

Summary
Ionotropic glutamate receptors (iGluRs) mediate the majority of excitatory neurotransmission in the brain. Their dysfunction is implicated in several neurological disorders, rendering iGluRs potential drug targets. Here, we performed a systematic analysis of the druggability of two major iGluR subfamilies, using molecular dynamics simulations in the presence of drug-like molecules. We demonstrate the applicability of druggability simulations by faithfully identifying known agonist and modulator sites on AMPA receptors (AMPARs) and NMDA receptors. Simulations produced the expected allosteric changes of the AMPAR ligand-binding domain in response to agonist. We also identified a novel ligand-binding site specific to the GluA3 AMPAR N-terminal domain (NTD), resulting from its unique conformational flexibility that we explored further with crystal structures trapped in vastly different states. In addition to providing an in-depth analysis into iGluR NTD dynamics, our approach identifies druggable sites and permits the determination of pharmacophoric features towards novel iGluR modulators.
Keywords: Ionotropic glutamate receptors, AMPA, NMDA, kainate receptors, druggability simulations, ligand binding, molecular dynamics, allosteric interactions
Graphical Abstract
eTOC Blurb
Lee et al. assess the druggability of the ionotropic glutamate receptor subfamilies, using molecular dynamics simulations in the presence of drug-like molecules and x-ray crystallography. The study presents a ligand-binding site in the GluA3 N-terminal domain and supports the role of conformational plasticity in modulating ligand binding.
Introduction
iGluRs are tetrameric cation channels that trigger depolarization of postsynaptic membrane upon activation by L-glutamate (Traynelis et al., 2010) and their signaling underlies learning and memory (Huganir and Nicoll, 2013; Kessels and Malinow, 2009; Paoletti et al., 2013). These receptors have been implicated in many pathological conditions including chronic pain, ischemia and epilepsy (Bowie, 2008), and in neurological disorders such as Parkinson’s, Alzheimer’s, and Huntington’s diseases, thereby sparking interest as potential drug targets (Bowie, 2008; Chang et al., 2012; Partin, 2015).
The four iGluR classes, N-methyl-D-aspartate receptors (NMDARs), α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPARs), kainate receptors (KARs), and delta receptors (Figure S1A), subtype-selectively assemble predominately as heterotetramers into receptors with distinct properties and functions (Herguedas et al., 2013; Traynelis et al., 2010). iGluRs have a shared architecture (Karakas et al., 2015; Mayer, 2016), comprised of an extracellular region (ECR), a transmembrane domain (TMD forming the ion channel), and an intracellular carboxyl-terminal domain (CTD) (Figure 1). The ECR consists of a distal N-terminal domain (NTD) that binds allosteric modulators in NMDARs (inset in Figure 1B), and a membrane-proximal ligand-binding domain (LBD) that binds glutamate (Figure 1A, inset; or glycine in GluN1 or GluN3 NMDAR subunits) (Traynelis et al., 2010). Both ECR domains are organized as pairs of dimers, which fold into clamshell-like structures that belong to the periplasmic-binding protein (PBP) superfamily.
Figure 1. iGluR structure and known ligand binding sites.

Crystal structures of two main iGluR subtypes (see Figure S1A). (A) GluA2 AMPAR structure (PDB ID: 4U5B) (Chen et al., 2014) highlights the NTD, LBD and TMD. Two equivalent chain pairs A/C and B/D are colored in orange and grey, respectively. The inset shows LBD dimer AD with ligand-binding sites for glutamate (agonist; green spheres) at the cleft between the D1 and D2 lobes, and allosteric modulators such as cyclothiazide (blue spheres) at the dimer interface are circled. (B) The structure of an allosterically-inhibited GluN1/N2B NMDAR (PDB ID: 4PE5) (Karakas and Furukawa, 2014) highlights the closely packed NTD, LBD and TMD. The GluN1 chains A/C and the GluN2B chains B/D are colored orange and grey, respectively. The inset shows a close-up view of the NTD heterodimer AB. The clamshell cleft divides each monomer into an upper lobe (UL) and a lower lobe (LL). The binding sites for the allosteric modulators ifenprodil (blue spheres), Zn2+ (from PDB ID: 3JPY) (Karakas et al., 2009) and polyamines (from functional data) (Mony et al., 2011) are indicated by the dashed circles.
Extensive functional and structural data show that channel gating is coupled to closure of the LBD clamshell upon agonist binding (Greger et al., 2017). In AMPARs, prolonged agonist binding also triggers desensitization where rearrangement within LBD dimers relieves the tension on the TMD to allow channel closure with agonist remaining bound (Armstrong et al., 2006; Hansen et al., 2007; Horning and Mayer, 2004; Sun et al., 2002). A number of positive allosteric modulators target the interface between LBD dimers (Figure 1A, inset; designated as D1), which results in dimer stabilization and attenuated desensitization (Jin et al., 2005; Partin, 2015; Sun et al., 2002).
In NMDARs, NTD ligands trigger profound changes in channel open probability and deactivation kinetics (Zhu and Paoletti, 2015). NMDAR NTD dimers exhibit loose packing between the lower lobes (LLs) that facilitate modulator binding (Mony et al., 2011) and enable packing against the LBD (Karakas et al., 2014), thereby mediating allosteric coupling between domain layers (Krieger et al., 2015; Tajima et al., 2016; Zhu and Paoletti, 2015). Although nonNMDARs harbor more rigid NTD dimers with tight packing of the LLs (Jin et al., 2009; Kumar and Mayer, 2010; Kumar et al., 2009; Kumar et al., 2011; Rossmann et al., 2011) and substantially looser connections between layers (Meyerson et al., 2014), AMPAR NTDs possess intrinsic conformational flexibilities that favor clamshell motions and enable transitions to NMDAR-like conformations, an observation that is particularly apparent for GluA3 (Dutta et al., 2012; Sukumaran et al., 2011). Moreover, interlayer dynamics between NTD and LBD are also observed in AMPARs (Dutta et al., 2015; Herguedas et al., 2016) raising the possibility of allosteric communication in the ECR of non-NMDA iGluRs.
These findings, together with the unique sequence divergence of the NTD, render it a potential drug target in contrast to the highly conserved LBD whose ligands show non-selective broad-range effects (Rogawski, 2011). We therefore assessed the druggability of iGluR NTDs using MD simulations with drug-like probes (Bakan et al., 2014; Bakan et al., 2011; Bakan et al., 2012). We show that sites for known NMDAR modulators are captured with good accuracy, as are those for agonists and modulators targeting the AMPAR LBD. Interestingly, probes prominently locate to the GluA3 AMPAR NTD LL dimer interface, in analogy to known modulator positions in NMDAR NTDs and in related metabotropic glutamate receptor (mGluR) and atrial natriuretic peptide receptor domains (Abe et al., 2003; Misono et al., 2011; Mony et al., 2011; Tsuchiya et al., 2002). Crystallography of the GluA3 NTD reveals an anion binding site between the lower lobes, in accord with our simulations, further highlighting the substantial flexibility of GluA3 NTD dimers. Together, our approach can identify and optimize modulator binding sites in iGluRs and beyond.
Results
We performed druggability simulations for three major iGluR subfamilies, AMPARs, NMDARs and KARs, in the presence of the probes listed in Figure S1. Before characterizing NTDs, we first verified the ability of our simulations to capture the well-known ligand-binding spectrum of the GluA2 LBD (Pohlsgaard et al., 2011).
Druggability simulations reveal acetate binding to the LBD cleft, and domain closure and stretching at the LBD-TMD linker
Our druggability simulations starting from the apo form of a GluA2 LBD monomer (PDB ID: 1FTO) (Armstrong and Gouaux, 2000) (runs 1 and 2, see Table S1) showed that an acetate (ACTT) ion readily located to the LBD cleft and remained bound throughout most of the simulations, favoring transient domain closure (Figure 2A). ACTT interacted first with R485 on the D1 lobe (a residue essential for agonist binding (Armstrong and Gouaux, 2000)), which drove its entry into the ligand-binding pocket and cleft closure was facilitated by the interaction with S654 on the D2 lobe (Figure 2A, bottom panel). This reveals the capability of our probes to mimic native ligands such as glutamate, which is also coordinated by R485 and S654 to trigger clamshell closure (Armstrong and Gouaux, 2000; Lau and Roux, 2007, 2011; Yu and Lau, 2017).
Figure 2. Probe binding and conformational changes in AMPAR LBD monomers or dimers.

