

SUMMARY
Critical to migration of tumor cells and endothelial cells is the proteolytic attack of membrane type 1 matrix metalloproteinase (MT1-MMP) upon collagen, growth factors, and receptors at cell surfaces. Lipid bilayer interactions of the substrate-binding hemopexin-like (HPX) domain of MT1-MMP were investigated by paramagnetic NMR relaxation enhancements (PREs), fluorescence, and mutagenesis. The HPX domain binds bilayers by blades II and IV on opposite sides of its β-propeller fold. The EPGYPK sequence protruding from both blades inserts among phospholipid head groups in PRE-restrained molecular dynamics simulations. Bilayer binding to either blade II or IV exposes the CD44 binding site in blade I. Bilayer association with blade IV allows the collagen triple-helix to bind without obstruction. Indeed, vesicles enhance proteolysis of collagen triple-helical substrates by the ectodomain of MT1-MMP. Hypothesized side-by-side MT1-MMP homodimerization would allow binding of bilayers, collagen, CD44, and head-to-tail oligomerization.
eTOC blurb
In order to migrate, tumor cells depend upon the protease known as membrane type 1 MMP. Marcink et al. have unraveled MT1-MMP positioning on lipid bilayers via opposite tips of its symmetric substrate-binding domain. The compatibility with binding of collagen, other substrates, and other protomers of MT1-MMP is discussed.
Graphical Abstract
INTRODUCTION
Membrane type 1 matrix metalloproteinase (MT1-MMP or MMP-14) was discovered as triggering invasion by tumor cells via proteolytic processing of proMMP-2 to MMP-2 on cell surfaces (Sato et al., 1994). MT1-MMP supports cell invasion in cancer metastasis, angiogenesis, and skeletal development (Itoh, 2015; Sabeh et al., 2009). It is the pericellular collagenase required for invasion of 3D collagen matrices by tumor cells and endothelial cells forming capillary tubes (Chun et al., 2004; Davis and Saunders, 2006; Fisher et al., 2009; Friedl and Wolf, 2008; Sabeh et al., 2004; Wolf et al., 2007). Cell motility dependent upon epidermal growth factor (EGF) receptor (ErbB) is promoted by MT1-MMP-catalyzed release of an EGF-like fragment from laminin 5 (Gilles et al., 2001; Koshikawa et al., 2000) and proteolysis of heparin-binding EGF (Koshikawa et al., 2010). Cell motility is also promoted by MT1-MMP shedding of cell adhesion molecules from cell surfaces (Itoh, 2015), e.g. hyaluronan receptor CD44 (Kajita et al., 2001), syndecan-1 (Endo et al., 2003), αV integrin (Deryugina et al., 2002), and intracellular adhesion molecule-1 (Sithu et al., 2007).
MT1-MMP can be internalized from the plasma membrane into endosomes and then be recycled back to the cell surface or to exosomes released from cells (Hakulinen et al., 2008). CD44 may recruit MT1-MMP to lamellipodia (Mori et al., 2002). During cell invasion, MT1-MMP-containing intracellular vesicles are trafficked to invadopodia by kinesin-1 recruited to the vesicles by its affinity for phosphatidic acid (Wang et al., 2017). MT1-MMP localizes to cholesterol-containing lipid rafts required for formation of invadopodia (Albrechtsen et al., 2011; Yamaguchi et al., 2009). MT1-MMP is exposed on the surface of extracellular vesicles such as exosomes (Hakulinen et al., 2008; Shimoda and Khokha, 2017).
The positioning of MT1-MMP upon membrane bilayers has the potential to influence its pivotal proteolytic activities. Its soluble extracellular domains comprise the catalytic domain (a zinc hydrolase), two flexible linkers, and the hemopexin-like (HPX) domain which adopts a β-propeller fold with four pseudo-symmetric blades (Bode, 1995). MT1-MMP is the only membrane type MMP with a crystal structure available for the HPX domain (Tochowicz et al., 2011). Both the HPX and catalytic domains were shown to bind peripherally to membrane mimics (Cerofolini et al., 2016; Van Doren et al., 2017). This suggests that the soluble domains may reside on membranes at least transiently, despite widespread depictions of them extending away from the membrane.
The soluble ectodomain of MT1-MMP (sMT1-MMP in which the second linker and transmembrane helix are removed) is active in collagenolysis and is monomeric, as is its HPX domain (Cerofolini et al., 2016; Collier et al., 2011; Tam et al., 2004; Zhao et al., 2015). PISA analysis of the small interfaces in the asymmetric unit of crystals of the HPX domain (PDB: 3C7X) predicts it to be a monomer in solution, though dimers in solution were also reported (Tochowicz et al., 2011). The HPX domain was dispensable for cellular collagenolysis by MT1-MMP, but membrane anchoring was required (Li et al., 2008). Other cellular assays implicated symmetric homodimerization about blades II and III in proMMP-2 activation and collagenolysis (Tochowicz et al., 2011). However, another study disputed this mode of homodimerization (Zarrabi et al., 2011). The discrepancies in reported requirements for the HPX domain in collagenolysis were attributed to differences in the constructs used (Amar et al., 2017; Zarrabi et al., 2011). The bulges emanating from blades I and IV were implicated in activation of proMMP-2, cell migration, lung metastasis, and formation of new blood vessels (Zarrabi et al., 2011). Dimerization of MT1-MMP occurs at the leading edge (invadopodia) but not trailing surfaces of migrating cells (Itoh et al., 2006; Itoh et al., 2011; Itoh et al., 2001; Lehti et al., 2002). Homodimerization via the transmembrane helix supports proMMP-2 activation (Itoh et al., 2008). The intracellular C-terminus also participates in dimerization (Lehti et al., 2002), potentially by its anchorage to the cytoskeleton in invadopodia via palladin (von Nandelstadh et al., 2014). MT1-MMP interactions with membranes could possibly couple with modes of its dimerization and association with other cell surface molecules.
Careful appraisal of the proximity of MT1-MMP to lipid bilayers is warranted by its relevance to the digestion of collagen, pro-growth factors, and receptors on cell membranes, as well as to targeting of therapeutics to the HPX domain of this protease (Basu et al., 2012; Remacle et al., 2012). We implemented two key tools in our investigation. First, nanodiscs have emerged as bilayer mimics preferred for structural and biophysical studies of membrane-associated proteins. Nanodiscs encircle disk-like bilayers with two chains of membrane scaffolding protein (MSP). This improves stability, homogeneity, planarity, and fluidity at high concentrations (Denisov and Sligar, 2016). Second, paramagnetic NMR has successfully docked GTPases peripherally to nanodiscs containing a paramagnetic ion conjugated to a lipid head group (Mazhab-Jafari et al., 2013). NMR has also measured soluble protein proximity to bilayer mimics containing the paramagnetic spin label on the fatty acyl chains within the bilayer, which provides detailed structural orientations upon fluid bilayer assemblies (Koppisetti et al., 2014; Lenoir et al., 2010; Lenoir et al., 2018; Marcink et al., 2017; Prior et al., 2015). We show NMR and fluorescence evidence that both blades II and IV, on opposite sides of the HPX domain, probably insert their protruding loop peptide sequences of EPGYPK in among the phospholipid head groups of model bilayers. With blade IV bound to the bilayer, the HPX domain appears free to bind the collagen triple-helix. Indeed, we observe that sMT1-MMP proteolysis of collagen-mimicking substrates is retained and enhanced in the presence of vesicles.
RESULTS
Nanodiscs Associate with Blades II and IV of the HPX Domain
The NMR spectra of equimolar mixtures of DMPC nanodiscs with the HPX domain from human MT1-MMP are enhanced by addition of NaCl to 200 to 300 mM (Fig. S1A). Lipid association can be monitored by rotational correlation time τc detected by 15N NMR relaxation (Prior et al., 2015; Thompson et al., 2017). The HPX domain interacting with the nanodiscs tumbles with τc around 21 ns at 30°C in 300 mM NaCl. This is almost twice the τc of the free state (Zhao et al., 2015) and 3- to 4-fold less than the τc of integral membrane proteins in nanodiscs of similar composition (Hagn et al., 2013). These observations and the sharpening of NMR peaks upon salt addition suggest that increased salt may weaken the association of nanodiscs with the HPX domain, presumably by diminishing the electrostatic attraction. Since the zeta potential (estimate of surface potential) of phosphatidylcholine (PC) vesicles is attenuated nearly to zero by 0.01 M NaCl or higher (Egawa and Furusawa, 1999), raising [NaCl] through the physiological range was not expected to alter the orienting influence of electrostatics at the bilayer-protein interface. High salt may shorten the residence time of the HPX domain upon a nanodisc, resulting in faster tumbling and better spectra of the HPX domain.
To locate binding sites for a lipid bilayer on the HPX domain, nanodiscs were prepared for paramagnetic NMR measurement of proximity by incorporating nitroxide spin-labeled 1-palmitoyl-2-stearoyl-(5-doxyl)-sn-glycero-3-phosphocholine at a mole fraction of 0.025 of the DMPC. The 5-doxyl substituent was positioned on the acyl chain under the phospholipid head groups whereas a 10-doxyl substituent was positioned deeper inside the bilayer. The rapid diffusion of the DPPC should move the doxyl group throughout the discoidal bilayer. The paramagnetic relaxation enhancements (PREs given as Γ2) emanating from the average depth of the spin label weakened 1H NMR peaks in the vicinity and were quantified at Ile, Leu, and Val methyl groups as exponential decay curves that could differ between paramagnetic (spin-labeled) and diamagnetic (unlabeled) nanodiscs (Fig. S1B–D). The most significant sites have large PREs from both 5-doxyl PC and 10-doxyl PC with Γ2 > 28 s−1 and localize to β-propeller blade II (Val380, Val389, and Leu395) or blade IV (Val473 and Val493) (Fig. 1A,B). Sites adjoining blade II have large PREs from only the more shallow spin label (5-doxyl PC), i.e. Ile357 in blade I and Leu442 in blade III; this behavior suggests that these sites may lie at the flanks of an interface with nanodiscs. The nanodiscs induced small amide chemical shift perturbations (CSPs) in all four blades (Figs. 1C,D and S1E) that agree with the explicit measurements of proximity that the PREs represent. The largest CSPs in blade II cluster around Glu396 of the β-bulge, including Asp376, Lys386, His387, Leu395, and Ile403 nearby (Fig. 1C). The CSPs of Gly398 and Tyr399 in the β-bulge are smaller. The largest CSPs in blade IV belong to Lys499 and Gln489 on opposite flanks of the β-bulge (Fig. 1C). The CSPs of Lys490 through Tyr497 spanning the β-bulge are small. A mixture of bilayer binding to opposite sides of the HPX domain agrees with negative staining electron microscopy evidence that addition of sMT1-MMP at low [NaCl] draws nanodiscs together into stacks (Fig. S2A,D).
