

Abstract
Atypical hemolytic uremic syndrome (aHUS) is a type of thrombotic microangiopathy (TMA) defined by thrombocytopenia, microangiopathic hemolytic anemia, and renal failure. aHUS is caused by uncontrolled complement activation in the alternative pathway (AP). A variety of genetic defects in complement-related factors or acquired autoantibodies to the complement regulators have been found in 50 to 60% of all cases. Recently, however, the classification and diagnosis of aHUS are becoming more complicated. One reason for this is that some factors, which have not been regarded as complement-related factors, have been reported as predisposing factors for phenotypic aHUS. Given that genotype is highly correlated with the phenotype of aHUS, careful analysis of underlying genetic abnormalities or acquired factors is needed to predict the prognosis or to decide an optimal treatment for the disease. Another reason is that complement dysregulation in the AP have also been found in a part of other types of TMA such as transplantation-related TMA and pregnancy-related complication. Based on these findings, it is now time to redefine aHUS according to the genetic or acquired background of abnormalities.
Here, we review the pathogeneses and the corresponding phenotypes of aHUS and complement-related TMAs.
Keywords: Atypical hemolytic uremic syndrome, Complement, Alternative pathway
Introduction
Thrombotic microangiopathy (TMA) is a pathological condition caused by the formation of microvascular thrombi, leading to thrombocytopenia, microangiopathic hemolytic anemia, and organ damage. Various hereditary or acquired etiologies are associated with TMA. The most common forms of TMAs are thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS) caused by the infection of Shiga-toxin producing Escherichia coli (STEC), named STEC-HUS. TTP arises from severe deficiency of a disintegrin-like and metalloprotease with thrombospondin type 1 motif, 13 (ADAMTS13)1–3), which is a specific cleaving protease of von Willebrand factor (VWF). Uncleaved VWF multimers due to the lack of ADAMTS13 activity promote thrombi formation in small vessels. STECHUS is the most frequent form of HUS, and predominantly found in children. Shiga-toxin binds globotriaosylceramide on target cell surface and leads to cytotoxicity including protein synthesis and apoptosis, and also induces the secretion of unusually large VWF multimers from endothelial cells4).
Historically, the term “atypical HUS (aHUS)” was used to describe HUS not caused by the infection of STEC. Therefore, a category of aHUS included not only complement-related TMA, but also TMAs caused by various factors, such as drug, malignancy, pregnancy, transplantation, etc. However, since 1980s, various clinical and experimental studies have shown that 50% to 60% cases of aHUS arise from inherited and/or acquired complement abnormalities in the alternative pathway (AP). According to these progresses, the term “aHUS” came to be used to only describe complement-mediated aHUS and was distinguished from TMAs with coexisting disease or triggers, which were named “secondary TMA” in the criteria of Japan5, 6). The classification of aHUS and secondary TMA, however, remains controversial. More recently, KDIGO controversies conference report has stated that the term “primary aHUS” was preferentially used instead of “aHUS”7). Moreover, in this conference report, “secondary TMAs” were re-included into the categories of aHUS, and the use of etiology-based terminology (e.g. pregnancy-aHUS, drug-aHUS) was introduced. To avoid confusion in clinical practice, consistency in the terminology is needed. In this article, the term “aHUS” is used to describe complement-related aHUS, and “secondary TMA” to indicate TMA caused by various underlying diseases or factors.
The Complement System
The complement system is an essential part of innate immunity, and is activated via three pathways: classical pathway, lectin pathway, and AP. Activation any one of these pathways eventually leads to the opsonization of pathogens, and the generation of anaphylatoxin and membrane attack complex (MAC).
Uncontrolled complement activation in the AP is strongly linked with the pathogenesis of aHUS. The activation of both classical and lectin pathway needs specific initiator, but the AP is activated spontaneously at a low level by the hydrolysis of complement factor C38, 9) (Fig. 1). C3, the most abundant complement protein in plasma, plays an important role in complement activation. The internal thioester bond in C3 is easily hydrolyzed by H2O, leading to the formation of C3(H2O). The interaction of C3(H2O) with complement factor B (CFB) followed by cleavage by complement factor D generates C3(H2O)Bb. This reaction occurs constantly and is called “tick-over”. The molecule of C3(H2O)Bb works as an initial fluid phase C3 convertase, which cleaves C3 into C3a and C3b. The C3b fragment can bind covalently to any surface via its thioester, and forms C3 convertase (C3bBb) in the presence of CFB and CFD. This reaction creates a feedforward loop that cleavages more C3 into C3a and C3b (“C3 amplification”). Furthermore, C3 convertase can recruit another C3b to form C3bBbC3b (C5 convertase) and generate C5a and C5b through C5 cleavage. The C5b molecule binds to C6, C7, C8 and C9 to create MAC (C5b-9), which cause a direct lysis of target by the formation of membrane pores. The anaphylatoxins, C3a and C5a have a strong chemotactic influence on phagocytes, and also cause an increase in vascular permeability via releasing histamine10).
Fig. 1.
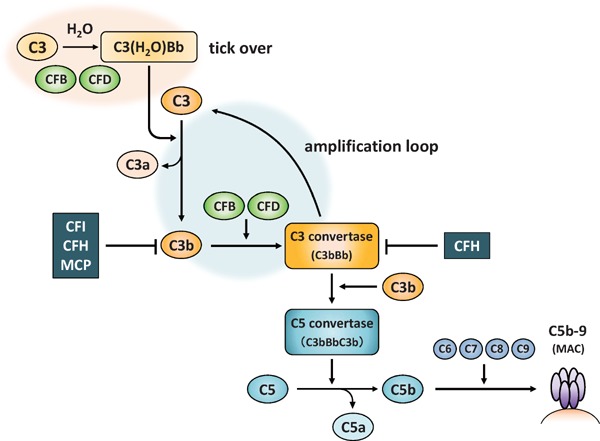
Alternative pathway of complement
CFB: complement factor B, CFD: complement factor D, CFI: complement factor I, CFH: complement factor H, MCP: membrane cofactor protein, MAC: membrane attack complex
Generally, C3(H2O)Bb or C3b is inactivated rapidly by various complement regulatory factors both in fluid phase and on host cell surface. On the other hand, once C3b binds to pathogens, which lack a complement regulatory protein, bound C3b preferentially reacts with CFB and CFD and causes the disruption of cell via the formation of MAC. In this manner, the complement attack occurs selectively on foreign substances, but not on the host cells.
Complement Dysregulation in the Patients with aHUs
In the AP, complement factor I (CFI) is a main protease for C3b inactivation. This regulatory process needs specific cofactor like complement factor H (CFH) and membrane cofactor protein (MCP). In the patients with aHUS, uncontrolled complement activation is caused by the loss of function variants in complement regulatory proteins or gain-of-function variants in complement activation factors. We here describe the detailed functions of complement-related factors and their abnormalities associated with aHUS (Table 1).
Table 1. Summary of the genetic or acquired complement abnormalities associated with aHUS.
| Genetic or acquired abnormalities | Frequency (%) | Main effect of mutant or acquired antibody | |
|---|---|---|---|
| Complement regulatory factors | CFH | 20%–30% |
|
| CFH/CFHR hybrid | - |
|
|
| CFI | 4%–8% |
|
|
| MCP | 8%–10% |
|
|
| anti-CFH antibody | 5%–20% | Inhibition of the complement regulatory function of CFH | |
| Complement activation factors | C3 | 4%–8% |
|
| CFB | < 1%–4% |
|
|
| Coagulation-related factors | THBD | 3%–5% |
|
| DGKE | 8% | Upregulation of prothrombotic factors and platelet activation** | |
| PLG | - | Reduced fibrinolytic activity** | |
| Other | INF2 | - | -** |
CFH: complement factor H, CFHR: complement factor H related, CFI: complement factor I, MCP: membrane cofactor protein, C3: complement component C3, CFB: complement factor B, THBD: thrombomodulin, DGKE: diacylglycerol kinase epsilon, PLG: plasminogen, INF2: inverted formin 2, TAFI: thrombin-activatable fibrinolysis inhibitor (plasma procarboxypeptidase B)
Some predisposing variants reside in N-terminal region of CFH show impaired cofactor activity for CFI.