(A) Clamshell motions in 120 ns druggability simulation of the apo LBD monomer (run 2 in Table S1) measured by the changes in the distance between the α-carbons of E402 in D1 and T686 in D2. The closest distance of approach (~ 7.5 Å) is comparable to that observed in the glutamate-bound structure (PDB 1FTJ), showing the ability of the unbound LBD monomers to sample closed conformers that are stabilized upon ligand binding. The overlay of the initial open (orange) conformation and a closed (green) form sampled at the times boxed in A highlights acetate binding to R485 and S654 (black arrows) and LBD closure (curled red arrows). (B) Cleft closure and linker separation in the 120 ns simulation of the LBD dimer (1FTO chains A and B, equivalent to chains A and D in whole receptors; run 3 in Table S1). The upper graph shows cleft closure for each subunit as in A and the lower graph shows the increase in the LBD-TMD linker distance represented by the inter-subunit P632 distance. The latter increases as the LBDs close (see overlay below) and pulls open the channel. (C) Probe molecules localized at the interface of the LBD dimer (from run 3 in Table S1) closely overlap with the experimentally observed positions of the allosteric modulators cyclothiazide (cyan balls/stick; from PDB ID: 1LBC; Sun et al., 2002) and (R,R)-2b (magenta balls/sticks; PDB ID: 4U5B; Chen et al., 2014). The zoom-out view shows the starting dimeric structure (PDB ID: 1FTO) with the subunits A and D in green and yellow. Subunit A is not displayed in the zoomed in panel for clarity. See also Figure S2.
Ligand-induced cleft closure results in an increase in the distance between the D2 lobes of the LBD dimer, which exerts strain on the LBD-TMD linkers, culminating in the opening of the channel gate (Armstrong and Gouaux, 2000; Chen et al., 2017; Mayer, 2016). Our simulation of the apo LBD dimer (run 3) also showed cleft closure (Figure 2B, top graph) and an increase in the distance between P632 residues in each of the D2 lobes at the top of the LBD-M3 linker (Figure 2B, bottom graph). We observed linker distance fluctuations between ~20 Å (starting value) and ~45 Å, similar to variations in the crystal structures (Pohlsgaard et al., 2011). Probe binding to the clefts was also observed in our LBD dimer simulations similar to that observed in the monomers. Overall, these data are in agreement with experimentally observed binding poses of agonists.
Probe molecules cluster at an allosteric site at the LBD dimer interface
AMPAR desensitization involves a rearrangement of the LBD dimer interface, leading to a ligand-bound, closed-channel conformation (Armstrong et al., 2006; Chen et al., 2017; Horning and Mayer, 2004; Meyerson et al., 2014; Sun et al., 2002; Twomey et al., 2017). Positive allosteric modulators such as cyclothiazide (Sun et al., 2002), aniracetam (Jin et al., 2005), and (R,R)-2b (Kaae et al., 2007) bind to and stabilize the dimer interface, leading to decreased desensitization (Partin, 2015). Here, we observed that probe molecules also concentrate at this modulator site. Indeed, their preferential positioning shows a good overlap with cyclothiazide and (R,R)-2b (Figures 2C and S2). Collectively, this comparative analysis shows that our method identifies the known binding sites of agonists and allosteric modulators on the LBD. These results encouraged us to explore potential ligand-binding sites in iGluR NTDs.
The NTDs of AMPARs, NMDARs, and KARs exhibit large variations in their ability to bind drug-like molecules at their dimeric interface
We first simulated the druggability of NTD monomers of nine iGluRs including the AMPAR, KAR and NMDAR subtypes (see Figure S1A). Simulations consistently identified binding events at the regions that form the NTD dimer interface. Such regions within 5 Å of the probes, termed ‘interfacial hotspots’ (HS), are illustrated for NTDs from NMDAR subunits GluN1 (Figure 3A) and GluN2B (Figure 3B), and AMPAR subunit GluA2 (Figure S3A-C). Table S2 lists the composition of probes at these HSs for all paralogs. Apart from the GluN1 LL, at least nine probes bound to the dimer interface in each case (Table S2), and among them we noted a dominance of non-charged/hydrophobic probes like isopropanol and isobutane. Hence, our approach enables detection of dimer association propensities for different subtypes of iGluR NTDs.
Figure 3. Capturing ligand-binding sites in NMDAR NTD monomer simulations.

(A-B) Probes bind at the dimer interface regions of GluN1 (A) and GluN2B run 2 (B), mimicking inter-subunit interactions, and binding of ifenprodil at the UL hotspot (HS) and polyamines at the LL HS. See probe-bound interfacial residues in other NTD monomer simulations in Figure S3A-C and Table S2. (C) A zoomed view into the GluN2B NTD cleft shows probe-binding sites for IPAM that are near the Zn2+-binding site (grey) (Karakas et al., 2009). (D) Zoomed views of the ULs of GluN1 and GluN2B show the overlap between the binding sites of the probes and that of ifenprodil. IBUT molecules preferentially bind to GluN1, and IPAM and BENZ to GluN2B. (E) Distribution of contacts between probe molecules and GluN1 (top) and GluN2B (bottom) residues. As shown in D, GluN1 P106-F113 make contacts with IBUT, and GluN2B E106-Q118 with IPAM and BENZ. See results for NMDAR NTD dimer in Figure S3D-F. (F) Close-up views of LLs show probe binding that mimics the polyamine-binding site. Residues reported to be important for polyamine binding (Mony et al., 2011) are colored white, and those observed in our simulations to interact with probes are displayed as sticks.
Druggability simulations provide insights into the interactions of NMDAR NTD with modulators
NMDAR NTDs bind allosteric ligands that modulate NMDARs in a subunit-selective fashion, including endogenous Zn2+ ions, polyamines and synthetic compounds, such as the phenylethanolamines ifenprodil and Ro25-6981 (Mony et al., 2009). Functional and structural studies have mapped binding sites to the clamshell cleft for Zn2+ (Choi and Lipton, 1999; Fayyazuddin et al., 2000; Karakas et al., 2009; Paoletti et al., 2000; Rachline et al., 2005; Romero-Hernandez et al., 2016), the UL dimer interface for phenylethanolamines (Karakas et al., 2011), and the LL dimer interface for polyamines (Mony et al., 2011) (Figure 1B). Our simulations of the GluN1 (Farina et al., 2011) and GluN2B NTD monomers (Karakas et al., 2009) captured all three sites (Figure 3) and provided further information on their molecular driving forces and specificities, as described below.
GluN2B NTD cleft closure is associated with negative allosteric modulation and is transmitted to the LBD via the LLs (Gielen et al., 2009; Mony et al., 2011; Tajima et al., 2016). Simulations of GluN2B monomers show a high affinity for positively-charged isopropylamine (IPAM) probes at the site that binds Zn2+ ions (Figure 3C). IPAMs bind near zinc-coordinating residues H127, E284, D102 and D265 (Karakas et al., 2009; Rachline et al., 2005).
Figure 3D shows probes localized near another modulator binding site, the phenylethanolamine site at the UL interface regions of GluN1 and GluN2B, which overlapped with ifenprodil (see also Table S2). GluN2B simulations indicated binding of IPAM and BENZ molecules at the site, and the overall binding affinity was ~ 54 nM akin to that reported for Ro25-6981 (Karakas et al., 2011). This site harbors a number of negatively charged residues (peaks at Glu106, Gln110, Asp113, Phe114, and Gln118 in Figure 3E lower graph), which strongly attract the positively charged IPAM. On the other hand, the ifenprodil-binding site on GluN1 lacks such negatively charged residues and instead contains Pro106, Tyr109, Thr110, and Phe113. In accord with the chemical properties of these residues, GluN1 simulations showed a cluster of hydrophobic (mainly IBUT) and a few polar (IPRO) probes at this site (Figure 3E upper graph). The higher affinity of GluN2B for IPAM is therefore explained by its specific sequence at the HS. Given that electrostatic interactions (that attract IPAMs) are stronger and long-ranged, GluN2B would drive the interaction with ifenprodil in line with its subunit-selectivity.
Finally, we observed a third cluster of aliphatic (IBUT) probes together with a few polar (IPRO and IMID) and charged (ACTT) probes in the GluN2B simulations near the LL polyamine site (Mony et al., 2011) (colored white in Figure 3F, right). Again, our simulations yielded a relatively small binding site on GluN1 (Figure 3F, left) in accordance with GluN2B driving subtype-selective polyamine potentiation and the site being coordinated by both subunits. It is also consistent with this region accepting a range of synthetic compounds with polar/charged character and the GluN1exon 5 splice insert locating to the LL dimer interface (Berger et al., 2013; Chandler et al., 1993; Traynelis et al., 1995).