Figure 1. NMR Assays Suggest that Nanodiscs Associate with Opposite Sides of the HPX Domain of MT1-MMP.

(A) Bilayer proximities of Ile/Leu/Val side chains were measured to DMPC nanodiscs encircled by MSP1D1 (Denisov et al., 2004) doped with an average of two nitroxide spin-labeled PC molecules per leaflet. The resulting PREs were plotted as Γ2 = R2, paramag – R2,diamag, the difference between methyl proton relaxation rates with and without doxyl-substituted PC. Γ2 values resulting from addition of 10-doxyl PC or 5-doxyl PC are plotted in pink triangles and green spheres, respectively. The symbols are filled where Γ2 > 30/s. The uncertainties indicated are those of the fits of the exponential decays and their differences.
(B) Ile/Leu/Val side chains with Γ2 > 30/s are marked on the crystal structure of the HPX domain (PDB: 3C7X) with spheres which are pink if near 10-doxyl PC and green if near 5-doxyl PC only. The blades of the β-propeller are colored blue for I, green for II, yellow for III, and pink IV, respectively, and throughout this article.
(C) Addition of the nanodiscs to the HPX domain in a 1:1 molar ratio induced the radial chemical shift perturbations (CSPs), quantified using eq. 2. CSPs exceeding 0.03 ppm are highlighted in orange.
(D) Sites of the larger CSPs (ΔωHN > 0.03 ppm) are plotted as spheres on the backbone.
See also Fig. S1.
Nanodisc Docking to Opposite Sides of the HPX Domain
The PREs and CSPs measured by NMR were separated into groups on opposite sides of the domain: one at blade II and the other at blade IV (Fig. 1B,D). Estimates of the average, apparent distances from the PRE-broadened methyl groups to the average depth of the 5-doxyl or 10-doxyl groups in the nanodisc bilayer level were calculated from the Γ2 values (Figs. 1A, S1D) and τc using eq. 3. Nanodisc-induced peak shifts (CSPs) and broadenings added supplementary but ambiguous restraints on proximity to the surface of the nanodisc (Table 1). These were used as loose distance restraints to the average, apparent depth within the bilayer of the spin label, suggested by the PREs it induced. These average, apparent depths were implemented with (i) ambiguous restraints in initial HADDOCK rigid body docking (Dominguez et al., 2003; Koppisetti et al., 2014; Prior et al., 2015) and (ii) distance restraints to a plane of similar depth in the bilayer during refinement by restrained molecular dynamics (MD) simulations, using NAMD and the CHARMM36 force field (Huang and MacKerell, 2013). From 4-ns segments of the restrained MD trajectories with blade II or IV at the nanodisc interface (Movies S1 and S2), 15 snapshots of the complexes of lowest energy and most consistency with the depth restraints were collected into structural ensembles (Fig. 2A,B,D,E with PDB IDs 6CM1 and 6CLZ). The RMSD values to the mean were around 1.18 and 1.24 Å for the backbone atoms (Table 1).
Table 1.
NMR Structural Statistics.
| Blade II in interface | Blade IV in interface | |
|---|---|---|
| PDB accession code | 6CM1 | 6CLZ |
| RMSD deviations a | ||
| Avg. RMSD (Å) for backbone atoms | 1.18 ± 0.28 | 1.27 ± 0.26 |
| Distance restraints | ||
| Lipid - protein methyl PREs, explicit | 3*7 | 3*4 |
| Lipid - amide shifts, broadenings, ambiguous | 11 | 3 |
| Restraint violations > 10% | ||
| Protein-lipid PREs | V380–10doxyl: 15±3%, 3.1±0.6A |
0 |
| Protein-lipid ambiguous residues | 0 | 0 |
| Structural quality | ||
| Procheck G-factor (all dihedrals) | −0.55 | −0.53 |
| Molprobity score b | 1.11 | 1.10 |
| Most favored Ramachadran plot residues (%)c | 84.7 | 85.3 |
| Allowed Ramachandran plot residues (%)c | 11.0 | 12.3 |
| Buried surface area (Å2)d | ||
| 1225 ± 90 | 1423 ± 68 | |
| CH3 groups with PREs from 5-doxyl DPCCe | ||
| 357δ, 380γ, 389γ 395δ, 442γ |
473γ, 493γ | |
| CH3 groups with PREs from 10-doxyl DPCCe | ||
| 357δ, 380γ, 389γ | 473γ, 493γ | |
| Amide groups perturbed by nanodiscs | ||
| 330 f, 342, 377, 379, 386, 387 f, 396, 422, 428 | 459, 489, 499 | |
calculated from the ensemble of 15 lowest energy structures
Combines the clash score, rotamers, and Ramachandran evaluation into a single score
From Procheck-NMR
Average buried surface area from the ensemble of 15 structures
Leu methyl groups are denoted δ and Val methyl groups γ
amide NMR peak undergoes both a shift and broadening
Figure 2. Modes of Nanodisc Binding to Blades II and IV.

(A, B) An ensemble of 15 HPX structural models positioning blade II upon a nanodisc model was drawn from a 4-ns MD trajectory restrained by NMR PRE-based distances and shown in Movie S1. The structures were selected for lowest energy and minimal violation of the NMR restraints. The viewing angle differs by 90° between panels. The inset displays the loop with sequence EPGYPK in contact with the nanodiscs, with polar residues purple and hydrophobic residues green.
(C) Partial insertion of the EPGYPK loop of blade II among the phospholipid head groups is shown, with lipid oxygen atoms pink and carbon white.
(D, E) The PRE distance-restrained ensemble of 15 HPX structural models positioning blade IV on a nanodisc model was selected from another 4-ns MD trajectory shown in Movie S2.
(F) Insertion of the EPGYPK loop of blade IV among the phospholipid head groups is shown.
See also Fig. S2.
The ensembles were assessed further by comparing the PREs suggested by their coordinates with the measured PREs. The back-calculated PREs appear correlated with the experimentally significant PREs, displaying approximate slope of 1.04 and Pearson’s r of 0.85 (Fig. S2E). The majority of simulated PREs underestimate the measured PREs. This might be related to the upper bounds on the distance restraints being generous to accommodate the averaging processes. The three largest measured PREs are exceeded by the back-calculated Γ2 values for these methyl groups (Fig. S2E). At the bilayer interface with blade II, back-calculated Γ2 values might suggest the spin-labeled acyl chain to approach more closely the side chains of Val389 and Leu395 by around 1.6 and 3.9 Å, respectively, than do Γ2 values from 1H NMR relaxation. The discrepancy for Val493 at the bilayer interface with blade IV corresponds to around 1.4 Å.
Protein-Bilayer Interfaces
In both complexes with nanodiscs (computationally docked using the NMR-based distance restraints), 13 residues contact phospholipid head groups throughout each ensemble. Central to the interface in both modes of binding is the outer β-strand and its β-bulge with sequence L/VEPGYPK, where the “YP” is recessed from the interface (Fig. 2). The TROSY spectra with and without nanodiscs manifest single amide NMR peaks of the Glu, Gly, and Lys residues in both β-bulges (Fig. S1E). The lack of peak doubling may suggest that the proline residues remain in the conformation of the free state in the crystal structure, which is trans for the first Pro and cis for the second Pro of each β-bulge. However, the Tyr amide peaks are too overlapped to judge this (Fig. S1E). In both interfaces with nanodiscs, the side chain of the first proline and the methylene group of the glycine insert far enough to contact fatty acyl chains and displace head groups to form a small bowl in the bilayer surface (Fig. 2C,F). The lipids around this bowl in the heart of the interface are more ordered in the MD simulations than those lipids that are more distant from the protein.
The net positive charge is greater in the blade IV interface where six side chains are positively charged and one negatively charged. In contrast, the blade II interface has four basic side chains and two acidic side chains in contact with the head groups. The positively charged side chains tend to be drawn into salt bridges to phosphoester linkages in the lipid head groups in the structural simulations. This is true of three of the four basic residues in the blade II interface and five of the six basic residues in the blade IV interface (Table 2). The interface centered at blade II includes apparent bilayer contacts of Lys362, the sequence 393ASLEPGYPK401 bulging from blade II, Glu405 and Arg408 of blade II, and Leu442 and Arg443 at the edge of blade III closest to blade II. The interface centered at blade IV includes apparent bilayer contacts with Lys454 and G458 at the near edge of blade III, the central bulging blade IV sequence 490KLKVEPGYPK499, and Lys482, Arg503, and Ser510 also from blade IV.
Table 2.
Potential Transient Salt Bridges between HPX Domain and Phospholipid Head Groups, Listing Minimum Frequencies in Each Ensemble
| Blade 2 interface | Salt bridae to DMPCa | |
| ARG 362 | HE | O12 (214) 7% |
| HH1 | O32b (204) 20% | |
| HH2 | O12 (214) 33% | |
| LYS 401 | HZ1 | O13 (213) 53% |
| HZ1 | O13 (215) 13% | |
| ARG 443 | HH2 | O14 (215) 80% |
| Blade 4 interface | ||
| LYS454 | HZ1 | O11/13/14 (195) 33% |
| LYS 482 | HZ1 | O14 (212) 47% |
| HZ1 | O14 (217) 80% | |
| LYS 490 | HZ1 | O13/14 (201) 27% |
| HZ2 | O13/22b (213) 66% | |
| LYS 499 | NZ1 | O11/13 (216) 54% |
| ARG 503 | HH1 | O12/13 (218) 80% |
Phosphate oxygen, except where noted. The number of the lipid molecule is listed in parentheses.