Pathologic mechanism of aHUS caused by DGKE, PLG, INF2 variants has not been well described.
Genetic Defect of Complement Regulatory Factors
Complement Factor H
CFH is a major complement regulatory factor in the AP, and consists of 20 short consensus proteins (SCRs), each of which has 60 amino residues. CFH functions not only as a cofactor for CFI converting C3b to inactivation form, but also as a decay-accelerating factor via competing with CFB in binding to C3b. This regulatory function depends on the N-terminal region of CFH (SCRs 1–4) containing C3bbinding site11). By contrast, C-terminal region (SCRs 19–20) contains both C3b12, 13) and surface glycan-binding site14, 15). Accordingly, CFH SCRs 19–20 are capable of binding to host surface like endothelial cells via glycosaminoglycans like heparin14) or sialic acid15) and can exert complement regulatory effect16).
Predisposing variants in CFH are the most frequent abnormalities in aHUS as they account for 20% to 30% of cases17–20). On the other hand, the frequency of genetic abnormalities in CFH is estimated to be less in Japan compared to that in Western countries and the US, at about 10%21, 22). The variants in CFH associated with aHUS are mostly heterozygous, are located in SCRs19–2018, 19), and seem to affect the protein function, but not a quantitative deficiency. Several studies have shown that the pathogenicity of these variants is due to impaired CFH binding to C3b, or heparin or sialic acid23, 24) expressed on host cell surface, leading to increased C3b and C5b-9 deposition onto host cells.
Complement Factor H Related (CFHR) Protein
The genes encoding the five CFHR (CFHR1–5) proteins reside in close proximity to CFH on chromosome 1q32. Each CFHR proteins have four to nine SCRs, which are high sequence identity to C-terminal region of CFH. Functionally, CFHR3, CFHR4, and CFHR5 proteins show the cofactor activity25, 26), whereas CFHR1 and CFHR2 proteins are likely to inhibit the formation of C5 convertase27), C3 convertase28), respectively. However, these complement regulatory effect are mostly observed at non-physiological concentrations. In addition to these findings, enhanced complement activation via CFHR proteins was also reported; CFHR1 and CFHR5 protein may compete with CFH for binding to bacterial ligands and C3b29, 30).
Although physiological functions of CFHR proteins remain incompletely characterized, the genetic deletions of these proteins are associated with aHUS. Due to an extremely high sequence identity in the CFH and CFHR gene family, various non-allele homologous recombinations can occur31). The homozygous deletion of CFHR1 is frequently found in the patients with anti-CFH antibodies as described in the following section. Hybrid protein consisting of CFH and CFHR were also predisposed to aHUS. For instance, a hybrid gene consisting of SCRs1–18 of CFH and SCRs4–5 of CFHR1 encodes a protein identical to S1191L/V1197A CFH mutant protein32). These changes cause the impaired control of complement activation on host cell surfaces due to the lack of CFH binding to C3b33). Another hybrid protein having SCRs1–4 of CFHR1 and SCRs19–20 of CFH competes with CFH for surface C3b binding34). So far, six different patterns of hybrid have been reported in aHUS31, 35, 36).
Complement Factor I
Serine protease of CFI works as a critical regulator of complement activation that cleaves C3b in the presence of specific cofactor like CFH and MCP. Generally, predisposing variants described in aHUS are heterozygous, and the frequency is reported to be 4% to 8%17, 20, 37), but no patients have been identified in Japan until now21, 22). The majority of variants are located in the exons, which encode the serine protease domain20). Predisposing variants result in impaired secretion of CFI or decreased proteolytic activity both in fluid phase and/or on cell surfaces38, 39).
Membrane Cofactor Protein (CD46)
MCP is a widely expressed transmembrane glycoprotein, and a cofactor for CFI-mediated cleavage of C3b on the cell surface. The extracellular N-terminal domains consist of four SCRs, which are responsible for C3b binding. The frequency of MCP variants in aHUS is reported to be 8% to 10%17–19), and 5% in Japan22). Most of predisposing variants related to aHUS are heterozygous and clustered in four extracellular SCRs region40). Generally, these variants reduce MCP expression, whereas some of them result in functional defect such as reduced C3b binding capacity and cofactor activity40).
Genetic Defect of Complement Activation Factors
Complement Component C3
C3 is a pivotal component in complement system and mainly synthesized by the liver. C3 variants have been detected in 4% to 8% patients with aHUS17, 18), but its frequency in Japan was much higher (31%)21, 22). The majority of predisposing variants is heterozygous and clusters on the surface predominantly around CFH binding site41). The functional analyses of C3 variants have shown that mutant C3 reduced the binding affinity for CFH and/or MCP, resulted in impaired CFI-mediated inactivation of C3b41–43). Moreover, one variant in C3 p.R139W has been reported to increase the binding affinity of C3 for CFB, leading to a hyperactive C3 convertase formation44).
In Japan, 32 of 104 patients with aHUS had predisposing variants in C322) and 24 of 32 patients belonging to 16 families had the same p.I1157T variant. This was located in the thioester containing domain of C3, and was likely to be resistant to inactivation by CFI in the presence of MCP43). A geographical distribution of the patients carrying this variant was found in a restricted area (Kansai district), especially in Mie21, 45). A prevalent C3 variant of p.R161W has also been reported in France44) and Nethrland46), the frequency of this variant was 42% (14 of 33 cases) and 78% (11 of 14 cases) of aHUS patients with C3 variants, respectively41).
Complement Factor B
CFB is a zymogen that carries the catalytic site for C3 convertase. CFB variants in aHUS are rare, accounting for only < 1%–4%20, 47). Pathogenic variants of CFB found in aHUS facilitate the formation of either hyperactive C3 convertase or convertase resistant to inactivation by complement regulatory factors47, 48). Patients carrying CFB variants generally show decreased C3 levels due to permanent activation of the AP. Marinozzi MC, et al. have performed detailed structural and functional analysis of CFB variants and suggested that 9 of 15 variants had no correlation with aHUS48). This finding suggests that the importance of the functional assessment of identified variants in aHUS patients.
Acquired Abnormalities Related to aHUS
Acquired autoantibodies against CFH have been identified in 5%–20% of patients with aHUS18, 49, 50) and 19% in Japan22). Interestingly, extremely high frequency of antibody-positive patients, an incidence rate of 56%, was reported in India51). Children aged 5 to 10 years old can be predominantly affected, but CFH antibodies are also found in adult52). Antibodies generally recognize the C-terminal region of CFH, and are demonstrated to inhibit the complement regulatory function of CFH on cell surface53–56).
Antibodies production against CFH is highly associated with the genetic deletion of CFHR1 protein49, 54, 57). Deletions are generally homozygous and sometimes accompanied with the genetic deletion of CFHR3 or CFHR449, 54). Of note, genetic deletion of CFHR1 is also found in healthy individuals, and its frequencies vary by the ethnic origin57–61). Therefore, it remains unclear how homozygous deletion of CFHR1 gene leads to anti-CFH antibodies production. Bhattacharjee A, et al.62) have proposed one hypothesis to this question. They have showed that CFH has autoantigenic epitope, which can be expressed when microbial molecules bind close to this area. CFHR1 protein has a structurally similar to autoangtigenic conformation of CFH and might function as an immune tolerance. Therefore, the loss of immune tolerance due to the absence of CFHR1 may attribute to autoimmune type of aHUS.