Taken together, these simulations (i) reveal multiple druggable sites on the NMDAR NTD surface consistent with binding phenylethanolamines, polyamines and Zn2+; (ii) highlight the role of GluN2B as a driver for drug binding and specificity; and (iii) elucidate specific features of probe molecules, both geometric and chemical, that exhibit differential affinity for various sites, which can guide the design of more selective pharmacophore models.
Relaxation along soft modes is a prerequisite for detecting druggable sites at the interface of NMDAR NTD dimers
Next, we explored the binding characteristics of the NMDAR NTD heterodimer (PDB ID: 3QEL). At first, we detected no significant probe entry to the ifenprodil-binding site within 60 ns (Figure S3D). A few probes were observed to bind to LL interface regions that are solvent-exposed despite dimerization consistent with polyamine binding (Mony et al., 2011; Traynelis et al., 1995), but the buried UL interface remained inaccessible. Similar results were seen using the apo heterodimer (PDB ID: 5B3J; data not shown).
To test whether the lack of probe entry was due to the closed-interface conformation used as input, we generated an ‘open’ dimer by allowing the structure to reconfigure along the soft modes predicted by the anisotropic network model (ANM) (Atilgan et al., 2001; Bahar et al., 2017), a technique that has proved useful in docking studies (Cavasotto et al., 2005; Floquet et al., 2006). We tested a mode that was confirmed experimentally to allosterically modulate glutamate binding in NMDARs (Zhu et al., 2013). This mode features (i) a rotation of the two subunits that rearranges the UL dimer interface, and (ii) a twisting and closure of the GluN1 NTD cleft that brings its LL into closer proximity with the GluN2B UL. The conformation generated along this mode (Figures S3E and S5F) formed an interface between the GluN1 LL and GluN2B UL where the Cα-distance between two marker residues, G200 (GluN1) and I329 (GluN2B) (that were cross-linked to confirm this motion (Zhu et al., 2013)), became 8.4 Å compared to 19.3 Å between equivalent residues in the starting structure (Figure S3E).
Druggability simulations starting from this open dimer showed probe entry through a newly formed opening at the base of the GluN1/N2B UL interface, enabling access to the ifenprodil-binding site. Probe distribution was similar to that seen in monomer simulations, where the region coordinating the phenolic group of ifenprodil attracted positively charged IPAMs (blue spheres in Figure S3F) while another IPAM mimicked the interactions of the piperidine group, together with an additional ACAM (pink sphere). Also, the residues that coordinate the binding site (sticks colored by chain) closely resembled those coordinating ifenprodil (magenta sticks) and other negative allosteric modulators observed in crystal structures and modeling studies (Burger et al., 2012; Karakas and Furukawa, 2014; Karakas et al., 2011). Overall, the overlap between our probe molecules and the experimentally observed allosteric inhibitor site underscores the utility of generating ANM-relaxed conformers for detecting buried drug-binding sites as shown previously (Bakan and Bahar, 2009; Floquet et al., 2006; Lee et al., 2017; Molina et al., 2009; Rueda et al., 2009; Taguchi and Kitao, 2016), and show that such an approach is relevant for inter-subunit interfaces.
The GluA3 AMPAR NTD reveals a high potential for drug-binding at the dimer interface
The above data validate our approach in identifying modulator-binding sites, which we next applied to currently unexplored targets. In contrast to NMDARs, no NTD modulators have yet been described for the AMPAR NTD, and whether this domain is a potential drug target is currently unclear.
When evaluating the druggability of the GluA1-GluA3 NTD dimers (GluA4 is similar to GluA2 (Dutta et al., 2012) and was therefore excluded), we observed probe binding mainly to the solvent-exposed surfaces of GluA1 and GluA2; whereas the GluA3 NTD exhibited high probe occupancy in the LL interface near the cleft (Figure 4A-C and S4). This unique behavior can be ascribed to the higher flexibility and looser packing of the GluA3 NTD dimer (Dutta et al., 2012; Herguedas et al., 2013; Rossmann et al., 2011; Sukumaran et al., 2011). Since GluA3 receptors have been implicated in various neurological disorders (Reinders et al., 2016; Yuan et al., 2015) and are enriched in specific regions of the brain (Gold et al., 1997; Rubio et al., 2017; Schwenk et al., 2014; Wang et al., 2011), we investigated the allosteric potential of this subunit in greater depth using X-ray crystallography and drugability simulations.
Figure 4. Distinctive ligand binding and conformational flexibility of GluA3 NTD dimers revealed by druggability simulations and X-ray crystallography.

(A-C) Conserved probe binding patterns observed in multiple druggability simulations of GluA1 (A), GluA2 (B) and GluA3 (C) NTD dimers. In contrast to GluA1 and GluA2, GluA3 features a high affinity ligand-binding site at the LL interface (see contact distributions in Figure S4). (D-H) Our crystal structures of the GluA3 NTD reveal unique dimeric states: one with phosphate bound between the pairs of R163 and R184 residues (D), which is comparable to the previously resolved open conformer (PDB ID: 3O21 dimer CD; see Figure S5); a super-open form (E); and a displaced dimer found in the same crystal as the phosphate bound form (G). Bottom views in F and H compare the PO43−-bound structure to the super-open and displaced structures, respectively. Phosphate binding is mimicked by dianionic methylphosphate binding in additional druggability simulations (see Figure S7).
GluA3 NTD crystal structures exhibit a spectrum of conformations
We trapped three conformational states of GluA3 NTD dimers, which we term: 1) phosphate-bound ‘parallel’, 2) ‘super-open’, and 3) displaced (Figures 4D-H and Figure S5 and Table S3). These conformers were predicted from our simulations (Dutta et al., 2012; Krieger et al., 2015; Sukumaran et al., 2011) and reveal two main motions – an opening of the LLs (Figures 4E-F and S5B), and a shearing motion, leading to a rotation of the two NTD monomers relative to one another (Figures 4G-H and S5C). This conformational spectrum is reminiscent of the allosterically active ligand-binding cores of mGluRs, GPCRs which harbor a Type-1 PBP domain that is closely related to NTDs and binds agonists in its clamshell cleft and cations at the dimer interface (Kunishima et al., 2000; Tsuchiya et al., 2002), although the extent of conformational change is smaller (Figure S5D-E). The displacement and closing of GluA3 NTD dimers also resembles the conformational change predicted by the ANM for the NMDAR dimer described above (see Figure S5F).
Anion binding modulates the extent of opening of the GluA3 NTD dimer interface, reminiscent of mGluRs
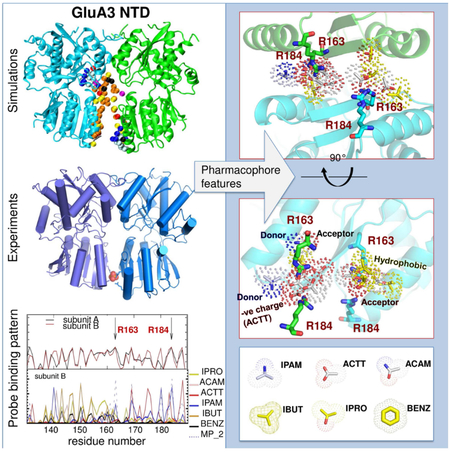
A hallmark of the GluA3 NTD is two pairs of arginines projecting into the base of the LL interface (Sukumaran et al., 2011), weakening the dimeric assembly (Rossmann et al., 2011). Here we capture a phosphate ion (PO43−) coordinated by the guanidinium groups of R163 and R184, thereby stabilizing a near-parallel arrangement of the NTD dimer (Figures 4D, F and S5B). A similar ‘parallel’ conformation was observed in our earlier GluA3 structures (PDB ID: 3O21 chains CD and PDB ID: 3P3W chains AC; see Figure S5B) (Sukumaran et al., 2011), but not in other AMPAR or KAR NTDs. This raises the possibility that other anions (such as ACTT in our simulations; Figures 4C, S7 and 6A-B) may stabilize a state unique to GluA3.
Figure 6. Pharmacophore models for two high-affinity pockets at the GluA3 NTD LL interface.

(A-B) Binding pocket formed by R163 and R184 from adjacent subunits and corresponding pharmacophore model shown from the bottom (A) and an open interface view (B). The probes represent the consensus types and poses sampled during the simulations. Hydrophobic probes are colored yellow and polar probes white, hydrogen bond donors are blue and acceptors red. Note the abundance of acetates, making it a primarily negatively charged site. (C-D) The hotspot around M150 and its pharmacophore model, colored as above. This site is hydrophobic (yellow).
