

Abstract
Background:
Desminopathy, a hereditary myofibrillar myopathy, mainly results from the desmin gene (DES) mutations. Desminopathy involves various phenotypes, mainly including different cardiomyopathies, skeletal myopathy, and arrhythmia. Combined with genotype, it helps us precisely diagnose and treat for desminopathy.
Methods:
Sanger sequencing was used to characterize DES variation, and then a minigene assay was used to verify the effect of splice-site mutation on pre-mRNA splicing. Phenotypes were analyzed based on clinical characteristics associated with desminopathy.
Results:
A splicing mutation (c.735+1G>T) in DES was detected in the proband. A minigene assay revealed skipping of the whole exon 3 and transcription of abnormal pre-mRNA lacking 32 codons. Another affected family member who carried the identical mutation, was identified with a novel phenotype of desminopathy, non-compaction of ventricular myocardium. There were 2 different phenotypes varied in cardiomyopathy and skeletal myopathy among the 2 patients, but no significant correlation between genotype and phenotype was identified.
Conclusions:
We reported a novel phenotype with a splicing mutation in DES, enlarging the spectrum of phenotype in desminopathy. Molecular studies of desminopathy should promote our understanding of its pathogenesis and provide a precise molecular diagnosis of this disorder, facilitating clinical prevention and treatment at an early stage.
Keywords: Desminopathy, Cardiomyopathy, Desmin gene, Splicing mutation
Introduction
Desminopathy is a largely heterogeneous group of conditions involving inherited or sporadic myofibrillar myopathy, also called desmin-related myopathy or desmin myopathy, among others. In terms of its mode of inheritance, the autosomal dominant form is predominant. Although the incidence and prevalence of desminopathy are currently unclear, the data reported thus far suggest that it is a rare disease with a prevalence of no more than 0.05%.[1] The disease onset ranges from childhood to late adulthood. Clinically, it is characterized as skeletal and cardiac myopathy.[2] The most typical symptoms manifest as progressive skeletal muscle weakness from the distal lower extremities and cardiomyopathy, as well as severe arrhythmia and respiratory problems.[3,4] It is progressive and may shift from its initial clinical presentation with age. The prognosis is poor, with a meta-analysis revealing a mortality rate of 26% (27/104) among desmin mutation carriers.[5] The causes of death include sudden cardiac death, heart failure, respiratory insufficiency, chest infection, and iatrogenic complications of cardiac treatment. The manifestation of cardiac disease is the major cause of premature death in desminopathy.[6] The pathology of myopathy is characterized by desmin-positive protein aggregates and degenerative changes of the myofibrillar apparatus.[1,7] Electron micrography of muscle tissue shows disruption of the cytoskeleton, and the accumulation of aggregated proteins and autophagy vacuoles.[7,8]
Desminopathy is caused by destruction of the structure of desmin, which is a class III intermediate filament protein. The mature desmin molecule contains 470 amino acids and is composed of alpha-helical rods consisting of 308 amino acid residues flanked by globular N-terminal (Head) and C-terminal (Tail) structures, which include four highly conversed alpha-helical subdomains (1A, 1B, 2A, 2B) linked by non-helical linkers.[2,5,9] Desmin is encoded by DES (OMIM #125660), which is located on chromosome 2q35 and consists of 9 exons within an 8.4-kb region.[10]DES mutations, including missense, non-sense, and splicing mutations, play a critical role among the pathogenic inherited factors associated with desminopathy.[11-13]
Over the past 2 decades, since the 1st discovery of a pathogenic mutation in DES,[14] over 60 DES mutations have been reported, predominantly in Caucasian populations and mostly missense mutations.[9] Phenotypes of cardiomyopathy mainly involved hypertrophic, dilated, restrictive, and arrhythmogenic right ventricular cardiomyopathies. Moreover, no splicing variations in DES have been studied in the Chinese population. Desminopathy with a phenotype of non-compaction of ventricular myocardium (NVM) has not been reported before.
Methods
Ethical approval
This study was approved by the Ethics Committee of Fuwai Hospital and carried out in accordance with the Declaration of Helsinki. Each participant provided written informed consent.