Ester carbonyl oxygen
The overall tilt of each mode of binding appears to differ. The binding mode at blade II has the collagen-binding face tilted modestly toward the bilayer, possibly due to electrostatic attractions of Arg374 and Lys378 (from the first β-hairpin loop of blade II) to phosphoester groups on that side. The short helix terminating blade I from Gly358 through Arg362 also packs with lipid head groups. On the opposite side of the blade II interface, hairpin loop residues Leu442 and Arg443 in blade III contact the head groups as well. In contrast, the mode of binding at blade IV tilts in the opposite direction with the collagen-binding face tipped away from the bilayer and the “exit” side tilting slightly towards the bilayer. (The exit side refers to the central channel which binds Cl− and Na+ ions (Tochowicz et al., 2011)). Electrostatic attractions on the exit side of Lys454 (in blade III) and Lys482 to phosphoesters (Table 2) might account for this apparent tilt. The central EPGYPK motif that inserts partly into the bilayer and the β-hairpin that presents Lys454 have counterparts with the blade II interface. However, the two other β-hairpin loops at the blade II interface have no apparent counterparts in the blade IV interface.
Fluorescent Lipid Interactions Probed by Mutations
Due to the pseudo four-fold symmetry of the β-propeller fold and the recent proposal that bilayer binding is centered upon blade III (Cerofolini et al., 2016), we examined bilayer binding by independent means. We sought to perturb bilayer binding by introducing site-directed Ser substitutions of basic residues placed strategically in the four blades of the β-propeller (Fig. 3A) of the sMT1-MMP (catalytic – linker – HPX) construct. We monitored FRET quenching of intrinsic Trp fluorescence by Pyrene-PE incorporated into small unilamellar vesicles (SUVs) (Ganguly et al., 2007; Tatulian, 2017). Addition of SUVs to wild-type sMT1-MMP attenuated Trp fluorescence emission by nearly 30% (Fig. 3B). Neither the K414S nor the K434S substitution in blade III interfered with fluorescence quenching by pyrene-labeled SUVs (Fig. 3B). Ser substitution for the Lys at the end of the EPGYPK motif of either blade II or IV, i.e. K401S or K499S, respectively, diminished the quenching of Trp fluorescence emission by SUVs containing the Pyrene-PE quencher (Fig. 3B). The R362S variant responded similarly, but to a lesser degree. The mutational evidence of electrostatic attraction of Arg362, Lys401, and Lys499 to SUVs corroborates the paramagnetic NMR-based proposal that lipid bilayers do recognize blades II and IV (Figs. 1, 2).
Figure 3. Basic Residues at β-Bulges from Blades II and IV Support Binding of Soluble MT1-MMP to Membrane Vesicles.

(A) Orange spheres identify the locations of Ser substitutions for Lys or Arg in the four blades.
(B) Quenching of Trp fluorescence from sMT1-MMP (orange with addition of doped SUVs and purple without) by addition SUVs composed of DMPC doped with Pyrene PE depends upon the point mutation. The background fluorescence from SUVs only was subtracted from the fluorescence of the mixtures as described (Ganguly et al., 2007). The uncertainties plotted are the SD of triplicate measurements. See also Fig. S3.
(C) The crystallographic structures of HPX domains of MMPs are superposed. Blade IV of MT1-MMP (green) is distinctive in projecting the EPGYPK loop, which is absent from the structures of soluble MMP-1 (red), MMP-2 (yellow), MMP-9 (pink), MMP-13 (purple), and MMP-12 (orange).
(D) Alignment of all human MMP sequences finds that the blade IV loop characteristic of membrane type MMPs is missing from the soluble MMPs. Parts of the corresponding loop from blade II can, however, be found in all MMPs.
See also Fig. S4.
Another approach led to a similar conclusion. The HPX domain was specifically labeled with a membrane-responsive fluor at single Cys substitutions (Koppisetti et al., 2014; Prior et al., 2015; Schulz et al., 2009). One site was placed in each of blades I, II, and IV, while three sites were selected in blade III (Fig. S3). Each single Cys was condensed with the iodoacetamide-substituted NBD fluorescent probe N,N’-dimethyl-N-(iodoacetyl)-N’-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)ethylenediamine) that responds to a hydrophobic environment with increased fluorescence emission (Schulz et al., 2009). Addition of SUVs to the IANBD conjugate at blade I (at D351C) or blade III (at N424C, N433C, or N446C) failed to increase significantly the fluorescence emission from the NBD (Fig. S3). Addition of SUVs to the HPX domain with IANBD conjugated to D385C in blade II or to D471C in blade IV increased the fluorescence emission significantly by 2.05-fold and 1.36 fold, respectively (Fig. S3). The greater increase from the label at D385C than from D471C is attributable to Asp385 being situated closer to the bilayer than is Asp471 in the structural models of Fig. 2.
MT-MMPs Are Distinguished by their EPGYPK/R Loops
A phylogenetic tree, constructed from each complement of MMP sequences obtained from 10 diverse vertebrate species, separates MT-MMPs from soluble MMPs (Fig. S4A). Evolutionary Trace analysis (Lichtarge et al., 1996) of this collection finds that most of the MT-MMPs containing a transmembrane helix (MT1-, MT2-, MT3- and MT5-MMP) also share in blades II and IV the hydrophobic PGYP loop motif flanked by polar residues (Figs. 3D, S4B). A majority of MMPs contain the GYP motif in blade II (Figs. 3D, S4B). In blade IV, MT1-MMP and most MT2-MMP sequences widen the motif to the full bilayer-binding EPGYPK peptide sequence, which is modified in MT3- and MT5-MMP sequences by an Arg replacement of the Lys (Figs. 3D, S4B). The crystal structure of the HPX domain of MT1-MMP differs from those of soluble MMPs in this loop bulging out from outer β-strand 4 of blade IV (Fig. 3C). The membrane-binding EPGYPK/R motif in blade IV appears to be a distinguishing characteristic of those MTMMPs that possess a transmembrane helix.
Vesicles Support Digestion of Collagen Triple-Helix
Streptococcal collagen–like protein 2 (Scl2) was engineered by inserting the collagenase-susceptible sequence from the α1 chain human collagen II (Fig. 4A), in a similar fashion as a collagen III sequence was inserted previously (Yu et al., 2012). Addition of SUVs kept sMT1-MMP proteolysis of the Scl2-collagen α1(II) linear from 1 to 6 h. Without vesicles, 30% less of the total substrate was digested at 6 h and 18 h (Fig. 4B,C). This might suggest the vesicles preserved the activity of the enzyme. For structural perspective, the coordinates of a collagen-model triple-helical peptide (THP) bound to the HPX domain (Zhao et al., 2015)(PDB: 2MQS) have been superimposed on those of the complexes with nanodiscs. If the binding is concurrent, blade IV is more likely to be oriented toward the lipid bilayer (Fig. 5A).
Figure 4. Vesicles Enhance Digestion of Engineered Streptococcus Collagen–Like Protein by the Ectodomain of MT1-MMP.

(A) The collagen mimic Scl2 comprises the trimerization domain V at the N-terminus and two S. pyogenes collagen-like CL domains. Six triplets from the α1 chain of human collagen II harboring the MMP-cleavable peptide bond (marked by ∧) were inserted between the CL domains.
(B) The time course of the digestion of Scl2-collagen α1(II) substrate (20 μM) by sMT1-MMP (20 nM) in the presence of SUVs (with 250 μM DMPC monomers) at 30°C was monitored by SDS-PAGE.
(C) The percentage of the Scl2-collagen α1(II) hydrolyzed in the absence or presence of the SUVs is plotted over time using circles or triangles, respectively. The uncertainties plotted are the SD of duplicate measurements.
See also Fig. S5.
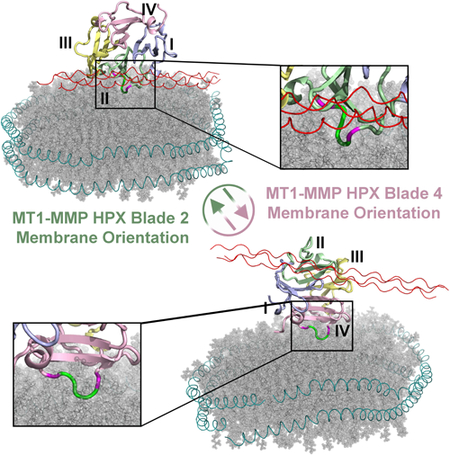
Figure 5. Hypothetical Assemblies of the HPX domain with Both the Collagen Triple-Helix and Lipid Bilayers.

(A) The solution structural model of the complex of a collagen triple-helical peptide with the HPX domain of MT1-MMP (Zhao et al., 2015) is superimposed on the PRE-based structural models presented in Fig. 2 that have blade IV in contact with the bilayer. The compatibility of bilayer and triple-helix binding favor this type of “ternary” complex.
(B) The superposition with experimental model with blade II in contact with the bilayer is plotted. The prospects of a steric clash between bilayer and triple-helix may disfavor formation of this type of “ternary” complex.
(C) The hypothesis of simultaneous binding of bilayers to both the blade II and IV interfaces and a collagen triple-helix to blade IV could be compatible with the “side-by-side” dimer in the crystallographic asymmetric unit of PDB: 3C7X. Green dots mark locations of outermost motifs of blades I and IV required for cell migration and proMMP-2 activation (Zarrabi et al., 2011).
We tested the effects of collagen-model THPs and vesicles upon the interactions of each with sMT1-MMP. Addition of SUVs enhanced the initial velocity of sMT1-MMP digestion of the fTHP-9 by 1.4-fold (Fig. S5A). Additions of the α1(I)772–786 THP to excess interfered in the proximity of sMT1-MMP to the SUVs, as observed by the FRET assay of quenching of Trp emission by Pyrene-PE in the SUVs (Fig. S5B). Competition of excess α1(I)772–786 THP and SUVs for binding blade II might be possible as illustrated (Fig. 5B).