Genetic Variants Unrelated to the Complement System in aHUs
Various factors not associated with complement system have been reported to lead the phenotypic aHUS. We here describe three coagulation-related proteins and one protein associated with nephrotic syndrome as the predisposing factors for aHUS.
Patients with aHUS having variants of thrombomodulin (THBD) were firstly described in 200963). Thrombomodulin is anticoagulant glycoprotein, but it also facilitates CFI-mediated C3b inactivation in the presence of cofactor like CFH, and enhances thrombin mediated activation of plasma procarboxypeptidase B (TAFI), which inactivates complement anaphylatoxins C3a and C5a63). The frequency of THBD variants in aHUS is estimated to be 3%–5%18, 63). Generally, predisposing variants are heterozygous and cluster in the lectin-like domain or the serine-threonine rich region63). These variants seem to induce excess activation of the AP by reduced CFI-mediated conversion of C3b into inactivated C3b and the activation of TAFI63). It is debatable whether thrombomodulin-associated aHUS can be classified as complement-associated.
Diacylglycerol kinase epsilon (DGKE), a lipid kinase family protein, shows anticoagulant effect via diacylglycerols-mediated activation of protein kinase C. In 2013, Lemaire, et al. have identified that the recessive DGKE variants in 13 aHUS patients belonging to 9 unrelated families64). Predisposing variants are generally homozygous or compound heterozygous, and sometimes reside in intronic region65). Although the data regarding the frequency was still limited, Schaefer F, et al. have reported that DGKE variants were identified in 8% of the 101 patients tested66). Underlying pathophysiologic mechanism of DGKE-associated aHUS is still unclear, but one possibility is that the loss of function of DGKE results in upregulation of prothrombotic factors and platelet activation. Although some aHUS patients showed the low levels of C367, 68), the relevance to complement system is still controversial69).
Plasminogen (PLG) is the inactivate precursor of plasmin and can degrade fibrin clot. One published paper has identified four genetic variants in the gene of PLG in four patients with aHUS70), and three of four variants were known as plasminogen deficiency-related variants71). Therefore, the authors have suggested that reduced fibrinolytic activity compromised the degradation of thrombi in aHUS. Hyvarinen S, et al. have reported that plasminogen was not likely to inhibit complement activation both on erythrocytes and endothelial cells, but it hindered platelet aggregation72).
More recently, Challis, et al. have performed whole-exome sequencing of patients with aHUS, who not responsive to anti-complement drug eculizumab, and identified the two variants in the gene of inverted formin 2 (INF2) in four patients belonging to two families73). INF2 is a formin protein that has actin polymerization and depolymerization activity, and the variants in this protein are known to cause familial autosomal dominant nephrotic syndrome74). Two missense variants of INF2 detected in aHUS resided in the diaphanous inhibitory domain, which was a mutational hot spot for focal segmental glomerulosclerosis. No efficacy of eculizumab treatment was reported so far, therefore the pathogenesis of INF2-mediated aHUS may be independent of complement activation.
Influence of Each Complement Abnormality on Clinical Characteristics and Prognosis of aHUs
The clinical manifestation and prognosis of aHUS are sometimes affected by each abnormality in aHUS-causing factors. Scaefer F, et al. have recently reported the phenotype and genotype correlation of 851 patients in the Grobal aHUS Registry, prior to eculizumab treatment66). The investigators have shown that the age at onset of initial aHUS was significantly affected by variants in MCP or CFI. Patients with CFH variants showed increased risk of end-stage renal disease (ESRD), whereas the patients with MCP variants were linked with longer ESRD-free survival, which has also been confirmed by previous reports18, 19, 37). Although the reports are limited, patients carrying DGKE variants generally presented aHUS with multiple relapsing episodes and proteinuria before the age of first year64, 65, 67, 75).
We have recently performed the epidemiological and genetic studies of Japanese patients with aHUS, and identified the following results22). The renal survival rate of the patients carrying MCP variant or anti-CFH antibodies was much better compared to CFH, C3, and the unidentified group. The risk of ESRD in the antibody-positive patients is likely to be significantly lower compared to previous report52), although the reason for this observation is still unclear. Another finding is that the different characteristics between the patients with C3 p.I1157T, a prevalent variant in Japan, and with other C3 variants. C3 p.I1157T were associated with high recurrence of aHUS compared to other C3 variants, but showed better renal outcomes both in the acute phase and during long-time follow-up. Furthermore, most of patients carrying C3 p. I1157T achieved remission with only supportive care or plasma therapy. These results have suggested that clinical presentation might be influenced differently according to individual variants even if the kind of defective protein is the same.
Underlying Complement Activation in Other Types of TMA
In recent years, several research groups have suggested that the genetic or acquired complement abnormalities in the AP were also related to the pathogenesis of secondary TMAs. Jodele S, et al.76) have analyzed six pediatric patients with hematopoietic stem-cell transplant (HSCT)-TMA, and revealed that five of six patients had the deletions in CFHR3 and CFHR1 gene, and three of six patients had anti-CFH antibodies. Another study performed by the same group revealed that an elevated soluble C5b-9 (sC5b-9), markers of terminal complement activation in the fluid phase, may be a good predictor of disease prognosis in the patients with HSCT-TMA77). The patients with HSCT-TMA presenting proteinuria and increased levels of sC5b-9 at the time of TMA diagnosis showed very poor survival (< 20% at 1 year), whereas HSCT-TMA patients having no proteinuria with normal serum C5b-9 had a survival of 100%.
Pregnancy can be a strong trigger for TMA. A retrospective study of 100 female patients with aHUS have shown that 21 (21%) patients developed aHUS related to pregnancy, and the onset of most cases were found in the postpartum period78). Therefore, the development of TMA during pregnancy would be one of important factors to suspect aHUS. HELLP (hemolysis, elevated liver enzyme, and low platelets) syndrome is one severe complication during pregnancy, and can be also classified into TMA. In 2008, Fakhouri F, et al. have identified the four variants in CFH, CFI, and MCP in 4 of 11 patients (36%) with HELLP syndrome79). In the subsequent larger study of 33 patients, three variants (one in MCP, two in CFI) have been found in three patients, but one of CFI variants had less evidence for uncontrolled complement activation80). More recently, Vaught AJ, et al.81) have performed the functional and genetic analysis of 13 HELLP syndrome patients and revealed that these patients showed significantly increased complement activation of AP compared with controls (62% vs 16%, p = 0.009). Furthermore, 46% of patients with HELLP syndrome had rare germline variants (minor allele frequency < 0.01) in the genes of CFHR1, CFHR5, C3, or homozygous genetic deletion of CFHRs.
The KDIGO controversies conference report has stated that the patients with pregnancy-associated or de novo transplantation associated TMA (aHUS) should have a full complement evaluation due to the high prevalence of rare genetic variants or autoantibodies described in these subgroups7). Although the number of reports is still limited, the genetic defects and/or complement activation in the AP have also been found in the secondary TMA patients related to hypertension, IgA nephropathy, or systemic lupus erythematosus, etc. Larger cohort study is needed to clarify the association between complement dysregulation and the pathogenesis of these secondary TMAs, but it is likely that this kind of coincidence should be extremely rare.
Detection of Complement Abnormalities to Diagnose aHUs
Atypical HUS is clinically diagnosed by exclusion of TTP, STEC-HUS, and secondary TMA. Unfortunately, there are no specific biomarkers to confirm the clinical diagnosis of aHUS. Differentiating aHUS from secondary TMA is sometimes most difficult because no biomarkers can distinguish them.
When aHUS is clinically suspected, a variety of specific protein-based assay are recommended as follows82); quantification of complement components and its regulators (C3, C4, CFH, CFI, MCP and CFB), the markers for complement activity (CH50 and AH50), and screening of anti-CFH autoantibodies. Decreased C3 levels, but not C4 may reflect the excess activation of the AP. However, even if these factors are within normal limits, the diagnosis of aHUS is not still ruled out. Genetic screening of genes known to cause aHUS (CFH, CFI, MCP, C3, CFB, THBD, DGKE, and CFHR) is important to confirm the clinical diagnosis of aHUS and to decide the treatment strategy as well as to estimate the outcome and prognosis. The drawback of genetic analysis is that it takes at least several weeks to obtain the result, and the known genetic abnormalities account for about 50% to 60% of aHUS cases.