Subjects
The proband (individual III 14), a 46-year-old male with symptoms of chest distress, shortness of breath, and syncope, was admitted to Fuwai Hospital, Beijing, China. The index patient had been diagnosed as having complete left bundle branch block at the age of 8 during a physical examination [Figure 1A], but did not receive any further treatment at that time. At the 1st medical consultation in September 2007, the patient presented symptoms of syncope, loss of consciousness, palpitation, and chest tightness, and an electrocardiogram showed 3rd-degree atrioventricular block (AVB) [Figure 1B]. Therefore, a pacemaker was implanted for symptom relief by ventricular pacing [Figure 1C]. He initially experienced fatigue of the distal extremities at age of 30 years. With disease progression, the symptom of fatigue spread from distal to proximal limbs. He was also admitted to a neurology department, but did not receive specific treatment there, and was diagnosed with desminopathy by neuropathologic examination of skeletal muscle from the right bicep. The pathologic results showed abnormal myofibril deposits, which were positive for desmin immunohistochemical staining, accompanied by myofibril hypertrophy and atrophy. Ultrastructural analysis of skeletal muscle revealed typical pathologic features of myopathy, such as myofibril hypertrophy, broken structure of desmin, and high-electron-density deposits. In recent years, the symptom of abdominal distension and diarrhea repeatedly developed and persisted for several days each time.
Figure 1.
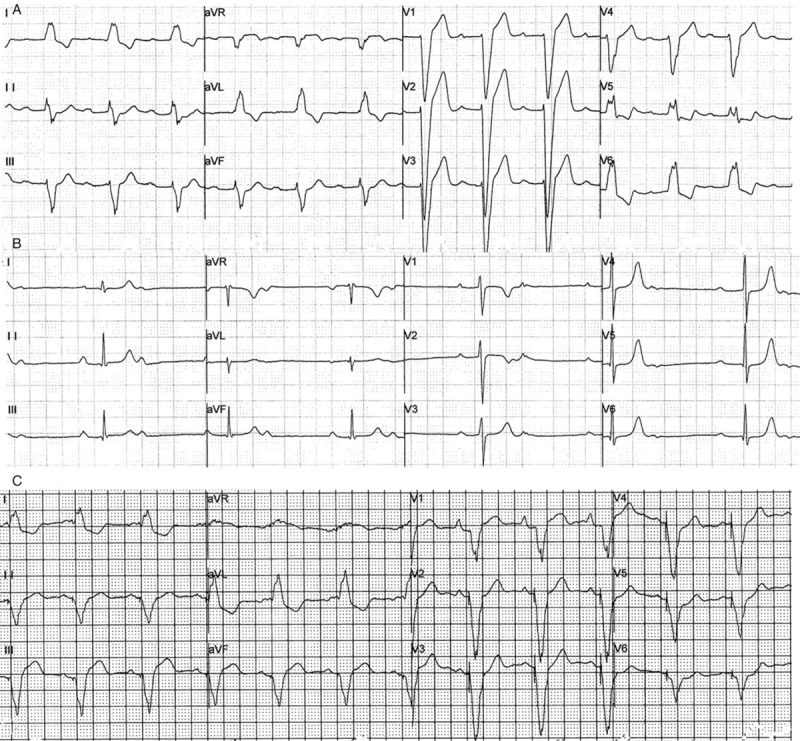
Electrocardiograph shows the aggravation of arrhythmia in the proband. (A) Complete left bundle branch block. (B) Third-degree atrioventricular. (C) Ventricular pacing after implanted a pacemaker.
During hospitalization, he underwent further examination including echocardiography, cardiac computed tomography (CT), and 24-h dynamic electrocardiography. A total of 9 family members (III 1, III 3, III 14, IV 1, IV 2, IV 3, IV 6, IV 8, and IV 9) were recruited in this study. The pedigree chart is shown in Figure 2A. Peripheral venous blood was collected from each subject for biochemical and genetic analyses. A clinical survey was also conducted for each participant.
Figure 2.
