

Summary
Urban particulate matter air pollution induces the release of pro-inflammatory cytokines including interleukin-6 (IL-6) from alveolar macrophages, resulting in an increase in thrombosis. Here we report that metformin provides protection in this murine model. Treatment of mice with metformin or exposure of murine or human alveolar macrophages to metformin prevented the particulate matter-induced generation of complex III mitochondrial reactive oxygen species (ROS), which were necessary for the opening of calcium release-activated channels (CRAC) and release of IL-6. Targeted genetic deletion of electron transport or CRAC channels in alveolar macrophages in mice prevented the particulate matter induced acceleration of arterial thrombosis. These findings suggest metformin as a potential therapy to prevent some of the premature deaths attributable to air pollution exposure worldwide.
eTOC blurb
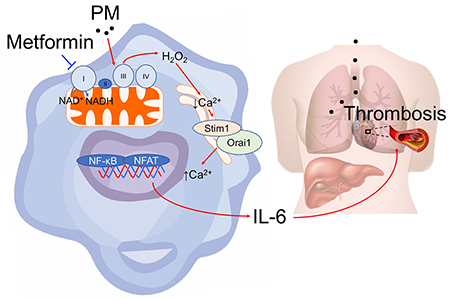
Air pollution exposure has been linked to a variety of poor health outcomes, including an increased risk of death attributable to ischemic cardiovascular events. Soberanes et al. find that metformin may hold promise as a therapy, as its capacity to act as a mitochondrial complex I inhibitor prevents accelerated thrombosis in a murine model of particulate matter inhalation.
Introduction
Exposure to particulate matter air pollution is a major public health concern. In the developed world, it is estimated that lifespan would be extended for 0.8 years for every 10 μg/m3 fall in the mean levels of particulate matter less than 2.5 microns in size, and exposure to PM was recently estimated to increase in all-cause mortality in the United States Medicare Population by 7% (Di et al., 2017; Pope et al., 2009). In the developing world, the large number of urban dwellers and the very high levels of PM suggest effects of particulate matter air pollution exposure on health are even more substantial (Europe, 2013). While air pollution exposure has been linked to a variety of poor health outcomes, the major driver of mortality is an increased risk of death attributable to ischemic cardiovascular events, primarily heart attacks and ischemic/thrombotic strokes (Pope et al., 2009).
Urban particulate matter air pollution consists of a core of ash or carbon decorated by organic molecules and metals that condense onto their surface during the combustion of fossil fuels (Nel, 2005). These latter features distinguish urban particulates from desert dust, volcanic ash and wood smoke. To cope with life in dusty or smoky environments, mammals have evolved efficient mechanisms to clear ambient particles. For example, alveolar macrophages and mucociliary clearance efficiently clear carbon-based nanomaterials from the lung to the larynx/feces with minimal or no inflammation (Duch et al., 2011; Semmler-Behnke et al., 2007). In contrast, we and others have shown that urban particulate matter air pollution induces the release of pro-inflammatory cytokines, including interleukin-6 (IL-6) from alveolar macrophages before the particles are cleared. Alveolar macrophage-produced IL-6 enters the circulation to induce the transcription of several coagulation factors in the liver, and augments the tendency toward arterial thrombosis in a murine model of stroke—findings lacking in mice deficient in IL-6 (Chiarella et al., 2014; Mutlu et al., 2007). Key findings of this model were recently confirmed in humans in an interventional trial of filtered compared with ambient air conducted in a region of China with high levels of ambient PM (Li et al., 2017). Accordingly, small molecules with acceptable risk profiles that can attenuate IL-6 release in response to PM in this model are predicted to lower the risk of arterial thrombosis in exposed populations.
In animal models, metformin, an FDA approved drug for type 2 diabetes mellitus, slows the growth of tumors, prolongs lifespan, and reduces the risk of ischemic cardiovascular events (Martin-Montalvo et al., 2013; Maruthur et al., 2016; Wheaton et al., 2014). As a result, metformin is one of a handful of small molecules that are being considered as potential therapies to delay the onset of aging phenotypes in healthy people (Longo et al., 2015). Disruption of mitochondrial signaling by inhibition of complex I of the mitochondrial electron transport chain is a mechanism by which metformin slows tumor growth (Liu et al., 2016; Wheaton et al., 2014). Because the release of IL-6 and other pro-inflammatory cytokines in response to PM may require the generation of mitochondrial ROS, we reasoned that metformin might reduce the risk of PM-induced thrombosis (Chiarella et al., 2014).
Results
Metformin prevents the increased susceptibility to arterial thrombosis after injury induced by exposure to PM.
We treated mice with a therapeutic dose of metformin in their drinking water (100 mg/kg/day) beginning 24 hours before exposure to concentrated ambient particulate matter (CAPs, PM2.5) air pollution via inhalation (8 hours per day on three consecutive days). Metformin reduced the PM-induced acceleration of the time to carotid occlusion after ferric chloride-induced injury (a model of ischemic stroke) to a level that was similar to those measured in mice exposed to filtered air (Fig. 1A and 1B). We have previously reported that the release of IL-6 from alveolar macrophages is necessary for this response (Mutlu et al., 2007). Accordingly, we measured the expression of the Il6 gene in alveolar macrophages, which was reduced in the metformin-treated animals (Fig. 1C). Metformin inhibited PM-induced IL-6 protein release in response to PM in alveolar macrophages ex vivo (Fig. 1D). Metformin also inhibited the PM-induced increase in IL-6 in bronchoalveolar lavage fluid, in alveolar macrophages obtained from mice 24 hours after PM was administered intratracheally, and in a murine alveolar macrophage cell line (MHS) (Fig. S1A-D).
Figure 1. Metformin prevents the release of IL-6 and enhanced tendency to thrombosis induced by exposure to particulate matter air pollution.

(A,B) Mice were administered metformin in the drinking water (150 mg/kg/day) then exposed to concentrated ambient particulate matter air pollution < 2.5 μm in diameter (CAPS) via inhalation in a versatile aerosol concentrator for eight hours daily on three consecutive weekdays. At the end of the third day, a standardized ferric chloride injury was induced in the carotid artery and the time to thrombosis was assessed using an ultrasonic probe placed on the artery distal to the injury (n=6, 5, 10 and 5 mice per condition, correspondingly, p<0.05 for comparison with filtered air controls). (C) Mice treated as in (A) were harvested for measurement of IL6 mRNA in alveolar macrophages (n=4 animals per condition, * p <0.05). (D) Mice were treated with a standardized PM from the US National Institute of Standards and Technology (NIST) 10 μg/animal, intratracheally, and 24 hours later the levels of IL-6 in the BAL fluid (ELISA) were measured (n=3 mice per condition, * p < 0.05 for indicated comparison). (E) Mice were treated with metformin in the drinking water for 24 hours and the levels of oxidized and reduced nicotinamide adenine dinucleotide (NAD+/NADH) were measured in BAL fluid macrophages by mass spectroscopy (n=5 animals per condition, * p<0.05). (F,G) Alveolar macrophages from BAL fluid were allowed to adhere overnight to glass coverslips, loaded with the mitochondrially localized oxidant sensitive dye MitoSOX (5 μM) and then exposed to PM containing perfusate (10 μg/ml) in the presence or absence of metformin (1 mM) on the stage of an epifluorescent microscope and oxidation of the dye was recorded from the same cellular region over time (n=3 mice per condition, * p<0.05). See also Figure S1.
Metformin inhibits electron transport and PM-induced mitochondrial ROS generation in alveolar macrophages.
We assessed the ability of systemically administered metformin to inhibit mitochondrial respiration in alveolar macrophages by measuring the ratio of oxidized-to-reduced nicotinamide adenine dinucleotide (NAD+/NADH) in alveolar macrophages immediately after their isolation. The ratio of NAD+/NADH was significantly reduced in primary alveolar macrophages from mice treated with metformin in their drinking water when measured by mass spectroscopy or a colorimetric test (Fig. 1E, and Fig. S1E). To determine the role of metformin on the generation of mitochondrially derived ROS, we continuously measured the fluorescence of an oxidant-sensitive mitochondrially localized dye (MitoSOX) in primary murine alveolar macrophages on the stage of an epifluorescent microscope. Oxidation of the dye was observed within 2 minutes of adding PM to the perfusate, and was reduced in cells pretreated with metformin before PM exposure (Fig. 1F,G).