Various biomarkers have been studied to reveal the underlying complement activation in aHUS. Increased levels of C5a and sC5b-9 have been detected in the acute phase of aHUS, but these markers do not clearly distinguish aHUS from other types of TMA46, 83, 84). Modified HAM test by using GPI-anchored protein-deficient cells showed high specificity in differentiating aHUS from TTP85). The hemolytic assay by using non-sensitized sheep red blood cells can be assessed cell-protected function of CFH21, 86, 87). It can be useful for rapid detection of CFH predisposing variants or anti-CFH autoantibodies, leading to defect C-terminal function of CFH. In addition to CFH-related variants, we have previously reported that C3 p. K1105Q positioned at the CFH binding interface also showed marked hemolysis in the hemolytic assay21).
Current Therapy for aHUs
A first-line treatment for aHUS had been empirically considered to be plasma treatment (plasma infusion and plasma exchange). This supplies functional complement regulatory factors and/or can remove the abnormal complement-related factors like mutant proteins or anti-CFH antibodies. Although the introduction of plasma therapy has decreased the mortality of patients with aHUS, 48% of pediatric and 67% of adult cases died or reached ESRD within 3-year follow-up18).
The treatment using complement-inhibiting drug, eculizumab, is now replacing plasma therapy as the gold-standard in the management of aHUS. Eculizumab, a monoclonal humanized antibody against C5, blocks the cleavage of C5 leading to prevent MAC formation, whereas an opsonization by C3b is unaffected. The efficacy and safety of this drug have been described in many reports88–91). In pediatric cases, eculizumab administration is recommended as a first-line therapy because children have a high risk of catheter-related complications and a low risk of secondary TMAs92). On the other hand, plasma therapy may be the first choice when eculizumab is unavailable or in adult cases having the possibility of secondary TMAs without the involvement of complement abnormalities. In autoimmune type of aHUS, combined therapy of concomitant immunosuppression and plasma therapy may allow better outcomes51, 52, 92), though some reports have recently implicated that eculizumab treatment might be effective in autoantibody-mediated aHUS. It is still to be elucidated what is the optimal treatment for autoantibody-mediated aHUS. Eculizumab might be inefficacious for aHUS patients having DGKE and INF2 variants64, 73).
In Japan, eculizumab was approved for treating complement-mediated aHUS in 2013, and its efficacy and safety have been established in both children68, 93, 94) and adults95–97). In the analysis of 118 Japanese patients with aHUS, 72% of patients received plasma therapy and 42% were treated with eculizumab22). The prognosis of Japanese patients was relatively favorable compared to Caucasian patients, and no significant differences were found between the outcomes of patients treated with eculizumab and without eciluzimab. This is probably due to the unique genetic background of the Japanese population, the predominance of C3 p.I1157T. Of note, it has been revealed that the C5 variant at position Arg885 impairs eculizumab efficacy, and was detected in 3.2% of paroxysmal nocturnal hemoglobinuria patients98). No patients have been reported in aHUS until now, but we should consider the screening of this polymorphism when eculizumab is inefficacious for aHUS patients.
Conclusions
Recent progress in the field of aHUS has revealed that the diverse factors were associated with aHUS. It remains unclear, however, whether the genetic variants unrelated to the complement system lead to uncontrolled complement activation or not. Actually, some patients with variants in DGKE or INF2 showed poor responses to anti-complement therapy, suggesting that specific treatment strategy may be needed according to individual abnormalities. Furthermore, the underlying complement abnormalities have also been detected in other types of TMAs, such as HSCT-TMA and pregnancy-related complication. However, it is also likely that this kind of coincidence should be extremely rare, and further studies are needed to elucidate the efficacy of complement inhibition for these subgroups with the confirmation underlying complement abnormalities by genetic and biological test.
Epidemiological and genetic studies of Japanese patients with aHUS have revealed that the distinct genetic background of Japan, high frequencies of C3 variants and low risk of carrying CFH variants. Moreover, a prevalent C3 p.I1157T is associated with favorable prognosis in spite of high recurrence rate. These data would be helpful to determine treatment strategies for Japanese patients with aHUS. Our institution, division of Nephrology and Endocrinology, the University of Tokyo Hospital, has been performing the cohort analysis of patients with aHUS. Consultation for diagnostic test of aHUS is available by e-mail (ahus-office@umin.ac.jp).
Acknowledgments and Notice of Grant Support
This study was supported by research grants from the Ministry of Health, Labour and Welfare of Japan and the Practical Research Project for Rare/Intractable Diseases from Japan Agency for Medical Research and Development, AMED (17ek0109254h0001).
Conflict of Interest
Honoraria: Hideki Kato and Masaomi Nangaku from Alexion Pharmaceuticals, Inc. Clinical research funding: Masaomi Nangaku from Alexion Pharmaceuticals, Inc.
References
- 1). Tsai HM, Lian EC: Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med, 1998; 339: 1585-1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2). Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, Yang AY, Siemieniak DR, Stark KR, Gruppo R, Sarode R, Shurin SB, Chandrasekaran V, Stabler SP, Sabio H, Bouhassira EE, Upshaw JD, Jr., Ginsburg D, Tsai HM: Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature, 2001; 413: 488-494 [DOI] [PubMed] [Google Scholar]
- 3). Kokame K, Matsumoto M, Soejima K, Yagi H, Ishizashi H, Funato M, Tamai H, Konno M, Kamide K, Kawano Y, Miyata T, Fujimura Y: Mutations and common polymorphisms in ADAMTS13 gene responsible for von Willebrand factor-cleaving protease activity. Proc Natl Acad Sci U S A, 2002; 99: 11902-11907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4). Huang J, Motto DG, Bundle DR, Sadler JE: Shiga toxin B subunits induce VWF secretion by human endothelial cells and thrombotic microangiopathy in ADAMTS13-deficient mice. Blood, 2010; 116: 3653-3659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5). Kato H, Nangaku M, Hataya H, Sawai T, Ashida A, Fujimaru R, Hidaka Y, Kaname S, Maruyama S, Yasuda T, Yoshida Y, Ito S, Hattori M, Miyakawa Y, Fujimura Y, Okada H, Kagami S: Clinical guides for atypical hemolytic uremic syndrome in Japan. Clin Exp Nephrol, 2016; 20: 536-543 [DOI] [PubMed] [Google Scholar]
- 6). Kato H, Nangaku M, Hataya H, Sawai T, Ashida A, Fujimaru R, Hidaka Y, Kaname S, Maruyama S, Yasuda T, Yoshida Y, Ito S, Hattori M, Miyakawa Y, Fujimura Y, Okada H, Kagami S: Clinical guides for atypical hemolytic uremic syndrome in Japan. Pediatr Int, 2016; 58: 549-555 [DOI] [PubMed] [Google Scholar]
