

SUMMARY
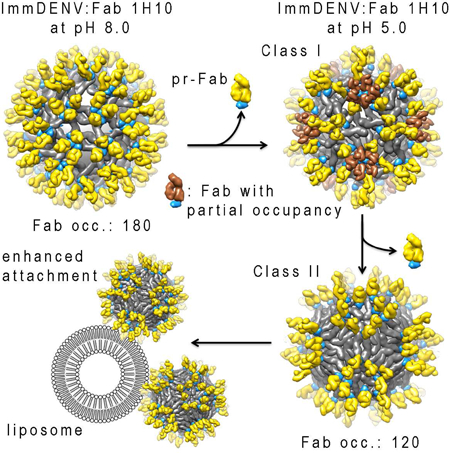
Dengue virus particles are released from cells in different maturation states. Fully immature DENV (immDENV) is generally non-infectious, but can become infectious when complexed with anti-precursor membrane (prM) protein antibodies. It is unknown how anti-prM antibody-coated particles can undergo membrane fusion since the prM caps the Envelope (E) protein fusion loop. Here, we determined cryoEM maps of the immDENV:anti-prM complex at different pH mimicking the extracellular (pH 8.0) or endosomal (pH 5.0) environments. At pH 5.0, there are two structural classes with fewer antibodies bound than at pH 8.0. These classes may represent different maturation states. Molecular simulations, together with the measured high affinity pr:antibody interaction (versus the weak pr:E interaction) and also the low pH cryoEM structures, suggest how antibody:pr complex can dislodge from the E protein at low pH. This exposes the E protein fusion loop enhancing virus interaction with endosomes.
Keywords: cryoEM, dengue virus, enhancement of infection, human antibody, immature dengue virus, maturation
Graphical Abstract
eTOC blurb
Wirawan et al. present structures of immature dengue virus complexed with anti-prM Fabs at pH 8.0 and 5.0. The two structural classes at pH 5.0 showed different maturation stages. These structures also suggest the mechanism on how antibody stimulate immature dengue virus attachment to liposome.
INTRODUCTION
Dengue virus (DENV), a major human pathogen (Bhatt et al., 2013), is a flavivirus along with West Nile, Zika and yellow fever viruses. There are four DENV serotypes (DENV1–4). It usually cause mild fever, but in some cases can lead to the severe dengue haemorrhagic fever or shock syndrome (Halstead, 2007).
The DENV particle contains a positive-sense RNA genome complexed with capsid proteins, which is surrounded by a lipid bilayer membrane. There are 180 copies each of the envelope (E) and the precursor-membrane (prM) proteins anchored on the lipid membrane of the immature DENV (immDENV) particles. The prM molecule is cleaved to form the membrane (M) protein in the mature DENV. The E protein contains three domains (DI, DII, and DIII) (Modis et al., 2005; Rey et al., 1995). The immunoglobulin-like DIII is thought to interact with host cell receptors (Rey et al., 1995). DII contains a hydrophobic fusion loop, which facilitates attachment of virus to the endosomal membrane (Modis et al., 2003, 2004). The prM protein is composed of the pr-molecule connected to the M protein ectodomain via a furin cleavage site (Li et al., 2008). The pr molecule caps the fusion loop of the E protein in the immDENV.
The immDENV particle, when first assembled in the endoplasmic reticulum, contains 60 spikes on its surface. Each spike consists of a trimer of the E:prM complex (Li et al., 2008; Zhang et al., 2003). The low pH trans-Golgi compartment (TGC) triggers the maturation process, causing the trimeric prM:E proteins spikes to arrange into dimers. This exposes the cleavage site on the prM, allowing processing by furin protease (Yu et al., 2008). At low pH, the cleaved pr remains associated to the E protein, preventing virus from fusing back into the cell (Yu et al., 2009). When the virus is released to the outside environment, the neutral pH causes the dissociation of the pr-molecule from the virus particle completing the virus maturation process (Yu et al., 2009). The maturation process is inefficient, as immDENV particles are frequently detected in DENV generated from in vitro cell cultures (Junjhon et al., 2010). Fully immDENV particles are not infectious in most cell lines e.g., mammalian Vero, BHK-21 cells, and C6/36 mosquito cells. However, all anti-prM and some anti-E antibodies, when complexed with immDENV can infect FcγR positive monocytic cells (Rodenhuis-Zybert et al., 2010). Large amount of anti-prM antibodies are detected in DENV immune individuals, suggesting that infectious immDENV:anti-prM antibody complexes may play an important role in pathogenesis (Dejnirattisai et al., 2010; Smith et al., 2012).
After internalization of the immDENV:antibody complex into cells, furin cleavage of the prM protein has been shown to be critical for infection (Rodenhuis-Zybert et al., 2010). While membrane bound furin has been detected in the endosome (Claus et al., 1998), it is unknown how the pr-molecule could dissociate from the E protein fusion loop allowing interaction with endosomal membrane, an essential step for fusion. This is because, in the normal DENV maturation pathway, a rise in pH (exposure to extracellular environment) is required for the dissociation of the cleaved pr from the virus particles (Yu et al., 2009), whereas in the endosome, pH is kept low. To understand this, we determined the cryo-electron microscopy (cryoEM) structures of the immDENV:Fab 1H10 (anti-prM human monoclonal antibody), at pH 8 and 5, mimicking conditions prior to cell entry and in the endosome, respectively.
The results from co-floatation assays of the fully immDENV:Fab 1H10 complex revealed enhanced attachment to liposomes at low pH. The low pH cryoEM complex structures showed a reduced number of Fab molecules on the virus surface compared with pH 8.0. The high affinity of the Fab 1H10 for pr at low pH versus the weak pr:E interaction suggests that the pr:Fab 1H10 complex dislodged from the E proteins together as a unit. The multiscale molecular dynamics (MD) simulations, showed the Fab:pr:E proteins experience significant steric hindrance during virus surface proteins reorganization at low pH. This likely results in the dissociation of the pr:Fab from some of the E proteins facilitating attachment of the virus to the endosomal membrane.
Results
Fab 1H10 enhances attachment of fully immDENV to liposomes
Due to the low levels of DENV3 growth in furin deficient LoVo cells, we could not obtain enough virus particles for the liposome co-floatation studies and since HMAb 1H10 is serotype cross-reactive (Smith et al., 2015), immDENV2 was used. HMAb 1H10 when complexed with immDENV particles can facilitate virus binding to monocytic THP-1 cells via interaction of antibody Fc region to the cell Fc receptor. The antibody may also aid the dissociation of pr-molecule from the E fusion loop, which facilitates virus attachment to the endosome. To investigate this, we exposed fully immDENV particle to anionic liposomes (composition similar to the late endosome), and performed co-floatation assays in a sucrose gradient (Zheng et al., 2010). Sample was loaded in the middle of the sucrose gradient. When virus particles interact with the liposomes, the virus will co-float with the liposomes at the top fractions after centrifugation.
ImmDENV at pH 8.0 without liposomes control showed virus settling in the middle fractions as expected (Figure 1A). When liposome was added to the immDENV particles at pH 8.0, the most intense band remains in the middle fraction, although the virus seemed to have a wider spread throughout the fractions. When Fab 1H10 was added to the immDENV and liposome at pH 8.0, the virus remains at the middle fractions. Uncomplexed immDENV at pH 5.0 showed co-floatation with the liposomes at the top four fractions, suggesting some direct interaction of the virus with liposomes (Figure 1A). When Fab 1H10 was added, interactions of the immDENV to liposome at pH 5.0 was enhanced, as the virus was detected only in the topmost fraction (Figure 1A).
Figure 1. Anti-prM HMAb 1H10 enhanced the attachment of immDENV to anionic liposome and enhanced infection of immDENV in monocytic THP-1 cell.
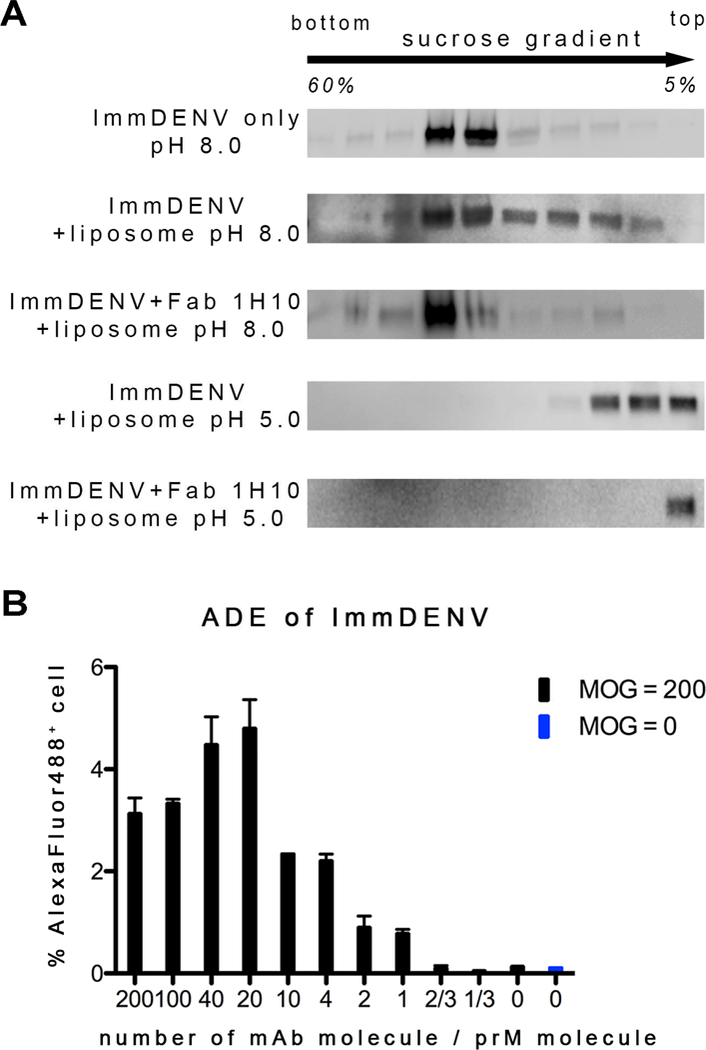
(A) Fab 1H10 enhanced attachment of the fully immDENV to anionic liposomes which composition mimic that of the endosome. Presence of virus was detected in different fractions of the sucrose gradient in a virus-liposome co-floatation assay by anti-E antibody. At all pH 8.0 conditions, with or without liposome and Fab, immDENV remained in the middle fractions where the viruses were loaded initially onto the gradient. At pH 5.0, immDENV co-floated with the liposomes in the top 4 fractions indicating some interactions with the liposomes. When Fab 1H10 was added to the virus-liposome mixture at pH 5.0, all immDENV was detected only in the topmost fraction indicating enhanced interaction with liposomes. The assay was repeated three times and showed consistent results.
(B) IgG 1H10 enhanced infection of immDENV3 in THP-1 cells at equal or higher than molar ratio of 1 antibody per viral prM molecule.
See also Figure S2.
CryoEM map of immDENV:Fab 1H10 complex at pH 8.0
CryoEM reconstruction requires samples to be homogenous, and therefore a saturated concentration of anti-prM antibody Fab fragments was added to immDENV. To ensure that the concentration of antibody used for cryoEM was biologically relevant, we performed infection of immDENV3:HMAb 1H10 complex to THP-1 monocytic cells. HMAb 1H10 was observed to enable immDENV3 infection at a molar ratio > 1 antibody per 1 prM molecules (Figure 1B). Therefore, a 1:1 molar ratio of Fab to prM molecules was chosen for cryo-electron microscopy (cryoEM) studies. This ratio was considered biologically relevant and is at saturating concentrations to the prM but with lower amounts of Fab in the background of the image compared to those at higher Fab ratio which also support immature virus infectivity (Figure 2A).
Figure 2. Samples for cryoEM studies and resolution of the cryoEM maps.
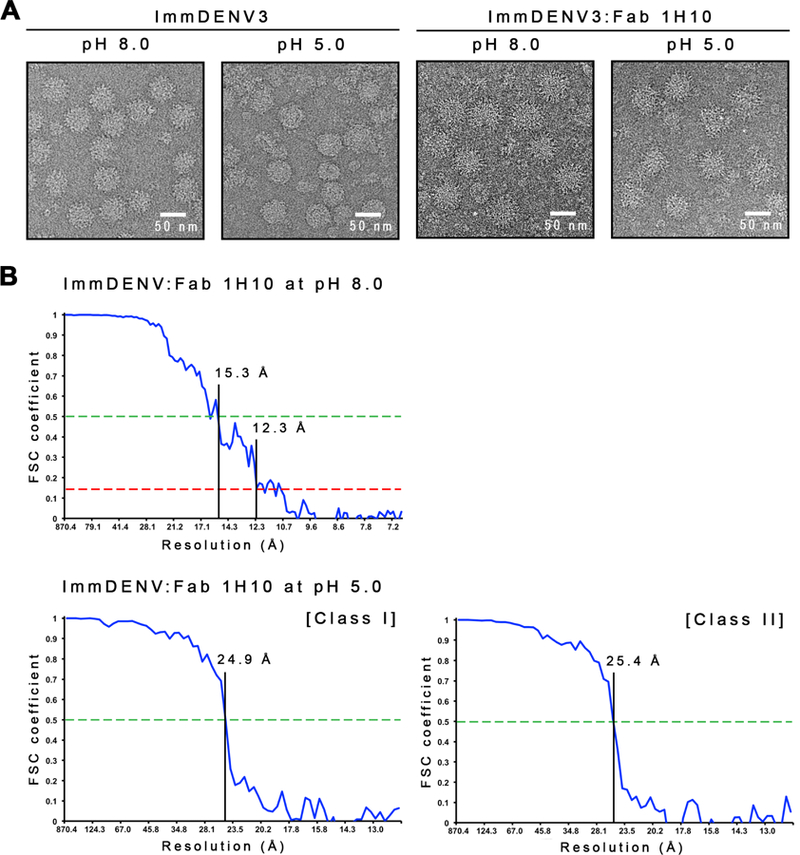
(A) Micrographs of the uncomplexed immDENV3 and the immDENV3:Fab 1H10 complexes at pH 8.0 and 5.0. The uncomplexed immDENV3 at pH 8.0 micrograph showed the sample contains mostly spiky fully immature virus. At pH 5.0, the uncomplexed virus becomes unstable and the surface of particles looked smoother. In the immDENV3:Fab 1H10 complex samples, at both pH 8.0 and 5.0, particles appear to have a bigger diameter and spikier, indicating the binding of Fab molecules.