Metformin prevents the generation of mitochondrial ROS to reduce PM-induced IL-6 release.
To determine whether metformin inhibits PM-induced mitochondrial ROS generation through its effects on complex I of the mitochondrial electron transport chain, we generated an alveolar macrophage cell line (MHS) stably transfected with lentiviral vectors encoding a yeast protein NDI1, which can transfer electrons from NADH to the ubiquinone pool without generating ROS and is insensitive to metformin (Fig. S2A,B) (Wheaton et al., 2014). In contrast, mammalian complex I generates ROS and is sensitive to metformin (Bridges et al., 2014). Compared with control-transfected cells, cells expressing NDI1 were resistant to metformin-induced inhibition of the basal oxygen consumption rate (OCR), PM-induced increases in mitochondrial ROS generation, and IL-6 production (Fig. 2A-C). A similar inhibition of PM-induced ROS generation and IL-6 release was observed in MHS cells treated with the complex I inhibitor piercidin A (Figure S2C,D). Unlike mammalian complex I, NDI1 is not capable of ROS generation, therefore, the ability to restore PM-induced mitochondrial ROS generation in NDI1 transfected cells suggested the generation of ROS downstream of complex I, most likely at complex III. Accordingly, we treated cells with a suppressor of superoxide production from mitochondrial complex III (S3QEL), which was identified through a chemical screen, and showed this prevented PM-induced ROS generation and IL-6 release (Fig. 2D,E) (Orr et al., 2015). In contrast, S1QEL, a suppressor of superoxide production from mitochondrial complex I identified in a similar screen, failed to prevent PM-induced ROS generation or IL-6 release (Fig. 2D,E) (Brand et al., 2016). The less selective mitochondrial antioxidant Mito-TEMPO and the untargeted combined superoxide dismutase/catalase mimetic EUK-134 also prevented PM-induced mitochondrial ROS and IL-6 generation (Fig. S2F,G). Together, these findings suggest that metformin inhibits mitochondrial ROS generation at complex III by reducing electron flow from the upstream complex I.
Figure 2. Metformin inhibits mitochondrial electron transport complex I to limit PM-induced ROS generation from complex III.

(A-C) A murine alveolar macrophage cell line (MHS) was stably transfected with a lentivirus encoding GFP and NDI1, a yeast protein capable of transferring electrons from NADH to complex II/III but incapable of ROS generation, or GFP alone. These cells were exposed to PM (10 μg/m3) in the presence or absence of metformin and oxygen consumption (Seahorse XF Analyzer) and the oxidation of MitoSOX were measured 4 hours later, and the release of IL-6 into the media was measured 24 hours later (minimum of 8 replicates per measurement, * p <0.05). (D,E) MHS cells were treated with a suppressor of superoxide production from complex III (S3QEL) or complex I (S1QEL) (both at 5 μM) and mitochondrial ROS generation was measured immediately after PM exposure as in (F) and IL-6 release was measured 4 hours later (n=3, * p <0.05). (F,G) MHS cells were treated with a selective activator of AMPK A-79662 (2 μM) or vehicle and phosphorylation of AMPK and PM-induced IL-6 release were measured. (H,I) MHS cells were treated with a dose of Antimycin A (10 μM) or mitochondrially targeted paraquat (5 μM) selected to restore mitochondrial ROS to levels similar to PM alone, or in the presence of low dose PM, for measurement of MitoSOX oxidation (as in (F)) and IL-6 release after 4 hours (n=3–6 per condition, * p <0.01). See also Figure S2.
Many of the biologic activities of metformin have been attributed to its ability to active AMPK independently or downstream of mitochondrial complex I inhibition (Ma et al., 2017). To determine if the inhibition of IL-6 generation by metformin was attributable to its ability to activate AMPK, we treated MHS cells with the AMPK activator A-769662. While both metformin and A-769662 activated AMPK, the AMPK activator had no effect on PM-induced IL-6 release from cells (Fig. 2F,G). Similarly, when mice were treated with A-769662 at a dose shown to mimic the effects of metformin in a model of high fat induced atherosclerosis, we observed no change in the PM-induced increase in IL-6 in BAL fluid (Fig S2H) (Ma et al., 2017). To determine whether mitochondrial ROS generation alone was sufficient to induce IL-6 release from alveolar macrophages, we treated MHS cells with Antimycin A, which blocks electron transport and increases ROS generation at complex III, or a modified version of paraquat that is targeted to the mitochondria (mito-paraquat), both titrated to induce similar oxidation of MitoSOX when compared to PM. (Fig. 2H, Fig. S2I,J). . Neither agent alone induced IL-6 release; however, the addition of either Antimycin A or mito-paraquat to a low dose of PM augmented IL-6 generation (Fig. 2I). These results suggest that PM enhances the generation of mitochondrial ROS at complex III, which are necessary but not sufficient for PM-induced IL-6 release.
Metformin inhibits PM-induced opening of Calcium Release Activated Ca2+ Channels
The rapidity of the oxidant response to PM prompted us to examine changes in intracellular calcium in response to PM. When primary alveolar macrophages were perfused with calcium free media and treated with PM, we observed a small increase in cytosolic calcium suggestive of endoplasmic reticulum calcium store depletion. Subsequent perfusion with calcium replete media was associated with a substantial increase in cytosolic calcium. Both of these responses were inhibited by metformin (Fig. 3A). The increase in cytosolic calcium suggested that PM might promote opening of calcium release activated Ca2+ channels (CRAC). Indeed, the PM-induced increase in cytosolic calcium was inhibited by the CRAC channel inhibitors, Synta-66 and 2- 2-Aminoethoxydiphenyl borate (2-ABP) as well as the calcium chelator, 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) (Fig. 3B). Treatment of primary murine alveolar macrophages with Synta-66 also attenuated PM-induced release of IL-6 (Fig 3C). Metformin did not inhibit store depletion or CRAC channel activation in response to thapsigargin (Fig S3A). Consistent with these findings, MHS cells stably transfected with lentiviruses encoding shRNAs targeting Stim1 or Orai1, both components of the CRAC channel, showed reductions in PM-induced increases in intracellular calcium in calcium replete media, and reductions in PM-induced IL-6 release, compared with control transfected cells (Fig. 3D-F and Fig. S3C,D). We have previously shown that PM-induced IL-6 release requires NF-κB activation and is augmented by cAMP response element binding protein (CREB) (Chiarella et al., 2014). In other systems, CRAC channel activation enhances inflammatory gene expression through the activation of Nuclear factor of activated T-cells (NFAT) (Jairaman et al., 2016; Jairaman et al., 2015). Consistent with this mechanism, pretreatment of MHS cells with the calcineurin inhibitor cyclosporine A attenuated PM-induced IL-6 release without affecting PM-induced ROS generation or calcium release (Fig. S3E-G).
Figure 3. Store operated calcium channels contribute to PM-induced IL-6 release.

(A,B). Primary alveolar macrophages isolated from wild-type mice were allowed to adhere to glass coverslips before loading with the calcium sensitive dye Fura-2 (2 μM) and treated with PM in calcium free followed by calcium replete (2 mM) media after 1 hour pretreatment with metformin (1 mM), Synta-66 (10 μM), 2-aminoethoxydiphenyl borate (2-ABP, 10 μM) or 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA, 10 μM) and the change in intracellular calcium concentration in response to extracellular calcium was recorded (n=3–4 mice per condition, * p < 0.05 for comparison with PM treated cells). (C) Primary alveolar macrophages from wild-type mice were pretreated for 1 hour with the CRAC channel inhibitor Synya-66 (10 μM) and then treated with PM (10 μg/cm2) and the level of IL-6 in the media was measured 24 hours later (n=3, * p<0.05). (D-F) MHS cells were stably transfected with lentiviruses encoding shRNA against Stim1, Orai1 or a scrambled shRNA and treated with vehicle or PM (10 μg/cm2) and (D,E) the change in intracellular calcium was measured as in (A), and (F) the level of IL-6 in the media was measured 24 hours after PM treatment. See also Figure S3.