In the absence of ligand, the parallel arrangement is less favorable, as a result of repulsion between R163 and R184, and consequently there are relatively few contacts between the LLs (local atomic contact density, LD, 6.7; (Bahadur et al., 2004; Herguedas et al., 2013; Sukumaran et al., 2011)). MD simulations in the presence and absence of PO43− show this parallel arrangement is stabilized by anion binding (Figure 5), with PO43− locking the LLs together and forming a number of stable contacts, while in the absence of PO43− the LLs exhibit relatively large motions. The GluA3 dimer in PO43−-free simulations samples a range of ‘super-open’ conformations as noted previously (Dutta et al., 2012), with LL residues exhibiting large fluctuations, while mostly intact UL interactions hold the monomers together.
Figure 5. MD simulations of the GluA3 NTD dimer in the presence and absence of phosphate reveal that it stabilizes the structure.

(A) Color-coded diagrams (from blue to red, with increasing root-mean-square fluctuations (RMSF) of α-carbons) observed over the course of 100 ns MD runs with (left; PO43− position shown by the red sphere) and without (right) phosphate bound to the NTD LL interface. The LLs are stabilized by phosphate binding. (B) RMSF profile of residues in each subunit, shown for both runs for comparison. LL residues (116-245 and 355-380) show large movement in the absence of phosphate, as compared to the phosphate-bound state, while the UL residues (1-115, 250-350) mostly remain stable, maintaining bound states. (C) The root-mean-square deviation (RMSD) from the starting conformer observed over the course of the simulations shows that phosphate removal leads to an excursion away from the starting structure followed by the stabilization of a state similar to the starting structure (black). In the presence of phosphate, the RMSD remains low throughout (violet).
We also captured this ‘super-open’ state by X-ray crystallography (Figure 4E and Figure S5A-B) - the structure exhibits a parallel arrangement of the NTD protomers, with the LLs splayed apart and all interface contacts lost. The center-of-mass distance for the LLs is 33.6 Å in the phosphate-bound dimer and increases to 37.3 Å in the super-open state. The latter is comparable to 37.9 Å for mGluR1 LLs (PDB ID: 1ISR). Therefore, GluA3 LLs can partially dissociate. This motion is modulated by anions, analogous to the closely related mGluR1 where gadolinium ions hold the LLs in a parallel conformation and modulate GPCR signaling (Abe et al., 2003; Tsuchiya et al., 2002).
Lastly, in the ‘displaced’ structure, a shearing motion between the NTDs leads to a substantial reconfiguration of the LL dimer interface (Figure 4G and H). New inter-LL contacts are mediated by symmetrical αE interactions involving F143, L146 and M150, along with several interactions between αE and β7, that are ‘out of reach’ in the parallel configuration (Figure S5A and C). Whereas in the parallel conformation R163 and R184 likely cause LL repulsion, these residues engage in additional interactions in the displaced conformation - R163 forms a salt bridge with E151 and R184 interacts with Q155 (Figure S5C). These interface contacts increase the LD to 44.5 (Figure S5A), a value which is usually obtained for NTD UL interfaces (Herguedas et al., 2013), indicating this is a stable, ‘low-energy’ state for the GluA3 NTD dimer.
Comparison of the dynamics of the parallel (phosphate-bound and phosphate-free), super-open, open, and displaced conformations further clarifies the conformational spectrum of GluA3 NTD dimers and shows that the displaced state is highly stable. Principal component analysis of the trajectories revealed that the phosphate-removed, super-open and open conformations explored large regions and exhibited transitions between states, while the displaced structure remained confined to the close vicinity of the starting conformer (Figure S6). Movements along the first two principal components (PCs), inter-dimer shear motions (PC1) and LL-LL opening (PC2), respectively (see Movies S1-S2), dominated the transitions. The phosphate-free and super-open systems underwent opening and closure motions along PC2; whereas open (PDB: 3O21 dimer CD) and super-open dimers revealed transitions along PC1. In fact, the open dimer reached structures in the basin of the displaced conformation. The parallel-to-displaced transition sampled by the crystal structures and simulations encompasses a shift of ~20 Å at helical termini facing the LBD (between PO43−-bound and displaced structures) (Figure 4H). An intermediate state (reached in the super-open simulation) is occupied by a ‘semi-displaced’ dimer (PDB ID: 3P3W) (Sukumaran et al., 2011), which resembles other AMPAR NTDs (Figure S5C) (Clayton et al., 2009; Dutta et al., 2012; Jin et al., 2009; Rossmann et al., 2011; Yao et al., 2011).
Overall, this LL flexibility yields a unique ability of GluA3 NTD dimers to capture ligands at their dimer interface. This behavior mirrors that of related signaling domains (e.g. in mGluRs) and supports the ligand-binding sites detected in our druggability simulations (Figure 4C). Additional runs with the phosphate derivative dianionic methylphosphate (MP_2) confirmed ligand binding to R184 in the super-open dimer (Figure S7A and D). Simulations of the open GluA3 (PDB 3O21 CD; Figure S7B and E) were similar to Figure 4C. In addition to the neighborhood of R163 and R184 that coordinate a negatively charged probe, another hotspot distinguished by its high probe-binding propensity is the vicinity of M150 (Figure S7). By contrast, probe access is severely restricted for the GluA2/A3 heterodimer (PDB ID: 5FWY AB) (Figure S7C and F), which forms tight dimers akin to GluA2 homodimers (Herguedas et al., 2016; Rossmann et al., 2011; Zhao et al., 2017).
Pharmacophore features of the GluA3 ligand site
Probes targeting the GluA3 LL interface can be divided into two groups. One is centered at the phosphate/anion binding site near R163 and R184 at the base of the interface and is enriched in acetates that shield the charge repulsion like PO43− (Figures 6A-B and S7). The second contains primarily hydrophobic molecules that bind in a pocket between the M150 residues from adjacent subunits (Figure 6C-D and S7). The probes that make large contributions to binding plus their effective affinities are listed in Table 1. Overall, this analysis suggests that there is a high potential for identifying GluA3-specific modulators with nanomolar affinities, and the pharmacophore models generated here could be used for screening small molecule libraries and refining hit compounds.
Table 1.
Druggability of GluA3 NTD LL interface pockets.
| LL Probe Cluster | ΔGB probe binding |
Affinity | Probe composition |
charge |
|---|---|---|---|---|
| Site 1 (near R163 & R184) |
−11.4 | 5.6 nM | 2 IPRO, 1 IPAM, 5 ACTT |
4e− |
| Site 2 (near M150) |
−13.08 | 0.34 nM | 5 IPRO, 2 ACAM, 1 BENZ |
0 |
Discussion
The unexpected similarity in the intrinsic dynamics of AMPARs and NMDARs, hinting at a potential allosteric role for non-NMDA iGluR NTDs (Dutta et al., 2015; Dutta et al., 2012; Herguedas et al., 2016), prompted us to evaluate the “druggability” of the AMPAR NTD. To achieve realistic detection of ligand-binding sites of iGluR NTDs and their maximal binding free energy, we performed MD simulations of these domains in the presence of drug-like probe molecules. We benchmarked this method by exploring the well-known ligand-binding landscape of the GluA2 LBD, where we found probe binding in the agonist-binding cleft and the allosteric modulator binding pocket at the dimer interface. Similarly, we observed interactions with known ligand sites in NMDAR NTDs including those for zinc, polyamines and phenylethanolamines, providing opportunities for designing higher-affinity compounds. Another result from our analysis is the accuracy with which the probe-binding hotspots overlap with known dimer interfaces on monomeric NTDs, opening new avenues for predicting protein-protein interfaces.
In AMPAR NTDs, we observe probe binding on the outer surfaces of the NTDs (Figures 4A-C) at sites that may interact with proteins involved in synaptic clustering of AMPARs (Garcia-Nafria et al., 2016; Greger et al., 2017; Watson et al., 2017). A similar role has been described for the delta iGluR and KAR NTDs where interaction with secreted proteins has been shown to be key for synaptic anchoring of these iGluRs and for signaling through the LBD and TMD (Elegheert et al., 2016; Matsuda et al., 2016; Yuzaki and Aricescu, 2017).