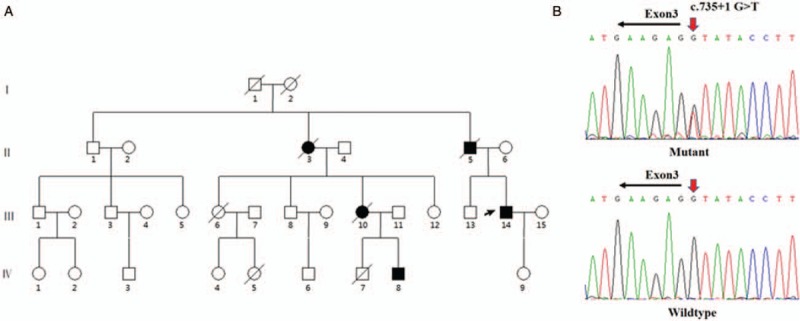
A splicing mutation in DES in a Chinese family with desminopathy. (A) Pedigree of this family. The black arrow indicated proband. (B) Sanger sequencing chromatogram shows a heterozygous c.735+1G>T splicing mutation in DES. DES: desmin gene.
Genetic testing
Genomic DNA was extracted from all subjects’ blood using the QIA amp DNA Blood Midi kit (QIAGEN, Hilden, Germany). Based on the patient's phenotype of desminopathy, DES was chosen as the target gene for analysis.[9] Therefore, polymerase chain reaction (PCR) was used to amplify all exons of the DES (NM_001927) using 9 pairs of primers (Supplementary material). All PCR products were sequenced using an ABI Prism 377 DNA sequencer (Applied Biosystems, Foster City, CA, USA). The identification of variants was performed using Chromas software (version 2.22, Technelysium, Brisbane, Australia).
In silico analysis
To predict the potential to affect splicing, the following splice-site prediction programs were used: (1) Splice Site Finder[15] (http://www.inter-activebiosoft-ware.com); (2) MAXENT[16] (http://genes.mit.edu/burgelab/maxent/Xmaxentscan-scoreseq.html); and (3) GENESPLICER[17] (http://www.interactivebiosoftware.com/doc/ala-mutvisual/2.5/splicing.html).
Minigene splicing assay
A minigene splicing assay was carried out to validate whether this variant affects splicing products. A splicing reporter minigene assay was established by PCR amplification of wild type (WT) and mutant genomic DNA sequences, as described previously.[18,19] The amplified target sequences contained exons 2 to 4 of DES, together with 520 base pairs (bp) from the 5’ and 3’ flanking intronic sequences. DES minigene fragments were digested with the restriction endonucleases BamHI and MluI (New England Bio Labs) and cloned into pCAS2 reporter vectors (provided by Mario Tosi, Rouen Institute for Biomedical Research, France). Sanger sequencing was performed to evaluate whether the WT and mutant type (MT) expression vectors had been constructed successfully. Two kinds of expression vector were transfected into HEK293T cells (Cell Resource Center, Peking Union Medical College, China). All cellular RNAs were extracted using TRIzol reagent (Life Technologies, Grand Island, NY). Reverse-transcription PCR (RT-PCR) was performed using the GoScript Reverse Transcription System (Promega Corporation, Madison, WI, USA). Complementary DNA sequences, including WT and MT, were amplified using the following primer pairs (forward primer: 5′-CTGACCCTGCTGACCCTCCT-3′; reverse primer: 5′-CTCTACCACGCCTTCTCAGCAA-3′). The resulting PCR products were separated by electrophoresis on a 1% agarose gel and analyzed by Sanger sequencing.
Results
Clinical features
In the recent events focused on here, upon hospitalization, 24-h dynamic electrocardiogram revealed atrial flutter, ventricular tachycardia, and AVB in the proband. Hence, he was treated with an implantable cardioverter defibrillator (ICD) to prevent sudden death. Echocardiography showed biatrium enlargement (left atrial diameter: 42 mm; right atrial diameter: 38 mm) and restriction of cardiac diastolic function (E/A>2) [Figure 3A]. CT scan also confirmed biatrium enlargement, but no abnormality of myocardial density [Figure 3B]. All the progress of desminopathy in the proband is shown in Table 1.
Figure 3.
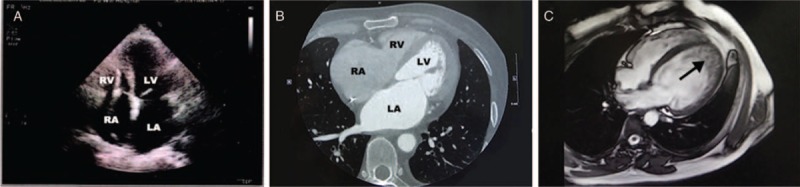
Imaging features of the patients. (A and B) Echocardiography and computed tomography show enlargement of left atrium (LA) and right atrium (RA) in the proband. (C) The black arrow indicated non-compaction of ventricular myocardium (individual VI 8) by magnetic resonance imaging.