Compatibility of Bilayer and Collagen Binding to Hypothesized HPX Dimers
The pericellular collagen digestion by MT1-MMP, characteristic of tumor cell invasion, requires membrane anchoring (Li et al., 2008) and could be facilitated by dimerization (Itoh et al., 2006). The isolated HPX domain at high concentration in physiological saline can form dimers (Tochowicz et al., 2011). Consequently, we considered the potential dimers and their compatibility with bilayer binding and collagen triple-helix binding, or lack thereof. For potential modes of dimerization with the largest of the small interfaces identified by PISA in the crystallographic asymmetric unit of the HPX domain (PDB: 3C7X), we considered the possibility of concurrent binding of a membrane to both protomers. A symmetric mode of dimerization, shaped like a “V”, is favored by one mutagenesis study (Tochowicz et al., 2011) and disputed by another (Zarrabi et al., 2011). The possibility of a “side-by-side” mode of dimerization appears to be compatible with mapping of interactions (Zarrabi et al., 2011), but has not been discussed. We computationally evaluated these two prospective modes of dimerization for compatibility with the measured proximity of the HPX domain to bilayers. We subjected each dimer to MD simulations with PRE-based distance restraints to a nanodisc (Table 1). We restrained one protomer via the PRE-based restraints which drew blade II to the bilayer. We simultaneously restrained the other protomer to the PREs placing blade IV at the bilayer. MD simulations of the symmetric dimer postulated (Tochowicz et al., 2011) using the same PRE-based restraints caused the partial unfolding of both blades II and IV (Fig. S6). The binding path of the collagen triple-helix (Zhao et al., 2015) clashes with the bilayer in the simulated assembly of the symmetric dimer with the nanodisc (Fig. S6A). Consequently, the symmetric dimer is probably not able to bind both collagen and membranes simultaneously via the HPX domain. On the other hand, the corresponding PRE-restrained MD simulation of the potential side-by-side dimer encountered neither structural perturbations (Movie S3) nor violations of the intermolecular distance restraints from Table 1 based on PREs. In the experimentally restrained simulation of the hypothetical side-by-side dimer, the protomer with blade IV at the bilayer presents an open binding site for the collagen triple-helix, without obstruction by the nanodisc (Fig. 5C and Movie S3).
DISCUSSION
MT-MMP HPX Domain Positioning at Membranes
These and recent measurements assert that the HPX domain of MT1-MMP occupies lipid bilayers at least transiently (Cerofolini et al., 2016; Van Doren et al., 2017). The Glu- and Gly-rich linker-2 sequence of ~25 residues connecting the HPX domain to the transmembrane helix of MT1-MMP (Itoh, 2015) appears long enough to reach the transmembrane helix for either mode of bilayer binding observed herein. With blade II bound, as few as about 14 of the linker-2 residues might suffice to reach the bilayer (Fig. S7A). With blade IV bound, the short distance to the bilayer could be spanned by 5 or 6 residues (Fig. S7B). Since the catalytic domain can also bind lipid bilayers (Van Doren et al., 2017), it may join the HPX domain transiently on bilayers, placing it strategically for proteolytic attack upon proteins anchored to plasma membranes or exosomes. This suggests that the common portrayal of the soluble domains of MT1-MMP radiating away from the membrane (Deryugina and Quigley, 2011; Turunen et al., 2017) should be revised. The recent structural question has been about the orientations between the soluble domains and bilayers, which is relevant to recognition of substrates and partners at plasma membranes.
The HPX domain of MT1-MMP was recently proposed to bind bilayered micelles (bicelles) primarily at blade III, based on the broadened NMR peaks of blade III but reaching as far as blade IV residues Lys490 and Glu494 (Cerofolini et al., 2016). The results herein confirm that Lys490 and Glu494 lie at an interface with bilayers. However, the previously hypothesized docking site does not agree with our results. Instead, the centering of the previous model on blade III results in greater steric conflict between the binding of bilayer and collagen. The previous modeling appears to rotate the HPX domain at least 50° away from the blade IV-centered complex herein (PDB: 6CLZ) and at least 150° from the blade II-centered complex herein (PDB: 6CM1). The negatively charged Ser-Glu sequence bulging from blade III is also unlikely to penetrate the phospholipid bilayer of DMPC vesicles and nanodiscs to the region of the phosphoesters and fatty acyl chains reached by the Pro-Gly motif from the EPGYPK sequence of blades II and IV. The fluorescence assays herein used 150 mM NaCl and pH 7.2 like previous NMR work (Cerofolini et al., 2016), while the new NMR results used 300 mM NaCl. These represent similar ionic conditions, as well as enhanced charge screening in PRE measurements. Two technical aspects of the previous work (Cerofolini et al., 2016) render it preliminary: First, the enrichment in CHAPS detergent of the 3:1 CHAPS:DMPC bicelles (1 – 2% w/v) does not mimic the membrane environment. They CHAPS-rich bicelles lack the integrity enjoyed by nanodiscs for structural and biophysical studies (Denisov and Sligar, 2016; Rouck et al., 2017). Second, reliance only upon NMR peak perturbations for interface mapping can be subject to systematic errors. The perturbations can propagate far beyond the interface (Arumugam et al., 1998; Takeda et al., 2003). That literature suggested the higher accuracy of more direct measures of interfacial contact.
The PRE NMR approach employed here, implicating blades II and IV as the interfaces (Figs. 1, 2), provides sensitive measures of intermolecular proximity in the 12 to 22 Å range, even in cases of partial occupation of the sites (Clore et al., 2007; Koppisetti et al., 2014; Prior et al., 2016). Site-specific labeling with a membrane-responsive fluor provides independent proximity information (Koppisetti et al., 2014; Prior et al., 2015; Schulz et al., 2009) corroborating blades II and IV being central to the interfaces with vesicles (Fig. S3). Site-directed mutants found that uncharged replacements of lysine side chains in blades II and IV interfered in association with vesicles, while those in blade III did not (Fig. 3A,B). This evidence for electrostatic attraction corroborates the appearance of multiple salt bridges from blade II or IV to lipid phosphoester linkages persisting through the NMR-restrained MD simulations (Table 2).
Potential Scope of Bilayer Binding by MMP HPX Domains
The membrane-binding EPGYP peptide motif of blade IV of the MT-MMPs possessing a transmembrane helix distinguishes them from most other MMPs (Figs. 3D, S4B). The known structures of soluble MMPs lack such a loop bulging from blade IV (Fig. 3C), rendering them unlikely to insert blade IV into lipid bilayers. However, the APGYP motif in blade IV of the GPI-anchored MT4-MMPs is similar enough to the EPGYP motif to hypothesize that MT4-MMPs may also be able to penetrate membrane surfaces. Likewise, the SPGFPK/M motif in blade II of soluble MMP-19 (groups 16 and 17 in Fig. S4B) might have the ability to insert among phospholipid head groups. The EPNYPK motif of blade II of soluble MMP-12 (Fig. 3D) also suggests the capacity for membrane association.
The MMP-12 HPX domain binds and kills bacteria using its KDEK loop in mice (KDDK in humans) located in blade II (Houghton et al., 2009). This β-hairpin loop is in close proximity to the EPNYPK loop which we hypothesize to bind bilayers. The 375KDGK378 peptide sequence of MT1-MMP shares homology with the bactericidal KDEK loop in blade II of MMP-12. Lys378 forms a salt bridge with the Glu396 of the EPGYP motif throughout the structural ensemble in which blade II of MT1-MMP is bound to the nanodisc. Asp376 and Lys378 of this loop in MT1-MMP approach a choline head group throughout the ensemble. Consequently, the corresponding bactericidal KDE/DK loop of MMP-12 can be hypothesized to contact bilayer or protein components of bacterial envelopes.
Membrane Positioning and Partner Interactions
Addition of a 20-fold molar excess of the THP over sMT1-MMP competes out about 25% of the association of sMT1-MMP with vesicles (Figure S5B). This partial competition can be explained by steric interference of bound THP with blade II binding of bilayers, along with retention of blade IV binding of bilayers (Fig. 5A,B). This suggests the orientation of Fig. 5A to be the more likely during collagenolysis.
In invadopodia, the anchorage of MT1-MMP to the actin cytoskeleton (von Nandelstadh et al., 2014) might promote its dimerization or oligomerization there. If homodimers of the HPX domain form on the surface of model membranes, the side-by-side positioning in the crystallographic asymmetric unit appears to be the hypothetical mode of homodimerization that is more compatible with the measured proximity to bilayers and the binding of the collagen triple-helix (Fig. 5C). If there is side-by-side dimerization of the HPX domain, it could place blade I of one protomer together with blade III of the next protomer in a head-to-tail fashion that could propagate to add more protomers. This possibility might foster the homo-oligomerization of MT1-MMP on cells that was reported (Itoh and Seiki, 2006; Lehti et al., 2002). Such head-to-tail homodimerization via blades I and III (Fig. 5C) would provide the accessibility of the outer β-strand and bulge of blade I needed for both proMMP-2 activation and homodimerization by MT1-MMP (Zarrabi et al., 2011).
The β-bulges on the surfaces of blades I and IV of MT1-MMP support tumor cell migration, invasion, and angiogenesis (Zarrabi et al., 2011). The β-bulge of blade I is exposed to binding partners when bound to bilayers, regardless of homodimerization of the HPX domain (Figs. 1B, 2D, 5C, S6A). Blade I is the site of heterodimerization with CD44 (Zarrabi et al., 2011). Since CD44 is a transmembrane proteoglycan and substrate of MT1-MMP, the positioning of the HPX domain on the plasma membrane by either blade II or IV could facilitate the association leading to shedding of CD44 to its soluble form, which results in activation of EGFR signaling in cell migration (Zarrabi et al., 2011). Alteration of the outer strand of blade IV interfered with proMMP-2 activation, cell migration, EGFR signaling, and metastasis, which was attributed to disruption of homodimerization (Zarrabi et al., 2011). However, the defects do not seem attributable directly to interference in homodimerization because the outer strand of blade IV is not part of either dimer interface suggested by crystallography. Nonetheless, the outer strand of blade IV now appears important for positioning of the HPX domain via its shallow insertion into membranes (Figs. 2D–F). This makes binding sites on the HPX domain accessible for interaction with key substrates in cell migration and metastasis, i.e. CD44, collagen (Fig. 5A), and presumably others such as syndecan-1.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Steven R. Van Doren (vandorens@missouri.edu).