Our data revealed extensive probe binding specifically at the GluA3 NTD LL dimer interface, a known allosteric site utilized in other receptors, such as mGluRs and natriuretic peptide receptors (Misono et al., 2011; Tsuchiya et al., 2002), and by NMDAR NTDs (Mony et al., 2011; Tajima et al., 2016). We further confirmed this finding experimentally with a GluA3 NTD structure coordinating PO43− between the NTD LLs. In aggregate, our results point to the GluA3 NTD as a potential drug target. However, although GluA3-containing AMPARs have received increasing attention recently (Gutierrez-Castellanos et al., 2017; Renner et al., 2017) with emerging therapeutic impacts (Davies et al., 2017; Reinders et al., 2016; Wu et al., 2007), allosteric modulation would require the existence of GluA3 homodimers. According to current models, AMPARs preferentially exist as heteromers containing the GluA2 subunit (which restricts Ca2+ flux) and are assembled from two pairs of heterodimers (Greger et al., 2017; Herguedas et al., 2016; Herguedas et al., 2013). Nevertheless, GluA2-lacking, Ca2+-permeable AMPARs exist in various types of interneurons (Geiger et al., 1995; Moga et al., 2003) and GluA3 subunits are enriched in the thalamus (Wang et al., 2011), auditory brain stem (Rubio et al., 2017), and cortex (Schwenk et al., 2014). Hence, further advances such as superresolution imaging and single cell transcriptomics/proteomics are expected to shed light on the exact composition of AMPARs in the brain. In fact, our results may enable the design of a fluorescent tracer molecule, capable of selectively identifying GluA3 homomers, which would be of therapeutic relevance (Davies et al., 2017; Wu et al., 2007).
Another question is whether and how the spectrum of GluA3 NTD dimer conformations seen in our simulations and structures would influence the rest of the receptor. First insights of how NTD dynamics couples to the downstream portions are emerging for NMDARs (Dutta et al., 2015; Krieger et al., 2015; Tajima et al., 2016; Zhu et al., 2016). The ‘displacement’ or ‘super-opening’ of the NTD LLs may lead to a further splaying of the inter-dimer LBD interface, with consequences for gating or interaction with auxiliary subunits (Mayer 2016).
In summary, our study opens an arena for the identification of drug-like molecules that may allosterically modulate GluA3 receptors or tracer molecules that would identify neuronal populations enriched in them, thereby leading to a better understanding of the AMPAR conformational spectrum and shedding light on the biology of this currently enigmatic AMPAR subunit.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ivet Bahar (bahar@pitt.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK293S GntI− (Reeves et al., 2002) stably producing GluA3 NTD (Rossman, EMBO, 2011) were grown at 37 °C in DMEM supplemented with 2.5% FBS, HEK293 cells are of female background. Cell lines were not authenticated.
METHOD DETAILS
Druggability molecular dynamics simulations
MD simulations and druggability analyses were performed as described previously (Bakan et al., 2012) using the probe set shown in Figure S1B (selected as representatives of drug fragments with different physicochemical properties), using the MD simulation package NAMD (Phillips et al., 2005) with the CHARMM22 force field for proteins (Mackerell et al., 2004), the TIP3P water model (Jorgensen et al., 1983), and CGENFF (Vanommeslaeghe and MacKerell, 2015) for the probes. A series of independent runs were performed using either known structures or alternative conformers generated by the ANM (Atilgan et al., 2001; Bahar et al., 2017) for different subtypes of NTDs and LBDs, either in monomeric or dimeric forms, with different compositions of probe molecules, as detailed in Table S1. We relaxed the systems using equilibration steps and performed NPT dynamics simulations with various durations, in the range 40 to 120 ns, each with 2 fs time step. Nose-Hoover constant pressure (1 bar) and temperature (300 K) were used. Some simulations without probes were run in GROMACS 5 (Abraham et al., 2015) also using CHARMM22 (Bjelkmar et al., 2010; Mackerell et al., 2004) and TIP3P water.
Druggability trajectory analysis
Trajectory analyses were performed using the method described earlier (Bakan et al., 2012), implemented in the DruGUI module of ProDy (Bakan et al., 2011;Bakan et al., 2014), a plugin for the molecular graphic progam VMD (Humphrey et al., 1996). All MD snapshots were superposed onto the reference PDB structure of the protein using Cα atoms and a cubical grid-based representation of the space. Grid edge size was set to 0.5 Å. Probe molecules having a non-hydrogen atom within 2.5 Å from protein atoms were considered to interact with the protein. For each probe type, the individual occupancy of grids was calculated using their central carbon atoms. We obtain occupancy of each probe for a given voxel. High occupancy voxels, called hot spots, within a distance less than 5.5 Å were merged and druggable sites were defined upon merger of at least six hot spots. Figures were made using VMD (Humphrey et al., 1996) and PyMOL (Schrödinger, 2015).
The binding energy of individual probes was calculated using the inverse Boltzmann relation, based on the ratio of the actual number of a given probe in a given grid cell on the protein surface, relative to that in solution, as described earlier (Bakan et al., 2012). Briefly, a grid-based method is used and the binding free energy of probe molecules at each grid point i is evaluated as ΔGi = − RT ln(ni/n0). Here, ni/n0 is the ratio of the observed density of probes in MD simulation at a given position on the protein surface (ni) to the average density in solution (n0), R is the gas constant and T is the absolute temperature (K). Binding events are accounted for by considering probe molecules as independent particles. The binding free energy map identifies interaction spots with low energy. The maximal binding affinities of probe hotspots were calculated as the sum of these binding energies, as described previously (Bakan et al., 2012). Evaluation of maximal achievable binding affinity utilizes a physics-based atomistic simulation and is therefore independent of any training set. The binding free energies have been evaluated in a fully automated way using the DruGUI module of ProDy, with the dissociation constant KD obtained from the binding free energy ΔGB = RT ln KD.
Anisotropic Network Model (ANM)
The ANM (Atilgan et al., 2001; Bahar et al., 2017) represents the protein as a network where each Cα atom defines the position of a node. The overall potential is represented as the sum of harmonic potentials between interacting nodes (Cα-Cα distance < 15Å). The force constants for the 3N×3N interactions are given by the elements of the Hessian matrix H. Collective motions of the protein are obtained in terms of the nonzero eigenvalues λk and eigenvectors uk of H (1 ≤ k ≤ 3N-6, where N is total number of nodes). The new conformation along the selected mode (k=2) was generated with ProDy (Bakan et al., 2014; Bakan et al., 2011) following where s was selected to scale the motion appropriately.
Expression, purification, crystallization and structure determination of GluA3 NTDs
GluA3 NTD (residues 1-380 of the mature protein) was produced in stably transfected HEK293S-GnTI− cells, as described previously (Rossmann et al., 2011). Briefly the purification consisted on cross-flow concentration and dialysis into 50 mM Tris pH 8; 1M NaCl, followed by affinity chromatography (His-Trap HP, GE Healthcare) and size exclusion chromatography on a S200 column (GE Healthcare). The sample was then treated with EndoH in 100mM sodium acetate (pH 5.2) in order to remove oligosaccharaides, followed by a size exclusion chromatography in 50 mM Hepes pH 7.4; 150 mM NaCl.
For crystallization experiments, the sample was concentrated to ~15 mg/ml. Crystals grew in two different forms, Form I in 6-12% PEG3350, 10% DMSO and 0.15 M Ammonium dihydrogen phosphate (pH 4.6), and Form II in 18% PEG3350 and 0.2 M ammonium citrate. Form I contained three protomers in the asymmetric unit with two of them assembling as the displaced structure and a second one forming a parallel dimer (PO43− bound) with a symmetry related protomer. Form II contained the super-open structure.
Data was collected at Diamond Light Source (I04 beam line) for crystal Form I and ESRF beamline ID14-4 for crystal Form II, to 1.96 Å resolution and 2.51 Å resolution respectively. For crystal Form I data were integrated, merged and reduced with Xia2 (Winter et al., 2013). For crystal Form II data were integrated with XDS (Kabsch, 2010) and scaled and merged with SCALA (Evans, 2006). Structures were solved by molecular replacement with PHASER (McCoy et al., 2007) using the GluA3 monomer (chain A of PDB 3O21) as a search model. Refinement was performed using the CCP4 suite of programs (Winn et al., 2011), using REFMAC5 (Murshudov et al., 2011) for coordinate refinement and COOT (Emsley et al., 2010) for manual modification and model building. Data collection parameters together with refinement and validation statistics are shown in Table S3.
QUANTIFICATION AND STATISTICAL ANALYSIS
All our systems were simulated in at least two independent runs as shown in Table S1. We show a good reproducibility as in Figure S2, S4, and S7D-F, where there are the correlation coefficients between the profiles for ligand binding frequencies as a function of residue number, obtained in independent runs for multiple subunits.