Exposure to particulate matter air pollution induces the opening of CRAC channels downstream of mitochondrial ROS generation.
Treatment of MHS cells with Synta-66 before PM administration or knockdown of Orai1 did not affect PM-induced MitoSOX oxidation, suggesting the effects of PM on mitochondrial ROS generation are independent of CRAC channel opening (Fig 4A). Metformin inhibited the PM-induced increase in cytosolic calcium in MHS cells transfected with a control vector, but not in MHS cells that stably expressed NDI1 (Fig 4B,C). These results suggest that metformin prevents CRAC channel opening in response to PM via its ability to inhibit mitochondrial ROS generation. Indeed, administration of S3QEL or the complex I inhibitor piercidin A prevented the PM-induced increases in cytosolic calcium (Fig 4D,E and Fig. S4A), further suggesting calcium signaling occurred downstream of mitochondrial ROS generation.
Figure 4. PM-induced CRAC channel activation occurs downstream of mitochondrial ROS generation.

(A) MHS cells were stably transfected with lentiviruses encoding shRNA against Stim1 and Orai1 or a scrambled shRNA and treated with vehicle or PM (10 μg/cm2) and the oxidation of MitoSOX dye was measured (B,C) MHS cells transfected with GFP or GFP-NDI1 were treated with PM with or without metformin (1 mM) and changes in intracellular calcium upon the addition of calcium replete media were measured. (D-F) MHS cells were treated with S3QEL (5 μM) 1 hour before treatment with PM (10 μg/cm3) and immediate changes in intracellular calcium and IL-6 release into the media after 24 hours were measured. (K-N) MHS cells were pretreated for 1 hour with the PLC inhibitor U73122 or the inactive control compound U73343 (both 200 nM) and PM induced changes in cytosolic calcium, IL-6 release (24 hours), and MitoSOX oxidation were measured. For all calcium measures, data from a representative experiment, where each line represents an individual cell region, and summary data from 3 independent replicates are shown. Thapsigargin (TG) (25 nM) was added to the cells at the end of each experiment (* p<0.05). See also Figure S4 and S5.
Calcium store depletion induced by mitochondrially generated ROS might result from the activation of phospholipase-C (PLC) in the membrane, the inhibition of sarco/endoplasmic reticulum Ca2+-ATPases or an increase in the leak of calcium from the endoplasmic reticulum to the cytosol. Accordingly, we treated MHS cells with the PLC inhibitor U73122 and found it prevented PM-induced store depletion, CRAC channel activation and IL-6 release while the inactive control compound had no effect (Fig. 4F-H) (Hofmann et al., 1999). These effects occurred downstream of mitochondrial ROS generation as PM-induced mitochondrial ROS generation was unaffected by U73122 (Fig. 4I). To determine whether mitochondrial ROS generation was sufficient to induce store depletion and activate CRAC channels, we treated cells with antimycin A or mito-paraquat, which alone did not activate CRAC channels (Fig. S4B,C).
Genetic deletion of mitochondrial electron transport prevents the increase in IL-6 in response to PM.
The intratracheal administration of PM did not induce the recruitment of inflammatory cells to the lung, suggesting tissue-resident alveolar macrophages are responsible for the acute effects of PM in vivo (Fig. S5A-B). This allowed us to determine the importance of mitochondrial ROS generation in the response to PM in vivo by generating mice lacking the nuclear-encoded mitochondrial transcription factor A (TFAM) in macrophages (CreCD11c/Tfamflox/flox). TFAM is anuclear encoded gene that is required for the transcription of genes encoded by the mitochondrial DNA, including 13 genes that encode necessary components of most complexes within the electron transport chain (Fisher et al., 1992). Flow sorting of myeloid cell populations of 8-12 week old CreCD11c/Tfamflox/flox and Tfamflox/flox mice showed similar numbers of alveolar macrophages and other myeloid cell populations in the lung and no obvious morphologic changes (Fig. S5C). Flow-sorted alveolar macrophages from CreCD11c/Tfamflox/flox exhibited reduced levels of Tfam mRNA while these levels were normal in neutrophils (Fig. 5A). When we treated CreCD11c/Tfamflox/flox mice with PM intratracheally, the acceleration of carotid thrombosis in response to PM was attenuated, and BAL fluid levels of IL-6 were reduced compared with Tfamflox/flox controls (Fig. 5B). To confirm our in vitro findings suggesting mitochondrial ROS were required for the generation of IL-6 in response to PM, we treated mice with Mito-TEMPO subcutaneously beginning one day before PM exposure. Treatment with Mito-TEMPO also prevented PM-induced thrombosis (Fig 5C). Consistent with these findings, IL-6 levels were reduced in primary alveolar macrophages isolated by BAL from CreCD11c/Tfamflox/flox mice when compared with those from Tfamflox/flox mice (Fig. 5D). Similarly, alveolar macrophages from CreCD11c/Tfamflox/flox mice failed to generate ROS or increase intracellular calcium in response to PM (Fig. 5E-H).
Figure 5. Mitochondrial electron transport is necessary for the PM-induced acceleration of carotid thrombosis.

(A-F) Mice deficient in Tfam, a nuclear encoded transcription factor necessary for mitochondrial DNA transcription, in alveolar macrophages (CreCD11c/Tfamflox/flox) were compared with control mice. (A) Levels of Tfam mRNA in flow sorted AMs and neutrophils. (B) Mice were treated with PBS or PM (10 μg, intratracheally) and 24 hours later, the time to cessation of carotid artery blood flow after a standardized ferric chloride injury was measured. (C) Wild-type C57Bl/6 mice were treated with the mitochondrially targeted antioxidant MitoTempo (0.7 mg/kg/day, intraperitoneally) 24 hours before and simultaneous with the administration of PM (10 μg/mouse) or PBS and the time to cessation of carotid artery blood flow after standardized ferric chloride injury was measured. (D) Primary alveolar macrophages from the indicated strains of mice were treated with PM and IL-6 levels in the media were measured 24 hours later (n=3, * P<0.05). (E,F) Primary alveolar macrophages from the indicated strains were loaded with MitoSOX and mitochondrial ROS generation was measured continuously after the administration of PM on the stage of an epifluorescent microscope (n= 4, * P <0.05). (G,H) Primary alveolar macrophages were loaded with Fura-2 for measurement of intracellular calcium levels after PM exposure in calcium free followed by calcium replete media. Alveolar macrophages isolated from 3 mice per group (* p<0.05). See also Figure S5.
Genetic deletion of Orai1 in alveolar macrophages prevents the increase in IL-6 in response to PM.
We used a similar strategy to examine the importance of CRAC channel activation in the response to PM in vivo. Orai1 was efficiently deleted in alveolar macrophages from CD11cCre/Orai1flox/flox animals compared with Orai1flox/flox controls (Fig. 6A). The acceleration of carotid thrombosis induced by the intratracheal administration of PM in Orai1flox/flox controls was absent in CD11cCre/Orai1flox/flox mice (Fig. 6B). Primary alveolar macrophages from CreCD11c/Orai1flox/flox mice showed reduced levels of IL-6 production and intracellular calcium release in response to PM compared with those from Orai1flox/flox mice (Fig. 6C-E). Consistent with these findings, the intratracheal administration of Synta-66 to mice simultaneously with the instillation of PM was associated with reduced levels of IL-6 in the BAL fluid 4 hours later (Fig. 6F). These findings are of interest as the loss of CRAC channels was previously reported to induce no phenotypic changes in alveolar macrophages (Gautier et al., 2012; Vaeth et al., 2015).
Figure 6. CRAC channel activation is necessary for PM-induced acceleration of carotid thrombosis.