METHOD DETAILS
Preparation of sMT1-MMP and its HPX Domain
Soluble MT1-MMP constructs were expressed and labeled as described (Zhao et al., 2015). Samples for NMR PRE experiments were grown in a 13C/1H-labeled deuterated E. coli culture using an established protocol (Tzeng et al., 2012). 50 mg/L of α-ketobutyrate and 100 mg/L of α-ketoisovalerate were added 1 h prior to induction for 13C/1H-labeling of isoleucine, valine, and leucine methyl groups in a 2H/15N background. The culture was optimized by the addition of 2% 2H/15N rich labeling media (ISOGRO, Sigma-Isotec). Post-induction growth was continued for 8 h and then cells were harvested and sonicated to collect the pellet of inclusion bodies.
Inclusion bodies containing the His-tagged HPX domain or sMT1-MMP were dissolved with 6 M GuHCl, dialyzed overnight with 6 M urea, and spun down to remove any precipitates. The His-tagged HPX domain was bound to a nickel-nitriloacetic acid (NTA) affinity column in 20 mM Tris-HCl (pH 7.2), 1 M NaCl, and 6 M urea and washed with 20 mM imidazole. Folding occurred while bound to the nickel-NTA affinity resin during a linear gradient to change the 6 M urea into a final buffer containing 20 mM Tris-HCl, 1 M NaCl, and 20 mM imidazole at pH 7.2. The flow rate was 1 ml/min. The HPX domain was eluted with 300 mM imidazole and dialyzed overnight to remove imidazole and NaCl.
His-tagged sMT1-MMP was purified in 6M urea using nickel-NTA affinity resin and then folded by stepwise denaturant removal by dialysis in 3 M, 1 M, and then 0 M urea containing 20 mM Tris-HCl (pH 7.2), 10 mM CaCl2, and 150 mM NaCl and later made active by the addition of 5 mM ZnCl2.
Preparation of Nanodiscs for NMR
Nanodiscs were formed with MSP1D1 scaffolding protein and 1,2-dimyristoyl-snglycero-3-phosphocholine (DMPC, Avanti) as described (Denisov et al., 2004; Schuler et al., 2013). Size exclusion chromatography separated the predominantly monodisperse nanodiscs from small amounts of self-associated nanodiscs. This employed either a Tosoh Haas TSK G4000 PWXL column (7.8-mm inner diameter × 30 cm) or a HiLoad 16/60 Superdex pg column (GE Healthcare). The nanodiscs were used immediately without freezing and concentrated in 1:1 molar ratio with the HPX domain using an Amicon Ultra-4 centrifugal filter (EMD Millipore) with 10,000 Da cutoff. When used, 5- or 10-doxyl DPPC was incubated with DMPC prior to forming nanodiscs in order to ensure full incorporation of the spin label. Samples were prepared for NMR typically in 20 mM Tris (pH 7.2) and 300 mM NaCl.
NMR Spectroscopy and Paramagnetic Relaxation Enhancements
Spectra were recorded using Bruker shaped tubes or 3 mm tubes to optimize sensitivity by mitigating the dielectric effects of the high [NaCl]. Spectra were acquired at 30°C on a Bruker Avance III 800 MHz Spectrometer with a TCI cryoprobe. Chemical shift assignments of NMR peaks of these complexes were analyzed by CCPNMR analysis (Vranken et al., 2005) and deposited in the BioMagResBank under accession codes 30425 and 30426, respectively.
PREs emanating from nitroxide spin-labels were recorded in the presence of 5-or 10-doxy-substituted PC. Five relaxation time points were collected at 4 ms intervals using a CPMG pulse train inserted into a13C HMQC (or 15N TROSY) pulse sequences as described (Koppisetti et al., 2014). The peak heights were normalized to those of the diamagnetic control using the signal to noise ratio of each spectrum. Curve fitting of the 5-point decays included the 6.8 ms of relaxation delays during the HSQC pulse sequence and prior to the CPMG pulse sequence (Fig. S1D). This decreased systematic fitting error, thereby increasing apparent accuracy of the quantification of the PREs. Exponential decays were measured and compared to diamagnetic controls without spin label in order to estimate the PRE as:
| (1) |
The amide chemical shift perturbations, measured from BEST-TROSY spectra (Lescop et al., 2010), are expressed as a radius with the 15N peak shifts normalized to the scale of 1H peak shifts:
| (2) |
Calculations to Dock the HPX domain to Bilayer Models
Due to the free diffusion of the DPPC in bilayers, the average depth of the 5-doxyl or 10-doxyl substituent in the nanodisc was used to represent this broad, average source of the PREs. The apparent averaged distance r between the methyl group detected by NMR and the plane representing the average depth of the nitroxide spin label was estimated by the approximate relationship:
| (3) |
where κ =1.23e-44 m6 s−2 and τc is the rotational correlation time which calibrates the relationship of the PRE to the distance r. In order to estimate τc, we measured the average of the amide 15N NMR transverse cross-correlation rates ηxy using the pulse sequence of (Liu and Prestegard, 2008) implemented as a exponentially decaying series of 1D spectra which we integrated and fitted to ηxy. This ηxy yielded the approximate τc using spectral density equations available (Lee et al., 2006). The uncertainty of each distance restraint was set to range from a lower bound of the closest possible contact of hard spheres up to an upper bound 10% above the apparent distance plus 2.9 Å of additional uncertainties from the depth of the doxyl spin label and ambiguity in proR vs. proS methyl groups of the Val and Leu side chains.
The HPX domain and phospholipids were docked as rigid bodies with HADDOCK 2.1 (de Vries et al., 2010) using the NMR-based estimates of proximity to the DMPC bilayer (Koppisetti et al., 2014). The PRE-derived distance restraints between affected methyl groups and the average depth of the 5-doxyl spin label were implemented in HADDOCK as ambiguous restraints satisfied by proximity to the C1E atom of an acyl chain of any of the DMPC molecules. Likewise, the distance restraints from PREs from the 10-doxyl spin label were satisfied by proximity to the C1J atom of an acyl chain of any of the DMPC molecules. The protein amide peaks with shifts (CSPs) or broadenings by the nanodiscs were restrained ambiguously to any lipid atom approaching within 20 Å in initial docking with HADDOCK.
The best-converged structural models from rigid body docking calculations in HADDOCK were used for MD refinement. Nanodiscs were modeled using the CHARMMM_GUI website (Jo et al., 2008) and aligned to the DMPC bilayer from rigid body docking. This complex was used as the starting structural model for refinement by restrained MD simulations in NAMD (Phillips et al., 2005) and analyzed through the VMD (Humphrey et al., 1996) and Pymol (DeLano, 2002) software packages. Distance restraints from the average depth of the 5-doxyl or 10-doxyl substituent of the spin-labeled PC were implemented in NAMD using an xy-plane collective variable named a colvar. The two xy-planes were chosen to pass through the average position of the fifth and tenth acyl carbons, respectively, among 30 DMPC molecules on the edges of the nanodiscs. In the colvar, a harmonic restraint with force constant of 1.0 was placed on the distance estimate to the PRE-broadened methyl group in the HPX domain. Ambiguous distances to amide groups based on CSPs and NMR line broadenings were restrained with colvars with force constant of 0.1 to within 22 Å of the xy-plane passing through the average position of the nitrogen of each choline head group of the same 30 lipids used for the PRE-based distance restraints. 2 ns of minimization and equilibration along with 4 ns of production time were run at 310K with rectangular boxed solvent and ions using NAMD 2.1 with CUDA GPU processing (Phillips et al., 2005). The lowest energy structures were selected from 1 ns segments of the equilibrated trajectories, provided the frames were separated by at least 20 ps. The trajectories were run for another 5 ns without restraints for a total of 11 ns at 310 K to verify stability of the assembly. Models of hypothetical dimers of the HPX domain docked with nanodiscs were prepared by alignment with the lowest energy structural models with blade II and IV at the interface. These starting dimer models were minimized and equilibrated for 4 ns. Lowest energy structures from the subsequent equilibrated 4 ns of the trajectory were used for visualization.
Back-calculation of PREs
The coordinates of the nanodisc-HPX domain ensembles were used to back-calculate Γ2 values for comparison with the Γ2 values from NMR. The distances between Ile/Leu/Val methyl groups and the average depth of the fifth or tenth acyl carbon of the DMPC molecules in the nanodisc were used as an imperfect representation of the fluctuating distances to the rapidly diffusible PC spin-labeled with a 5-doxyl or 10-doxyl group, respectively. These distances were used to back-calculate Γ2 values using eq. 3. Back-calculated Γ2 values were plotted against measured Γ2 values deemed experimentally significant by their amplitude exceeding the uncertainties by at least 2-fold. The error bars of back-calculated Γ2 values in these plots reflect the variability of the distances in the 15-member structural ensembles. Uncertainties in measured Γ2 are derived from the curve fits. The back-calculated Γ2 values might be overestimated by not accounting for the averaging with sample populations with spin-labeled PC distant due to its diffusion within the nanodisc or due to dissociation of the HPX domain. The closest approaches are nonetheless dominant in determining Γ2 because of its proportionality to r−6 (eq. 3).
Intrinsic Tryptophan Quenching Assays of Membrane Proximity
Single site-directed mutations were made in sMT1-MMP via QuikChange with PCR master mix (Agilent Technologies). SUVs were prepared by suspending DMPC monomers in 20 mM Tris (pH 7.2), with or without addition of lipid probes. After an hour of incubation at 37°C, the hydrated lipids were subjected to multiple freeze-thaw cycles in liquid nitrogen, forming large unilamellar vesicles. Subsequent sonication of these large unilamellar vesicles led to the formation of SUVs. Binding of sMT1-MMP (1 μM) to SUVs of DMPC was determined through the decrease of the intrinsic tryptophan emission (λex = 285 nm, λem = 315 nm) in the absence and presence of a lipid probe. 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(1-pyrenesulfonyl) lipid (Pyrene-PE, Avanti) was incorporated as a quencher of protein intrinsic emission. All experiments were performed on a BioTek Synergy MX plate reader. Emission was measured after an incubation period and background fluorescence was corrected through subtraction of just Pyrene-PE liposomes in the buffer composed of 20 mM Tris (pH 7.2), 150 mM NaCl, 10 mM CaCl2, and 100 μM ZnCl.