(B) [Top] Fourier Shell Correlation (FSC) curve of immDENV:Fab 1H10 at pH 8.0 cryoEM map determined by gold-standard FSC procedure. The 12 Å resolution of the map is estimated by using FSC 0.143 correlation cut-off. [Bottom] Fourier shell correlation curve of class I and II immDENV:Fab 1H10 at pH 5.0 cryoEM maps. The FSC curve was plotted from two reconstructed maps of two half-datasets of the final iteration step and the resolution was estimated by using 0.5 cut-off. Class I and II pH 5.0 complexed structures were determined to a resolution of ~25Å, suggesting the structures were flexible.
See also Figure S1.
The cryoEM structure of immDENV:Fab 1H10 at pH 8.0 was determined to 12 Å resolution using the Fourier shell correlation gold standard procedure at a cutoff at 0.143 (Figures 2B, 3A and S1A). The map was fitted with the cryoEM structure of immDENV and a Fab homology model (Figure S1A). All three possible binding sites in the icosahedral asymmetric unit were observed to be bound by Fab 1H10 (Figures 3A and 3B). The virus structure was not altered by Fab binding.
Figure 3. The 12 Å resolution cryoEM structure of immDENV3:Fa b 1H10 complex at pH 8.0.
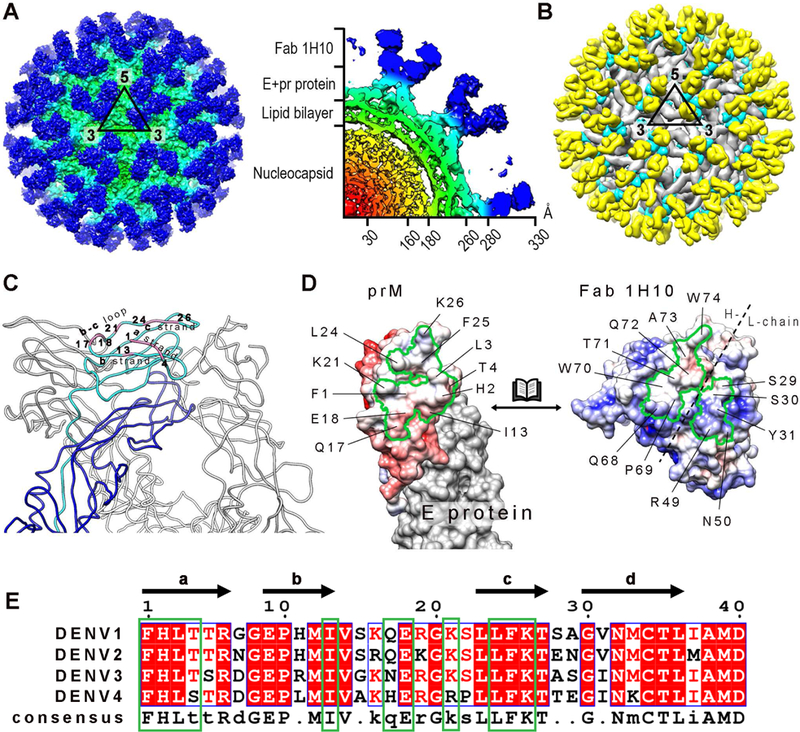
(A) Surface of the cryoEM map of immDENV3:Fab 1H10 complex (left) and a quarter of the center-section of the map (right). The map is colored according to its corresponding radius (red: 0–30 Å, orange: 31–160 Å, yellow: 161– 180 Å, green: 181–260 Å, cyan: 261–280Å and blue : >281 Å).
(B) The structure of immDENV3:Fab 1H10 which was fitted into the cryoEM map. E, prM proteins and Fab surfaces are colored in grey, cyan and yellow, respectively. We observed 180 copies of Fabs binding to prM molecules on the virus surface.
(C) The 1H10 epitope (pink) on prM consists of a and c strands, and the b-c loop. One of the E:prM complexes within a trimeric spike is colored with the two proteins in blue and cyan, respectively while the other two E:prM complexes are colored in grey.
(D) Open-book representation of the surface potential of the prM:Fab interacting interfaces. The boundaries of the epitope and the paratope are marked by black solid lines. Positive and negative charges are colored in blue and red, respectively.
(E) Sequence comparison of the Fab 1H10 epitope (green box) across four DENV serotypes show high similarities consistent with the ability of antibody to cross-react with all serotypes. White and red letters represent identical residues, whereas black letter indicates non-conserved residues.
See also Figures S1 and S9.
Since the resolution of the map did not permit observation of side chain densities, the interacting interface were identified by observing pairs of Cα atoms with less than 8Å distance between them. The epitope on the pr-molecule comprised of the a and c strands, and the b-c loop (Figure 3C) and they showed complementary electrostatic charges to the Fab paratope (Figure 3D). A positively charged patch from light chain of Fab 1H10 is interacting with a negatively charged patch on pr molecule. The rest of the interface between the prM and Fab are made of hydrophobic interactions. Sequence comparison of the epitope with the other DENV serotypes showed ~90% identity, consistent with the ability of the antibody to cross-react (Figure 3E). The E and prM proteins of DENV2 strain used in the liposome co-sedimentation assay shares 68% and 71% sequence identity, respectively, to the DENV3 strain used in the cryoEM experiments (Figure S2).
CryoEM maps of two classes of immDENV:Fab 1H10 complex particles at pH 5.0
ImmDENV:Fab 1H10 complex was first formed at pH 8.0 and then pH was lowered to 5.0, prior to freezing on cryoEM grids. The cryoEM maps of two structural classes of particles, representing 35% and 49%, respectively, from the total particles number, were both determined to ~25Å resolution (Figures 2B, 4A, S1B and S1C). Poor resolution of the maps suggested that the structures were flexible, however, the Fab densities were clearly observed.
Figure 4. Class I and II cryoEM structures of immDENV3:Fab 1H10 complex at pH 5.0.
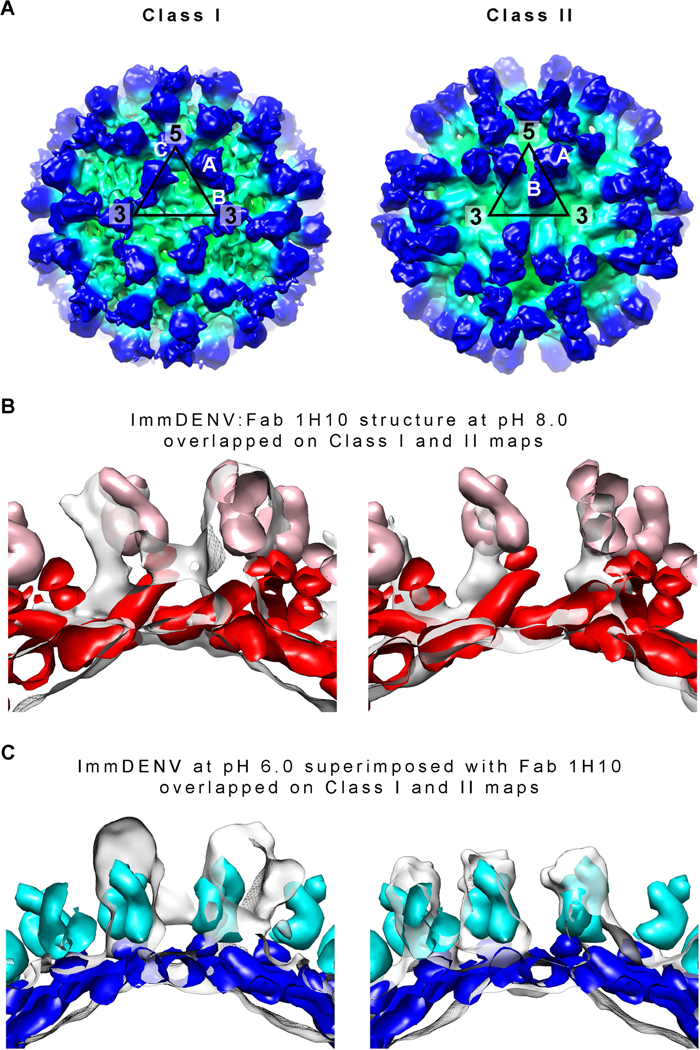
(A) CryoEM maps of the Class I (left) and Class II (right) particles of immDENV3:Fab 1H10 complex at pH 5.0. The map is colored according to its radius (red: 0–30Å, orange: 31–160Å, yellow: 161–180Å, green: 181–260Å, cyan: 261–280Å and blue: >281Å).
(B and C) Comparison of the class I and II cryoEM maps with two models – (B) structures of immDENV:Fab 1H10 complex at pH 8.0 (the structure before maturation), and (C) immDENV2 at pH 6.0 (Yu et al., 2009) (structure after completion of maturation) superimposed with the pr:Fab 1H10 structure. The Fabs and the prM:E molecules of the pH 8.0 model are colored in pink and red, respectively, while that of the pH 6.0 model are in light blue and blue. The height of densities (transparent grey surface) corresponding to the E protein:pr:Fab 1H10 complex in the class I and class II cryoEM maps is similar to that of the virus:Fab 1H10 pH 8.0 and pH 6.0 models, respectively. This suggests that the class I and II structures may represent the early and late stages of the low pH-induced structural change during the maturation process, respectively.
See also Figures S1 and S3
To interpret the maps, we first determined the appropriate initial model for fitting. There are two possible initial models, (1) the cryoEM structure of immDENV:Fab 1H10 at pH 8.0, (2) Fab homology model superimposed onto the uncomplexed immDENV at pH 6.0 (Yu et al., 2009) (the structure at the end of the maturation process). Since the Fab densities of the cryoEM maps are the clearest, they can be used as a guide for comparison of the maps to the initial starting models. Comparison between the top views of the initial models showed that the positions of the Fab molecules A and B are approximately the same, even though their E protein organizations are completely different (Figure S3A). However, from the side view, the prM:E complex is “ standing up” in the immDENV at pH 8.0 model while that at pH 6.0 is “lying down”, indicating that the heights of the prM:E:Fab complex from the base of the E protein layer are very different (Figure S3B), i.e. 155Å and 115Å at pH 8.0 and pH 6.0, respectively. This suggests comparison of the height of the prM:E:Fab in the model structures with the cryoEM densities could be used to determine the appropriate initial fitting model. One of the Fab densities in the asymmetric unit in both Class I and II cryoEM maps is at approximately the same position, as the Fab molecule A in both initial models (Figure S3A), thus we used this Fab molecule for comparison. We aligned the E protein layer of both models with its corresponding densities in the cryoEM maps (Figures 4B and 4C), and the results suggest that the class I and II structures likely are closer to the pH 8.0 and pH 6.0 model structures, respectively. Hence, class I and II structures may represent the early and late stages of the maturation process, respectively.
The final fit for the class I map (Figure S1B) showed that all of the E protein molecules mainly undergo translations from the immDENV:Fab 1H10 at pH 8.0 structure (Figures 5, S4A and S4B). In the immDENV:Fab 1H10 at pH 8.0 structure, the DIIIs of all E proteins are on a lower plane, while their fusion loops are on a higher plane pointing outwards. The three individual E proteins within an asymmetric unit are colored in red, blue, and green in Figure 5. The DIII of the red E protein in the pH 8.0 complex structure is positioned close to the 5-fold vertex whereas the red E protein of the class I pH 5.0 complex structure undergoes translation and rotation resulting in its DIII positioned closer to the 3-fold vertex. The pr:Fab on the red molecule is clustered around the 5-fold vertex. The blue E protein molecule also is translated slightly away from the 3-fold vertex, compared with the pH 8.0 complex structure. The green E protein molecule moves such that its DIII is positioned much closer to the icosahedral 2-fold vertex. Both the red and the blue E protein molecules are found at a lower plane than the green molecules (Figure S4B). The Fab densities around the 5-fold vertex cannot fit all 5 molecules of Fab that were complexed with the red E proteins, suggesting partial Fab occupancies (Figure 5). This indicates that in the class I pH 5.0 complex structure, some Fabs have dislodged from the red E protein molecules.
Figure 5. The arrangement of the surface proteins of the class I and II immDENV:Fab 1H10 complex at low pH shows dissociation of some Fab:pr and identifies four stages of the maturation process.