Table 1.
Desminopathy progression with age in the proband

The proband's father (II 5), who had been implanted with a pacemaker at the age of 42, had also experienced cardiomyopathy, severe arrhythmia, and muscle weakness and died of heart and renal failure 14 years later. Moreover, both the proband's aunt (II 3) and his cousin (III 10) had died after giving birth to their 4th (III 12) and 2nd (VI 8) children at the ages of 34 and 29 years, respectively. In addition, his nephew (VI 8) had been diagnosed with NVM by magnetic resonance imaging [Figure 3C] and 3rd-degree AVB with the symptoms of chest distress and edema of lower extremity, which were treated with a pacemaker when he was 22 years old. Currently 2 patients were detected with elevated serum creatine kinase level (normal range: 0–200 IU/L), 399 IU/L (III 14) and 320 IU/L (VI 8), respectively. These abnormal clinical features were not observed in others in this family.
Analysis of DES mutation
The results of Sanger sequencing of DES are shown in Figure 2B. The proband and his nephew (VI 8) were heterozygous for a nucleotide G>T transversion in the donor splice site of intron 3 (c.735+1G>T). Among the rest of the subjects, there were no carriers of this mutation.
In silico analysis predicted the splice-site mutation (c.735+1G>T), using splicing prediction software including Splice Site Finder, MAXENT, and GENESPLICER. It also indicated that it was likely to influence splicing products of pre-mRNA.
Exon skipping identified by minigene assay
To examine the functional effect of the c.735+1G>T mutation in DES on pre-mRNA splicing, a mutant DES minigene was transfected into HEK293T cells for transcription. Upon 1% agarose gel electrophoresis, transcripts generated by WT and MT minigenes were located near the 500-bp marker (DL2000; Takara, Japan) in the gel. The RT-PCR sequencing results showed that the WT and MT minigenes produced 524- (expected size) and 428-bp bands, respectively [Figure 4B]. By sequencing the 2 different products, the WT transcript was shown to contain exons arranged sequentially from exon 2 to exon 4, while in the MT transcript, exon 2 was followed by exon 4, but with exon 3 missing. This deletion of exon 3 resulted in a reduction of 96 bp (32 codons) in the length of the MT transcript [Figure 4C].
Figure 4.
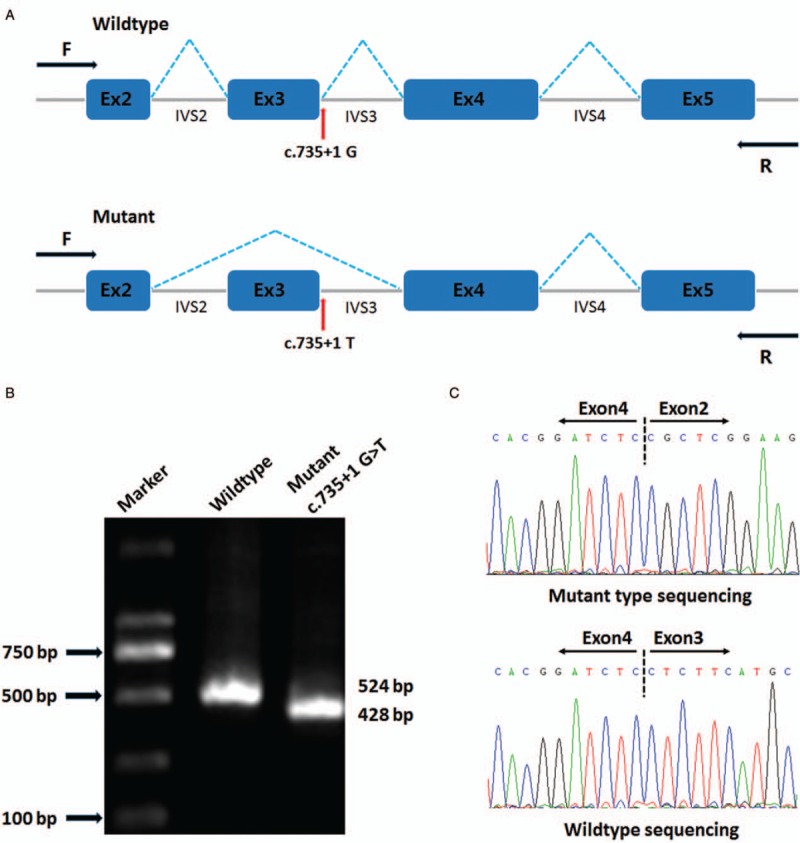
Splicing skipping was identified by a minigene assay. (A) Schematic diagram of the abnormal splicing process. (B) Reverse-transcription polymerase chain reaction (RT-PCR) products were separated by electrophoresis. (C) Sanger sequencing of RT-PCR products identified exon 3 skipping in the mutant type.