Site-Directed Fluorescence to Probe Bilayer Insertion
Site directed cysteine mutations were introduced via QuikChange with PCR master mix (Agilent). Conjugation of the fluor sensitive probe IANBD (Invitrogen) to the single Cys proceeded by incubating a 10-fold excess of IANBD to overnight at 25°C in a vacuum chamber. Unreacted IANBD was removed by desalting with Sephadex G-25 resin (GE Healthcare). Completion of conjugation was monitored by absorbance.
The fluorescence assays used 10 nM of the IANBD-conjugated sMT1-MMP with SUVs that were 250 μM in DMPC monomers. The fluorochrome was excited at 478 nm and detected at 541 nm in a BioTek Synergy MX plate reader. Emission was measured after 1 h of incubation with the SUVs in the assay buffer of 20 mM Tris (pH 7.2), 150 mM NaCl. The emission with SUVs was normalized by the emission without SUVs.
Preparation and Proteolysis of Bacterial Scl2 Collagen-like Substrate
The recombinant bacterial collagen with its domain structure of V-CL-CL and inserted sequence from type II collagen is illustrated in Figure 4A. The six GXY triplets with the sequence GPPGPQG^LAGQRGIVGLP from the human collagen α1(II) chain were inserted between two collagen-like (CL) domains of the Streptococcal Scl2 protein, with the caret sign denoting the site for hydrolysis by collagenolytic MMPs. This six-triplet sequence corresponds to residues 769–786 of the triple-helical region in human type II collagen α1 chain. An N-terminal octa-His tag was included for purification. It is followed by the natural trimerization V domain of the bacterial collagen. The expression construct was obtained by inserting the annealed oligonucleotide encoding the MMP-cleavable collagen α1(II) sequence between the DNA sequences of the two CL domains using SmaI and ApaI restriction sites. The inserted human MMP-cleavable site is flanked by bacterial collagen sequences of GKD-GKD-GQP-GKP on the N-terminal side and GPRGEQ-GPT-GPT on the C-terminus (Yu et al., 2012). The resulting DNA sequence was confirmed by sequencing. Recombinant protein was expressed using the cold-shock vector system in the E. coli BL21 strain, as reported (An et al., 2014).
The sMT1-MMP was activated for use in proteolysis assays by introducing the Zn2+ and Ca2+ ions required for activity. 20 nM sMT1-MMP was incubated at 30°C for 30 min in 20 mM Tris (pH 7.2), 150 mM NaCl, 10 mM CaCl2, and 100 μM ZnCl both in the presence and absence of SUVs (250 μM in DMPC monomers). The proteolysis reactions were started by adding the aforementioned recombinant bacterial Scl II -collagen α1(II) substrate to 20 μM to the activated sMT1-MMP (20 nM). Aliquots were removed from the proteolysis mixture at several time points and analyzed by SDS-PAGE. Intact bands migrating with similar mobility as the 74 kDa marker were analyzed using ImageJ software. Background noise was subtracted and the initial time point provided the reference intensity.
Kinetics of sMT1-MMP Hydrolysis of Collagen-model Triple-Helical Peptide
The proenyzme form of sMT1-MMP (pro-sMT1-MMP) was obtained from R&D Systems (catalog # 918-MP) and activated by incubation with 1 mg/mL of activated rhTrypsin-3 (Sigma-Aldrich) for 1 h at 37˚C in TS buffer, i.e., 50 mM Tris (pH 7.5), 50 mM NaCl, 10 mM CaCl2, 0.05% Brij-35. After MT1-MMP activation, the remaining trypsin-3 activity was quenched by addition of 1 mM AEBSF (R&D Systems) and incubation for 15 min at room temperature. Activated enzyme was aliquoted and stored at −80˚C for further use. The triple-helical substrate fTHP-9 [(Gly-Pro-Hyp)5-Gly-Pro-Lys(Mca)-Gly-Pro-Gln-Gly^Cys(Mob)-Arg-Gly-Gln-Lys(Dnp)-Gly-Val-Arg-(Gly-Pro-Hyp)5-NH2, where Hyp = 4-hydroxyproline, Mca = (7-methoxycoumarin-4-yl) acetyl, Mob = 4-methoxybenzyl, and Dnp = 2,4-dinitrophenyl] was synthesized as described previously (Minond et al., 2004).
Enzyme kinetics were measured in a BioTek H1 plate reader running Gen5 2.09 software using λ excitation = 324 nm and λ emission = 393 nm as described previously (Lauer-Fields et al., 2008; Palmier and Van Doren, 2007). To allow the proper folding of the peptide triple-helix, substrate fTHP-9 was diluted in TS Buffer and left overnight at 4 °C. The next day, 1 mM DMPC stock was prepared in T S buffer for the assay and further diluted into 50 μl reactions over a series of concentrations. The sMT1-MMP was added to each tube containing DMPC and incubated for 1 h at room temperature. All enzymatic assays were performed in a black, low-volume 384 well plates. 10 μL of activated sMT1-MMP + DMPC (or sMT1-MMP + TS buffer as control) was added to each well and then 5 μL of substrate fTHP-9 was dispensed into each well. Final enzyme and substrate concentrations were 1.8 nM and 1.7 μM, respectively. The assays were performed in the absence and presence of SUVs ranging from 100 to 800 μM DMPC monomers. The plate was immediately read to obtain fluorescence in relative fluorescence units (RFUs). The fluorescence was monitored continuously for 180 min to determine initial reaction rates. The kinetic protocol at 25 °C used 30 s of shaking followed by reading each well every 30 s. Plates were stored at ambient temperature for 24 h before a final reading. All measurements were performed in triplicate. The effects of the SUVs upon relative initial velocities were compared.
Assays of Competition between THP and SUV Binding to sMT1-MMP
A similar THP, but without fluor or quencher, was synthesized with the sequence of ((Gly-Pro-Hyp)4-Gly-Pro-Gln-Gly-Ile-Ala-Gly-Gln-Arg-Gly-Val-Val-Gly-Leu-Hyp-(Gly-Pro-Hyp)4-Gly-Tyr-NH2)3 as described (Lauer-Fields et al., 2009). It is designated α1(I)772–786 THP because it corresponds to residues 772–786 of the type I collagen α1 chain. α1(I)772–786 THP was added to excess (1, 2, or 4 μM) over the sMT1-MMP (200 nM) used in assays of membrane proximity measured by intrinsic tryptophan quenching by Pyrene-PE, performed as described above. The SUVs had a DMPC monomer concentration of 250 μM and the Pyrene-PE quencher present at 5 μM.
Negative Stain Electron Microscopy
500 nM sMT1-MMP was mixed 1:1 with nanodiscs composed of DMPC and MSP1D1 in 20 mM Tris (pH 7.2), 10 mM CaCl2, and 100 μM ZnCl. 5 μl of this sample was placed immediately on freshly glow-discharged, carbon-coated copper 200-mesh grids (Pelco Easiglow, 15 mA, 45 s, 0.39 mBar), blotted with Whatman #1 filter paper to remove excess liquid and stained with 5 μl Nano-W (Nanoprobes). Nanodiscs alone (665 nM) were applied similarly to other grids for comparison. These grids were then imaged on a JEOL JEM 1400 Transmission Electron Microscope and analyzed further using ImageJ.
Evolutionary Trace Analysis
Sequences spanning the HPX-like domains of MMPs were acquired and aligned from Blastp https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins, using the blastp algorithm and spanning 10 diverse species of vertebrates. After the removal of redundancies and truncated sequences, 102 aligned sequences were submitted to the Evolutionary Trace server at http://mordred.bioc.cam.ac.uk/. The resulting phylogenetic tree was subdivided into 15 trace levels that separate the branches of the tree by progressively high sequence identity within each branch. Residues conserved within a subfamily at a selected trace level were recorded.
QUANTIFICATION AND STATISTICAL ANALYSIS
The error bars plotted in Fig. 1A are derived from the uncertainty of the fits and propagation of the error through the differences taken. In Fig. 3B and 4C, the triplication and duplication, respectively, used to estimate each mean and SD is given in the legends. Table 1 lists for the 15-member ensembles the uncertainties of the atomic coordinates, the violation of a distance restraint across an ensemble, and the areas buried in each bilayer-protein interface. Table 2 quantifies the proportion of the structural models from each 15-member ensemble displaying each apparent salt bridge.
DATA AND SOFTWARE AVAILABILITY
The coordinates of the complexes of the HPX domain with DMPC-containing nanodiscs are deposited in the RCSB Protein Data Bank under accession codes 6CM1 and 6CLZ for the ensembles with blade II or IV in the interface, respectively. The corresponding NMR peak assignments are deposited in the BioMagResBank under accession codes 30426 and 30425, respectively.
Supplementary Material
Movie S1. Related to Fig. 2. MD Trajectory with Blade II Associated with Nanodisc.
Movie S2. Related to Fig. 2. MD Trajectory with Blade IV Associated with Nanodisc.
Movie S3. Related to Fig. 5. MD Trajectory of Hypothetical Dimer with Blades II and IV Interacting with Nanodisc.
Highlights.