(A,B) Mice deficient in Orai1, a necessary component of CRAC channels, in alveolar macrophages (CreCD11c/Orai1flox/flox) were compared with control mice. (A) Levels of Orai1 mRNA in flow sorted AMs and neutrophils. (B) Mice were treated with PBS or PM (10 μg, it) and 24 hours later, the time to cessation of carotid artery blood flow after a standardized ferric chloride injury was measured in Orai1flox/flox mice and CreCD11c/Orai1flox/flox mice. (C) Primary alveolar macrophages were treated with PM and the levels of IL-6 were measured 24 hours later (n=3, * P<0.05). (D,E) Primary alveolar macrophages were loaded with Fura-2 for measurement of intracellular calcium levels after PM exposure in calcium free followed by calcium replete media (n=3, * P<0.05). (F) Wild-type mice were treated with Synta-66 (10 μM) intratracheally or vehicle simultaneous with the intratracheal instillation of PM (10 μg/mouse) and the levels of IL-6 in the BAL were measured 6 hours later (n=3, * P<0.05).
Metformin prevents PM induced ROS generation, CRAC channel activation and IL-6 production in human macrophages.
Our data suggest that PM induces the generation of mitochondrial ROS, which augment IL-6 release by activating CRAC channels. To confirm the potential importance of these findings in humans, we measured mitochondrial ROS generation and calcium levels in primary human alveolar macrophages obtained from healthy donors after treatment with metformin and or Synta-66. Consistent with our findings in murine alveolar macrophages, metformin inhibited mitochondrial ROS generation, calcium release and IL-6 release after PM exposure (Fig. 7A-D). As our results suggested that metformin acts to limit mitochondrial ROS and calcium-mediated activation of IL-6, we reasoned it would likely affect other processes in alveolar macrophages triggered by exposure to PM. Therefore, mice were treated in the drinking water with metformin for 24 hours before we instilled PM intratracheally. Alveolar macrophages were flow-sorted from whole lung homogenates 24 hours later for transcriptomic analysis (RNA-Seq) and metabolomic analysis. Metformin treatment resulted in significant changes in the measured metabolites in alveolar macrophages including reductions in glycolytic and TCA intermediates, as well as high energy phosphates, with little change in amino acids (Fig. S6). While the overall change in metabolites was highly significant (P <0.001 by ANOVA), individual metabolites did not reach significance after corrections for multiple comparisons (FDR p<0.05) (Table S1). Metformin treatment alone had a relatively minor effect on gene expression in alveolar macrophages in the steady state (476 differentially expressed genes, FDR p<0.05), but the response to PM exposure differed substantially when compared with untreated animals (1313 differentially expressed genes, FDR p<0.05) (Fig. 7E,F, Table S1). To better understand the changes induced by PM and metformin, we performed k-means clustering of the 1285 differentially expressed genes identified by an ANOVA-like test, FDR p<0.001 (Fig. 7G, Supplementary Table S1). Genes down-regulated after PM exposure (Cluster 1) include those related to signal transduction and cellular developmental process. Consistent with our findings, the genes up-regulated in response to PM and relatively downregulated by metformin (Cluster 2) included ROS biosynthetic process, neutrophil chemotaxis, and the acute phase response. Genes that were down-regulated upon administration of PM and metformin (Cluster 4) were associated with platelet aggregation and cell division (spindle assembly) (Griss et al., 2015).
Figure 7. Metformin prevents PM-induced IL-6 release from primary human alveolar macrophages and modifies transcriptional response to PM in murine alveolar macrophages.

(A) Flow sorted alveolar macrophages were isolated from biopsies from donor lungs obtained at the time of lung transplantation and allowed to adhere to glass coverslips for 4-8 hours. The cells were loaded with MitoSOX (5 μM) and then pretreated with saline, metformin (1 mM) or Synta-66 (10 μM) prior to treatment with PM (10 μg/cm2) for measurement of mitochondrial ROS generation. (B,C) Alveolar macrophages were allowed to adhere to glass coverslips prior to loading with Fura-2 (2 μM) and then treated with metformin (1 mM) or saline 1 hour before treatment with PM and intracellular calcium levels were measured in calcium free followed by calcium replete (1 mM) media. Thapsigargin (TG, 25 nM) was added at the end of the experiment. (B) Representative time-series plots and (C) composite data from four individuals is shown. (D) Primary human alveolar macrophages were treated with metformin (1 mM) 1 hour before treatment with PM (10 μg/cm2) and IL-6 levels in the media were measured 4 hours later. (E-G) Mice were treated with metformin in the drinking water for 24 hours before and after instilling PM (10 μg /mouse) intratracheally and alveolar macrophages flow-sorted from whole lung homogenates 24 hours later were subjected to transcriptional profiling via RNA-seq. (E) Schematic of the experimental design. Numbers indicate differentially expressed genes (FDR p < 0.05). (F) Volcano plots demonstrating up- and down-regulated genes in mice treated with metformin before and after exposure to PM, numbers indicate up- and down-regulated genes (FDR p < 0.05). (G) k-means clustering identifies metformin-specific clusters of genes. Differentially expressed genes (1285 genes, identified using ANOVA-like test implemented in edgeR package, FDR p < 0.001) in alveolar macrophages exposed to PM in the presence and absence of metformin pretreatment in vivo were subjected to k-means clustering (number of genes per cluster is shown on the left). Representative gene names and GO processes for each cluster are shown on the right side. (H,I) Chaperones are enriched in cluster 3. Gene Set Enrichment Analysis shows enrichment for chaperone genes after treatment with metformin that is more significant in alveolar macrophages from PM exposed mice. (J) MHS cells stably transfected with a lentivirus encoding NDI1 or a control lentivirus (GFP) were treated with PM and the levels of mRNA encoding the indicated genes were measured 24 hours later using RT-qPCR (n=3, * P<0.05). See also Figure S6.
Surprisingly, many of the genes that were upregulated in response to PM and metformin treatment compared with PM treatment alone (Cluster 3) were related to the unfolded protein response, negative regulation of intracellular signal transduction, and regulation of the response to stress. These findings suggest metformin induces a stress response that could indirectly suppress inflammatory signaling. To explore this hypothesis further, we examined the expression of a curated list of chaperone proteins in our data set, which showed that many genes identified as human chaperones were upregulated in alveolar macrophages from metformin and PM treated animals (Figure 7H). We confirmed upregulation of chaperone genes using Gene Set Enrichment analysis (enrichment score 0.32, FDR p = 0.017, Figure 4I)) (Subramanian et al., 2005) (Brehme et al., 2014). Using MHS cells transfected with NDI1, we showed that the increase in selected chaperones in metformin- and PM-treated cells was dependent on the ability of metformin to inhibit complex I (Fig. 7J.). These data suggest the intriguing hypothesis that complex I inhibition by metformin may also attenuate inflammatory signaling pathways indirectly by triggering global cellular stress response pathways that protect against macromolecular damage.
Discussion
We found that the commonly used drug metformin can attenuate PM-induced IL-6 release from alveolar macrophages and reduce the resulting increase in the risk of arterial thrombosis after injury. Metformin acted as a complex I inhibitor in alveolar macrophages to reduce mitochondrial ROS from complex III of the mitochondrial electron transport chain in response to PM. In alveolar macrophages from mice and humans, PM-induced mitochondrial ROS generation caused endoplasmic reticulum calcium store depletion, and the opening of store operated calcium channels, which augmented IL-6 release. Mitochondrial ROS alone were insufficient to induce store depletion or IL-6 release but acted in concert with particles to augment these responses. Our results provide genetic evidence in vivo to support the importance of signaling by mitochondrial ROS and CRAC channels in the release of IL-6 from alveolar macrophages, and in the accelerated thrombosis after carotid artery injury induced by particulate matter air pollution exposure.
In a murine alveolar macrophage like cell line, we could restore PM-induced signaling events by overexpressing the yeast protein NDI1, which can transfer electrons from NADH to the ubiquinone pool, but cannot generate ROS and is insensitive to metformin (Seo et al., 1998). Using small molecules that suppress superoxide production specifically at Complex III or Complex I, we show that NDI1 restores electron flux to complex III to restore mitochondrial ROS generation. These data are consistent with previous studies showing that metformin inhibits the generation of ROS induced by reverse electron transport (Batandier et al., 2006; Bridges et al., 2014). An interesting aspect of our study is the relationship between mitochondrial ROS and calcium release. Our findings using inhibitors implicate the activation of PLC in the store depletion induced by mitochondrial ROS upon PM exposure, but additional studies are required to better understand these pathways.