- 7). Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Fremeaux-Bacchi V, Kavanagh D, Nester CM, Noris M, Pickering MC, Rodriguez de Cordoba S, Roumenina LT, Sethi S, Smith RJ: Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int, 2017; 91: 539-551 [DOI] [PubMed] [Google Scholar]
- 8). Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT: Complement System Part II: Role in Immunity. Front Immunol, 2015; 6: 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9). Lukawska E, Polcyn-Adamczak M, Niemir ZI: The role of the alternative pathway of complement activation in glomerular diseases. Clin Exp Med, 2018; [DOI] [PubMed] [Google Scholar]
- 10). Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Kohl J: The role of the anaphylatoxins in health and disease. Mol Immunol, 2009; 46: 2753-2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11). Gordon DL, Kaufman RM, Blackmore TK, Kwong J, Lublin DM: Identification of complement regulatory domains in human factor H. J Immunol, 1995; 155: 348-356 [PubMed] [Google Scholar]
- 12). Sharma AK, Pangburn MK: Identification of three physically and functionally distinct binding sites for C3b in human complement factor H by deletion mutagenesis. Proc Natl Acad Sci U S A, 1996; 93: 10996-11001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13). Jokiranta TS, Hellwage J, Koistinen V, Zipfel PF, Meri S: Each of the three binding sites on complement factor H interacts with a distinct site on C3b. J Biol Chem, 2000; 275: 27657-27662 [DOI] [PubMed] [Google Scholar]
- 14). Blackmore TK, Hellwage J, Sadlon TA, Higgs N, Zipfel PF, Ward HM, Gordon DL: Identification of the second heparin-binding domain in human complement factor H. J Immunol, 1998; 160: 3342-3348 [PubMed] [Google Scholar]
- 15). Blaum BS, Hannan JP, Herbert AP, Kavanagh D, Uhrin D, Stehle T: Structural basis for sialic acid-mediated self-recognition by complement factor H. Nat Chem Biol, 2015; 11: 77-82 [DOI] [PubMed] [Google Scholar]
- 16). Ferreira VP, Herbert AP, Hocking HG, Barlow PN, Pangburn MK: Critical role of the C-terminal domains of factor H in regulating complement activation at cell surfaces. J Immunol, 2006; 177: 6308-6316 [DOI] [PubMed] [Google Scholar]
- 17). Osborne AJ, Breno M, Borsa NG, Bu F, Fremeaux-Bacchi V, Gale DP, van den Heuvel LP, Kavanagh D, Noris M, Pinto S, Rallapalli PM, Remuzzi G, Rodriguez de Cordoba S, Ruiz A, Smith RJH, Vieira-Martins P, Volokhina E, Wilson V, Goodship THJ, Perkins SJ: Statistical Validation of Rare Complement Variants Provides Insights into the Molecular Basis of Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy. J Immunol, 2018; 200: 2464-2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18). Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, Daina E, Fenili C, Castelletti F, Sorosina A, Piras R, Donadelli R, Maranta R, van der Meer I, Conway EM, Zipfel PF, Goodship TH, Remuzzi G: Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol, 2010; 5: 1844-1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19). Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaime F, Dragon-Durey MA, Ngo S, Moulin B, Servais A, Provot F, Rostaing L, Burtey S, Niaudet P, Deschenes G, Lebranchu Y, Zuber J, Loirat C: Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol, 2013; 8: 554-562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20). Noris M, Bresin E, Mele C, Remuzzi G. Genetic Atypical Hemolytic-Uremic Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews(R). Seattle (WA): University of Washington, Seattle; GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.; 1993–2017 [Google Scholar]
- 21). Yoshida Y, Miyata T, Matsumoto M, Shirotani-Ikejima H, Uchida Y, Ohyama Y, Kokubo T, Fujimura Y: A novel quantitative hemolytic assay coupled with restriction fragment length polymorphisms analysis enabled early diagnosis of atypical hemolytic uremic syndrome and identified unique predisposing mutations in Japan. PLoS One, 2015; 10: e0124655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22). Fujisawa M, Kato H, Yoshida Y, Usui T, Takata M, Fujimoto M, Wada H, Uchida Y, Kokame K, Matsumoto M, Fujimura Y, Miyata T, Nangaku M: Clinical characteristics and genetic backgrounds of Japanese patients with atypical hemolytic uremic syndrome. Clin Exp Nephrol, 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23). Manuelian T, Hellwage J, Meri S, Caprioli J, Noris M, Heinen S, Jozsi M, Neumann HP, Remuzzi G, Zipfel PF: Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J Clin Invest, 2003; 111: 1181-1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24). Hyvarinen S, Meri S, Jokiranta TS: Disturbed sialic acid recognition on endothelial cells and platelets in complement attack causes atypical hemolytic uremic syndrome. Blood, 2016; 127: 2701-2710 [DOI] [PubMed] [Google Scholar]
- 25). McRae JL, Duthy TG, Griggs KM, Ormsby RJ, Cowan PJ, Cromer BA, McKinstry WJ, Parker MW, Murphy BF, Gordon DL: Human factor H-related protein 5 has cofactor activity, inhibits C3 convertase activity, binds heparin and C-reactive protein, and associates with lipoprotein. J Immunol, 2005; 174: 6250-6256 [DOI] [PubMed] [Google Scholar]
- 26). Csincsi AI, Kopp A, Zoldi M, Banlaki Z, Uzonyi B, Hebecker M, Caesar JJ, Pickering MC, Daigo K, Hamakubo T, Lea SM, Goicoechea de Jorge E, Jozsi M: Factor H-related protein 5 interacts with pentraxin 3 and the extracellular matrix and modulates complement activation. J Immunol, 2015; 194: 4963-4973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27). Heinen S, Hartmann A, Lauer N, Wiehl U, Dahse HM, Schirmer S, Gropp K, Enghardt T, Wallich R, Halbich S, Mihlan M, Schlotzer-Schrehardt U, Zipfel PF, Skerka C: Factor H-related protein 1 (CFHR-1) inhibits complement C5 convertase activity and terminal complex formation. Blood, 2009; 114: 2439-2447 [DOI] [PubMed] [Google Scholar]
- 28). Skerka C, Chen Q, Fremeaux-Bacchi V, Roumenina LT: Complement factor H related proteins (CFHRs). Mol Immunol, 2013; 56: 170-180 [DOI] [PubMed] [Google Scholar]
- 29). Fritsche LG, Lauer N, Hartmann A, Stippa S, Keilhauer CN, Oppermann M, Pandey MK, Kohl J, Zipfel PF, Weber BH, Skerka C: An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD). Hum Mol Genet, 2010; 19: 4694-4704 [DOI] [PubMed] [Google Scholar]
- 30). Goicoechea de Jorge E, Caesar JJ, Malik TH, Patel M, Colledge M, Johnson S, Hakobyan S, Morgan BP, Harris CL, Pickering MC, Lea SM: Dimerization of complement factor H-related proteins modulates complement activation in vivo. Proc Natl Acad Sci U S A, 2013; 110: 4685-4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31). Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, Kavanagh D, Noris M, Pickering M, Sanchez-Corral P, Skerka C, Zipfel P, Smith RJ: Atypical aHUS: State of the art. Mol Immunol, 2015; 67: 31-42 [DOI] [PubMed] [Google Scholar]