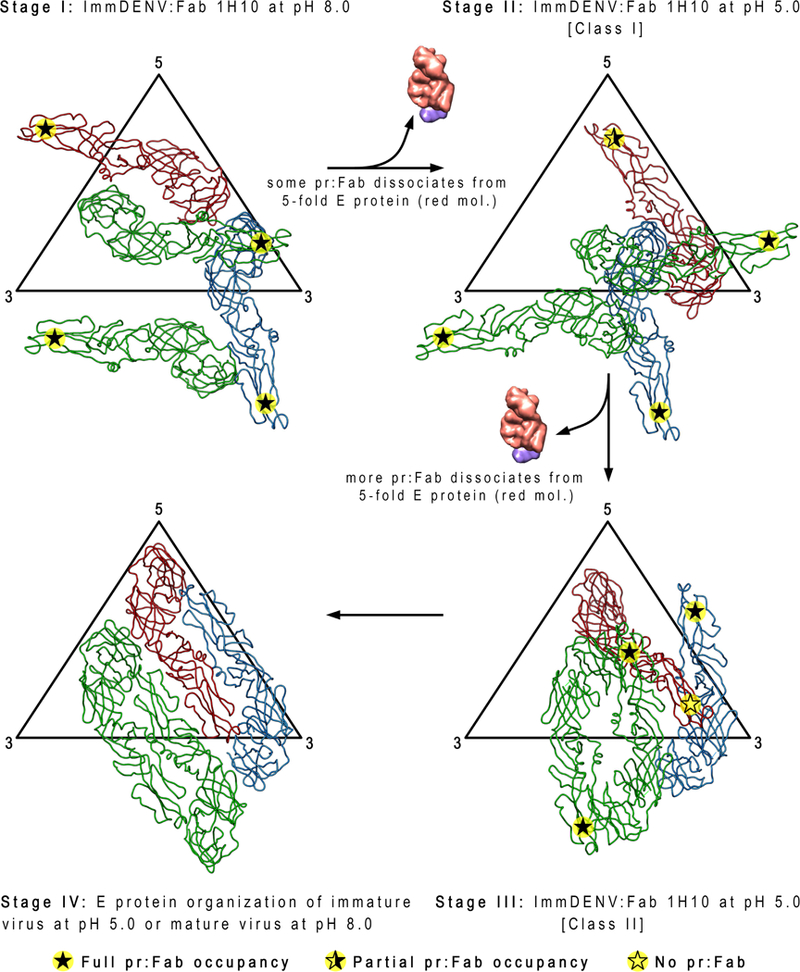
At pH 8.0, Fab 1H10 is bound to all pr-molecules (indicated by filled star). The transition from pH 8.0 to pH 5.0 in the class I structure causes some of the pr:Fab 1H10 complexes to dissociate from the E protein red molecule (indicated by half-filled star). Further structural rearrangements of the E proteins in the class II pH 5.0 structures result in all pr:Fab complexes dissociating from the same E protein red molecules (indicated by open star). This suggests that the red molecule likely experiences more clashes compared with the other E proteins (green and blue) in the asymmetric unit during maturation. The class I and II structures may also represent intermediate steps of the maturation process (stage II and III, respectively) that are partially stabilized by Fab binding. The class I structure (stage II) exists in a state closer to stage I of the maturation process, while the class II (stage III) structure is closer to the completion of the maturation process (stage IV). Comparison of the class I (stage II) structure with the immDENV:Fab 1H10 complex at pH 8.0 (stage I) shows mainly translational movements of all three individual E proteins in an asymmetric unit. On the other hand, in the class II structure (stage III), the E proteins nearly form two dimers (red:blue, green:green molecules) that were previously observed in the immature virus at pH 6.0 (Yu et al., 2009) or the mature virus pH 8.0 (Kostyuchenko et al., 2013) structures (stage IV).
See also Figure S4.
The final fit for the class II map (Figure S1C) showed that the red and blue E protein molecules almost form a dimer, and so do the two green E protein molecules (Figures 5 and S4C). In this structure, the densities corresponding to the Fab molecules bound to the red E protein molecules are missing and therefore, only 120 copies of Fab were observed on the virus surface.
Maturation structural transitions simulated using targeted molecular dynamics
We first ran equilibrium molecular dynamics simulations of a near-atomistic model of immDENV at acidic pH, containing all surface proteins with transmembrane (TM) domains embedded within a lipid bilayer membrane. This computational method, based on Newton’s equations of motion, allowed us to assess the per-residue interaction energies between E and pr proteins, prior to dissociation. As shown in Figure S5, three distinctive interacting regions on the E proteins were observed, in proximity to: i) the N67 glycosylation site; ii) the fusion peptide region (residues D98-S112); and iii) H242-K245. This is illustrated in Figure S5, in which a snapshot the E:pr complex is colored by average interaction energy. The former two sites of interaction involved largely hydrophobic contacts, the affinities of which would thus not be expected to be pH-dependent (Figure S6). The latter, on the other hand, was primarily electrostatic in origin; in particular, an observed pr:E interaction energy of up to ~ −15 kcal mol−1 encompassed contacts between H242 on the E protein and D63 on pr, further supported by a salt bridge between K244 on the E protein and D65 on pr. The interactions of this complex are expected to become less stable upon deprotonation of H242 at neutral pH, in agreement with previous studies supporting the possible role of this residue as a pH sensor (Chaudhury et al., 2015; Li et al., 2008). However, because of the low number of interaction sites between the pr and the E proteins, their affinity is likely weak at all pH conditions.
To investigate the structural motions required for maturation, we next conducted targeted molecular dynamics (TMD) studies. This method allows us to track relevant conformational changes by applying forces that gradually bias the initial protein structures towards the target conformation (Schlitter et al., 1993). Our explicitly-solvated simulations of the full virus contained ~1.5 million particles, and were hence computationally very intensive. We therefore focused on simulating the transition between stages I to II, and III to IV (Figure 5), rather than the drastic structural changes between stages II to III. Representative snapshots (Figure 6A) and a movie (Movie S1) of the E and prM proteins in the TMD simulations of the virus are shown during the stage I-II (Figure 5) transition. The root mean squared deviation (RMSD) of E proteins with respect to their stage II structure gradually decreased from ~50 Å to <10Å (Figure S7A) as a result of the targeted bias. By tracking the number of pr:E interactions over the course of the simulation, it was observed that most of the pr molecules dissociated from the red E protein molecules (Figures 6B and 6C) during the early part of the stage I-II transition (~frame 20 onwards, Figures 6B and S7B). This dissociation occurs at the point where the E-prM protomers within the trimeric spikes start to move apart from each other (Figure 6C). During this process, the blue E protein molecule approached the adjacent pr:red E proteins, dislodging the pr molecule from the red E protein and subsequently covering the pr binding site on the red E protein (Figure 6C). This extent of pr binding site occlusion on the red E protein by neighboring E proteins was not observed for the blue or green molecules. During the simulation, we also observed partial dissociation of pr proteins from the blue E protein molecules (Figures 6A and S7B). This finding was attributed to the displacement of the blue pr molecules by the neighboring red pr molecules, while the latter is dissociated from their partner E proteins. However, the blue pr molecules remained close to the associated E proteins and could readily rebind, forming stable complexes until the end of the simulation experiment (Figure S7B). The cryoEM structure suggests that some pr molecules are able to rebind to the red E proteins at stage II, as partial occupancy was detected, although this binding event was not evident during the TMD simulations.
Figure 6. Molecular insights into the stage I-II transition using molecular dynamics simulations.

(A) Representative snapshots extracted from targeted molecular dynamics simulations and their corresponding simulation frame number. E proteins from molecular simulations are colored in lighter colors than those of the Stage I and II cryoEM structures. Level of pr-Fab occupancies are shown as filled or partially filled stars.
(B) Pr molecules interacting with the red E proteins were blocked by the neighboring blue E protein molecules within a trimeric spike during the structural rearrangement from stage I to II. Plot showing number of pr molecules on the red E proteins that are blocked by neighboring blue E protein molecules during the structural arrangement (blue line) against frame number. The number of dissociated pr proteins from the E proteins (right y-axis) is also shown in brown. The grey dashed lines indicate the frames of the snapshots in panel (A).
(C) Snapshots of the motions of pr:E complexes within a trimeric spike at different frames, showing clashes (black outlined arrow in frames 25 and 85) mainly occurring between the pr:blue E protein complex, with the pr molecule on the red E protein leading to the dissociation of the pr from the red E protein molecule. The E proteins from the molecular simulations are colored in lighter shades than those from Stage I and II cryoEM structures.
(D) Plot of number of Fab-Fab clashes between the Fab on the pr-blue E protein and that on the pr-red E protein (red line) throughout the simulation. At around frame 25, severe clashes between the Fab:pr:E red molecules and Fab:pr:E blue molecules were observed when Fabs were superimposed onto the simulated pr-E virus molecule.
See also Figures S5, S6, S7 and S8.
To further verify that the pr proteins bound to the red E protein molecules indeed have a higher likelihood to dissociate from the E protein than those bound to the blue or green molecules, three additional, independent simulations were carried out. In each simulation, we applied a TMD bias to either red, blue or green molecules alone. Consistent with the earlier findings, the fastest pr dissociation occurred for those bound to the red E protein molecules (Figure S7C and S7D) compared with the other E proteins.
TMD simulations focused on the rearrangement of E proteins (Figure 7 and Movie S2) between stages III and IV in the absence of pr (Figure 5) showed a smooth structural transition, with minimal steric clashes. The structural changes (RMSDs) along the transition pathway with respect to final and initial coordinates are shown in Figure S7E. The RMSDs with respect to final coordinates decreased from ~20 Å to < 5 Å. Collectively, the simulations indicate that the two classes of immDENV:Fab 1H10 at pH 5.0 cryoEM structures are biologically meaningful intermediates representing the low pH induced DENV maturation process.
Figure 7. Possible E protein structural rearrangement between stage III-IV determined using molecular dynamics simulations.
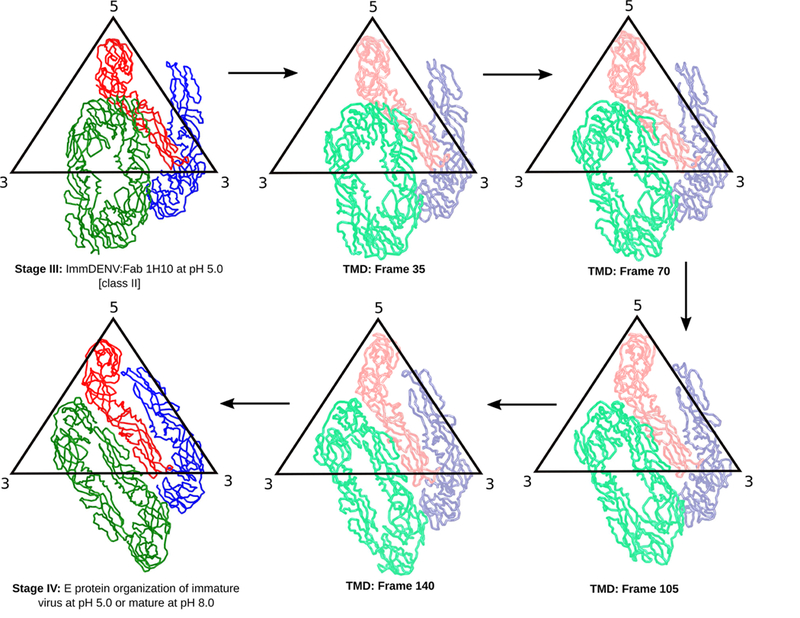
E protein movements from targeted molecular dynamics simulations are shown at different frame numbers along the conformational transition pathway.
See also Figure S7.
HMAb and Fab 1H10 remain bound to prM under different pH conditions
The cryoEM maps showed that there are fewer copies of Fabs on the low pH complex class I and class II structures, compared with the pH 8.0 complex structure. Since the resolution of acidic pH maps is low, we were unable to determine structurally whether the Fab alone or the Fab:pr complex detached from the E proteins on the virus surface. If the Fab alone detaches from the virus, this likely would be due to changes in the Fab affinity for pr when the pH is reduced. Therefore, we evaluated the binding property of HMAb or Fab 1H10 to prM molecule at different pH conditions using ELISA, biolayer interferometry (BLI) analysis and hydrogen deuterium exchange mass spectrometry (HDX-MS).
We characterized binding of the IgG and Fab 1H10 to the immDENV particle using ELISA. Two types of ELISAs were performed. The first tested the binding of IgG or Fab to immDENV particles coated on plates, during which the pH of the buffers used was kept constant throughout the experiment. The second ELISA mimics the cryoEM experiment and the infection conditions, in which the immDENV:Fab 1H10 complex was formed at pH 8.0, and the pH was then lowered to one of the conditions corresponding to that of the early (pH 6.0) or late (pH 5.0) endosomes.
In the first ELISA experiment, where the pH was kept constant, the antibody and Fab binding to immDENV was not reduced at any tested pH conditions (pH 8.0 to pH 5.0) (Figure 8A). This is also observed at all IgG concentrations tested (2, 5 and 10 μg/ml). In the second experiment, we modified the ELISA so that the Fab 1H10 was added to the immDENV coated on plates at pH 8.0, and then exposed to various pH conditions. We compared all conditions to the amount of Fab bound to immDENV when kept consistently at pH 8.0 (Figure 8A). The results suggest that the Fab does not detach from the prM once it is bound, irrespective of pH conditions. Only Fab was tested, as it matched the conditions used in the cryoEM study.
Figure 8. Fab 1H10 remains bound to pr-molecule at pH range of 5.0 to 8.0.