Blades II and IV insert into lipid head groups a loop characteristic of some MT-MMPs
Basic side chains at the bilayer binding sites are drawn to lipid phosphoesters
Vesicles enhance digestion of collagen-like substrates by the MT1-MMP ectodomain
Binding of bilayers, collagen, CD44, and possibly dimerization are compatible
Acknowledgments
This work was supported by NIH grant R01 CA098799. B.A. was supported by R01 GM60048. The 800 MHz spectrometer was purchased by NIH grant S10 RR022341 and funds from the University of Missouri. We are most grateful to Barbara Brodsky for the bacterial collagen substrate, Tommi A. White for providing training in negative staining EM, and Alexander W.E. Franz and Linda L. Randall for detailed comments on the manuscript and support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
REFERENCES
- Albrechtsen R, Stautz D, Sanjay A, Kveiborg M, and Wewer UM (2011). Extracellular engagement of ADAM12 induces clusters of invadopodia with localized ectodomain shedding activity. Exp. Cell Res 317, 195–209. [DOI] [PubMed] [Google Scholar]
- Amar S, Smith L, and Fields GB (2017). Matrix metalloproteinase collagenolysis in health and disease. Biochim. Biophys. Acta 1864, 1940–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An B, Abbonante V, Yigit S, Balduini A, Kaplan DL, and Brodsky B (2014). Definition of the Native and Denatured Type II Collagen Binding Site for Fibronectin Using a Recombinant Collagen System. J. Biol. Chem 289, 4941–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam S, Hemme CL, Yoshida N, Suzuki K, Nagase H, Berjanskii M, Wu B, and Van Doren SR (1998). TIMP-1 contact sites and perturbations of stromelysin 1 mapped by NMR and a paramagnetic surface probe. Biochemistry 37, 9650–9657. [DOI] [PubMed] [Google Scholar]
- Basu B, Correa de Sampaio P, Mohammed H, Fogarasi M, Corrie P, Watkins NA, Smethurst PA, English WR, Ouwehand WH, and Murphy G (2012). Inhibition of MT1-MMP activity using functional antibody fragments selected against its hemopexin domain. Int. J. Biochem. Cell Biol 44, 393–403. [DOI] [PubMed] [Google Scholar]
- Bode W (1995). A helping hand for collagenases: the haemopexin-like domain. Structure 3, 527–530. [DOI] [PubMed] [Google Scholar]
- Cerofolini L, Amar S, Lauer JL, Martelli T, Fragai M, Luchinat C, and Fields GB (2016). Bilayer Membrane Modulation of Membrane Type 1 Matrix Metalloproteinase (MT1-MMP) Structure and Proteolytic Activity. Sci Rep 6, 29511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TH, Sabeh F, Ota I, Murphy H, McDonagh KT, Holmbeck K, Birkedal-Hansen H, Allen ED, and Weiss SJ (2004). MT1-MMP-dependent neovessel formation within the confines of the three-dimensional extracellular matrix. J. Cell Biol 167, 757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM, Tang C, and Iwahara J (2007). Elucidating transient macromolecular interactions using paramagnetic relaxation enhancement. Curr. Opin. Struct. Biol 17, 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier IE, Legant W, Marmer B, Lubman O, Saffarian S, Wakatsuki T, Elson E, and Goldberg GI (2011). Diffusion of MMPs on the surface of collagen fibrils: the mobile cell surface-collagen substratum interface. PLoS One 6, e24029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GE, and Saunders WB (2006). Molecular balance of capillary tube formation versus regression in wound repair: role of matrix metalloproteinases and their inhibitors. J. Investig. Dermatol. Symp. Proc 11, 44–56. [DOI] [PubMed] [Google Scholar]
- de Vries SJ, van Dijk M, and Bonvin AM (2010). The HADDOCK web server for data-driven biomolecular docking. Nat Protoc 5, 883–897. [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002). The PyMOL Molecular Graphics System. (Palo Alto, CA, DeLano Scientific; ). [Google Scholar]
- Denisov IG, Grinkova YV, Lazarides AA, and Sligar SG (2004). Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J. Am. Chem. Soc 126, 3477–3487. [DOI] [PubMed] [Google Scholar]
- Denisov IG, and Sligar SG (2016). Nanodiscs for structural and functional studies of membrane proteins. Nat Struct Mol Biol 23, 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deryugina EI, and Quigley JP (2011). The Role of Matrix Metalloproteinases in Cellular Invasion and Metastasis In Extracellular Matrix Degradation, Parks WC, and Mecham RP, eds. (Berlin: Springer-Verlag; ), pp. 145–191. [Google Scholar]
- Deryugina EI, Ratnikov BI, Postnova TI, Rozanov DV, and Strongin AY (2002). Processing of integrin alpha(v) subunit by membrane type 1 matrix metalloproteinase stimulates migration of breast carcinoma cells on vitronectin and enhances tyrosine phosphorylation of focal adhesion kinase. J. Biol. Chem 277, 9749–9756. [DOI] [PubMed] [Google Scholar]
- Dominguez C, Boelens R, and Bonvin AM (2003). HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc 125, 1731–1737. [DOI] [PubMed] [Google Scholar]
- Egawa H, and Furusawa K (1999). Liposome adhesion on mica surface studied by atomic force microscopy. Langmuir 15, 1660–1666. [Google Scholar]
- Endo K, Takino T, Miyamori H, Kinsen H, Yoshizaki T, Furukawa M, and Sato H (2003). Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J. Biol. Chem 278, 40764–40770. [DOI] [PubMed] [Google Scholar]
- Fisher KE, Sacharidou A, Stratman AN, Mayo AM, Fisher SB, Mahan RD, Davis MJ, andDavis GE (2009). MT1-MMP- and Cdc42-dependent signaling co-regulate cell invasion and tunnel formation in 3D collagen matrices. J. Cell Sci 122, 4558–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, and Wolf K (2008). Tube travel: the role of proteases in individual and collective cancer cell invasion. Cancer Res 68, 7247–7249. [DOI] [PubMed] [Google Scholar]
- Ganguly B, Banerjee J, Elegbede AI, Klocke DJ, Mallik S, and Srivastava DK (2007). Intrinsic selectivity in binding of matrix metalloproteinase-7 to differently charged lipid membranes. FEBS Lett 581, 5723–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles C, Polette M, Coraux C, Tournier JM, Meneguzzi G, Munaut C, Volders L, Rousselle P, Birembaut P, and Foidart JM (2001). Contribution of MT1-MMP and of human laminin-5 γ2 chain degradation to mammary epithelial cell migration. J. Cell Sci 114, 2967–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagn F, Etzkorn M, Raschle T, and Wagner G (2013). Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J. Am. Chem. Soc 135, 1919–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakulinen J, Sankkila L, Sugiyama N, Lehti K, and Keski-Oja J (2008). Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J. Cell. Biochem 105, 1211–1218. [DOI] [PubMed] [Google Scholar]
- Houghton AM, Hartzell WO, Robbins CS, Gomis-Ruth FX, and Shapiro SD (2009). Macrophage elastase kills bacteria within murine macrophages. Nature 460, 637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, and MacKerell AD Jr. (2013). CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem 34, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD: visual molecular dynamics. J. Mol. Graph 14, 33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- Itoh Y (2015). Membrane-type matrix metalloproteinases: Their functions and regulations. Matrix Biol 44–46, 207–223. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Ito N, Nagase H, Evans RD, Bird SA, and Seiki M (2006). Cell surface collagenolysis requires homodimerization of the membrane-bound collagenase MT1-MMP. Mol. Biol. Cell 17, 5390–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Ito N, Nagase H, and Seiki M (2008). The second dimer interface of MT1-MMP, the transmembrane domain, is essential for ProMMP-2 activation on the cell surface. J. Biol. Chem 283, 13053–13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Palmisano R, Anilkumar N, Nagase H, Miyawaki A, and Seiki M (2011). Dimerization of MT1-MMP during cellular invasion detected by fluorescence resonance energy transfer. Biochem. J 440, 319–326. [DOI] [PubMed] [Google Scholar]
- Itoh Y, and Seiki M (2006). MT1-MMP: a potent modifier of pericellular microenvironment. J. Cell. Physiol 206, 1–8. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Takamura A, Ito N, Maru Y, Sato H, Suenaga N, Aoki T, and Seiki M (2001). Homophilic complex formation of MT1-MMP facilitates proMMP-2 activation on the cell surface and promotes tumor cell invasion. EMBO J 20, 4782–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer V, and Im W (2008). CHARMM-GUI: a web-based graphical user interface for CHARMM. [DOI] [PubMed]
- Kajita M, Itoh Y, Chiba T, Mori H, Okada A, Kinoh H, and Seiki M (2001). Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J. Cell Biol 153, 893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppisetti RK, Fulcher YG, Jurkevich A, Prior SH, Xu J, Lenoir M, Overduin M, and Van Doren SR (2014). Ambidextrous binding of cell and membrane bilayers by soluble matrix metalloproteinase-12. Nature communications 5, 5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshikawa N, Giannelli G, Cirulli V, Miyazaki K, and Quaranta V (2000). Role of cell surface metalloprotease MT1-MMP in epithelial cell migration over laminin-5. J. Cell Biol 148, 615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshikawa N, Mizushima H, Minegishi T, Iwamoto R, Mekada E, and Seiki M (2010). Membrane type 1-matrix metalloproteinase cleaves off the NH 2-terminal portion of heparin-binding epidermal growth factor and converts it into a heparin-independent growth factor. Cancer Res 70, 6093–6103. [DOI] [PubMed] [Google Scholar]
- Lauer-Fields JL, Chalmers MJ, Busby SA, Minond D, Griffin PR, and Fields GB (2009). Identification of specific hemopexin-like domain residues that facilitate matrix metalloproteinase collagenolytic activity. J. Biol. Chem 284, 24017–24024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer-Fields JL, Minond D, Chase PS, Baillargeon PE, Saldanha SA, Stawikowska R, Hodder P, and Fields GB (2008). High throughput screening of potentially selective MMP-13 exosite inhibitors utilizing a triple-helical FRET substrate. Bioorg. Med. Chem [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Hilty C, Wider G, and Wuthrich K (2006). Effective rotational correlation times of proteins from NMR relaxation interference. J. Magn. Reson 178, 72–76. [DOI] [PubMed] [Google Scholar]