DATA AND SOFTWARE AVAILABILITY
The PDB accession number for GluA3 PO43−-bound and displaced NTD dimers is 6FPJ, and that for the super-open dimer is 6FLR. The displaced dimer is formed from subunits A and C from 6FPJ and the phosphate-bound dimer can be assembled from subunit B and its counterpart B’ in another asymmetric unit. Likewise, the super-open dimer can be assembled from subunit A and its counterpart A’. The software used for druggability simulations and their analysis is a plugin for VMD that is available for download along with installation instructions at http://prody.csb.pitt.edu/tutorials/drugui_tutorial/intro.html, and software and instructions for ANM calculations can be found at http://prody.csb.pitt.edu and http://anm.csb.pitt.edu.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tris | Sigma | Cat # T1503 |
| Sodium chloride | Fisher Chemical | Cat # 10598630 |
| EndoH | Roche | Cat # 11643053001 |
| Sodium acetate | Fisher Chemical | Cat # 10794761 |
| Hepes | Sigma | Cat # H3375 |
| PEG3350 | Hampton Research | Cat # HR2-527 |
| Ammonium dihydrogen phosphate | Sigma | Cat # 467782 |
| Fetal Bovine Serum | Gibco | Cat # 10270106] |
| DMEM | Life Technologies Ltd | Cat # 31966047 |
| Ammonium citrate | Sigma | Cat # 09834 |
| Deposited Data | ||
| Crystal structure of GluA3 NTD | This paper | PDB: 6FPJ |
| Crystal structure of GluA3 NTD | This paper | PDB: 6FLR |
| Crystal structure of GluA3 NTD | (Sukumaran et al., 2011) | PDB: 3O21 |
| Crystal structure of GluA3 NTD | (Sukumaran et al., 2011) | PDB: 3P3W |
| Crystal structure of GluA1 NTD | (Yao et al., 2011) | PDB: 3SAJ |
| Crystal structure of GluA2 NTD | (Rossmann et al., 2011) | PDB: 3HSY |
| Crystal structure of GluA2/A3 NTD heteromer | (Herguedas et al., 2016) | PDB: 5FWY |
| Crystal structure of GluA4 NTD | (Dutta et al., 2012) | PDB: 4GPA |
| Crystal structure of GluK2 NTD | (Kumar et al., 2009) | PDB: 3H6G |
| Crystal structure of GluK3 NTD | (Kumar et al., 2010) | PDB: 3OLZ |
| Crystal structure of GluK5 NTD | (Kumar et al., 2011) | PDB: 3OM1 |
| Crystal structure of GluN1 NTD | (Farina et al., 2011) | PDB: 3Q41 |
| Crystal structure of GluN2B NTD | (Karakas et al., 2009) | PDB: 3JPY |
| Crystal structure of GluN1/N2B NTD heteromer | (Karakas et al., 2011) | PDB: 3QEL |
| Crystal structure of GluA2 LBD | (Armstrong and Gouaux 2000) | PDB: 1FTO |
| Experimental Models: Cell Lines | ||
| HEK293S-GnTI- cells stably expressing GluA3 NTD | (Rossmann et al., 2011) | N/A |
| Software and Algorithms | ||
| DruGUI | (Bakan et al., 2012) | http://prody.csb.pitt.edu/drugui/ |
| ProDy | (Bakan et al., 2011) | http://prody.csb.pitt.edu/ |
| VMD | (Humphrey et al., 1996) | http://www.ks.uiuc.edu/Research/vmd/ |
| PyMOL | (Schrödinger, 2015) | https://www.pymol.org/ |
| NAMD | (Phillips et al., 2005) | http://www.ks.uiuc.edu/Research/namd/ |
| Gromacs 5 | (Abraham et al., 2015) | www.gromacs.org/ |
| Xia2 | (Winter et al., 2013) | https://xia2.github.io/ |
| XDS | (Kabsch, 2010) | http://xds.mpimfheidelberg.mpg.de/ |
| SCALA | (Evans, 2006) | http://www.ccp4.ac.uk/html/scala.html |
| PHASER | (McCoy et al., 2007) | http://www.phaser.cimr.cam.ac.uk/index.php/Phaser_Crystallographic_Software |
| CCP4 suite | (Winn et al., 2011) | http://www.ccp4.ac.uk/ |
| REFMAC5 | (Murshudov et al., 2011) | http://www.ccp4.ac.uk/html/refmac5.html |
| COOT | (Emsley et al., 2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
Highlights.
Known agonist and modulator sites were confirmed on AMPA and NMDA receptors
A ligand-binding site was identified in the AMPAR GluA3 NTD
Ligand binding is dependent on conformational flexibility
Additional crystal structures support the role of GluA3 NTD conformation plasticity
Acknowledgments
Support from NIH grants P41GM103712 and P30DA035778 is acknowledged by IB. IHG acknowledges funding from the Medical Research Council (MC_U105174197) and the Biotechnology and Biological Sciences Research Council (BB/N002113/1). The authors thank Diamond Light Source for beamtime (proposal mx6641) and the staff of beamline I04, for assistance with crystal testing and data collection. We also thank the European Synchrotron Radiation Facility, Grenoble, France and the local contact there for providing assistance in using beamline ID14-4.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Lead contact
Declaration of Interests
The authors declare no competing interests.
References
- Abe H, Tateyama M, and Kubo Y (2003). Functional identification of Gd3+ binding site of metabotropic glutamate receptor 1alpha. FEBS Lett 545, 233–238. [DOI] [PubMed] [Google Scholar]
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, and Lindahl E (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25. [Google Scholar]
- Armstrong N, and Gouaux E (2000). Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron 28, 165–181. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Jasti J, Beich-Frandsen M, and Gouaux E (2006). Measurement of conformational changes accompanying desensitization in an ionotropic glutamate receptor. Cell 127, 85–97. [DOI] [PubMed] [Google Scholar]
- Atilgan AR, Durell SR, Jernigan RL, Demirel MC, Keskin O, and Bahar I (2001). Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys J 80, 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahadur RP, Chakrabarti P, Rodier F, and Janin J (2004). A dissection of specific and non-specific protein-protein interfaces. J Mol Biol 336, 943–955. [DOI] [PubMed] [Google Scholar]
- Bahar I, Jernigan RL, and Dill KA (2017). Protein Actions: Principles and Modeling (Garland Science). [Google Scholar]
- Bakan A, and Bahar I (2009). The intrinsic dynamics of enzymes plays a dominant role in determining the structural changes induced upon inhibitor binding. Proc Natl Acad Sci U S A 106, 14349–14354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakan A, Dutta A, Mao W, Liu Y, Chennubhotla C, Lezon TR, and Bahar I (2014). Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics 30, 2681–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakan A, Meireles LM, and Bahar I (2011). ProDy: protein dynamics inferred from theory and experiments. Bioinformatics 27, 1575–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakan A, Nevins N, Lakdawala AS, and Bahar I (2012). Druggability Assessment of Allosteric Proteins by Dynamics Simulations in the Presence of Probe Molecules. J Chem Theory Comput 8, 2435–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger ML, Pohler T, Schadt O, Stanger M, Rebernik P, Scholze P, and Noe CR (2013). Exploring the polyamine regulatory site of the NMDA receptor: a parallel synthesis approach. ChemMedChem 8, 82–94. [DOI] [PubMed] [Google Scholar]
- Bjelkmar P, Larsson P, Cuendet MA, Hess B, and Lindahl E (2010). Implementation of the CHARMM Force Field in GROMACS: Analysis of Protein Stability Effects from Correction Maps, Virtual Interaction Sites, and Water Models. J Chem Theory Comput 6, 459–466. [DOI] [PubMed] [Google Scholar]
- Bowie D (2008). Ionotropic glutamate receptors & CNS disorders. CNS Neurol Disord Drug Targets 7, 129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger PB, Yuan H, Karakas E, Geballe M, Furukawa H, Liotta DC, Snyder JP, and Traynelis SF (2012). Mapping the binding of GluN2B-selective N-methyl-D-aspartate receptor negative allosteric modulators. Mol Pharmacol 82, 344–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavasotto CN, Kovacs JA, and Abagyan RA (2005). Representing receptor flexibility in ligand docking through relevant normal modes. J Am Chem Soc 127, 9632–9640. [DOI] [PubMed] [Google Scholar]
- Chandler P, Pennington M, Maccecchini ML, Nashed NT, and Skolnick P (1993). Polyamine-like actions of peptides derived from conantokin-G, an N-methyl-D-aspartate (NMDA) antagonist. J Biol Chem 268, 17173–17178. [PubMed] [Google Scholar]
- Chang PK, Verbich D, and McKinney RA (2012). AMPA receptors as drug targets in neurological disease--advantages, caveats, and future outlook. Eur J Neurosci 35, 1908–1916. [DOI] [PubMed] [Google Scholar]
- Chen L, Durr KL, and Gouaux E (2014). X-ray structures of AMPA receptor-cone snail toxin complexes illuminate activation mechanism. Science 345, 1021–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhao Y, Wang Y, Shekhar M, Tajkhorshid E, and Gouaux E (2017). Activation and Desensitization Mechanism of AMPA Receptor-TARP Complex by Cryo-EM. Cell 170, 1234–1246 e1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YB, and Lipton SA (1999). Identification and mechanism of action of two histidine residues underlying high-affinity Zn2+ inhibition of the NMDA receptor. Neuron 23, 171–180. [DOI] [PubMed] [Google Scholar]
- Clayton A, Siebold C, Gilbert RJ, Sutton GC, Harlos K, McIlhinney RA, Jones EY, and Aricescu AR (2009). Crystal structure of the GluR2 amino-terminal domain provides insights into the architecture and assembly of ionotropic glutamate receptors. J Mol Biol 392, 1125–1132. [DOI] [PubMed] [Google Scholar]