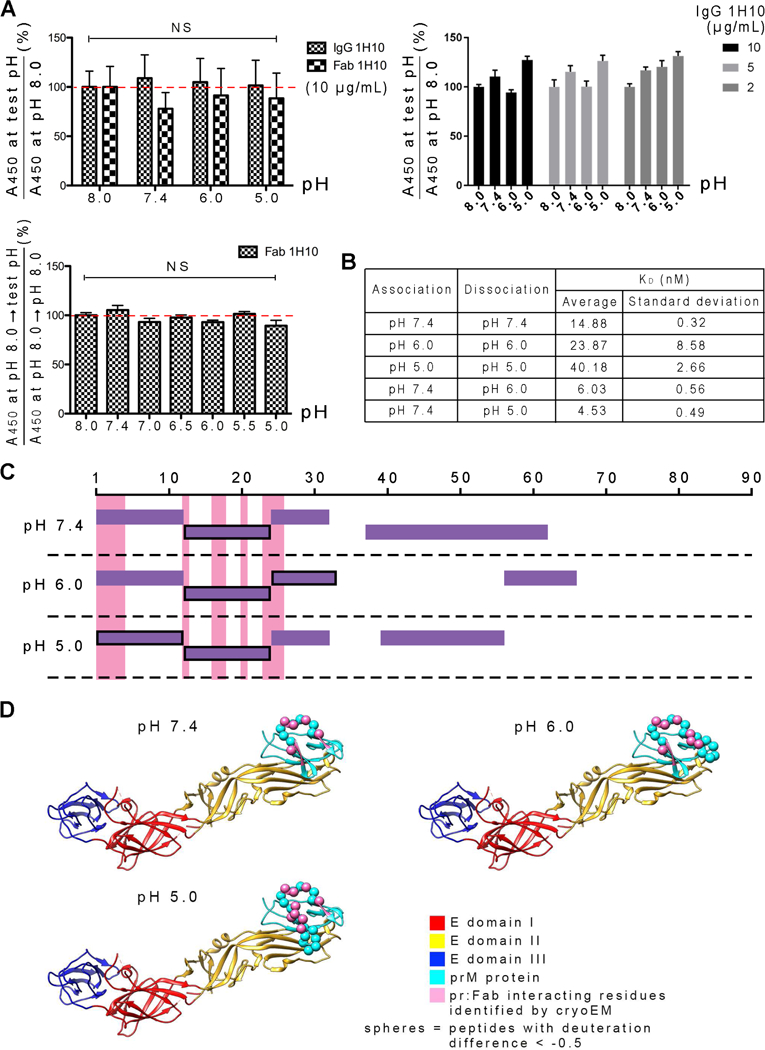
(A) ELISA assay showing the IgG and Fab bind equally well to immDENV over a pH range of 5.0 to 8.0 when pH is kept constant throughout the assay (Top panels). The same is observed when binding of Fab was done at pH 8.0 and then washed with other pH buffers (bottom panel). NS indicates results are not-significantly different analyzed by one-way ANOVA.
(B) Kinetics of HMAb 1H10 binding to recombinant prM:E linked protein determined by BLI analysis. When association and dissociation of the prM:E protein to the captured IgG were done at a constant pH, all are in the nM range suggesting high affinity interactions at all pH conditions. When association was done at pH 7.4 and then dissociation at either pH 6.0 or 5.0, results suggest that once the antibody is bound at neutral pH, exposure of the complex to low pH does not result in the detachment of HMAb 1H10 from the prM:E protein. The values summarized in (A) and (B) are average and standard deviation from three independent experiments.
(C) The Fab 1H10-pr interacting interface is largely the same when Fab-prM:E complex samples that were formed at pH 7.4 were then incubated at pH 5.0, 6.0 or 7.4, determined by HDX-MS. Plot of the detectable pr peptides from the prM:E:Fab complex samples that were also obtained in the uncomplexed prM:E protein controls. The peptides that have deuterium exchange difference of <−0.5 when subtracting between the Fab complexed and the uncomplexed prM-E at the same pH conditions are boxed with a black line. These peptides indicate the regions that are more buried on the pr-molecule upon Fab binding, consistent with the cryoEM epitope (pink). Fab remains bound to the epitope at all pH values.
(D) HDX-MS identified epitope (spheres) shown on the structure of prM:E complex with that identified by cryoEM (pink).
We also determined the kinetics of the binding of HMAb 1H10 to the recombinant prM:E complex using BLI analysis. The prM and E proteins in the prM:E complex were fused together via a polypeptide linker (Li et al., 2008). This prM:E construct was used due to difficulties in producing correctly folded protein when prM was expressed alone. IgG was attached to an anti-human Fc biosensor tip and dipped into wells containing recombinant prM:E protein diluted in various pH buffers. This association step was then followed by a dissociation step in which the biosensor tip was dipped into wells with buffer at the same pH used for the association. The results showed that, when compared across the pH conditions, the affinity (KD) of IgG for prM was ranked in this descending order: pH 7.4, pH 6.0, pH 5.0. Despite this finding, the affinities at each of the pH conditions were in the nM range, indicating high affinity interactions between HMAb 1H10 and prM:E (Figure 8B). In the second experiment, the association was carried out at pH 7.4 with the dissociation in either pH 6.0 or 5.0 buffers. The affinity of the antibody to prM:E becomes even stronger when the dissociation step was done at low pH (6.0 and 5.0) than at pH 7.4. The results showed that, once HMAb 1H10 is bound to prM at neutral pH, the antibody will remain bound in the low pH environment of the endosome.
HDX-MS was next used to determine the interaction interface between Fab 1H10 and prM:E proteins at pH 5.0, 6.0 or 7.4. HDX-MS measures the deuterium and hydrogen exchange at the exposed backbone regions of proteins. After incubation of protein with deuterated water, the deuterated protein is digested and then the molecular weights of the resultant peptides are measured by mass spectrometry.
To prepare the sample for HDX-MS analysis, we first mixed Fab 1H10 with prM:E at pH 7.4 and then exposed the complex to pH 6.0 or 5.0. We compared the peptide profiles between the deuterated uncomplexed prM:E with that of the prM:E:Fab 1H10 complex. Deuterated peptides from the prM:E:Fab 1H10 sample were subtracted with equivalent peptides from the uncomplexed prM:E sample. The pr peptides with a difference of < −0.5 deuteron indicate buried regions of the protein covered by the Fab molecule (Figures 8C and 9A). Using this technique, the interaction site was identified to peptide resolution (Figures 8C and 8D). A limitation of this technique is that data cannot be obtained for all peptides. For the pr-molecule, the peptide coverage was 74%, 46% and 55% at pH 7.4, 6.0 and 5.0, respectively (Figure 8C). The coverage for the M portion of the prM protein was much poorer, i.e. 66%, 27% and 0% at pH 7.4, 6.0 and 5.0, respectively (Figure 9B). The coverage for the E protein was also not complete, i.e. 71%, 43% and 43% at pH 7.4, 6.0 and 5.0, respectively (Figure 9B). There was especially very poor peptide coverage for half of the DII of the E protein (residues 55 to 130) (Figure 9B). The peptides from the M portion of the prM and the E proteins that could be detected largely showed very low deuteron differences at all pH conditions when compared with their uncomplexed prM:E at the respective pH, suggesting minimal antibody induced changes (difference of < −0.5 deuteron) (Figure 9A). One exception was at pH 6.0, in which peptides spanning residues 31 to 42 (peptide 31–42), and 45–67 on DII showed <−0.5 deuteron difference (Figure 9A). These sites are physically distant from the antibody-binding site, and it is unclear how the antibody could induce changes in this region.
Figure 9. The binding footprint of Fab 1H10 on recombinant prM:E protein was the same at pH 7.4, 6.0, and 5.0 as determined by HDX-MS.
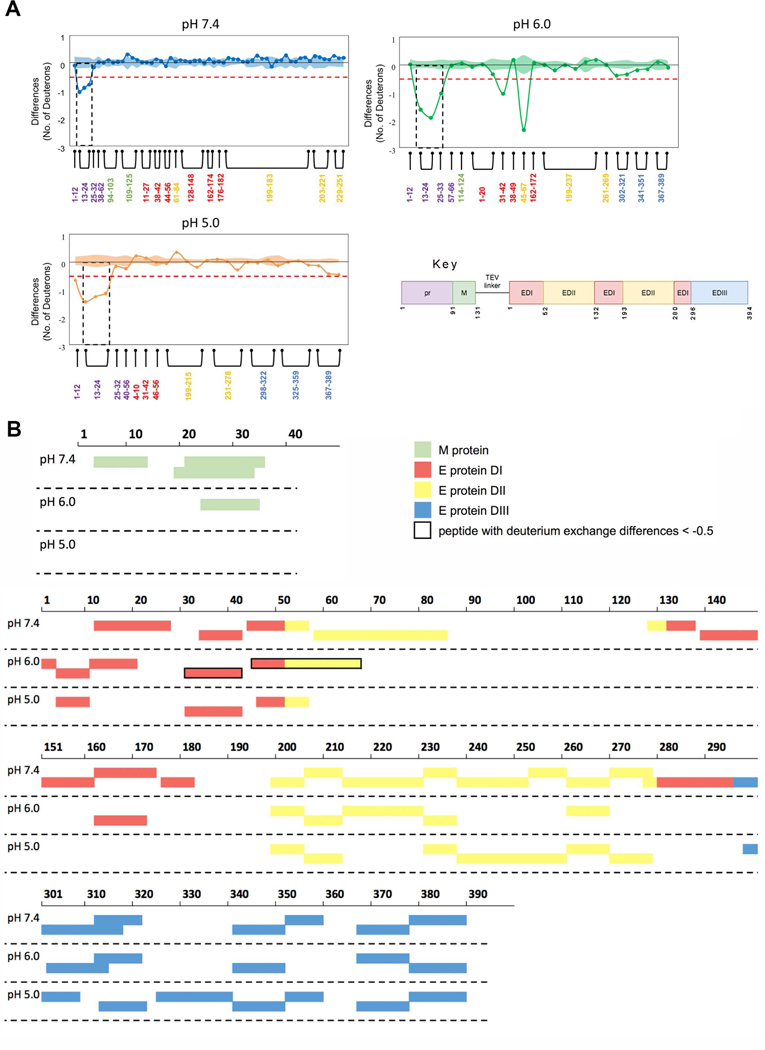
(A) Differences in the number of deuterons between pepsin proteolyzed fragments of prM:E protein and prM:E:Fab1H10 complex after 30 sec of deuterium exchange at pH 7.4, pH 6.0 and pH 5.0. Each node represents a pepsin proteolyzed peptide and is listed according to its position from the N-terminus. Differences in deuterium exchange below −0.5 deuterons are considered significant (red dashed line). Standard error for each peptide at each pH condition is indicated by the shaded regions along the X-axis and peptide residue numbers are colored according to the legend in each difference plot. The standard error for each peptide represent standard deviations observed across all the HDX-MS measurements from three independent HDX-MS experiments and the standard error for a given peptide represent the sum of the sigma standard deviations of each of the two conditions being compared.
(B) Plot of the M and E protein peptides detected in both prM:E and prM:E:Fab 1H10 complex samples. The peptides that have deuterium exchange difference of < −0.5 deuterons are boxed in thick black outline. These regions are more buried upon Fab binding, however comparison across pHs is difficult as there are no common peptides with exactly the same length detected.
The peptide coverage of the pr-molecule for residues 1 to 32 was complete for all pH conditions, and these residues span the epitope identified by the cryoEM study (Figure 8C). This allowed us to determine the epitope bound by the Fab across pH conditions. The HDX-MS results showed that the part of the epitope identified by cryoEM that binds most tightly to Fab at pH 7.4 involves a peptide spanning residues 13 to 24 (peptide 13–24) (< −0.5 deuteron difference) (Figure 9A). In the pH 6.0 and pH 5.0 conditions, the same peptide had similar deuterium difference as pH 7.4. In addition, at pH 6.0 and pH 5.0, both peptides 25–33, and 1–12 also had < −0.5 deuteron difference. Overall, the data shows that the 1H10 epitope on the pr-molecule identified by HDX-MS and cryoEM is consistent, and the Fab stays bound at all pH conditions. The HDX-MS results showed tight interaction between Fab 1H10 and pr at the peptide spanning residues 13–24 across pH 7.4 to pH 5.0, which is located at the center of the epitope identified by cryoEM (Figure 8D). At lower pH conditions, tight interactions also were detected for peptides at the periphery of the epitope (Figure 8D).
In conclusion, the results from the ELISAs, BLI and HDXMS showed that Fab 1H10 and prM do not dissociate from each other when pH is decreased.
Discussion
In the previously reported subnanometer-resolution structures of the immDENV at pH 8.0 and mature DENV, the start and end positions of all E proteins on DENV during maturation were determined (stage I and IV, respectively in Figure 5) (Kostyuchenko et al., 2013). The three individual E proteins in an asymmetric unit undergo large rotations ranging from ~137o to ~300o. Without intermediate structures, it is hard to understand how the immDENV E proteins can move so drastically. Even with the current state of the art molecular dynamics approaches, it is highly challenging to simulate the entire structural transition of the whole maturation process. Here we showed two new stages of maturation - stages II and III and their E protein organization are closer to that of the previously identified stage I and IV structures, respectively. Hence, we assigned the E proteins coloring in class II and III by observing for the minimal movement from the original stage I and IV structures, respectively. These intermediate structures allow us to simulate the motions of the E proteins occurring between stages I-II (Figure 6A), and III-IV (Figure 7) without clashes, further strengthening the notion that the low resolution class I and II low-pH cryoEM structures are plausible maturation intermediates.