- 32). Venables JP, Strain L, Routledge D, Bourn D, Powell HM, Warwicker P, Diaz-Torres ML, Sampson A, Mead P, Webb M, Pirson Y, Jackson MS, Hughes A, Wood KM, Goodship JA, Goodship TH: Atypical haemolytic uraemic syndrome associated with a hybrid complement gene. PLoS Med, 2006; 3: e431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33). Heinen S, Sanchez-Corral P, Jackson MS, Strain L, Goodship JA, Kemp EJ, Skerka C, Jokiranta TS, Meyers K, Wagner E, Robitaille P, Esparza-Gordillo J, Rodriguez de Cordoba S, Zipfel PF, Goodship TH: De novo gene conversion in the RCA gene cluster (1q32) causes mutations in complement factor H associated with atypical hemolytic uremic syndrome. Hum Mutat, 2006; 27: 292-293 [DOI] [PubMed] [Google Scholar]
- 34). Eyler SJ, Meyer NC, Zhang Y, Xiao X, Nester CM, Smith RJ: A novel hybrid CFHR1/CFH gene causes atypical hemolytic uremic syndrome. Pediatr Nephrol, 2013; 28: 2221-2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35). Valoti E, Alberti M, Tortajada A, Garcia-Fernandez J, Gastoldi S, Besso L, Bresin E, Remuzzi G, Rodriguez de Cordoba S, Noris M: A novel atypical hemolytic uremic syndrome-associated hybrid CFHR1/CFH gene encoding a fusion protein that antagonizes factor H-dependent complement regulation. J Am Soc Nephrol, 2015; 26: 209-219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36). Challis RC, Araujo GS, Wong EK, Anderson HE, Awan A, Dorman AM, Waldron M, Wilson V, Brocklebank V, Strain L, Morgan BP, Harris CL, Marchbank KJ, Goodship TH, Kavanagh D: A De Novo Deletion in the Regulators of Complement Activation Cluster Producing a Hybrid Complement Factor H/Complement Factor H-Related 3 Gene in Atypical Hemolytic Uremic Syndrome. J Am Soc Nephrol, 2016; 27: 1617-1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37). Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, Mele C, Bresin E, Cassis L, Gamba S, Porrati F, Bucchioni S, Monteferrante G, Fang CJ, Liszewski MK, Kavanagh D, Atkinson JP, Remuzzi G, International Registry of R, Familial HT : Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood, 2006; 108: 1267-1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38). Kavanagh D, Richards A, Noris M, Hauhart R, Liszewski MK, Karpman D, Goodship JA, Fremeaux-Bacchi V, Remuzzi G, Goodship TH, Atkinson JP: Characterization of mutations in complement factor I (CFI) associated with hemolytic uremic syndrome. Mol Immunol, 2008; 45: 95-105 [DOI] [PubMed] [Google Scholar]
- 39). Nilsson SC, Kalchishkova N, Trouw LA, Fremeaux-Bacchi V, Villoutreix BO, Blom AM: Mutations in complement factor I as found in atypical hemolytic uremic syndrome lead to either altered secretion or altered function of factor I. Eur J Immunol, 2010; 40: 172-185 [DOI] [PubMed] [Google Scholar]
- 40). Richards A, Kathryn Liszewski M, Kavanagh D, Fang CJ, Moulton E, Fremeaux-Bacchi V, Remuzzi G, Noris M, Goodship TH, Atkinson JP: Implications of the initial mutations in membrane cofactor protein (MCP; CD46) leading to atypical hemolytic uremic syndrome. Mol Immunol, 2007; 44: 111-122 [DOI] [PubMed] [Google Scholar]
- 41). Schramm EC, Roumenina LT, Rybkine T, Chauvet S, Vieira-Martins P, Hue C, Maga T, Valoti E, Wilson V, Jokiranta S, Smith RJ, Noris M, Goodship T, Atkinson JP, Fremeaux-Bacchi V: Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome. Blood, 2015; 125: 2359-2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42). Fremeaux-Bacchi V, Miller EC, Liszewski MK, Strain L, Blouin J, Brown AL, Moghal N, Kaplan BS, Weiss RA, Lhotta K, Kapur G, Mattoo T, Nivet H, Wong W, Gie S, Hurault de Ligny B, Fischbach M, Gupta R, Hauhart R, Meunier V, Loirat C, Dragon-Durey MA, Fridman WH, Janssen BJ, Goodship TH, Atkinson JP: Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood, 2008; 112: 4948-4952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43). Martinez-Barricarte R, Heurich M, Lopez-Perrote A, Tortajada A, Pinto S, Lopez-Trascasa M, Sanchez-Corral P, Morgan BP, Llorca O, Harris CL, Rodriguez de Cordoba S: The molecular and structural bases for the association of complement C3 mutations with atypical hemolytic uremic syndrome. Mol Immunol, 2015; 66: 263-273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44). Roumenina LT, Frimat M, Miller EC, Provot F, Dragon-Durey MA, Bordereau P, Bigot S, Hue C, Satchell SC, Mathieson PW, Mousson C, Noel C, Sautes-Fridman C, Halbwachs-Mecarelli L, Atkinson JP, Lionet A, Fremeaux-Bacchi V: A prevalent C3 mutation in aHUS patients causes a direct C3 convertase gain of function. Blood, 2012; 119: 4182-4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45). Matsumoto T, Fan X, Ishikawa E, Ito M, Amano K, Toyoda H, Komada Y, Ohishi K, Katayama N, Yoshida Y, Matsumoto M, Fujimura Y, Ikejiri M, Wada H, Miyata T: Analysis of patients with atypical hemolytic uremic syndrome treated at the Mie University Hospital: concentration of C3 p.I1157T mutation. Int J Hematol, 2014; 100: 437-442 [DOI] [PubMed] [Google Scholar]
- 46). Volokhina EB, Westra D, van der Velden TJ, van de Kar NC, Mollnes TE, van den Heuvel LP: Complement activation patterns in atypical haemolytic uraemic syndrome during acute phase and in remission. Clin Exp Immunol, 2015; 181: 306-313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47). Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, Carreras L, Arranz EA, Garrido CA, Lopez-Trascasa M, Sanchez-Corral P, Morgan BP, Rodriguez de Cordoba S: Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci U S A, 2007; 104: 240-245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48). Marinozzi MC, Vergoz L, Rybkine T, Ngo S, Bettoni S, Pashov A, Cayla M, Tabarin F, Jablonski M, Hue C, Smith RJ, Noris M, Halbwachs-Mecarelli L, Donadelli R, Fremeaux-Bacchi V, Roumenina LT: Complement factor B mutations in atypical hemolytic uremic syndrome-disease-relevant or benign? J Am Soc Nephrol, 2014; 25: 2053-2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49). Jozsi M, Licht C, Strobel S, Zipfel SL, Richter H, Heinen S, Zipfel PF, Skerka C: Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood, 2008; 111: 1512-1514 [DOI] [PubMed] [Google Scholar]
- 50). Schaefer F, Ardissino G, Ariceta G, Fakhouri F, Scully M, Isbel N, Lommele A, Kupelian V, Gasteyger C, Greenbaum LA, Johnson S, Ogawa M, Licht C, Vande Walle J, Fremeaux-Bacchi V: Clinical and genetic predictors of atypical hemolytic uremic syndrome phenotype and outcome. Kidney Int, 2018; 94: 408-418 [DOI] [PubMed] [Google Scholar]
- 51). Sinha A, Gulati A, Saini S, Blanc C, Gupta A, Gurjar BS, Saini H, Kotresh ST, Ali U, Bhatia D, Ohri A, Kumar M, Agarwal I, Gulati S, Anand K, Vijayakumar M, Sinha R, Sethi S, Salmona M, George A, Bal V, Singh G, Dinda AK, Hari P, Rath S, Dragon-Durey MA, Bagga A: Prompt plasma exchanges and immunosuppressive treatment improves the outcomes of anti-factor H autoantibodyassociated hemolytic uremic syndrome in children. Kidney Int, 2014; 85: 1151-1160 [DOI] [PubMed] [Google Scholar]