Inhibition of electron transport by metformin activates AMPK, which is proposed as a mechanism for its antidiabetic effects. Our finding that mitochondrially targeted antioxidants mimicked the effects of metformin in vitro and in vivo argue against a direct role for electron transport inhibition or AMPK activation in this protection. Furthermore, when we treated primary alveolar macrophages or mice with a direct activator of AMPK, PM-induced IL-6 release was unaffected. Unbiased transcriptional profiling of alveolar macrophages from metformin treated animals using RNA-Seq showed only small differences between metformin treated and untreated animals, however, there were marked differences in their response to PM. Specifically, alveolar macrophages from metformin treated animals showed a significant upregulation of chaperone genes involved in proteostasis in response to PM when compared with untreated control animals. Using an alveolar macrophage cell line, we showed this chaperone response could be attributed to metformin’s ability to inhibit complex I. It is possible that this response is further attributable to the activation of AMPK, although others have reported that AMPK activation results in downregulation of proteostasis genes (Dai et al., 2015).
Our findings may have implications for humans. For example, in a recent study of people residing in an area of China with high levels of PM exposure, treatment with a respiratory filter to reduce PM exposure resulted in decreased levels of oxidative stress, C-reactive protein and fibrinogen (both transcriptional targets of IL-6), and catecholamines, thereby validating key components of the mouse model we used in this study (Li et al., 2017). Our finding that metformin prevented PM-induced IL-6 release from human alveolar macrophages supports a similar trial with metformin in high risk individuals, particularly given its low cost and safety. In addition, the finding that metformin reduces PM-induced IL-6 release may partially explain the association between metformin use and reduced levels of CRP and cardiovascular risk observed in patients with Type II diabetes (Maruthur et al., 2016). Consistent with this hypothesis, IL-6 and C-reactive protein (CRP) have both been identified as independent risk factors for the development of ischemic cardiovascular disease, and the administration of an inhibitor of IL-1β reduced IL-6 levels, CRP and cardiovascular risk in a recent randomized clinical trial (Ridker, 2016; Ridker et al., 2017). Furthermore, the unexpected finding that metformin activated proteostasis genes and inhibited inflammation might explain some of the protection against age-related phenotypes in mice administered metformin and provide an additional rationale for proposed studies to administer metformin to humans to prevent the accumulation of age-related phenotypes (Barzilai et al., 2016).
Limitations of Study.
First, the dose of metformin we used in mice achieves plasma/tissue concentrations similar to those measured in humans on standard treatment doses for Type II diabetes (Chandel et al., 2016), but it is not known if metformin is effectively taken up by human alveolar macrophages in vivo. Second, we used urban particulate matter obtained from the air around our laboratories in Chicago, and from the National Institute of Standards and Technology from air in Washington, D.C., both of which have been characterized (Mutlu et al., 2018; Poster et al., 1999). As the epidemiologic link between PM exposure and CV disease is not dependent on geography, our results are likely to be broadly applicable (Lelieveld et al., 2015). Nevertheless, it is possible PM from some regions may act via additional mechanisms. Third, the Cre driver we used to delete Tfam and Orai1 in alveolar macrophages also targets dendritic cells in the lung. This is perhaps less important for Tfam, which only affects mitochondrial biogenesis, as tissue-resident alveolar macrophages persist for months without input from the bone marrow, while both CD11b+ and CD103+ dendritic cells turn over relatively rapidly (1-2 weeks) in the lung (Misharin et al., 2014; Misharin et al., 2017). Fourth, the loss of TFAM in macrophages will impair ATP production, depolarize the mitochondrial membrane and reduce the supply of metabolic intermediates, among other effects. Therefore, our results using these animals should be interpreted in the context of our in vitro and in vivo studies. Fifth, our data using the inhibitor U73122 implicate activation of PLC by mitochondrial ROS when PM are present in the depletion of calcium from ER stores and activation of CRAC channels. The precise molecular targets of mitochondrial ROS and PM in this pathway, however, are not known. Finally, while we show the administration of PM results in rapid (<2 minutes) generation of mitochondrial ROS, dissecting the precise molecular mechanisms by which this occurs will require further study.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: Scott Budinger (s-buding@northwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
All studies using samples obtained from human subjects were approved by the Northwestern University Institutional Review Board. All human subjects provided written informed consent prior to enrolment into the study. The following inclusion criteria were applied: donor lung is suitable for transplant, recipient provided written informed consent, sufficient amount of tissue was provided for isolation of alveolar macrophages. Information about donor’s sex, heath status, previous exposure to metformin or other drugs is not available.
Mouse models.
All animal experiments and procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee at Northwestern University. C57BL/6J mice were bred in our facility and our colonies are refreshed yearly with mice purchased from the Jackson Laboratory. CreCD11c mice were purchased from the Jackson Laboratory and bred in house. Tfamfl/fl mice were generated by Ozgene as we have previously described (Hamanaka et al., 2013). Orai1fl/fl mice were provided by Amgen and generated as described (Somasundaram et al., 2014). All experiments were performed with littermate controls. Number of animals per group was determined based on our previous publications. Ten-to sixteen-week-old male mice were used for experiments. Investigators were not blinded to the group allocation. Mice were housed at the Center for Comparative Medicine at Northwestern University, in microisolator cages, with standard 12 hour light/darkness cycle, ambient temperature 23 °C and were provided standard rodent diet (Envigo/Teklad LM-485) and water ad libitum.
Murine model of PM exposure.
Inhalational exposure to PM2.5 CAPs was performed as previously described (Chiarella et al., 2014). Briefly, mice were housed 8 hours per day for 3 consecutive days in a chamber connected to a Versatile Aerosol Concentration and Exposure System (VACES). We exposed control mice to filtered air in an identical chamber connected to the VACES in which a Teflon filter was placed on the inlet valve to remove all particles. We estimated ambient PM2.5 concentrations as the mean of reported values from the 4 EPA monitoring locations closest to our location. The mean concentration in the PM exposure chamber was 118.3 ± 5.21 μg/m3. For intratracheal exposure experiments in mice, we used an urban PM collected from ambient air in Washington, DC (National Institute of Standards and Technology standard reference material, SRM 1649a). We instilled either PM suspended in 50 μl of sterile PBS (vortexed prior to instillation) or PBS (control).
Murine model of arterial thrombosis induced by FeCl3 injury to the carotid artery.
This technique has been previously described in detail (Chiarella et al., 2014). Briefly, mice were anesthetized and the left carotid artery was dissected and isolated from the surrounding tissue with paraffin; the adventitia of the artery was treated with Whatman filter paper of a standard size (generated with a mouse ear punch device) soaked in freshly prepared 10% FeCl3. The carotid blood flow was continuously measured using Transonic TS420 Transit-Time Perivascular Flowmeter (Transonic Systems). The application of FeCl3 led to a 2- to 3 mm-long carotid thrombus.
Murine alveolar macrophages and cell lines.
The murine alveolar macrophage cell line (MH-S, ATCC CRL-2019, originating from male BALB/c mouse) was cultured in RPMI medium in 10% FBS supplemented with 10 μM β-mercaptoethanol at 37 °C and 5% CO2. Cell culture and generation of MHS cells with stable knockdown of Orai1, Stim1 or a scrambled shRNA control were performed using the Sigma mission lentiviral packaging mix (Sigma-Aldrich) with the following catalog numbers: ORAI1 (TRCN0000125405), STIM1 (TRCN0000193400), non-target control (SHC312). Transformed cells were cultured with 10 μg/ml puromycin for 2 passages and reduction in the expression of the target gene was assessed using western blot or RT-PCR.