Our targeted simulations indicated that in the absence of Fab molecules, the rearrangement of the immDENV surface protens during the early part of the transition between stages I and II (Figure 5) results in pr dissociating from the red E protein (Figure 6B and 6C). Three independent targeted simulations, in which biases were applied either to red (5-fold), blue (3-fold) or green (2-fold) molecules alone, in each case revealed that the fastest pr dissociation occurred for those bound to the red E protein molecules (Figure S7C and S7D) compared with the other E proteins. In addition, to enforce the entire transition, a sequential biasing pathway was required to bypass steric clashes around half-way through the process (Figures 6 and S8). Whilst we cannot completely rule out the possibility that other transition pathways may exist, these would have to be highly asymmetric to avoid clashes due to the bulky Fab molecule. Irrespective, the key finding from out targeted studies was that, irrespective of biasing strategy, the pr proteins bound to the red E protein molecules consistently have a higher likelihood to dissociate from the E protein than those bound to the blue or green molecules. Furthermore, when Fab is superimposed onto the simulated frames, more extensive clashes were detected at an even earlier time point/frame (Figure 6D) compared to the uncomplexed model (Figure 6B). This is consistent with the results from the co-floatation assay, which showed the uncomplexed immDENV at low pH can interact with liposome, and when Fab is added to immDENV, enhanced attachment to liposome was detected (Figure 1A). These results suggest that the weak dissociation of pr from E can occur at low pH, and this dissociation is enhanced upon Fab binding.
The results from ELISAs, BLI and HDX-MS experiments consistently showed that the Fab 1H10 affinity for pr-molecule remains strong (nM affinity) at low pH conditions. A previous study (Zheng et al., 2010) using surface plasmon resonance consistently showed weak interaction between the pr and E proteins at pH 6.0 to 8.0. The disparity in affinity between the Fab to pr binding, and pr to E binding suggests that Fab:pr complex, as a unit, may dissociate from the E protein when experiencing virus surface protein clashes induced at low pH. In the class I structure, the red E protein molecule in the asymmetric unit showed reduced Fab occupancies, while in the class II structure, all Fabs on the red molecules were missing (Figure 5). This suggests that the red E protein experiences the most steric hindrance during the low pH structural rearrangement compared with the green and blue molecules. This is also supported by the molecular simulations, which showed that steric hindrance between surface proteins induces mainly pr dissociation from the red E protein molecule (Figure 6). The calculated per-residue interaction energies further indicated that the low pH E:pr protein complex is stabilized in several regions that include salt bridges H242 – D63 and K244 – D65, supporting previous proposals for mechanisms of pH sensing during maturation (Figure S5).
The cryoEM structures suggest that some pr likely dissociates from the red E protein when moving from stage I to II, but all will dissociate during the transition from stages II to III.
Since we did not add furin protease to the low pH complex mixture for the cryoEM studies, the lack of some Fab densities in the low pH complex maps might be due to some of the Fab 1H10-pr complexes detaching from the E proteins and “flopping” on the virus surface, leading to the Fab-pr densities being “averaged out” during the reconstruction process. Superposition of Fab 1H10 onto the simulated molecular structures during the stage I-II transition also showed significant Fab-Fab steric clashes between the Fab:pr:E protomers within a trimeric spike (Figure 6D). A majority of the Fab-Fab clashes involved those on the red E molecules, consistent with the loss of this Fab (and likely together with its associated pr) observed in the class II cryoEM map (Figure 5). Therefore, we propose that Fab could help in dislodging the pr from the E protein by increasing the steric hindrance during virus surface reorganization.
The structural work showed that some prM:Fab complexes have detached from the virus surface, which may enhance the exposure of the furin cleavage site on prM. Even though the liposome co-floatation assay was performed in the absence of furin protease, the attachment of the immDENV:Fab 1H10 complex to the liposome at pH 5.0 was enhanced, compared with the immDENV only. It has been shown previously that furin cleavage is important for the infectivity of immDENV:anti-prM antibody complexes (Rodenhuis-Zybert et al., 2010). Since furin is membrane bound (Teuchert et al., 1999), it is likely that only the site where the virus interacted with the endosomal membrane, are processed by furin protease allowing the completion of the virus-endosomal membrane fusion process.
The ability of pr-molecules to interfere with the interaction of recombinant E protein (rE) with liposomes at pH conditions ranging from 4.50 to 6.25 has been reported previously (Zheng et al., 2010). At pH 5.75 and pH 6.25, mimicking the mildly acidic conditions in the TGC, the addition of pr-molecules blocked rE protein interaction with liposomes. At lower pH (i.e. pH 4.5 and pH 5.0), mimicking the stronger acidic condition of the late endosome, the rE protein was able to interact with liposomes (Zheng et al., 2010). Based on this, the authors suggested that the pr-molecules interact with E protein and remain on the newly synthesized immDENV in the TGC (pH 6.0) (Zheng et al., 2010) during transportation out of the cell. However, when fully immDENV enters another cell, in the late endosome, due to the stronger acidic condition (pH 5.0), the pr-molecule dissociates from the E proteins, thereby exposing the E protein fusion loop, enabling immDENV to fuse (Zheng et al., 2010). This would suggest that by simply lowering the pH to 5.0 alone, pr can uniformly dissociate from all E proteins. However, in our class I and II cryoEM structures of the immDENV:Fab 1H10 complex at pH 5.0, even though there are less Fab densities on the virus surface compared with the pH 8.0 structure, there were still two Fabs bound on each asymmetric unit, indicating most pr-molecules remain on the virus surface. Our low pH cryoEM structures combined with molecular simulations suggest that steric hindrance plays an important role in inducing pr dissociation from a specific E protein molecule across all asymmetric units on the virus.
The Fab 1H10 molecule may stabilize the E protein organization at certain stages of the maturation process of the immDENV at low pH. Our previous subnanometer resolution structures of the immature and mature virus at pH 8.0 (Kostyuchenko et al., 2013) combined with the class I and II immDENV:Fab 1H10 complex at pH 5.0 structures represent four stages (stage I – IV) of the maturation process. The class I low pH complex structure is likely at the early stages of maturation (stage II) as, the structure only involves mostly translation from the maturation stage I immDENV:Fab 1H10 at pH 8.0 structure (Figures 5 and 6A, and Movie S1). The class II structure, on the other hand, is closer to late stage of maturation (stage III), as the E protein organization is more similar to the fully mature virus (Kostyuchenko et al., 2013), and the uncomplexed immature virus at pH 6.0 (Yu et al., 2009) structures (stage IV) (Figures 5 and 7, and Movie S2). More transitional stages in between the class I and II cryoEM structures would need to be determined to fully understand the maturation process.
CryoEM structures of immDENV complexed with an anti-prM Fab fragment of a murine monoclonal antibody (MMAb) 2H2 at pH 7.0 and 6.0 have been solved previously (Wang et al., 2013). The structure at pH 7.0 showed that Fab 2H2 binds to an epitope that overlaps with that of HMAb 1H10 (Figures S9A and S9B). Contrary to immDENV:Fab 1H10 complex structures at pH 5.0, the immDENV:Fab 2H2 complex structure at pH 6.0 is identical to that at pH 7.0. One possible explanation for this discrepancy is the difference in the pH of the samples used for cryoEM studies. Although the uncomplexed immDENV was observed to undergo structural changes at pH 6.0 (Yu et al., 2009), the same pH may not be able to induce the structural changes of E proteins for maturation when the virus forms a complex with anti-prM Fabs. This could occur because the affinity for binding of pr molecule to E protein is slightly higher at pH 6.0 compared with 5.0 (Zheng et al., 2010), and the prM:E:Fab complex may thus form a rigid body at pH 6.0, restricting movement of E proteins.
DENV can adopt diverse morphologies including mature, expanded mature, partially immature and fully immature virus forms, and each of them can infect cells under the right circumstances (Lok, 2016). Hence, each of these morphologies should be considered in studies of pathogenesis and immunity. The mature and the expanded mature virus can attach to and infect cells readily. The immDENV particle, on the other hand, requires formation of a complex with either anti-prM antibodies or some anti-E protein antibodies (Rodenhuis-Zybert et al., 2011; Rodenhuis-Zybert et al., 2010) or DC-SIGN molecules (Richter et al., 2014) to become infectious. The mechanisms by which these different molecules initiate infection by immDENV could vary, and thus be of interest for further investigation. The partially immDENV form of virus may be able to infect without forming any antibody complex, since they may have more mature virus portion on their surface. However, there is a gradient of immaturity among these partially immature viruses, and it is unclear at what point along this gradient the formation of the anti-prM complex becomes more important. Anti-prM antibodies constitute a major component of antibodies in the anti-DENV human immune response. Also in tissue culture, highly immDENV particles are often detected (Junjhon et al., 2010; Murray et al., 1993; van der Schaar et al., 2007). These findings signify the importance of investigating how anti-prM antibody could enhance immDENV infectivity. The anti-prM antibody likely enhances the attachment of the immDENV to Fcγ-positive cell types such as monocytes and macrophages. Here, we showed the mechanism by which the anti-prM antibody also could help in the dissociation of pr-molecule from the E proteins on immDENV, thereby enhancing attachment of virus to the endosomal membrane which may increase the efficiency of the initial stages of the fusion process. We also showed two other transitional structures during maturation. These findings fill in the gaps in the understanding of the role of anti-prM antibodies in the pathogenesis of immDENV and should help to devise ways to therapeutically target this virus morphology.
STAR METHODS
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shee-Mei Lok (sheemei.lok@duke-nus.edu.sg).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
C6/36 mosquito cells, baby hamster kidney strain 21 (BHK-21) fibroblast cells, and LoVo cells were from the American Type Culture Collection (ATCC). THP-1 monocytic cells were from Prof. Eng Eong Ooi laboratory (Duke-NUS, Singapore).
S2 drosophila cells stably expressing recombinant prM-E protein were generated using the Drosophila Expression System (Invitrogen).
Viruses
DENV3 (strain D3/SG/05K863DK1/2005) and DENV2 (strain D2/SG/07K3598DK2/2007) were from Prof. Eng Eong Ooi laboratory (Duke-NUS, Singapore).
METHOD DETAILS
Cell lines maintenance and propagation of viruses
C6/36 mosquito cells, baby hamster kidney strain 21 (BHK-21) fibroblast cells, and THP-1 monocytic cells were grown in RPMI 1640 supplemented with 1% L-glutamine, 25 mM HEPES (HyClone) and 10% fetal bovine serum. LoVo cells were cultured in Ham’s F12 Nutrient Mixture (ThermoFisher Scientific) supplemented with 10% fetal bovine serum (ThermoFisher Scientific) and 25 mM HEPES (ThermoFisher Scientific). C6/36 cells were grown at 29°C while B HK-21, THP-1, and LoVo cells were grown at 37°C.
DENV3 (strain D3/SG/05K863DK1/2005) stock was used to infect C6/36 cells at a multiplicity of infection (MOI) of 10 when the cells were about 80% confluent. At 2 hr post infection (hpi), the inoculum was replaced with maintenance medium containing 2% fetal bovine serum and a final concentration of 20 mM NH4Cl. The virus supernatant was harvested 48 hpi. This virus was used for the infection of THP-1 cells and for cryoEM studies.
DENV2 (strain D2/SG/07K3598DK2/2007) virus supernatant was prepared by infecting LoVo cells at MOI of 10 with DENV2 stock for 2 hr. Following which, the inoculum was replaced with maintenance medium containing 2% fetal bovine serum and 25 mM HEPES. The virus supernatant was harvested 24 hpi. DENV2 was used on the LoVo cells infection instead of DENV3 because DENV2 could be grown to higher titres. This virus was used for the liposome co-floatation assay.
Viral titres were quantified in terms of plaque forming unit (pfu) / mL by plaque assay and genome copy number (GCP) / mL by one-step real time quantitative PCR
Preparation of virus for cryoEM
DENV3 (strain D3/SG/05K863DK1/2005) virus supernatant from 10 cell-stacks of C6/36 cells was precipitated with 8% (w/v) PEG8000 at 4 °C overnight. The virus was then pelleted down at 14,300 x g for 1 hr and re-suspended in NTE buffer (12 mM Tris, pH 8.0, 120 mM NaCl, 1 mM EDTA). This is followed by purification through a 24% (w/v) sucrose cushion, re-suspended in NTE buffer, and further purification by continuous 10–30% (w/v) potassium tartrate-glycerol gradient. The collected virus band was buffer exchanged to NTE buffer and concentrated to a final volume of about 50 to 100 µL in an Amicon Ultra Centrifugal Filter with molecular weight cut-off of 100 kDa (Merck Millipore). The purity and final concentration of the virus sample was estimated by comparing the E protein band intensity on Coomassie stained 4–15% SDS-PAGE gel against band intensities of bovine serum albumin (BSA) standards.
Preparation of virus for ELISA
DENV3 (strain D3/SG/05K863DK1/2005) virus supernatant from C6/36 cells was precipitated with 8% (w/v) PEG8000 at 4 °C over night, pelleted down at 14,300 x g for 1 hr and re-suspended in NTE buffer.
Preparation of virus for co-floatation assay
DENV2 (strain D2/SG/07K3598DK2/2007) virus supernatant from LoVo cells was directly pelleted by ultracentrifugation using SW41 rotor (Beckman) at 145,000 x g for 2 hrs and the pellet was then re-suspended in NTE buffer.
Preparation of liposomes for co-floatation assay
Liposomes were made using the following lipid composition; DOPC, DOPE, DOPS, BMP and cholesterol in a molar ratio of 4:1:1:2:2 (Zaitseva et al., 2010). After the lipid solutions were mixed and freeze dried under vacuum, the lipid powder was rehydrated using either buffer at pH 8.0 (12 mM Tris, 120 mM NaCl, 1 mM EDTA) or at pH 5.0 (12 mM Tris, 120 mM NaCl, 1 mM EDTA, 50 mM MES). The lipid suspension was then subjected to 5 freeze-thaw cycles in liquid nitrogen and water bath. This was followed with extrusion through 100 nm filter at least 40 times to ensure homogeneity of liposome diameters. The size of the liposome was then checked with dynamic light scattering, Zetasizer Nano S (Malvern). The liposomes were kept on ice and used within the same day.