- Davies B, Brown LA, Cais O, Watson J, Clayton AJ, Chang VT, Biggs D, Preece C, Hernandez-Pliego P, Krohn J, et al. (2017). A point mutation in the ion conduction pore of AMPA receptor GRIA3 causes dramatically perturbed sleep patterns as well as intellectual disability. Hum Mol Genet 26, 3869–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A, Krieger J, Garcia-Nafria J, Lee J, Greger IH, and Bahar I (2015). Cooperative dynamics in intact AMPA and NMDA glutamate receptors – similarities and subfamily-specific differences. Structure 23, 1692–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A, Shrivastava IH, Sukumaran M, Greger IH, and Bahar I (2012). Comparative Dynamics of NMDA- and AMPA-Glutamate Receptor N-Terminal Domains. Structure 20, 1838–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elegheert J, Kakegawa W, Clay JE, Shanks NF, Behiels E, Matsuda K, Kohda K, Miura E, Rossmann M, Mitakidis N, et al. (2016). Structural basis for integration of GluD receptors within synaptic organizer complexes. Science 353, 295–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P (2006). Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62, 72–82. [DOI] [PubMed] [Google Scholar]
- Farina AN, Blain KY, Maruo T, Kwiatkowski W, Choe S, and Nakagawa T (2011). Separation of domain contacts is required for heterotetrameric assembly of functional NMDA receptors. J Neurosci 31, 3565–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayyazuddin A, Villarroel A, Le Goff A, Lerma J, and Neyton J (2000). Four residues of the extracellular N-terminal domain of the NR2A subunit control high-affinity Zn2+ binding to NMDA receptors. Neuron 25, 683–694. [DOI] [PubMed] [Google Scholar]
- Floquet N, Marechal JD, Badet-Denisot MA, Robert CH, Dauchez M, and Perahia D (2006). Normal mode analysis as a prerequisite for drug design: application to matrix metalloproteinases inhibitors. FEBS Lett 580, 5130–5136. [DOI] [PubMed] [Google Scholar]
- Garcia-Nafria J, Herguedas B, Watson JF, and Greger IH (2016). The dynamic AMPA receptor extracellular region: a platform for synaptic protein interactions. J Physiol 594, 5449–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, and Monyer H (1995). Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron 15, 193–204. [DOI] [PubMed] [Google Scholar]
- Gielen M, Siegler Retchless B, Mony L, Johnson JW, and Paoletti P (2009). Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature 459, 703–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold SJ, Ambros-Ingerson J, Horowitz JR, Lynch G, and Gall CM (1997). Stoichiometries of AMPA receptor subunit mRNAs in rat brain fall into discrete categories. J Comp Neurol 385, 491–502. [DOI] [PubMed] [Google Scholar]
- Greger IH, Watson JF, and Cull-Candy SG (2017). Structural and Functional Architecture of AMPA-Type Glutamate Receptors and Their Auxiliary Proteins. Neuron 94, 713–730. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Castellanos N, Da Silva-Matos CM, Zhou K, Canto CB, Renner MC, Koene LMC, Ozyildirim O, Sprengel R, Kessels HW, and De Zeeuw CI (2017). Motor Learning Requires Purkinje Cell Synaptic Potentiation through Activation of AMPA-Receptor Subunit GluA3. Neuron 93, 409–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Yuan H, and Traynelis SF (2007). Structural aspects of AMPA receptor activation, desensitization and deactivation. Curr Opin Neurobiol 17, 281–288. [DOI] [PubMed] [Google Scholar]
- Herguedas B, Garcia-Nafria J, Cais O, Fernandez-Leiro R, Krieger J, Ho H, and Greger IH (2016). Structure and organization of heteromeric AMPA-type glutamate receptors. Science 352, aad3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herguedas B, Krieger J, and Greger IH (2013). Receptor heteromeric assembly-how it works and why it matters: the case of ionotropic glutamate receptors. Prog Mol Biol Transl Sci 117, 361–386. [DOI] [PubMed] [Google Scholar]
- Horning MS, and Mayer ML (2004). Regulation of AMPA receptor gating by ligand binding core dimers. Neuron 41, 379–388. [DOI] [PubMed] [Google Scholar]
- Huganir RL, and Nicoll RA (2013). AMPARs and synaptic plasticity: the last 25 years. Neuron 80, 704–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD: visual molecular dynamics. J Mol Graph 14, 33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- Jin R, Clark S, Weeks AM, Dudman JT, Gouaux E, and Partin KM (2005). Mechanism of positive allosteric modulators acting on AMPA receptors. J Neurosci 25, 9027–9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R, Singh SK, Gu S, Furukawa H, Sobolevsky AI, Zhou J, Jin Y, and Gouaux E (2009). Crystal structure and association behaviour of the GluR2 amino-terminal domain. EMBO J 28, 1812–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983). Comparison of simple potential functions for simulating liquid water. J Chem Phys 79, 926–935. [Google Scholar]
- Kaae BH, Harpsoe K, Kastrup JS, Sanz AC, Pickering DS, Metzler B, Clausen RP, Gajhede M, Sauerberg P, Liljefors T, et al. (2007). Structural proof of a dimeric positive modulator bridging two identical AMPA receptor-binding sites. Chem Biol 14, 1294–1303. [DOI] [PubMed] [Google Scholar]
- Kabsch W (2010). Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, and Furukawa H (2014). Crystal structure of a heterotetrameric NMDA receptor ion channel. Science 344, 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Regan MC, and Furukawa H (2015). Emerging structural insights into the function of ionotropic glutamate receptors. Trends Biochem Sci 40, 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Simorowski N, and Furukawa H (2009). Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Simorowski N, and Furukawa H (2011). Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature 475, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, and Malinow R (2009). Synaptic AMPA receptor plasticity and behavior. Neuron 61, 340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger J, Bahar I, and Greger IH (2015). Structure, Dynamics, and Allosteric Potential of Ionotropic Glutamate Receptor N-Terminal Domains. Biophys J 109, 1136–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar J, and Mayer ML (2010). Crystal structures of the glutamate receptor ion channel GluK3 and GluK5 amino-terminal domains. J Mol Biol 404, 680–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar J, Schuck P, Jin R, and Mayer ML (2009). The N-terminal domain of GluR6-subtype glutamate receptor ion channels. Nat Struct Mol Biol 16, 631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar J, Schuck P, and Mayer ML (2011). Structure and assembly mechanism for heteromeric kainate receptors. Neuron 71, 319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, Nakanishi S, Jingami H, and Morikawa K (2000). Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 407, 971–977. [DOI] [PubMed] [Google Scholar]
- Lau AY, and Roux B (2007). The free energy landscapes governing conformational changes in a glutamate receptor ligand-binding domain. Structure 15, 1203–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau AY, and Roux B (2011). The hidden energetics of ligand binding and activation in a glutamate receptor. Nat Struct Mol Biol 18, 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Feng Z, Xie XQ, and Bahar I (2017). Allosteric Modulation of Intact gamma-Secretase Structural Dynamics. Biophys J 113, 2634–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackerell AD Jr., Feig M, and Brooks CL 3rd. (2004). Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem 25, 1400–1415. [DOI] [PubMed] [Google Scholar]
- Matsuda K, Budisantoso T, Mitakidis N, Sugaya Y, Miura E, Kakegawa W, Yamasaki M, Konno K, Uchigashima M, Abe M, et al. (2016). Transsynaptic Modulation of Kainate Receptor Functions by C1q-like Proteins. Neuron 90, 752–767. [DOI] [PubMed] [Google Scholar]
- Mayer ML (2016). Structural biology of glutamate receptor ion channel complexes. Curr Opin Struct Biol 41, 119–127. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson JR, Kumar J, Chittori S, Rao P, Pierson J, Bartesaghi A, Mayer ML, and Subramaniam S (2014). Structural mechanism of glutamate receptor activation and desensitization. Nature 514, 328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misono KS, Philo JS, Arakawa T, Ogata CM, Qiu Y, Ogawa H, and Young HS (2011). Structure, signaling mechanism and regulation of the natriuretic peptide receptor guanylate cyclase. FEBS J 278, 1818–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moga DE, Janssen WG, Vissavajjhala P, Czelusniak SM, Moran TM, Hof PR, and Morrison JH (2003). Glutamate receptor subunit 3 (GluR3) immunoreactivity delineates a subpopulation of parvalbumin-containing interneurons in the rat hippocampus. J Comp Neurol 462, 15–28. [DOI] [PubMed] [Google Scholar]