Mouse primary alveolar macrophages were isolated by bronchoalveolar lavage performed in euthanized mice with 3 ml of PBS with 1 mM EDTA. Only male mice were used as a source of alveolar macrophages. The lavage was centrifuged at 300 g for 10 minutes and resuspended in RPMI supplemented with 10% FBS and plated in a density of 100,000 cells/cm2. Cell purity was analyzed by flow cytometry and was confirmed to be >95%.
METHOD DETAILS
IL-6 measurements.
The production of IL-6 in broncheoalveolar lavage fluid on the lung of mice exposed to particulate matter, and In vitro IL-6 production in human alveolar macrophages was determined by ELISA using a kit (ThermoFisher cat# KHC0061 and KMC0061) as previously described (Mutlu et al., 2007)
Measurement of the NAD+/NADH ratio and metabolomics in primary alveolar macrophages.
The NAD+/NADH ratio in alveolar macrophages was measured using two methods. For both methods, mice were treated with metformin in their drinking water for three days and BAL macrophages were harvested by gently BAL through a surgically placed tracheostomy tube (6 sequential lavages) in PBS containing (0.5 mM EDTA). The cell pellet was washed twice in PBS and cells were counted (Cellometer K2, Nexcelon Bioscience). In the first method, 100,000 cells were used for measurement of the NAD+/NADH using an assay kit from Abcam (ab65348) according to the manufacturer’s instructions. In the second method, 100,000 cells were resuspended in 75 μL in PBS solution and snap frozen in liquid nitrogen. using. The cell solution was then thawed and centrifuged for 15 min at 20,000g, 4 °C. and filtered using a 10-kDa-molecular weight cutoff filter and then split into two. The first half was used to determine total NAD+ (NAD+NADH), and the second half to determine NADH after heating the samples at 333 K for 30 min. The supernatant was collected for LCMS analysis by High-Performance Liquid Chromatography and High-Resolution Mass Spectrometry and Tandem Mass Spectrometry (HPLC-MS/MS). Specifically, system consisted of a Thermo Q-Exactive in line with an electrospray source and an Ultimate3000 (Thermo) series HPLC consisting of a binary pump, degasser, and auto-sampler outfitted with a Xbridge Amide column (Waters; dimensions of 4.6 mm × 100 mm and a 3.5 μm particle size). The mobile phase A contained 95% (vol/vol) water, 5% (vol/vol) acetonitrile, 20 mM ammonium hydroxide, 20 mM ammonium acetate, pH = 9.0; B was 100% Acetonitrile. The gradient was as following: 0 min, 15% A; 3 min, 45% A; 10 min, 60% A; 10.1-11 min, 75% A; 11.1 min, 15% A; 11.1-15 min, 15% A with a flow rate of 400 μL/min. The capillary of the ESI source was set to 275 μC, with sheath gas at 45 arbitrary units, auxiliary gas at 5 arbitrary units and the spray voltage at 4.0 kV. In positive/negative polarity switching mode, a selective iron monitoring method for target irons was used and MS1 data was collected at a resolution of 70,000. The automatic gain control (AGC) target was set at 1 × 106 and the maximum injection time was 200 ms. The target ions were subsequently fragmented for confirmation purpose, using the higher energy collisional dissociation (HCD) cell set to 30% normalized collision energy in MS2 at a resolution power of 17,500. Sample volumes of 25 μl were injected. Data acquisition and analysis were carried out by Xcalibur 4.0 software and Tracefinder 2.1 software, respectively (both from Thermo Fisher Scientific).
Measurement of mitochondrial ROS using MitoSOX and intracellular calcium imaging with Fura-2.
Live cell imaging was used to determine changes in intracellular calcium concentration and mitochondrial superoxide production, using the fluorescent probes MitoSOX (2 μM) and Fura-2 (10 μM). Cells were plated in glass plates for 24 hr in HBSS medium, then washed and incubated in Hank’s buffer containing 2 μM MitoSOX-Red and 1μg/ml of Fura-2 for 30 min at 37 °C in a 5% CO2 atmosphere. Cells were washed and incubated in Hank’s buffer for 30 minutes in the dark, then mounted in the Olympus DSU Spinning Disc Confocal microscope attached to a perfusion system and perfused with 1 ml/min of HBSS media in a 5% CO2 atmosphere at 37 °C for the duration of the experiment. Investigators were not blinded to the group allocation and treatment. Single-cell [Ca2+]cyt measurements were done according to the protocol described previously (McNally et al., 2012). Image acquisition and analysis was performed using Slidebook (Denver, CO). For data analysis, regions of interest were drawn around single cells, background subtracted, and the F340/F380 intensity ratios were determined for each time point. The F340/F380 intensity ratios were converted to [Ca2+]cyt using the formula:
| (1) |
Where R is the F340/F380 fluorescence intensity ratio and Rmax (= 9.645) and Rmin (= 0.268) were determined by in vitro calibration of Fura-2 pentapotassium salt. β (= 20.236) was determined from the Fmin/Fmax ratio at 380 nm, and Kd is the apparent dissociation constant of Fura-2 binding to Ca2+ (135 nmol).
Isolation of human alveolar macrophages
A small biopsy of donor lung tissue was obtained at the time of lung transplantation surgery and used for isolation via FACSorting (Bharat et al., 2016). Briefly, lung tissue was infiltrated with mixture of collagenase and DNase I and digested at 37°C for 30 min, chopped into 2-3 mm pieces with fine scissors and digested for another 15 min. The resulting single cell suspension was passed through the 40 μμm filter, centrifuged and red blood cells were lysed using BD Pharm Lyse buffer (BD Pharmingen). Following live/dead staining with eFluor506 viability dye (eBioscience/Affymetrix) and incubation with Fc-blocking reagent (Biolegend) cells were incubated with mixture of fluorochrome conjugated antibodies for 30 min. Alveolar macrophages were sorted on BD SORP FACS Aria III instrument in RHLCCC Flow Cytometry Core facility with 100 um nozzle, at 40 psi using MACS buffer as a capture media. Alveolar macrophages were identified as singlets/CD45+/live/CD15−/HLA-DR++/CD206++/CD169+.
The cell pellet was washed and resuspended in 10 ml of RPMI medium with 10% FBS supplemented with penicillin, streptomycin, and amphotericin B. The cells were then counted (hemacytometer; Trypan Blue), and 100,000 cells were plated on Primaria Cell Culture 12-well plates (Corning). The cells were used 24 hours after plating.
Transcriptome profiling via mRNA-seq.
Mouse alveolar macrophages were isolated via FACSorting at indicated time points. Approximately 100,000 cells were sorted into MACS buffer, immediately pelleted and lysed in RLT Plus buffer supplemented with 2-mercaptoethanol (Qiagen). RNA was isolated using RNeasy Plus kit with genomic DNA removal step. RNA quality was assessed on TapeStation 4200 instrument (Agilent), all samples had RNA integrity number (RIN) over 7. RNA-seq libraries were prepared from 100 ng of total RNA, starting with poly(A) enrichment and followed by NEB Next RNA Ultra I chemistry. Libraries were quantified and assessed on Qubit fluorimeter (Invitrogen) and TapeStation 4200, correspondingly, multiplexed and sequenced on NextSeq 500 instrument (Illumina), 75 bp, single end reads, to the average sequencing depth of 6×106 reads per sample. Over 94% of reads had Q score over 30. Reads were demultiplexed and mapped to mm10 version of the mouse genome using TopHat2 aligner and mapped to the genomic features using HTSeq and counts processed using edgeR package to estimate differentially expressed genes. FDR p value less than 0.05 was used to identify differentially expressed genes. K-means clustering was performed using GENE-E. Gene ontology analysis was performed using GOrilla on two unranked gene lists. The RNA-seq dataset is available at GEO: GSE98731.