- 52). Dragon-Durey MA, Sethi SK, Bagga A, Blanc C, Blouin J, Ranchin B, Andre JL, Takagi N, Cheong HI, Hari P, Le Quintrec M, Niaudet P, Loirat C, Fridman WH, Fremeaux-Bacchi V: Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome. J Am Soc Nephrol, 2010; 21: 2180-2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53). Jozsi M, Strobel S, Dahse HM, Liu WS, Hoyer PF, Oppermann M, Skerka C, Zipfel PF: Anti factor H autoantibodies block C-terminal recognition function of factor H in hemolytic uremic syndrome. Blood, 2007; 110: 1516-1518 [DOI] [PubMed] [Google Scholar]
- 54). Moore I, Strain L, Pappworth I, Kavanagh D, Barlow PN, Herbert AP, Schmidt CQ, Staniforth SJ, Holmes LV, Ward R, Morgan L, Goodship TH, Marchbank KJ: Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood, 2010; 115: 379-387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55). Strobel S, Hoyer PF, Mache CJ, Sulyok E, Liu WS, Richter H, Oppermann M, Zipfel PF, Jozsi M: Functional analyses indicate a pathogenic role of factor H autoantibodies in atypical haemolytic uraemic syndrome. Nephrol Dial Transplant, 2010; 25: 136-144 [DOI] [PubMed] [Google Scholar]
- 56). Strobel S, Abarrategui-Garrido C, Fariza-Requejo E, Seeberger H, Sanchez-Corral P, Jozsi M: Factor H-related protein 1 neutralizes anti-factor H autoantibodies in autoimmune hemolytic uremic syndrome. Kidney Int, 2011; 80: 397-404 [DOI] [PubMed] [Google Scholar]
- 57). Abarrategui-Garrido C, Martinez-Barricarte R, Lopez-Trascasa M, de Cordoba SR, Sanchez-Corral P: Characterization of complement factor H-related (CFHR) proteins in plasma reveals novel genetic variations of CFHR1 associated with atypical hemolytic uremic syndrome. Blood, 2009; 114: 4261-4271 [DOI] [PubMed] [Google Scholar]
- 58). Hageman GS, Hancox LS, Taiber AJ, Gehrs KM, Anderson DH, Johnson LV, Radeke MJ, Kavanagh D, Richards A, Atkinson J, Meri S, Bergeron J, Zernant J, Merriam J, Gold B, Allikmets R, Dean M: Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: characterization, ethnic distribution and evolutionary implications. Ann Med, 2006; 38: 592-604 [PMC free article] [PubMed] [Google Scholar]
- 59). Zipfel PF, Edey M, Heinen S, Jozsi M, Richter H, Misselwitz J, Hoppe B, Routledge D, Strain L, Hughes AE, Goodship JA, Licht C, Goodship TH, Skerka C: Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet, 2007; 3: e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60). Dragon-Durey MA, Blanc C, Marliot F, Loirat C, Blouin J, Sautes-Fridman C, Fridman WH, Fremeaux-Bacchi V: The high frequency of complement factor H related CFHR1 gene deletion is restricted to specific subgroups of patients with atypical haemolytic uraemic syndrome. J Med Genet, 2009; 46: 447-450 [DOI] [PubMed] [Google Scholar]
- 61). Leban N, Abarrategui-Garrido C, Fariza-Requejo E, Aminoso-Carbonero C, Pinto S, Chibani JB, Khelil AH, Sanchez-Corral P: Factor H and CFHR1 polymorphisms associated with atypical Haemolytic Uraemic Syndrome (aHUS) are differently expressed in Tunisian and in Caucasian populations. Int J Immunogenet, 2012; 39: 110-113 [DOI] [PubMed] [Google Scholar]
- 62). Bhattacharjee A, Reuter S, Trojnar E, Kolodziejczyk R, Seeberger H, Hyvarinen S, Uzonyi B, Szilagyi A, Prohaszka Z, Goldman A, Jozsi M, Jokiranta TS: The major autoantibody epitope on factor H in atypical hemolytic uremic syndrome is structurally different from its homologous site in factor H-related protein 1, supporting a novel model for induction of autoimmunity in this disease. J Biol Chem, 2015; 290: 9500-9510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63). Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, Del-Favero J, Plaisance S, Claes B, Lambrechts D, Zoja C, Remuzzi G, Conway EM: Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med, 2009; 361: 345-357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64). Lemaire M, Fremeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M, Fakhouri F, Taque S, Nobili F, Martinez F, Ji W, Overton JD, Mane SM, Nurnberg G, Altmuller J, Thiele H, Morin D, Deschenes G, Baudouin V, Llanas B, Collard L, Majid MA, Simkova E, Nurnberg P, Rioux-Leclerc N, Moeckel GW, Gubler MC, Hwa J, Loirat C, Lifton RP: Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet, 2013; 45: 531-536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65). Mele C, Lemaire M, Iatropoulos P, Piras R, Bresin E, Bettoni S, Bick D, Helbling D, Veith R, Valoti E, Donadelli R, Murer L, Neunhauserer M, Breno M, Fremeaux-Bacchi V, Lifton R, Remuzzi G, Noris M: Characterization of a New DGKE Intronic Mutation in Genetically Unsolved Cases of Familial Atypical Hemolytic Uremic Syndrome. Clin J Am Soc Nephrol, 2015; 10: 1011-1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66). Schaefer F, Ardissino G, Ariceta G, Fakhouri F, Scully M, Isbel N, Lommele A, Kupelian V, Gasteyger C, Greenbaum LA, Johnson S, Ogawa M, Licht C, Vande Walle J, Fremeaux-Bacchi V: Clinical and genetic predictors of atypical hemolytic uremic syndrome phenotype and outcome. Kidney Int, 2018; [DOI] [PubMed] [Google Scholar]
- 67). Westland R, Bodria M, Carrea A, Lata S, Scolari F, Fremeaux-Bacchi V, D'Agati VD, Lifton RP, Gharavi AG, Ghiggeri GM, Sanna-Cherchi S: Phenotypic expansion of DGKE-associated diseases. J Am Soc Nephrol, 2014; 25: 1408-1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68). Miyata T, Uchida Y, Ohta T, Urayama K, Yoshida Y, Fujimura Y: Atypical haemolytic uraemic syndrome in a Japanese patient with DGKE genetic mutations. Thromb Haemost, 2015; 114: 862-863 [DOI] [PubMed] [Google Scholar]
- 69). Bruneau S, Neel M, Roumenina LT, Frimat M, Laurent L, Fremeaux-Bacchi V, Fakhouri F: Loss of DGKepsilon induces endothelial cell activation and death independently of complement activation. Blood, 2015; 125: 1038-1046 [DOI] [PubMed] [Google Scholar]
- 70). Bu F, Maga T, Meyer NC, Wang K, Thomas CP, Nester CM, Smith RJ: Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J Am Soc Nephrol, 2014; 25: 55-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71). Schuster V, Hugle B, Tefs K: Plasminogen deficiency. J Thromb Haemost, 2007; 5: 2315-2322 [DOI] [PubMed] [Google Scholar]
- 72). Hyvarinen S, Jokiranta TS: Minor Role of Plasminogen in Complement Activation on Cell Surfaces. PLoS One, 2015; 10: e0143707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73). Challis RC, Ring T, Xu Y, Wong EK, Flossmann O, Roberts IS, Ahmed S, Wetherall M, Salkus G, Brocklebank V, Fester J, Strain L, Wilson V, Wood KM, Marchbank KJ, Santibanez-Koref M, Goodship TH, Kavanagh D: Thrombotic Microangiopathy in Inverted Formin 2-Mediated Renal Disease. J Am Soc Nephrol, 2017; 28: 1084-1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74). Brown EJ, Schlondorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, Higgs HN, Henderson JM, Pollak MR: Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet, 2010; 42: 72-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75). Sanchez Chinchilla D, Pinto S, Hoppe B, Adragna M, Lopez L, Justa Roldan ML, Pena A, Lopez Trascasa M, Sanchez-Corral P, Rodriguez de Cordoba S: Complement mutations in diacylglycerol kinase-epsilon-associated atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol, 2014; 9: 1611-1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76). Jodele S, Licht C, Goebel J, Dixon BP, Zhang