Discussion
In the present study, we investigated a desminopathy-associated pedigree and identified a novel phenotype with desminopathy and the possibility of pathogenicity of a splice-site mutation using Sanger sequencing and minigene assay. Our results demonstrated that the proband and his nephew (VI 8) are carriers of a heterozygous splice-site mutation (c.735+1G>T) of DES, for which functional assessment revealed abnormality of the RNA splicing process and exon 3 skipping. However, clinical phenotypes were totally different in the 2 affected individuals. These findings are significant because they not only provide possible genetic pathogenic evidence for desminopathy associated with a splice site of DES, but also enlarge the spectrum of phenotype.
Desmin is a critical component of the extra-sarcomeric cytoskeleton. Its biologic functions include structural and functional alignment and anchoring of myofibrils, as well as the positioning of cell organelles and conduction of cell signaling pathways.[1] An abnormal structure of desmin could thus produce a range of dysfunctions. DES, a single-copy gene, encodes desmin and is mainly expressed in muscle cells. DES mutation is the most common etiologic factor of desminopathy at the genetic level. Pathogenic mutations can impact upon the mRNA transcription and translation of desmin, generating structurally and functionally abnormal copies of the desmin protein. In addition to damage to the cytoskeletal network, mutant desmin impairs protein interactions, cell signaling cascades, mitochondrial functions, and protein quality control mechanisms.[1] In the case of DES splicing mutation, this causes defects in the function of desmin, which may increase the fragility of myofibrils and impair their contraction.[2]
Several lines of evidence indicated that DES splice-site mutations were responsible for the desminopathy exhibited by our patients. First, a heterozygous splice-site mutation in DES was identified by Sanger sequencing. Moreover, in silico analysis predicted that this mutation would affect splicing at the pre-mRNA level. Third, by expressing genomic DNA fragments, we showed that this kind of mutation inactivated the splice site and caused exon 3 skipping [Figure 4A]. Exon 3 consists of 96 bp and encodes a total of 32 codons. Although deletion of exon 3 would not disrupt the reading frame, it would result in the translation of a truncated desmin protein lacking 32 amino acids. The heptad repeat pattern is in turn disrupted by this deletion, preventing normal formation of the coiled-coil structure.[20] Park et al[21] reported that splice-site mutant desmin is expressed but not functional in SW13 (vim–) cells. This is supported by functional analysis indicating that desmin lacking 32 amino acids was incapable of forming a filamentous network in SW13 (vim–) cells. This mutant desmin abnormally aggregated in patients’ affected muscles.[21]
Exon skipping is a relatively common finding in inherited diseases, which is often caused by point mutations in the highly conserved AG splice acceptor and GT splice donor sites.[22,23] Many different pathogenic desmin mutations have already been reported, most of which are missense mutations mainly located within the 2B helical subdomain and the tail of the α-helical rod in desmin.[1,5,9] In 2000, a splicing defect (c.735+3A>G) resulting in the deletion of exon 3 in DES was reported for the first time.[2] Subsequently, a series of splice-site mutations were revealed. DES mutations in the c.735+1 locus have been studied in Caucasians,[3,13,20,24] but none of research was identified in functional experiment. To date, all splicing mutations in DES have been reported to be located in the highly conversed acceptor and donor splice sites flanking exon 3 [Table 2], which results in the deletion of 32 amino acids from Asp214 to Glu245, instead of in-frame fusion between exon 2 and exon 4.[20] Unlike most missense mutations affecting the highly conserved YRKLLEGEE motif of the 2B helix,[21,25] all of the reported splicing mutations only disrupt the structure of the conversed 1B subdomain in desmin. Cetin et al found a special splice-site mutation (c.1289–2 >G) in DES, which causes a limb girdle muscular dystrophy phenotype instead of desminopathy.[26] Therefore, research performed to date indicates that nearly all pathogenic splice-site mutations in DES are gathered upstream or downstream of exon 3.