Metabolomic analysis
Mice were treated with metformin in their drinking water for three days and BAL macrophages were harvested by gently BAL through a surgically placed tracheostomy tube (6 sequential lavages) in PBS containing (0.5mM EDTA). The cell pellet was washed twice in PBS and cells were counted (Cellometer K2, Nexcelon Bioscience). The samples were dried using SpeedVac. 50 μl of 50% acetonitrile was added to the tube for reconstitution following by overtaxing for 30 sec. Samples solution was then centrifuged for 15 min @ 20,000g, 4 °C. Supernatant was collected for LCMS analysis. Samples were analyzed by High-Performance Liquid Chromatography and High-Resolution Mass Spectrometry and Tandem Mass Spectrometry (HPLC-MS/MS). Specifically, system consisted of a Thermo Q-Exactive in line with an electrospray source and an Ultimate3000 (Thermo) series HPLC consisting of a binary pump, degasser, and auto-sampler outfitted with a Xbridge Amide column (Waters; dimensions of 4.6 mm × 100 mm and a 3.5 μm particle size). The mobile phase A contained 95% (vol/vol) water, 5% (vol/vol) acetonitrile, 20 mM ammonium hydroxide, 20 mM ammonium acetate, pH = 9.0; B was 100% Acetonitrile. The gradient was as following: 0 min, 15% A; 2.5 min, 30% A; 7 min, 43% A; 16 min, 62% A; 16.1-18 min, 75% A; 18-25 min, 15% A with a flow rate of 400 μL/min. The capillary of the ESI source was set to 275 °C, with sheath gas at 45 arbitrary units, auxiliary gas at 5 arbitrary units and the spray voltage at 4.0 kV. In positive/negative polarity switching mode, an m/z scan range from 70 to 850 was chosen and MS1 data was collected at a resolution of 70,000. The automatic gain control (AGC) target was set at 1 × 106 and the maximum injection time was 200 ms. The top 5 precursor ions were subsequently fragmented, in a data-dependent manner, using the higher energy collisional dissociation (HCD) cell set to 30% normalized collision energy in MS2 at a resolution power of 17,500. The sample volumes of 25 μl were injected. Data acquisition and analysis were carried out by Xcalibur 4.0 software and Tracefinder 2.1 software, respectively (both from Thermo Fisher Scientific).
Phenotyping and isolation of the immune cells in the murine lungs via flow cytometry and cell sorting.
Identification and isolation of the immune cells by flow cytometry and cell sorting were performed as described previously (Misharin et al., 2013). Briefly, the mice were euthanized and the lungs were perfused through the right ventricle with 10 ml of HBSS with Ca2+ and Mg2+, dissected and infiltrated with collagenase and DNase I, chopped into 2-3 mm fragments, transferred into C-tubes (Miltenyi) and subjected to mechanical disintegration using GentleMACS instrument (Miltenyi). The resulting single cell suspension was filtered through 40 um filter, and subjected to CD45-enrichment using corresponding magnetic microbeads (Miltenyi), stained with eFluor506 viability dye, followed by the mixture of fluorescently labeled antibodies. Cell counts were obtained on K2 cell counter (Nexcelom) using acridine orange to discriminate nucleated cells from debris and propidium iodide to discriminate dead cells. Data were acquired on BD LSR II instrument, cell sorting was performed BD SORP FACS Aria III instrument, 100 um nozzle, 40 psi pressure.
Quantitative RT-PCR
We isolated total RNA from mouse lungs, sorted cells or cell cultures using a commercially available system (TRIzol; Invitrogen) and performed qRT-PCR reactions using IQ SYBR Green superscript analyzed on a Bio-Rad IQ5 Real-Time PCR Detection System using the following primer sequences: IL-6 (5’-TTCCATCCAGTTGCCTTCTTGG-3’, 5’-TTCTCATTTCCACGATTTCCCAG-3’); TFAM (5′-CCAAAAAGACCTCGTTCAGC-3′, 5′-ATGTCTCCGGATCGTTTCAC-3’); mRPL19 (5′-GAAGGTCAAAGGGAATGTGTTCAA-3′, 5′-TTTCGTGCTTCCTTGGTCTTAGA-3′)
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
We report all data as mean ± SEM. We subjected all data to 1-way ANOVA. When ANOVA indicated a significant difference, we explored individual differences with 2-tailed Student’s t test using Bonferroni’s correction for multiple comparisons (Prism 6; Graphpad). Statistical methods for RNA-Seq analysis above. For metabolomics, 154 detected metabolites in all samples were analyzed using two way ANOVA followed by multiple comparisons using the two stage linear step up procedure of Benjamini, Krieger and Yekutieli with a FDR q value of 0.1. For some assays (RNA-seq, quantitative RT-PCR, murine model of arterial thrombosis) power and sample size were estimated based on our previous work, the observed data met or exceeded these criteria. The statistical parameters and criteria for significance can be found in the figure legends.
DATA AND SOFTWARE AVAILABILITY
The RNA-seq dataset, containing raw and processed data, is available at GEO: GSE98731.
Supplementary Material
Table S1. RNA-Seq of flow sorted alveolar macrophages from mice treated with PM (10 μg/mouse, i.t.) in the presence or absence of metformin. Refers to Figure 7.
Highlights.
Metformin prevents IL-6 dependent thrombosis induced by urban particulate matter
Metformin inhibits mitochondrial complex I to prevent ROS-mediated IL-6 release
Metformin inhibits mitochondrial ROS to prevent CRAC channel activation
Mitochondrial ROS and CRAC channel inhibition in vivo prevent thrombosis
Acknowledgments:
Authors declare no conflict of interest.
RNA-Seq data have been deposited to GEO, accession number GSE98731. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE98731
Grant Funding
AVM: NHLBI HL135124, NIH NIAMS AR061593, ATS/Scleroderma Foundation Research Grant, DOD grant PR141319, and BD Bioscience Immunology Research Grant. PAR: HL076139. AB: NIH HL125940 and the Thoracic Surgery Foundation, Society of University Surgeons, John H. Gibbon Jr. Research Scholarship from American Association of Thoracic Surgery. BDJ: HL128867, Parker B. Francis Research Opportunity Award. JIS: NIH AG049665, HL048129, HL071643, HL085534. GMM: NIH ES015024 and ES025644 and ES026718. KR: NIH HL079190 and HL124664. GRSB: NIH ES013995, HL071643, AG049665, VA BX000201, DOD PR141319. HP: NIH AR064546, AG049665 HL134375 and the Mabel Greene Myers Chair. NSC: NIH Grants AG049665, HL071643, CA197532. Core Facilities (Flow Cytometry and Genomics NCI Cancer Center Support Grant P30 CA060553, the Feinberg School of Medicine, the Center for Genetic Medicine, and Feinberg’s Department of Biochemistry and Molecular Genetics, the Office of the Provost, the Office for Research, and Northwestern Information Technology and maintained and developed by Feinberg IT and Research Computing Group.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: The authors declare no competing interests.