K, Sivakumaran TA, Davies SM, Pluthero FG, Lu L, Laskin BL: Abnormalities in the alternative pathway of complement in children with hematopoietic stem cell transplant-associated thrombotic microangiopathy. Blood, 2013; 122: 2003-2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77). Jodele S, Davies SM, Lane A, Khoury J, Dandoy C, Goebel J, Myers K, Grimley M, Bleesing J, El-Bietar J, Wallace G, Chima RS, Paff Z, Laskin BL: Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: a study in children and young adults. Blood, 2014; 124: 645-653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78). Fakhouri F, Roumenina L, Provot F, Sallee M, Caillard S, Couzi L, Essig M, Ribes D, Dragon-Durey MA, Bridoux F, Rondeau E, Fremeaux-Bacchi V: Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol, 2010; 21: 859-867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79). Fakhouri F, Jablonski M, Lepercq J, Blouin J, Benachi A, Hourmant M, Pirson Y, Durrbach A, Grunfeld JP, Knebelmann B, Fremeaux-Bacchi V: Factor H, membrane cofactor protein, and factor I mutations in patients with hemolysis, elevated liver enzymes, and low platelet count syndrome. Blood, 2008; 112: 4542-4545 [DOI] [PubMed] [Google Scholar]
- 80). Crovetto F, Borsa N, Acaia B, Nishimura C, Frees K, Smith RJ, Peyvandi F, Palla R, Cugno M, Tedeschi S, Castorina P, Somigliana E, Ardissino G, Fedele L: The genetics of the alternative pathway of complement in the pathogenesis of HELLP syndrome. J Matern Fetal Neonatal Med, 2012; 25: 2322-2325 [DOI] [PubMed] [Google Scholar]
- 81). Vaught AJ, Braunstein EM, Jasem J, Yuan X, Makhlin I, Eloundou S, Baines AC, Merrill SA, Chaturvedi S, Blakemore K, Sperati CJ, Brodsky RA: Germline mutations in the alternative pathway of complement predispose to HELLP syndrome. JCI Insight, 2018; 3: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82). Angioi A, Fervenza FC, Sethi S, Zhang Y, Smith RJ, Murray D, Van Praet J, Pani A, De Vriese AS: Diagnosis of complement alternative pathway disorders. Kidney Int, 2016; 89: 278-288 [DOI] [PubMed] [Google Scholar]
- 83). Noris M, Galbusera M, Gastoldi S, Macor P, Banterla F, Bresin E, Tripodo C, Bettoni S, Donadelli R, Valoti E, Tedesco F, Amore A, Coppo R, Ruggenenti P, Gotti E, Remuzzi G: Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood, 2014; 124: 1715-1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84). Cataland SR, Holers VM, Geyer S, Yang S, Wu HM: Biomarkers of terminal complement activation confirm the diagnosis of aHUS and differentiate aHUS from TTP. Blood, 2014; 123: 3733-3738 [DOI] [PubMed] [Google Scholar]
- 85). Gavriilaki E, Yuan X, Ye Z, Ambinder AJ, Shanbhag SP, Streiff MB, Kickler TS, Moliterno AR, Sperati CJ, Brodsky RA: Modified Ham test for atypical hemolytic uremic syndrome. Blood, 2015; 125: 3637-3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86). Sanchez-Corral P, Gonzalez-Rubio C, Rodriguez de Cordoba S, Lopez-Trascasa M: Functional analysis in serum from atypical Hemolytic Uremic Syndrome patients reveals impaired protection of host cells associated with mutations in factor H. Mol Immunol, 2004; 41: 81-84 [DOI] [PubMed] [Google Scholar]
- 87). Roumenina LT, Roquigny R, Blanc C, Poulain N, Ngo S, Dragon-Durey MA, Fremeaux-Bacchi V: Functional evaluation of factor H genetic and acquired abnormalities: application for atypical hemolytic uremic syndrome (aHUS). Methods Mol Biol, 2014; 1100: 237-247 [DOI] [PubMed] [Google Scholar]
- 88). Nurnberger J, Philipp T, Witzke O, Opazo Saez A, Vester U, Baba HA, Kribben A, Zimmerhackl LB, Janecke AR, Nagel M, Kirschfink M: Eculizumab for atypical hemolytic-uremic syndrome. N Engl J Med, 2009; 360: 542-544 [DOI] [PubMed] [Google Scholar]
- 89). Zuber J, Le Quintrec M, Krid S, Bertoye C, Gueutin V, Lahoche A, Heyne N, Ardissino G, Chatelet V, Noel LH, Hourmant M, Niaudet P, Fremeaux-Bacchi V, Rondeau E, Legendre C, Loirat C: Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant, 2012; 12: 3337-3354 [DOI] [PubMed] [Google Scholar]
- 90). Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, Bingham C, Cohen DJ, Delmas Y, Douglas K, Eitner F, Feldkamp T, Fouque D, Furman RR, Gaber O, Herthelius M, Hourmant M, Karpman D, Lebranchu Y, Mariat C, Menne J, Moulin B, Nurnberger J, Ogawa M, Remuzzi G, Richard T, Sberro-Soussan R, Severino B, Sheerin NS, Trivelli A, Zimmerhackl LB, Goodship T, Loirat C: Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med, 2013; 368: 2169-2181 [DOI] [PubMed] [Google Scholar]
- 91). Licht C, Ardissino G, Ariceta G, Cohen D, Cole JA, Gasteyger C, Greenbaum LA, Johnson S, Ogawa M, Schaefer F, Vande Walle J, Fremeaux-Bacchi V: The global aHUS registry: methodology and initial patient characteristics. BMC Nephrol, 2015; 16: 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92). Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, Coppo R, Emma F, Johnson S, Karpman D, Landau D, Langman CB, Lapeyraque AL, Licht C, Nester C, Pecoraro C, Riedl M, van de Kar NC, Van de Walle J, Vivarelli M, Fremeaux-Bacchi V, International HUS : An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol, 2016; 31: 15-39 [DOI] [PubMed] [Google Scholar]
- 93). Ohta T, Urayama K, Tada Y, Furue T, Imai S, Matsubara K, Ono H, Sakano T, Jinno K, Yoshida Y, Miyata T, Fujimura Y: Eculizumab in the treatment of atypical hemolytic uremic syndrome in an infant leads to cessation of peritoneal dialysis and improvement of severe hypertension. Pediatr Nephrol, 2015; 30: 603-608 [DOI] [PubMed] [Google Scholar]
- 94). Ito N, Hataya H, Saida K, Amano Y, Hidaka Y, Motoyoshi Y, Ohta T, Yoshida Y, Terano C, Iwasa T, Kubota W, Takada H, Hara T, Fujimura Y, Ito S: Efficacy and safety of eculizumab in childhood atypical hemolytic uremic syndrome in Japan. Clin Exp Nephrol, 2016; 20: 265-272 [DOI] [PubMed] [Google Scholar]
- 95). Okumi M, Omoto K, Unagami K, Ishida H, Tanabe K: Eculizumab for the treatment of atypical hemolytic uremic syndrome recurrence after kidney transplantation associated with complement factor H mutations: a case report with a 5-year follow-up. Int Urol Nephrol, 2016; 48: 817-818 [DOI] [PubMed] [Google Scholar]
- 96). Yamaguchi M, Hori M, Hiroshi N, Maruyama S: Postpartum atypical hemolytic uremic syndrome with complement factor H mutation complicated by reversible cerebrovascular constriction syndrome successfully treated with eculizumab. Thromb Res, 2017; 151: 79-81 [DOI] [PubMed] [Google Scholar]
- 97). Nakamura H, Anayama M, Makino M, Makino Y, Tamura K, Nagasawa M: Atypical Hemolytic Uremic Syndrome Associated with Complement Factor H Mutation and IgA Nephropathy: A Case Report Successfully Treated with Eculizumab. Nephron, 2018; 138: 324-327 [DOI] [PubMed] [Google Scholar]
- 98). Nishimura J, Yamamoto M, Hayashi S, Ohyashiki K, Ando K, Brodsky AL, Noji H, Kitamura K, Eto T, Takahashi T, Masuko M, Matsumoto T, Wano Y, Shichishima T, Shibayama H, Hase M, Li L, Johnson K, Lazarowski A, Tamburini P, Inazawa J, Kinoshita T, Kanakura Y: Genetic variants in C5 and poor response to eculizumab. N Engl J Med, 2014; 370: 632-639 [DOI] [PubMed] [Google Scholar]