholesterolemia: case series and review of the literature. Case Rep Transplant, 2011; 2011: 154908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40). Page MM, Ekinci EI, Jones RM, Angus PW, Gow PJ, O'Brien RC: Liver transplantation for the treatment of homozygous familial hypercholesterolaemia in an era of emerging lipid-lowering therapies. Intern Med J, 2014; 44: 601-604 [DOI] [PubMed] [Google Scholar]