Virus – liposome co-floatation assay
Virus samples of immDENV2 grown in LoVo cells were incubated with Fab 1H10 for 30 min at 37°C and pH 8.0 before addition of liposome. The immDENV:Fab 1H10 complex and liposome mixture was then adjusted to pH 5.0 by the addition of 1 M MES or maintained at pH 8.0 and incubated for another 5 min at 37°C. The following samples were included as control: uncomplexed virus without liposome and uncomplexed virus mixed with liposome at pH 8.0 and pH 5.0. The samples were then mixed with sucrose solution to a total volume of 1500 µL and a final concentration of 30% sucrose (weight/volume). 1500 µL each of 60%, 50%, 40%, 30% solution containing the sample, 20%, 10%, and 5% sucrose solutions were layered on top of each other (Liao et al., 2010; Sanchez-San Martin et al., 2008; Zheng et al., 2010). All sucrose solutions were of the same pH as the sample and were made with either buffer at pH 8.0 (12 mM Tris, 120 mM NaCl, 1 mM EDTA) or pH 5.0 (12 mM Tris, 120 mM NaCl, 1 mM EDTA, 50mM MES). Sucrose gradients were centrifuged for 2 hr at 175,000 rpm at 4°C in a swing rotor with zero deceleration. Fractions of 500 µL were collected, precipitated with trichloroacetic acid (TCA) as previously described (Link and LaBaer, 2011). The TCA precipitated samples were then used for SDS-PAGE followed by western blotting analysis.
Western blotting
The western blots were blocked in 5% BSA in PBS-T for one hour then incubated overnight at 4°C with primary antibody, mouse anti- E IgG 4G2 at 1 µg/mL. This was followed by three rounds of PBS-T washes and an hour of incubation at room temperature with secondary antibody, goat HRP conjugated anti-mouse IgG (H+L) (Invitrogen) at 1:2000 dilution. The blots were then developed using ECL substrate (GE Healthcare) and ChemiDoc Touch Imaging System (Bio-Rad) was used for the western blot imaging.
Quantification of immDENV genome-containing particles (GCP)
As immDENV does not form plaques in most cells, we quantify the immDENV by conducting RT-PCR. ImmDENV RNA was extracted from purified immDENV sample using QIAamp™ Viral RNA Mini Kit (Qiagen) according to manufacturer’s instructions. The one-step real-time quantitative PCR (qPCR) was done using the BioRad iTaq Universal Probes One-Step Kit (Bio-Rad) according to manufacturer’s instructions. DENV specific primers used are forward: 5’-CGWTCTGTGCCTGGAWTGATG-3’ and reverse: 5’-AAGGACTAGAGGTTAKAGGAGACCC-3’ as previou sly published (Warrilow et al., 2002). The fluorescent probe used has the following sequence 5’-TCTGGTCTTTCCCAGCGTCAATATGCTGTT-3’ with FAM at 5’ en d, Zen quencher in the middle and Iowa Black FQ quencher at 3’ end. The 10 µL reaction contained 5 µL of 2x reaction mix, 0.25 µL of reverse transcriptase, 0.5 µL each of forward and reverse primer, 1.75µL of nuclease free water and 2uL of viral RNA or RNA standard dilutions. The thermocycler setup was 10 min at 50°C for rever se transcription reaction, 3 min at 95°C for polymerase activation and DNA denaturation, 40 cycles of 15 sec at 95°C for DNA denaturation and 30 sec at 60°C for primer anne aling and extension. The Cq value of the viral RNA sample was compared against the standard curve generated using the RNA standard dilutions to determine the GCP/mL in the sample. To create a positive sense RNA standard for the real-time qPCR, a DNA template (IDT) was used with the MegaScript™ T7 RNA transcription kit. After transcription, the RNA standard was purified using the RNeasy™ Mini Kit (Qiagen) and quantified using Nanodrop™. The DNA template contained T7 promoter region at the 5’ end of sequence complementary to the target region in the one-step real-time qPCR. The sequence of the DNA template is 5’- TAA TAC GAC TCA CTA TAG CGT TCT GTG CCT GGA ATG ATG CTG AGG AGA CAG CAG GAT CTC TGG TCT CTC CCA GCG TCA ATA TGC TGT TTT TGT TTG CGG GGG GTC TCC TCT AAC CTC TAG TCC TT-3’.
Enhancement of infection assay using THP-1 cells
After the immDENV titre (GCP/mL) was determined by one-step real-time qPCR, the concentration of prM molecule in the sample was calculated based on the assumption that each virus particle has 180 copies of prM molecule. HMAb 1H10 was diluted to achieve various molar ratios of antibody to prM molecule. The virus sample was incubated for 1 hr at 37°C with varying dilutions o f HMAb 1H10 in cell culture medium containing 2% FBS. Virus or virus-antibody complexes were added to 1 × 105THP-1 cells at multiplicity of GCP (MOG) of 200 for virus grown in C6/36 treated with NH4Cl 2 hpi. To determine the proportion of infected cells, cells were harvested at 72 hpi, fixed with 4% formaldehyde, permeabilized with 0.1% saponin and stained with AlexaFluor 488 conjugated anti-E HMAb 4.8A (5 µg/mL). HMAb 4.8A was conjugated with fluorescent dye using the Alexa Fluor® 488 Antibody Labeling Kit. The cells were finally analyzed using a MacsQuant® VYB Flow Cytometer (Miltenyi Biotec). The data gating and analysis were done using the FlowJo™ software (FlowJo, LLC). One-way analysis of variance (ANOVA) was used for statistical analysis of the data using GraphPad Prism software. Graphed values are presented as mean ± SD.
CryoEM data acquisition and processing
As the amount of immDENV particles grown in LoVo cells is too low to allow cryoEM studies, we prepared the immDENV3 by growing the virus in C6/36 cells in the presence of ammonium chloride. The level of immaturity of the immDENV3 sample were assessed by cryoEM, and found to contain mostly immature virus (Figure 2A). The immDENV3 was mixed with Fab 1H10 at a molar ratio of one Fab molecule per E protein (Figure 2A). E protein concentration of was determined by SDS-PAGE gel by comparing to BSA standards. For the low pH sample, a final concentration of 50 mM of MES buffer at pH 5.0 was added to the pre-formed immDENV3:Fab 1H10 complex at pH 8.0 complex. These samples were then incubated at 37°C for 30 min followed by 2 hr at 4°C. A 2.5 µL of the sample was then applied onto an ultra-thin carbon-coated lacey carbon copper grid and then blotted with filter paper for 2 sec before plunge freezing in liquid ethane using the FEI Vitrobot Mark IV. Image acquisitions were done at liquid nitrogen temperature using a FEI Titan Krios electron microscope connected to a 4K x 4K FEI Falcon I direct electron detector, under the following conditions: 300 kV, a nominal magnification of 47,000, an electron dose of 18 e-/Å2 and a defocus range of −1 to −4 µm. The final pixel size of the micrographs was 1.7 Å. Micrographs showing severe drift and astigmatism were discarded. Particles from each micrograph were boxed manually using e2boxer tool in EMAN2 software package (Tang et al., 2007). A total of contrast transfer function (CTF) parameters for each micrograph were determined using the program CTFFIND3 (Mindell and Grigorieff, 2003).
CryoEM image reconstruction of immDENV3:Fab 1H10 sample at pH 8.0
The 6Å resolution structure of immDENV1 (PDB 4B03) was used as a starting model for the cryoEM image reconstruction. Twenty-five cycles of iterative orientation search was carried out using multipath simulated annealing protocol (MPSA) (Liu et al., 2007). At each cycle, 3D reconstruction was conducted using the make3d program from EMAN (Ludtke et al., 1999). The final 12Å resolutio n map was reconstructed using 2,886 particles. The resolution of the final map was determined by gold standard Fourier shell correlation procedure using cut-off value of 0.143 (Figure 2B). The resolution of the map (~12 Å) is consistent with the clearly resolved trans-membrane helical densities in the cryoEM map (Figure S1A).
CryoEM image reconstruction of immDENV3:Fab 1H10 sample at pH 5.0
The dataset for the DENV3:Fab 1H10 complex at pH 5.0 contained heterogeneous population of particles. Hence, two-dimensional classification (RELION) (Scheres, 2012) was used to first discard broken or irregular shaped particles. The particles in the class averages that seem ordered and round were then selected for further processing involving three-dimensional classification and refinement in RELION. Icosahedral symmetry was used during the classification and map reconstruction processes. Three classes of particles were then selected; models were made from each class and served as initial models for orientation search in MPSA reconstruction. During the image reconstruction process, the first two models showed similar reconstructed maps, thus the particles classified under these two models were combined. The reconstruction was continued with the first and third reference models. The final reconstructed maps of two classes of particles in the population: class I and class II were reconstructed from 2,216 and 3,106 particles, respectively, representing 35.1 % and 49.2 %, respectively, from the total selected particles. The resolution of class I and II maps were both determined to ~25Å. The resolution of the final map was determined by plotting the Fourier shell correlation coefficient between two reconstructed maps of two half-datasets of the final iteration step using cut-off value of 0.5 (Figure 2B). Although icosahedral symmetry was imposed to the two particle classes, the two classes produced interpretable maps. Nevertheless, some local flexibility in the structure may exist. This can lead to deviation from icosahedral symmetry that in turn it lowers the resolution. The non-icosahedral structural classes will not produce an interpretable map using this method. We present here the two structural classes that are icosahedral to a good extent.
Interpretation of the cryoEM map of immDENV3:Fab 1H10 complex at pH 8.0
The cryoEM structure of immDENV1 (PDB 4B03)(Kostyuchenko et al., 2013) was used to fit the cryoEM immDENV3:Fab 1H10 complex at pH 8.0 map and the fit was optimized using the “fit in map” function in Chimera (Pettersen et al., 2004). The amino acid residues of the fitted DENV1 E and prM proteins were then mutated to those of DENV3. Further refinement of the fitting was done using molecular dynamics flexible fitting (MDFF) (Trabuco et al., 2009) in VMD (Humphrey et al., 1996) and NAMD (Phillips et al., 2005). A weighting factor of 0.5 was used to include the contribution of the cryoEM map in the overall potential energy of the molecular dynamics simulation, and symmetry restraints were applied to avoid clashes between neighboring protein molecules. The simulation was carried with 20,000 steps of minimization followed by 100,000 steps of molecular dynamics. The final structure was visually inspected for any clashes using Chimera. The final coordinate is free from misfit and clashes (Figure S1A).
Model fitting of the class I and class II cryo-EM maps of immDENV:Fab 1H10 complex at pH 5.0
Two models were used in the fitting of the class I and II cryoEM maps i.e. the structure of immDENV:Fab 1H10 complex at pH 8.0 and the structure of immDENV2 (PDB ID 3IYA) at pH 6.0 (Yu et al., 2009) with Fab 1H10 superimpose on all pr-molecules (Figures S3A and S3B). To decide which model should be used for fitting of the class I and II maps, the two models were superimposed onto each of the maps. The superposition of the two structures onto the class I and II maps showed that the height between the densities of E:pr:Fab molecules of class I map is similar to the model of the immDENV3-Fab 1H10 complex at pH 8.0 model (Figure 4B), whereas class II map issimilar to the model of the immDENV2-Fab 1H10 model at pH 6.0 (Figure 4C).
The immDENV3-Fab 1H10 pH 8.0 structural model was superimposed onto the class I map and the fit was initialized by first moving the red E protein molecules (mol) complexed with pr and Fab 1H10 as a rigid body (refer to Figure 5). Since the Fab densities are the clearest, they were used as a guide for fitting. The red E protein mol:pr:Fab had to be rotated ~37° clockwise and tra nslated in order to put the Fab molecule into the protruding Fab densities C located around the 5-fold vertex (Figure 4A, left panel). Following this, blue E protein mol:pr:Fab was translated so that the Fab is fitted into Fab A density. Lastly, the green E protein mol:pr:Fab was translated and the Fab fitted into the Fab density B located near the 3-fold vertex (Figure 4A, left panel). The final fitting shows no clashes and all densities are interpreted (Figure S1B).
The immDENV2 at pH 6.0 with Fab 1H10 model was superimposed onto the class II map. Fitting of class II map was started by fitting the Fab molecule of the blue E protein mol:pr:Fab (located near to 3-fold vertices) into Fab density A (Figure 4A, right panel). It required ~20° rotation clockwise with on ly slight translation of the E protein blue mol to fit the Fab molecules into densities and at the same time avoiding clashes between neighboring blue E protein mol. Subsequently, the green E protein mol:pr:Fab (located on 2-fold vertices) was rotated ~23° clock wise and the Fab on this E protein was fitted into the Fab densities B (Figure 4A, right). The red E protein mol:pr complex was fitted into the remaining densities while avoiding clashes with neighboring red E protein mol and the adjacent E proteins. The Fab molecule on red E protein mol was removed from the model because there are no Fab densities observed for this molecule (Figure S1C).
Simulation setup and energetics analysis
The structure of stage I (immDENV:Fab 1H10 at pH 8.0, Figure 5) was determined by cryoEM as described in the previous section. For stage II (immDENV:Fab 1H10 class I at pH 5.0, Figure 5), because the TM regions were not resolved by cryoEM, we used the coordinates of immDENV1 (PDB 4B03) and superimposed the resolved ecto-domains into the cryoEM map, using the align function in PyMOL v1.8.4.0 (Schrodinger, 2015). With this approach, the green molecule had its TM helices located unphysically far away from the lipid bilayer, at a higher plane than the TM regions of red and blue molecules, and overlapping with their ectodomains. We resolved this discrepancy by running steered MD simulations of a single E protein chain with a harmonic force pulling the TM region away from the ectodomain by ~30Å. We then replaced the green molecules with this more “extend ed” E protein conformation. Once the full virus structural models were generated, we aligned the virus E proteins of stage II to stage I using the SVDSuperimposer function in BioPython (Cock et al., 2009). The corresponding transformation matrix for the alignment was used to move the prM molecules, generating aligned models for the full complex. These aligned stage I and stage II pre-processed structures were then used for setting up the TMD simulations.