- Molina G, Vogt A, Bakan A, Dai W, Queiroz de Oliveira P, Znosko W, Smithgall TE, Bahar I, Lazo JS, Day BW, et al. (2009). Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat Chem Biol 5, 680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mony L, Kew JN, Gunthorpe MJ, and Paoletti P (2009). Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. Br J Pharmacol 157, 1301–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mony L, Zhu S, Carvalho S, and Paoletti P (2011). Molecular basis of positive allosteric modulation of GluN2B NMDA receptors by polyamines. EMBO J 30, 3134–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, and Vagin AA (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, and Zhou Q (2013). NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14, 383–400. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Perin-Dureau F, Fayyazuddin A, Le Goff A, Callebaut I, and Neyton J (2000). Molecular Organization of a Zinc Binding N-Terminal Modulatory Domain in a NMDA Receptor Subunit. Neuron 28, 911–925. [DOI] [PubMed] [Google Scholar]
- Partin KM (2015). AMPA receptor potentiators: from drug design to cognitive enhancement. Curr Opin Pharmacol 20C, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005). Scalable molecular dynamics with NAMD. J Comput Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlsgaard J, Frydenvang K, Madsen U, and Kastrup JS (2011). Lessons from more than 80 structures of the GluA2 ligand-binding domain in complex with agonists, antagonists and allosteric modulators. Neuropharmacology 60, 135–150. [DOI] [PubMed] [Google Scholar]
- Rachline J, Perin-Dureau F, Le Goff A, Neyton J, and Paoletti P (2005). The micromolar zinc-binding domain on the NMDA receptor subunit NR2B. J Neurosci 25, 308–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves PJ, Callewaert N, Contreras R, and Khorana HG (2002). Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc Natl Acad Sci U S A 99, 13419–13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinders NR, Pao Y, Renner MC, da Silva-Matos CM, Lodder TR, Malinow R, and Kessels HW (2016). Amyloid-beta effects on synapses and memory require AMPA receptor subunit GluA3. Proc Natl Acad Sci U S A 113, E6526–E6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner MC, Albers EH, Gutierrez-Castellanos N, Reinders NR, van Huijstee AN, Xiong H, Lodder TR, and Kessels HW (2017). Synaptic plasticity through activation of GluA3-containing AMPA-receptors. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA (2011). Revisiting AMPA receptors as an antiepileptic drug target. Epilepsy Curr 11, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Hernandez A, Simorowski N, Karakas E, and Furukawa H (2016). Molecular Basis for Subtype Specificity and High-Affinity Zinc Inhibition in the GluN1 -GluN2A NMDA Receptor Amino-Terminal Domain. Neuron 92, 1324–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmann M, Sukumaran M, Penn AC, Veprintsev DB, Babu MM, and Greger IH (2011). Subunit-selective N-terminal domain associations organize the formation of AMPA receptor heteromers. EMBO J 30, 959–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio ME, Matsui K, Fukazawa Y, Kamasawa N, Harada H, Itakura M, Molnar E, Abe M, Sakimura K, and Shigemoto R (2017). The number and distribution of AMPA receptor channels containing fast kinetic GluA3 and GluA4 subunits at auditory nerve synapses depend on the target cells. Brain Struct Funct 222, 3375–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda M, Bottegoni G, and Abagyan R (2009). Consistent improvement of cross-docking results using binding site ensembles generated with elastic network normal modes. J Chem Inf Model 49, 716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger L (2015). The PyMOL Molecular Graphics System, Version 1.8. [Google Scholar]
- Schwenk J, Baehrens D, Haupt A, Bildl W, Boudkkazi S, Roeper J, Fakler B, and Schulte U (2014). Regional diversity and developmental dynamics of the AMPA-receptor proteome in the mammalian brain. Neuron 84, 41–54. [DOI] [PubMed] [Google Scholar]
- Sukumaran M, Rossmann M, Shrivastava I, Dutta A, Bahar I, and Greger IH (2011). Dynamics and allosteric potential of the AMPA receptor N-terminal domain. EMBO Journal 30, 972–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Olson R, Horning M, Armstrong N, Mayer M, and Gouaux E (2002). Mechanism of glutamate receptor desensitization. Nature 417, 245–253. [DOI] [PubMed] [Google Scholar]
- Taguchi J, and Kitao A (2016). Dynamic profile analysis to characterize dynamics-driven allosteric sites in enzymes. Biophys Physicobiol 13, 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima N, Karakas E, Grant T, Simorowski N, Diaz-Avalos R, Grigorieff N, and Furukawa H (2016). Activation of NMDA receptors and the mechanism of inhibition by ifenprodil. Nature 534, 63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Hartley M, and Heinemann SF (1995). Control of proton sensitivity of the NMDA receptor by RNA splicing and polyamines. Science 268, 873–876. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R, et al. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya D, Kunishima N, Kamiya N, Jingami H, and Morikawa K (2002). Structural views of the ligand-binding cores of a metabotropic glutamate receptor complexed with an antagonist and both glutamate and Gd3+. Proc Natl Acad Sci U S A 99, 2660–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC, Yelshanskaya MV, Grassucci RA, Frank J, and Sobolevsky AI (2017). Structural bases of desensitization in AMPA receptor-auxiliary subunit complexes. Neuron 94, 569–580 e565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K, and MacKerell AD Jr. (2015). CHARMM additive and polarizable force fields for biophysics and computer-aided drug design. Biochim Biophys Acta 1850, 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liu H, and Zhang ZW (2011). Elimination of redundant synaptic inputs in the absence of synaptic strengthening. J Neurosci 31, 16675–16684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JF, Ho H, and Greger IH (2017). Synaptic transmission and plasticity require AMPA receptor anchoring via its N-terminal domain. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G, Lobley CMC, and Prince SM (2013). Decision making in xia2. Acta Crystallogr D Biol Crystallogr 69, 1260–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Arai AC, Rumbaugh G, Srivastava AK, Turner G, Hayashi T, Suzuki E, Jiang Y, Zhang L, Rodriguez J, et al. (2007). Mutations in ionotropic AMPA receptor 3 alter channel properties and are associated with moderate cognitive impairment in humans. Proc Natl Acad Sci U S A 104, 18163–18168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao G, Zong Y, Gu S, Zhou J, Xu H, Mathews II, and Jin R (2011). Crystal structure of the glutamate receptor GluA1 N-terminal domain. Biochem J 438, 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A, and Lau AY (2017). Energetics of Glutamate Binding to an Ionotropic Glutamate Receptor. J Phys Chem B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Low CM, Moody OA, Jenkins A, and Traynelis SF (2015). Ionotropic GABA and Glutamate Receptor Mutations and Human Neurologic Diseases. Mol Pharmacol 88, 203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzaki M, and Aricescu AR (2017). A GluD Coming-Of-Age Story. Trends Neurosci 40, 138–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Lomash S, Chittori S, Glasser C, Mayer ML, and Schuck P (2017). Preferential assembly of heteromeric kainate and AMPA receptor amino terminal domains. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, and Paoletti P (2015). Allosteric modulators of NMDA receptors: multiple sites and mechanisms. Curr Opin Pharmacol 20C, 14–23. [DOI] [PubMed] [Google Scholar]
- Zhu S, Stein RA, Yoshioka C, Lee CH, Goehring A, McHaourab HS, and Gouaux E (2016). Mechanism of NMDA Receptor Inhibition and Activation. Cell 165, 704–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Stroebel D, Yao CA, Taly A, and Paoletti P (2013). Allosteric signaling and dynamics of the clamshell-like NMDA receptor GluN1 N-terminal domain. Nat Struct Mol Biol 20, 477–485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The PDB accession number for GluA3 PO43−-bound and displaced NTD dimers is 6FPJ, and that for the super-open dimer is 6FLR. The displaced dimer is formed from subunits A and C from 6FPJ and the phosphate-bound dimer can be assembled from subunit B and its counterpart B’ in another asymmetric unit. Likewise, the super-open dimer can be assembled from subunit A and its counterpart A’. The software used for druggability simulations and their analysis is a plugin for VMD that is available for download along with installation instructions at http://prody.csb.pitt.edu/tutorials/drugui_tutorial/intro.html, and software and instructions for ANM calculations can be found at http://prody.csb.pitt.edu and http://anm.csb.pitt.edu.