References
- Barzilai N, Crandall JP, Kritchevsky SB, and Espeland MA (2016). Metformin as a Tool to Target Aging. Cell metabolism 23, 1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batandier C, Guigas B, Detaille D, El-Mir MY, Fontaine E, Rigoulet M, and Leverve XM (2006). The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. Journal of bioenergetics and biomembranes 38, 33–42. [DOI] [PubMed] [Google Scholar]
- Bharat A, Bhorade SM, Morales-Nebreda L, McQuattie-Pimentel AC, Soberanes S, Ridge K, DeCamp MM, Mestan KK, Perlman H, Budinger GR, et al. (2016). Flow Cytometry Reveals Similarities Between Lung Macrophages in Humans and Mice. American journal of respiratory cell and molecular biology 54, 147–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Goncalves RLS, Orr AL, Vargas L, Gerencser AA, Jensen MB, Wang YT, Melov S, Turk CN, Matzen JT, et al. (2016). Suppressors of Superoxide-H(2)O(2) Production at Site I(Q) of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell metabolism 24, 582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, Orton K, Villella A, Garza D, Vidal M, et al. (2014). A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell reports 9, 1135–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges HR, Jones AJ, Pollak MN, and Hirst J (2014). Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. The Biochemical journal 462, 475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, Avizonis D, Reczek CR, Weinberg SE, Menz S, Neuhaus R, Christian S, Haegebarth A, Algire C, and Pollak M (2016). Are Metformin Doses Used in Murine Cancer Models Clinically Relevant? Cell metabolism 23, 569–570. [DOI] [PubMed] [Google Scholar]
- Chiarella SE, Soberanes S, Urich D, Morales-Nebreda L, Nigdelioglu R, Green D, Young JB, Gonzalez A, Rosario C, Misharin AV, et al. (2014). beta(2)-Adrenergic agonists augment air pollution-induced IL-6 release and thrombosis. The Journal of clinical investigation 124, 2935–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai S, Tang Z, Cao J, Zhou W, Li H, Sampson S, and Dai C (2015). Suppression of the HSF1-mediated proteotoxic stress response by the metabolic stress sensor AMPK. The EMBO journal 34, 275–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Q, Wang Y, Zanobetti A, Wang Y, Koutrakis P, Choirat C, Dominici F, and Schwartz JD (2017). Air Pollution and Mortality in the Medicare Population. New England Journal of Medicine 376, 2513–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duch MC, Budinger GRS, Liang YT, Soberanes S, Urich D, Chiarella SE, Campochiaro LA, Gonzalez A, Chandel NS, Hersam MC, et al. (2011). Minimizing Oxidation and Stable Nanoscale Dispersion Improves the Biocompatibility of Graphene in the Lung. Nano Letters 11, 5201–5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Europe, W.H.O.R.O.f. (2013). In Review of evidence on health aspects of air pollution - REVIHAAP Project: Technical Report (Copenhagen: WHO Regional Office for Europe (c) World Health Organization 2013.). [PubMed] [Google Scholar]
- Fisher RP, Lisowsky T, Parisi MA, and Clayton DA (1992). DNA wrapping and bending by a mitochondrial high mobility group-like transcriptional activator protein. The Journal of biological chemistry 267, 3358–3367. [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, et al. (2012). Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 13, 1118–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griss T, Vincent EE, Egnatchik R, Chen J, Ma EH, Faubert B, Viollet B, DeBerardinis RJ, and Jones RG (2015). Metformin Antagonizes Cancer Cell Proliferation by Suppressing Mitochondrial-Dependent Biosynthesis. PLoS Biol 13, e1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka RB, Glasauer A, Hoover P, Yang S, Blatt H, Mullen AR, Getsios S, Gottardi CJ, DeBerardinis RJ, Lavker RM, et al. (2013). Mitochondrial reactive oxygen species promote epidermal differentiation and hair follicle development. Science signaling 6, ra8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, and Schultz G (1999). Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397, 259. [DOI] [PubMed] [Google Scholar]
- Jairaman A, Maguire CH, Schleimer RP, and Prakriya M (2016). Allergens stimulate store-operated calcium entry and cytokine production in airway epithelial cells. Scientific reports 6, 32311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jairaman A, Yamashita M, Schleimer RP, and Prakriya M (2015). Store-Operated Ca2+ Release-Activated Ca2+ Channels Regulate PAR2-Activated Ca2+ Signaling and Cytokine Production in Airway Epithelial Cells. Journal of immunology (Baltimore, Md. : 1950) 195, 2122–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelieveld J, Evans JS, Fnais M, Giannadaki D, and Pozzer A (2015). The contribution of outdoor air pollution sources to premature mortality on a global scale. Nature 525, 367. [DOI] [PubMed] [Google Scholar]
- Li H, Cai J, Chen R, Zhao Z, Ying Z, Wang L, Chen J, Hao K, Kinney PL, Chen H, et al. (2017). Particulate Matter Exposure and Stress Hormone Levels: A Randomized, Double-Blind, Crossover Trial of Air Purification. Circulation 136, 618–627. [DOI] [PubMed] [Google Scholar]
- Liu X, Romero IL, Litchfield LM, Lengyel E, and Locasale JW (2016). Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell metabolism 24, 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD, Antebi A, Bartke A, Barzilai N, Brown-Borg HM, Caruso C, Curiel TJ, de Cabo R, Franceschi C, Gems D, et al. (2015). Interventions to Slow Aging in Humans: Are We Ready? Aging Cell 14, 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A, Wang D, An Y, Fang W, and Zhu H (2017). Comparative transcriptomic analysis of mice liver treated with different AMPK activators in a mice model of atherosclerosis. Oncotarget 8, 16594–16604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, et al. (2013). Metformin improves healthspan and lifespan in mice. Nature communications 4, 2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruthur NM, Tseng E, Hutfless S, Wilson LM, Suarez-Cuervo C, Berger Z, Chu Y, Iyoha E, Segal JB, and Bolen S (2016). Diabetes Medications as Monotherapy or Metformin-Based Combination Therapy for Type 2 DiabetesA Systematic Review and Meta-nalysisDiabetes Medications as Monotherapy or Metformin-Based Combination Therapy.Annals of Internal Medicine N/A, N/A-N/A. [DOI] [PubMed] [Google Scholar]
- McNally BA, Somasundaram A, Yamashita M, and Prakriya M (2012). Gated regulation of CRAC channel ion selectivity by STIM1. Nature 482, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK 3rd, erdnikovs S, Filer A, Clark AR, Buckley CD, et al. (2014). Nonclassical Ly6C(−) monocytes drive the development of inflammatory arthritis in mice. Cell reports 9, 591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GRS, and Perlman H (2013). Flow Cytometric Analysis of the Macrophages and Dendritic Cell Subsets in the Mouse Lung. American journal of respiratory cell and molecular biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, Chen C-I, Anekalla KR, Joshi N, Williams KJN, et al. (2017). Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. The Journal of Experimental Medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutlu GM, Green D, Bellmeyer A, Baker CM, Burgess Z, Rajamannan N, Christman JW, Foiles N, Kamp DW, Ghio AJ, et al. (2007). Ambient particulate matter accelerates coagulation via an IL-6 dependent pathway. J. Clin. Invest 117, 2952–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nel A (2005). Atmosphere. Air pollution-related illness: effects of particles. Science 308, 804–806. [DOI] [PubMed] [Google Scholar]
- Orr AL, Vargas L, Turk CN, Baaten JE, Matzen JT, Dardov VJ, Attle SJ, Li J, Quackenbush DC, Goncalves RL, et al. (2015). Suppressors of superoxide production from mitochondrial complex III. Nature chemical biology 11, 834–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope CA III, Ezzati M, and Dockery DW (2009). Fine-Particulate Air Pollution and Life Expectancy in the United States. N Engl J Med 360, 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM (2016). From C-Reactive Protein to Interleukin-6 to Interleukin-1. Moving Upstream To Identify Novel Targets for Atheroprotection 118, 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. (2017). Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. New England Journal of Medicine 377, 1119–1131. [DOI] [PubMed] [Google Scholar]
- Semmler-Behnke M, Takenaka S, Fertsch S, Wenk A, Seitz J, Mayer P, Oberdorster G, Kreyling WG, Semmler-Behnke M, Takenaka S, et al. (2007). Efficient elimination of inhaled nanoparticles from the alveolar region: evidence for interstitial uptake and subsequent reentrainment onto airways epithelium. Environmental Health Perspectives 115, 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo BB, Kitajima-Ihara T, Chan EK, Scheffler IE, Matsuno-Yagi A, and Yagi T (1998). Molecular remedy of complex I defects: rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proc Natl Acad Sci U S A 95, 9167–9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaram A, Shum AK, McBride HJ, Kessler JA, Feske S, Miller RJ, and Prakriya M (2014). Store-Operated CRAC Channels Regulate Gene Expression and Proliferation in Neural Progenitor Cells. The Journal of Neuroscience 34, 9107–9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaeth M, Zee I, Concepcion AR, Maus M, Shaw P, Portal-Celhay C, Zahra A, Kozhaya L, Weidinger C, Philips J, et al. (2015). Ca2+ Signaling but Not Store-Operated Ca2+ Entry Is Required for the Function of Macrophages and Dendritic Cells Journal of immunology (Baltimore, Md. : 1950) 195, 1202–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, et al. (2014). Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 3, e02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. RNA-Seq of flow sorted alveolar macrophages from mice treated with PM (10 μg/mouse, i.t.) in the presence or absence of metformin. Refers to Figure 7.
Data Availability Statement
The RNA-seq dataset, containing raw and processed data, is available at GEO: GSE98731.