We used a previously resolved virus structure for stage IV (mature virus at pH 8.0) (Kostyuchenko et al., 2013), and modelled the complete E protein into the cryoEM structure of stage III (immDENV:Fab 1H10 class II at pH 5.0, Figure 5), using the E protein coordinates from stage IV as template. As described above, the structural model for stage IV was then aligned to that of stage III. The E and prM proteins (complete with TM regions) were used to model the transition between stages I and II. For the stage III to IV transition, due to the experimentally observed pr peptide cleavage, we only included the E proteins in our simulation. We modeled the viral membrane in both systems, using the protocol developed in our previous work (Marzinek et al., 2016). Initially, equally-spaced phospholipids were placed on the surface of an ideal sphere of vesicle diameter ~400 Å (according to cryo-EM observations; see Figure 3A).The chosen vesicle composition was a mixture of molecules of POPC (PC), POPE (PE), and POPS (PS) in a 6:3:1 ratio as determined experimentally (Zhang et al., 2012). Each protein-lipid system was centered in a cubic box (~600×600×600 Å) followed by conjugate gradient minimization in vacuum. All simulations were conducted using the NAMD package (Phillips et al., 2005) together with the coarse-grained MARTINI 2.2 force field (Marrink et al., 2007; Monticelli et al., 2008). All proteins were treated using standard MARTINI 2.2 parameters, with default bonds, angles, and dihedrals applied in order to preserve secondary structure. The acidic pH for the transition from stage I to II was assumed to result in protonated histidines and hence these were treated in their ionized state; other amino acids were assigned their default protonation states. In the transition from stage III to IV (pH=8.0), default protonation states were assigned to all amino acids. Subsequently ~1,300,000 MARTINI water beads were added into the box in explicit-solvent simulations, out of which ~10% corresponded to antifreeze particles. Chloride particles were added to neutralize the overall system charge. Each system was subject to ~300,000 steps equilibration in the NPT ensemble (N - constant number of atoms, P – constant pressure and T - constant temperature) with position restraints on the protein backbone beads. All simulations employed stochastic (Langevin) dynamics with 4 fs time step and 1 ps−1 damping coefficient to maintain the temperature at 310K. The Nosé-Hoover Langevin piston pressure control was used with isotropic pressure of 1 bar, oscillation period of 2 ps and a 1 ps damping time scale (Feller et al., 1995). A 12 Å cut-off distance was used for electrostatics and van der Waals interactions with a smooth switching function at 9 Å. A 14 Å cut-off distance between pairs for inclusion in pair lists was employed. Periodic boundaries were applied in all directions. All simulations were and 20 CPUs (Intel® Xeon® CPU E5–2680 v2 @ 2.8 GHz) each. All simulation snapshots and videos were made using VMD software (Humphrey et al., 1996).
The per-residue interaction energies (Figure_S5) of the E:pr complex were extracted from unrestrained equilibrium simulation of 400,000 steps in the NPT ensemble and involved immature DENV (stage I) at acidic pH. Protein contacts that were found with a 2 nm cut-off distance from one another were subject to interaction energy calculations. The interaction energy corresponded to the sum of short-ranged electrostatic and van der Waals interactions and was averaged over 180 E protein and 180 pr protein chains.
Targeted molecular dynamics (TMD) simulations
In TMD, steering forces are applied to bias the simulation so that the protein conformations tend towards the target structure (Schlitter et al., 1993). We applied the bias in the form of a harmonic potential that is dependent upon the gradient of the root mean squared distance (RMSD) to the target structure. At each step of the simulation, the RMSD between current coordinates and the target structure is calculated. The target RMSD is linearly interpolated from the initial (at the first step) to the final RMSD (at the last step). The force constant in the harmonic potential is scaled by the number of particles which undergo the transition.
Each TMD simulation employed 1,000,000 steps in the NPT ensemble. The RMSD harmonic force constant was set to 50,000 kcal mol−1 Å2 biasing ~70 to 220,000 particles. In all transitions the bias was applied only to the E proteins. Initially, for the the entire simulation time. However when the coordinates were ~25–35 Å RMSD away from the target structure, significant clashes between domains III of the E proteins around the 3- and 5-fold symmetry vertices were observed. Hence we modified the transition, and instead enforced a sequential series of biases on the E proteins, for: i) all chains for RMSD from ~50 to 35 Å, ii) blue and green molecules for RMSD from ~35 to 25 Å, and finally iii) all chains for RMSD from ~25 Å until t he last frame of the simulation (Figure 6A). In order to assess the influence of pulling velocity on the transition pathway we also employed a twice longer biased simulation (of 2,000,000 steps) with the same sequential series of pulling. Very similar results were achieved in terms of number of pr molecules on red E proteins blocked by blue E proteins as well as number of clashes between Fab on red E proteins and Fab on blue E proteins (Figure S8). In addition, in order to assess the affinity of interactions in the Fab:pr:E complex, three additional independent simulations were performed. Each employed TMD harmonic biases to either red (5-fold), blue (3-fold) or green (2-fold) molecules alone. For the stage III to IV transition, all E proteins were biased throughout the course of the entire simulation (i.e. from an RMSD of ~20 Å at the beginning, reaching ~4 Å at the end, Figure S7E).
Evaluating pr-E dissociation and pr site occlusion
A pr molecule was assumed to have dissociated from its corresponding E protein if residues 1–79 of pr formed less than 70 contacts with the E protein. This threshold of 70 was estimated from the contact counts from the pr:E interactions for the stage I structure. A contact was defined as two backbone atoms approaching to within 10 Å of each other.
The ideal pr:E binding was mapped onto all E proteins to determine the optimal pr position if its binding site is not occluded. This process was done by superimposing (with the SVDSuperimposer function in BioPython) the pr:E complex onto the all E proteins throughout the simulations, using residues 64–73, 101–107, 115–118 and 243–247 (contact residues on the E protein). The pr site was then deemed to be occluded if the resultant optimal pr protein clashed with any neighboring E proteins. A clash was detected if at least five C-alpha atoms present in the modeled pr approached to within 6 Å of a C-alpha atom from a neighboring E protein.
Modeling Fab 1H10 clashes
From the stage I to II TMD simulations of E and prM proteins, we modeled the positions of 180 Fab 1H10 molecules by superimposing the pr:Fab 1H10 complex structure onto the 180 prM proteins throughout the simulation trajectory. As described earlier, this was performed using the SVDSuperimposer function in BioPython. Once the coordinates of the Fab 1H10 were calculated, we evaluated the number of pairwise Fab clashes present as the TMD simulation progressed. A clash between two Fab 1H10 molecules was detected as described before.
Sequence and electrostatic potential surface analyses.
The amino acid residues sequence alignment was done using MultAlin, multiple sequence alignment webserver (Corpet, 1988). The aligned amino acid sequences were forwarded to ESPript 3.0 (Robert and Gouet, 2014) to render the sequence similarities (Figures 3E and S2). The electrostatic potential surfaces were calculated using the PDB2PQR server and APBS software (Baker et al., 2001; Dolinsky et al., 2004; Jurrus et al., 2018).
Enzyme-linked immunosorbent binding assay (ELISA)
ELISA was used to compare the binding affinity between the IgG and the Fab of 1H10 to whole virus particles coated on a plate. Fifty µL of virus suspension in NTE were coated on ELISA plates and incubated overnight at 4°C. The plate was blocked with 300 µL of 5% skim milk at room temperature for 2 hr. To test for effect of pH upon binding affinity, the IgG or Fab was diluted in NTE buffers with 50 mM MES at various pH values to final concentration of 10 µg/mL. The final pH of the NTE-MES buffers was adjusted with HCl and measured with a pH meter. The virus-coated wells were then incubated with 50 µL of diluted IgG or Fab 1H10 for 1 hr at 37°C. Afte r washing, the plate was incubated with peroxidase-conjugated goat anti-human IgG (1:1,000, Invitrogen) or peroxidase-conjugated goat anti-mouse IgG (1:1,000, Sigma) for 1 hr at 37°C. This step was followed by further washes with NTE-T (NTE and 0.01% Tween20) and incubations with 50 µL of tetramethylbenzidine (Invitrogen). The reaction was stopped by the addition of 50 µL of 1 M HCl. The absorbance at 450 nm was read using a microplate reader (Tecan). The data was analyzed and plotted using Prism (GraphPad). The experiment was also repeated with different dilutions of IgG 1H10, namely 10 µg/mL, 5 µg/mL, and 2 µg/mL to determine if the concentration of IgG 1H10 would affect the binding to coated virus particle.
A modified ELISA experiment was carried out to determine whether exposure to different pH conditions would alter Fab-virus interaction after the Fab had been allowed to interact with the virus particles at pH 8.0. The experiment was conducted by diluting the Fab in NTE buffer at pH 8.0 and adding them to the virus-coated wells. After 1 hrincubation at 37°C, the plate was washed with NTE-T. NTE-MES buffers at different pH values was then added to the wells, following which, the plate was incubated for 30 min at 37°C. The plate was then washed with NTE-MES buf fers at different pH’s with 0.1% Tween20 added. After washing, the plate was incubated with peroxidase-conjugated goat anti-human IgG (1:1,000, Invitrogen), developed, read and analyzed as mentioned in the previous paragraph.
Expression and purification of recombinant prM:E protein
The cloned the prM:E recombinant protein DNA vector contains prM (residues 1 to 130), a linker containing a Tobacco Etch Virus (TEV) protease recognition site (GENLYFQG), and sE (residues 1 to 394) (dengue virus 2, strain 16681) (Li et al., 2008). The furin cleavage site of prM (87-RREKR-91) was also mutated to 87-STEKS-91. The vector was then co-transfected with 1 µg pCoHygro vector into Drosophila S2 cells (Invitrogen) by the standard calcium phosphate precipitation method. Transfected S2 cells were selected in the presence of 300 µg/mL Hygromycin-B for one month to generate stable cell line. Drosophila S2 stable cell lines were scaled up and cultured in serum-free medium until the cells have reached a density of 5×106 cells/mL. The and the medium harvested 4 to 6 days after induction. Culture supernatant was then 1288 clarified from cellular debris by centrifugation followed by filtration through 0.45 µm pore size filter. Immunoaffinity column containing AminoLink™ Plus Coupling Resin (Thermo Scientific) coupled to anti-prM mAb 2H2 were loaded with the clarified culture supernatant. The recombinant protein was eluted with 100 mM glycine-HCl buffer at pH 2.7 followed by addition of 1 M Tris at pH 9.0 to neutralize the eluate. Eluted protein was then dialyzed into 10 mM Tris and 150 mM NaCl at pH 7.5 using SnakeSkin ™ dialysis tubing with 3.5 kDa molecular weight cut-off. The protein was further purified using a Superdex75 (GE Healthcare) gel-filtration column. The purified protein was concentrated to 1–3 mg/mL using an Amicon® Ultra Centrifugal Fil ter with 10kDa molecular weight cut-off.
BioLayer Interferometry analysis (BLI)
BLI experiments and analysis were performed on Octet® RED96 system (forteBIO). After equilibration of the biosensor tips for 60 sec in PBS-T buffer (phosphate-buffered saline and 0.005% Tween20). HMAb 1H10 was captured on Anti-Human Fc Capture biosensor tip by dipping the tips in wells containing HMAb 1H10 diluted to a final concentration of 5 µg/mL in PBS-T buffer for 300 sec or until 1 nm of each biosensor tip was coated. The biosensor tips were then allowed to equilibrate in PBS-T for 120 sec before dipping them for 300 sec in wells containing serially-diluted recombinant prM:E in PBS-T (at pH 7.4) or PBS-T with 50 mM MES (at pH 6.0) or PBS-T with 50 mM MES (at pH 5.0). After association step with the prM:E, the biosensor tips were dipped in wells with buffers at respective pH for 300 sec to measure the dissociation rate of the prM:E from the IgG. The biosensor tips were then regenerated by 3 cycles of 5 sec each in 10 mM glycine at pH 2.5 and PBS-T at pH 7.4.
Hydrogen Deuterium Exchange – Mass Spectrometry (HD X-MS)
The samples for HDX-MS analysis were made by incubating the prM:E control and prM:E:Fab 1H10 complex first at pH 7.4 for complex formation and then subsequently adjusting the pH to pH 7.4, 6.0, and 5.0. The sample buffer for pH 7.4 was 10 mM Tris and 150 mM NaCl while the sample buffers for the pH 5.0 and 6.0 were prepared with 10 mM Tris, 150 mM NaCl, and 10 mM MES. The D2O labeling solution was made by dehydrating the buffers using Savant™ SpeedVac™ Concentrator and done by mixing 4 µL of protein solution (15 µM) with 36 µL of D2O labeling solution (final D2O content was 90% and final protein concentration was 1.5 µM in labeling solution). For negative control, the prM:E protein was mixed with H2O buffer. The samples were incubated for a predetermined set of times under each pH condition at 28°C with triplicates at each time point. After each specific incubation time, 10 µL of quenching buffer (7.5 M guanidine chloride and 2% TFA) was added so as to lower the pH to 2.5, quench the HDX reaction, and denature the protein. The quenched protein mixture was immediately injected into an online Poroszyme®Immobilised Pepsin Cartridge (ThermoScientific) that is directly connected to the Acquity Ultra Performance Liquid Chromatography system (Waters) to separate the peptides. The separated peptides were directly injected into the SYNAPT G2-Si Mass Spectrometry system (Waters). Each measurement was made in triplicate. The data obtained were then analyzed with DynamX™ HDX Analysis Software. In short, peptide identification was automatically performed by the software and incorrectly assigned peptide spectra were cleaned up manually. The deuterium uptake percentage at different pH was then calculated and compared with the average uptake percentage for samples in H2O buffer.