- Lehti K, Lohi J, Juntunen MM, Pei D, and Keski-Oja J (2002). Oligomerization through hemopexin and cytoplasmic domains regulates the activity and turnover of membrane-type 1 matrix metalloproteinase. J. Biol. Chem 277, 8440–8448. [DOI] [PubMed] [Google Scholar]
- Lenoir M, Coskun U, Grzybek M, Cao X, Buschhorn SB, James J, Simons K, and Overduin M (2010). Structural basis of wedging the Golgi membrane by FAPP pleckstrin homology domains. EMBO Rep 11, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenoir M, Ustunel C, Rajesh S, Kaur J, Moreau D, Gruenberg J, and Overduin M (2018). Phosphorylation of conserved phosphoinositide binding pocket regulates sorting nexin membrane targeting. Nature communications 9, 993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescop E, Kern T, and Brutscher B (2010). Guidelines for the use of band-selective radiofrequency pulses in hetero-nuclear NMR: example of longitudinal-relaxation-enhanced BEST-type 1H-15N correlation experiments. J. Magn. Reson 203, 190–198. [DOI] [PubMed] [Google Scholar]
- Li XY, Ota I, Yana I, Sabeh F, and Weiss SJ (2008). Molecular dissection of the structural machinery underlying the tissue-invasive activity of membrane type-1 matrix metalloproteinase. Mol. Biol. Cell 19, 3221–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtarge O, Bourne HR, and Cohen FE (1996). An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol 257, 342–358. [DOI] [PubMed] [Google Scholar]
- Liu Y, and Prestegard JH (2008). Direct measurement of dipole-dipole/CSA cross-correlated relaxation by a constant-time experiment. J. Magn. Reson 193, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcink TC, Koppisetti RK, Fulcher YG, and Van Doren SR (2017). Mapping Lipid Bilayer Recognition Sites of Metalloproteinases and Other Prospective Peripheral Membrane Proteins In Matrix Metalloproteases: Methods and Protocols, Galea CA, ed. (New York, NY: Springer New York; ), pp. 61–86. [DOI] [PubMed] [Google Scholar]
- Mazhab-Jafari MT, Marshall CB, Stathopulos PB, Kobashigawa Y, Stambolic V, Kay LE, Inagaki F, and Ikura M (2013). Membrane-dependent modulation of the mTOR activator Rheb: NMR observations of a GTPase tethered to a lipid-bilayer nanodisc. J. Am. Chem. Soc 135, 3367–3370. [DOI] [PubMed] [Google Scholar]
- Minond D, Lauer-Fields JL, Nagase H, and Fields GB (2004). Matrix metalloproteinase triple-helical peptidase activities are differentially regulated by substrate stability. Biochemistry 43, 11474–11481. [DOI] [PubMed] [Google Scholar]
- Mori H, Tomari T, Koshikawa N, Kajita M, Itoh Y, Sato H, Tojo H, Yana I, and Seiki M (2002). CD44 directs membrane-type 1 matrix metalloproteinase to lamellipodia by associating with its hemopexin-like domain. EMBO J 21, 3949–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmier MO, and Van Doren SR (2007). Rapid determination of enzyme kinetics from fluorescence: overcoming the inner filter effect. Anal. Biochem 371, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005). Scalable molecular dynamics with NAMD. J Comput Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior SH, Byrne TS, Tokmina-Roszyk D, Fields GB, and Van Doren SR (2016). Path to Collagenolysis: COLLAGEN V TRIPLE-HELIX MODEL BOUND PRODUCTIVELY AND IN ENCOUNTERS BY MATRIX METALLOPROTEINASE-12. J. Biol. Chem 291, 7888–7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior SH, Fulcher YG, Koppisetti RK, Jurkevich A, and Van Doren SR (2015). Charge-Triggered Membrane Insertion of Matrix Metalloproteinase-7, Supporter of Innate Immunity and Tumors. Structure 23, 2099–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remacle AG, Golubkov VS, Shiryaev SA, Dahl R, Stebbins JL, Chernov AV, Cheltsov AV, Pellecchia M, and Strongin AY (2012). Novel MT1-MMP small-molecule inhibitors based on insights into hemopexin domain function in tumor growth. Cancer Res 72, 2339–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouck JE, Krapf JE, Roy J, Huff HC, and Das A (2017). Recent advances in nanodisc technology for membrane protein studies (2012–2017). FEBS Lett 591, 2057–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeh F, Ota I, Holmbeck K, Birkedal-Hansen H, Soloway P, Balbin M, Lopez-Otin C, Shapiro S, Inada M, Krane S, et al. (2004). Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J. Cell Biol 167, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeh F, Shimizu-Hirota R, and Weiss SJ (2009). Protease-dependent versus -independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. The Journal of Cell Biology 185, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E, and Seiki M (1994). A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 370, 61–65. [DOI] [PubMed] [Google Scholar]
- Schuler MA, Denisov IG, and Sligar SG (2013). Nanodiscs as a new tool to examine lipid-protein interactions. Methods Mol. Biol 974, 415–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TA, Choi MG, Raychaudhuri S, Mears JA, Ghirlando R, Hinshaw JE, and Prinz WA (2009). Lipid-regulated sterol transfer between closely apposed membranes by oxysterol-binding protein homologues. J. Cell Biol 187, 889–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda M, and Khokha R (2017). Metalloproteinases in extracellular vesicles. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1864, 1989–2000. [DOI] [PubMed] [Google Scholar]
- Sithu SD, English WR, Olson P, Krubasik D, Baker AH, Murphy G, and D’Souza SE (2007). Membrane-type 1-matrix metalloproteinase regulates intracellular adhesion molecule-1 (ICAM-1)-mediated monocyte transmigration. J. Biol. Chem 282, 25010–25019. [DOI] [PubMed] [Google Scholar]
- Takeda M, Terasawa H, Sakakura M, Yamaguchi Y, Kajiwara M, Kawashima H, Miyasaka M, and Shimada I (2003). Hyaluronan recognition mode of CD44 revealed by cross-saturation and chemical shift perturbation experiments. J. Biol. Chem 278, 43550–43555. [DOI] [PubMed] [Google Scholar]
- Tam EM, Moore TR, Butler GS, and Overall CM (2004). Characterization of the distinct collagen binding, helicase and cleavage mechanisms of matrix metalloproteinase 2 and 14 (gelatinase A and MT1-MMP): the differential roles of the MMP hemopexin c domains and the MMP-2 fibronectin type II modules in collagen triple helicase activities. J. Biol. Chem 279, 43336–43344. [DOI] [PubMed] [Google Scholar]
- Tatulian SA (2017). Chapter Nine - Interfacial Enzymes: Membrane Binding, Orientation, Membrane Insertion, and Activity In Methods Enzymol, Michael HG, ed. (Academic Press; ), pp. 197–230. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Ramachandran S, Case LB, Tolbert CE, Tandon A, Pershad M, Dokholyan NV, Waterman CM, and Campbell SL (2017). A Structural Model for Vinculin Insertion into PIP2-Containing Membranes and the Effect of Insertion on Vinculin Activation and Localization. Structure 25, 264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tochowicz A, Goettig P, Evans R, Visse R, Shitomi Y, Palmisano R, Ito N, Richter K, Maskos K, Franke D, et al. (2011). The dimer interface of the membrane type 1 matrix metalloproteinase hemopexin domain: crystal structure and biological functions. J. Biol. Chem 286, 7587–7600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen SP, Tatti-Bugaeva O, and Lehti K (2017). Membrane-type matrix metalloproteases as diverse effectors of cancer progression. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1864, 1974–1988. [DOI] [PubMed] [Google Scholar]
- Tzeng SR, Pai MT, and Kalodimos CG (2012). NMR studies of large protein systems. Methods Mol. Biol 831, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Doren SR, Marcink TC, Koppisetti RK, Jurkevich A, and Fulcher YG (2017). Peripheral membrane associations of matrix metalloproteinases. Biochim. Biophys. Acta [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Nandelstadh P, Gucciardo E, Lohi J, Li R, Sugiyama N, Carpen O, and Lehti K (2014). Actin-associated protein palladin promotes tumor cell invasion by linking extracellular matrix degradation to cell cytoskeleton. Mol. Biol. Cell 25, 2556–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, and Laue ED (2005). The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhang F, He J, Wu P, Tay LWR, Cai M, Nian W, Weng Y, Qin L, Chang JT, et al. (2017). Binding of PLD2-Generated Phosphatidic Acid to KIF5B Promotes MT1-MMP Surface Trafficking and Lung Metastasis of Mouse Breast Cancer Cells. Dev. Cell 43, 186–197.e187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, Stack MS, and Friedl P (2007). Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol 9, 893–904. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Takeo Y, Yoshida S, Kouchi Z, Nakamura Y, and Fukami K (2009). Lipid Rafts and Caveolin-1 Are Required for Invadopodia Formation and Extracellular Matrix Degradation by Human Breast Cancer Cells. Cancer Res 69, 8594–8602. [DOI] [PubMed] [Google Scholar]
- Yu Z, Visse R, Inouye M, Nagase H, and Brodsky B (2012). Defining requirements for collagenase cleavage in collagen type III using a bacterial collagen system. J. Biol. Chem 287, 22988–22997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrabi K, Dufour A, Li J, Kuscu C, Pulkoski-Gross A, Zhi J, Hu Y, Sampson NS, Zucker S, and Cao J (2011). Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer cell migration. J. Biol. Chem 286, 33167–33177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Marcink TC, Sanganna Gari RR, Marsh BP, King GM, Stawikowska R, Fields GB, and Van Doren SR (2015). Transient collagen triple helix binding to a key metalloproteinase in invasion and development. Structure 23, 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Related to Fig. 2. MD Trajectory with Blade II Associated with Nanodisc.
Movie S2. Related to Fig. 2. MD Trajectory with Blade IV Associated with Nanodisc.
Movie S3. Related to Fig. 5. MD Trajectory of Hypothetical Dimer with Blades II and IV Interacting with Nanodisc.
Data Availability Statement
The coordinates of the complexes of the HPX domain with DMPC-containing nanodiscs are deposited in the RCSB Protein Data Bank under accession codes 6CM1 and 6CLZ for the ensembles with blade II or IV in the interface, respectively. The corresponding NMR peak assignments are deposited in the BioMagResBank under accession codes 30426 and 30425, respectively.