Since deuteration rate is highly dependent on pH, the incubation timings in D2O labeling solution for samples at pH 5.0 and 6.0 were extrapolated using the equations described previously (Coales et al., 2010; Li et al., 2014). Briefly, the intrinsic HDX rate is slowed down by 107.5-x-fold when the pH is lowered from pH 7.5 to pH x where x is 7.4, 6.0 or 5.0. (x = 7.0, 6.5, 6.0, 5.5, 5.0). Hence, for comparable deuteration of amide backbone at all pH conditions, we performed the labelling at same temperature of 28°C for all the different pH conditions. The labelling reactions were also done for different incubation timings according to the pH of the sample, such as:
| Labelling time at various pH | ||
|---|---|---|
| pH 7.4 | pH 6.0 | pH 5.0 |
| 30 sec | 15 min 48 sec | 2 hr 38 min |
| 1 min | 31 min 37 sec | 5 hr 16 min |
| 5 min | 2hr 38 min | 1 day 2 hr (or 26 hr) |
| 10 min | 5 hr 16 min | 2 day 4 hr (or 52 hr) |
QUANTIFICATION AND STATISTICAL ANALYSIS
Where appropriate, statistical details are given in the methods section and figure legends.
Supplementary Material
Highlights:
ImmDENV3 complexed with anti-prM antibody structures at neutral and acidic pH.
Two structural classes at acidic pH represent DENV maturation intermediates.
Structures show how the antibody helps overcome barrier for immature virus to fuse.
MD simulates maturation structural transitions and antibody assisted removal of pr.
ACKNOWLEDGEMENTS
This study has been supported by the following grants awarded to S-M.L.: NRF Investigatorship (NRF-NRFI2016–01), to J.E.C.: US NIH grant U54 AI057157 (the Region IV Southeast Regional Center of Excellence in Emerging Infections and Biodefense), and to S.A.S: US NIH grant K08 AI103038. S-M.L, P.J.B, C.S.V, and G.A thank MOE Tier 3 (MOE2012-T3-008) for support. The DENV2 and 3 strains were kindly provided by Eng-Eong Ooi (Duke-NUS Medical School). The HMAb 4.8A used in the THP-1 infection assay was kindly provided by John Schieffelin. We thank the NSCC and BMSI (A*STAR) for computing facilities. We thank Aravinda M. de Silva (The University of North Carolina at Chapel Hill) for helpful discussions and Jiaqi Wang, Xin-Ni Lim and Valerie S.Y. Chew (DUKE-NUS Medical School) for help with fusion assay experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
Structure coordinates and cryoEM density map accession numbers
The cryoEM maps of immDENV3:Fab 1H10 complex at pH 8.0 and class I and II immDENV3:Fab 1H10 at pH 5.0 have been deposited in Electron Microscopy Databank under accession codes EMD-9649, EMD-9650, and EMD-9651, respectively. The coordinates of immDENV3:Fab 1H10 complex at pH 8.0 and class I and II immDENV3:Fab 1H10 at pH 5.0 were deposited in Protein Data Bank under accession codes 6IDI, 6IDK, and 6IDL, respectively.
References
- Baker NA, Sept D, Joseph S, Holst MJ, and McCammon JA (2001). Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, et al. (2013). The global distribution and burden of dengue. Nature 496, 504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury S, Ripoll DR, and Wallqvist A (2015). Structure-based pKa prediction provides a thermodynamic basis for the role of histidines in pH-induced conformational transitions in dengue virus. Biochem Biophys Rep 4, 375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claus V, Jahraus A, Tjelle T, Berg T, Kirschke H, Faulstich H, and Griffiths G (1998). Lysosomal Enzyme Trafficking between Phagosomes, Endosomes, and Lysosomes in J774 Macrophages. ENRICHMENT OF CATHEPSIN H IN EARLY ENDOSOMES. Journal of Biological Chemistry 273, 9842–9851. [DOI] [PubMed] [Google Scholar]
- Coales SJ, E SY, Lee JE, Ma A, Morrow JA, and Hamuro Y (2010). Expansion of time window for mass spectrometric measurement of amide hydrogen/deuterium exchange reactions. Rapid Commun Mass Spectrom 24, 3585–3592. [DOI] [PubMed] [Google Scholar]
- Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, et al. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpet F (1988). Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 16, 10881–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejnirattisai W, Jumnainsong A, Onsirisakul N, Fitton P, Vasanawathana S, Limpitikul W, Puttikhunt C, Edwards C, Duangchinda T, Supasa S, et al. (2010). Cross-reacting antibodies enhance dengue virus infection in humans. Science 328, 745–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky TJ, Nielsen JE, McCammon JA, and Baker NA (2004). PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res 32, W665–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller SE, Zhang Y, Pastor RW, and Brooks BR (1995). Constant pressure molecular dynamics simulation: The Langevin piston method. The Journal of Chemical Physics 103, 4613–4621. [Google Scholar]
- Halstead SB (2007). Dengue. The Lancet 370, 1644–1652. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD: visual molecular dynamics. J Mol Graph 14, 33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- Junjhon J, Edwards TJ, Utaipat U, Bowman VD, Holdaway HA, Zhang W, Keelapang P, Puttikhunt C, Perera R, Chipman PR, et al. (2010). Influence of pr-M cleavage on the heterogeneity of extracellular dengue virus particles. J Virol 84, 8353–8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurrus E, Engel D, Star K, Monson K, Brandi J, Felberg LE, Brookes DH, Wilson L, Chen J, Liles K, et al. (2018). Improvements to the APBS biomolecular solvation software suite. Protein Sci 27, 112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyuchenko VA, Zhang Q, Tan JL, Ng TS, and Lok SM (2013). Immature and mature dengue serotype 1 virus structures provide insight into the maturation process. J Virol 87, 7700–7707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Rodnin MV, Ladokhin AS, and Gross ML (2014). Hydrogen-deuterium exchange and mass spectrometry reveal the pH-dependent conformational changes of diphtheria toxin T domain. Biochemistry 53, 6849–6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Lok SM, Yu IM, Zhang Y, Kuhn RJ, Chen J, and Rossmann MG (2008). The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science 319, 1830–1834. [DOI] [PubMed] [Google Scholar]
- Liao M, Sanchez-San Martin C, Zheng A, and Kielian M (2010). In vitro reconstitution reveals key intermediate states of trimer formation by the dengue virus membrane fusion protein. J Virol 84, 5730–5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link AJ, and LaBaer J (2011). Trichloroacetic acid (TCA) precipitation of proteins. Cold Spring Harb Protoc 2011, 993–994. [DOI] [PubMed] [Google Scholar]
- Liu X, Jiang W, Jakana J, and Chiu W (2007). Averaging tens to hundreds of icosahedral particle images to resolve protein secondary structure elements using a Multi-Path Simulated Annealing optimization algorithm. J Struct Biol 160, 11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok SM (2016). The Interplay of Dengue Virus Morphological Diversity and Human Antibodies. Trends Microbiol 24, 284–293. [DOI] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, and Chiu W (1999). EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128, 82–97. [DOI] [PubMed] [Google Scholar]
- Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, and de Vries AH (2007). The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B 111, 7812–7824. [DOI] [PubMed] [Google Scholar]
- Marzinek JK, Holdbrook DA, Huber RG, Verma C, and Bond PJ (2016). Pushing the Envelope: Dengue Viral Membrane Coaxed into Shape by Molecular Simulations. Structure 24, 1410–1420. [DOI] [PubMed] [Google Scholar]
- Mindell JA, and Grigorieff N (2003). Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 142, 334–347. [DOI] [PubMed] [Google Scholar]
- Modis Y, Ogata S, Clements D, and Harrison SC (2003). A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci U S A 100, 6986–6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modis Y, Ogata S, Clements D, and Harrison SC (2004). Structure of the dengue virus envelope protein after membrane fusion. Nature 427, 313–319. [DOI] [PubMed] [Google Scholar]
- Modis Y, Ogata S, Clements D, and Harrison SC (2005). Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J Virol 79, 1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monticelli L, Kandasamy SK, Periole X, Larson RG, Tieleman DP, and Marrink SJ (2008). The MARTINI Coarse-Grained Force Field: Extension to Proteins. J Chem Theory Comput 4, 819–834. [DOI] [PubMed] [Google Scholar]
- Murray JM, Aaskov JG, and Wright PJ (1993). Processing of the dengue virus type 2 proteins prM and C-prM. J Gen Virol 74 (Pt 2), 175–182. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005). Scalable molecular dynamics with NAMD. J Comput Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey FA, Heinz FX, Mandl C, Kunz C, and Harrison SC (1995). The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature 375, 291–298. [DOI] [PubMed] [Google Scholar]
- Richter MK, da Silva Voorham JM, Torres Pedraza S, Hoornweg TE, van de Pol DP, Rodenhuis-Zybert IA, Wilschut J, and Smit JM (2014). Immature dengue virus is infectious in human immature dendritic cells via interaction with the receptor molecule DC-SIGN. PLoS One 9, e98785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert X, and Gouet P (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42, W320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenhuis-Zybert IA, Moesker B, da Silva Voorham JM, van der Ende-Metselaar H, Diamond MS, Wilschut J, and Smit JM (2011). A fusion-loop antibody enhances the infectious properties of immature flavivirus particles. J Virol 85, 11800–11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenhuis-Zybert IA, van der Schaar HM, da Silva Voorham JM, van der Ende-Metselaar H, Lei HY, Wilschut J, and Smit JM (2010). Immature dengue virus: a veiled pathogen? PLoS Pathog 6, e1000718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-San Martin C, Sosa H, and Kielian M (2008). A stable prefusion intermediate of the alphavirus fusion protein reveals critical features of class II membrane fusion. Cell Host Microbe 4, 600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlitter J, Engels M, Krüger P, Jacoby E, and Wollmer A (1993). Targeted Molecular Dynamics Simulation of Conformational Change-Application to the T ↔ R Transition in Insulin. Molecular Simulation 10, 291–308. [Google Scholar]
- Schrodinger LLC (2015). The PyMOL Molecular Graphics System, Version 1.8. [Google Scholar]
- Smith SA, Nivarthi UK, de Alwis R, Kose N, Sapparapu G, Bombardi R, Kahle KM, Pfaff JM, Lieberman S, Doranz BJ, et al. (2015). Dengue Virus prM-Specific Human Monoclonal Antibodies with Virus Replication-Enhancing Properties Recognize a Single Immunodominant Antigenic Site. J Virol 90, 780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SA, Zhou Y, Olivarez NP, Broadwater AH, de Silva AM, and Crowe JE Jr. (2012). Persistence of circulating memory B cell clones with potential for dengue virus disease enhancement for decades following infection. J Virol 86, 2665–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, and Ludtke SJ (2007). EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157, 38–46. [DOI] [PubMed] [Google Scholar]
- Teuchert M, Schäfer W, Berghöfer S, Hoflack B, Klenk H-D, and Garten W (1999). Sorting of Furin at the Trans-Golgi Network. Journal of Biological Chemistry 274, 8199–8207. [DOI] [PubMed] [Google Scholar]
- Trabuco LG, Villa E, Schreiner E, Harrison CB, and Schulten K (2009). Molecular dynamics flexible fitting: a practical guide to combine cryo-electron microscopy and X-ray crystallography. Methods 49, 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Schaar HM, Rust MJ, Waarts BL, van der Ende-Metselaar H, Kuhn RJ, Wilschut J, Zhuang X, and Smit JM (2007). Characterization of the early events in dengue virus cell entry by biochemical assays and single-virus tracking. J Virol 81, 12019–12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Li L, Pennington JG, Sheng J, Yap ML, Plevka P, Meng G, Sun L, Jiang W, and Rossmann MG (2013). Obstruction of dengue virus maturation by Fab fragments of the 2H2 antibody. J Virol 87, 8909–8915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrilow D, Northill JA, Pyke A, and Smith GA (2002). Single rapid TaqMan fluorogenic probe based PCR assay that detects all four dengue serotypes. J Med Virol 66, 524–528. [DOI] [PubMed] [Google Scholar]
- Yu IM, Holdaway HA, Chipman PR, Kuhn RJ, Rossmann MG, and Chen J (2009). Association of the pr peptides with dengue virus at acidic pH blocks membrane fusion. J Virol 83, 12101–12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu IM, Zhang W, Holdaway HA, Li L, Kostyuchenko VA, Chipman PR, Kuhn RJ, Rossmann MG, and Chen J (2008). Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 319, 1834–1837. [DOI] [PubMed] [Google Scholar]
- Zaitseva E, Yang ST, Melikov K, Pourmal S, and Chernomordik LV (2010). Dengue virus ensures its fusion in late endosomes using compartment-specific lipids. PLoS Pathog 6, e1001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Hunke C, Yau YH, Seow V, Lee S, Tanner LB, Guan XL, Wenk MR, Fibriansah G, Chew PL, et al. (2012). The stem region of premembrane protein plays an important role in the virus surface protein rearrangement during dengue maturation. J Biol Chem 287, 40525–40534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Corver J, Chipman PR, Zhang W, Pletnev SV, Sedlak D, Baker TS, Strauss JH, Kuhn RJ, and Rossmann MG (2003). Structures of immature flavivirus particles. EMBO J 22, 2604–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng A, Umashankar M, and Kielian M (2010). In vitro and in vivo studies identify important features of dengue virus pr-E protein interactions. PLoS Pathog 6, e1001157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.