Table 2.
Splice-site mutations resulting in deletion of 32 codons encoded by exon 3 in desmin gene
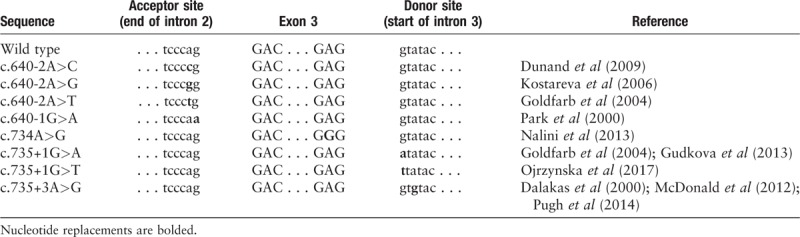
When performing a combined analysis of genotype and phenotype, we were unable to identify a significant correlation between them. In this study, 2 patients were identified to have a splicing mutation, as determined by sequencing analysis.[27-31] Based on an analysis of their phenotypes, we speculated that 3 other patients (II 3, II 5, and IV 10) who had died probably carried the same pathogenic mutation. With few exceptions, the phenotypic manifestations in members of the same family with desminopathy were concordant.[32] Interestingly, 2 surviving patients shared the same mutation, yet they presented with different cardiac and skeletal muscle involvement, restrictive cardiomyopathy (RCM), and skeletal myopathy developing with age (the proband) and NVM alone (VI 8). Despite isolated cardiomyopathy, we could not predict whether this young patient (VI 8) will experience progressive skeletal myopathy in the future. We cannot rule out a possible phenotype of skeletal myopathy followed by cardiomyopathy. Hence, it is necessary to follow-up the progression of desminopathy in this case. In addition, three patients suffered from severe arrhythmia, necessitating the implantation of a pacemaker device, at 36 years of age in the proband, at 42 years of age in the proband's father (II 5), and at 18 years of age in the proband's nephew (VI 8). With the development of desminopathy, the index patients also underwent the implantation of an ICD against ventricular tachycardia. Therefore, diagnosis and therapy for severe arrhythmia earlier should be pay high attention to prevent sudden death. Furthermore, the proband occasionally manifested diarrhea, so we suspect that this splicing mutation had affected his gastrointestinal smooth muscle.
DES mutations are pleiotropic, causing different phenotypes such as various cardiomyopathies, skeletal myopathies, smooth muscle myopathy, and respiratory dysfunction, even among mutations located in close vicinity to each other.[9] Moreover, 2 kinds of splicing mutation in the same nucleotide have been reported. Specifically, a variant (c.735+1G>A) causing exon 3 skipping was described in association with the 1st case of desminopathy with a transition from a hypertrophic to a restrictive and dilated phenotype.[3] In addition, Ojrzynska et al[13] reported a patient carrying a pathogenic variant (c.735+1G>T) who was diagnosed with progressive skeletal muscle weakness and RCM. Adding to these previous findings, our study shows a family affected by desminopathy with RCM or NVM and with or without skeletal myopathy. Ripoll-Vera et al[33] concluded that RCM is the most common echocardiographic pattern in desminopathy.
It is quite complex why DES mutations are pleiotropic, generating variable phenotypes. Phenotypic differences can probably be explained by several reasons. Dominant mutations show a wide phenotypic variability which probably results from interactions between desmin, chaperones and other intermediate filaments.[32,34] Alpha-B-crystallin serves as such a chaperone for desmin, but there were no mutation in alpha-B-crystallin associated with desminopathy in this family. The location of DES mutations exert a significant influence on phenotypic characteristics.[32]DES mutations mainly gather in 2B helix which more than 50% of the know DES mutations have found in this region.[32] Mutations in the 2B domain were predominant in patients with an isolated neurologic phenotype, whereas head and tail domain mutations were predominant in patients with an isolated cardiac phenotype.[5] Gene dosage effects resulted from heterozygous, homozygous, double heterozygous, and compound heterozygous mutations are also at play.[9] The desmin degradation pathways were regulated by ubiquitin-proteasome and autophagy system.[35,36] Different protein degradation pathways will cause pathologic desmin aggregation in different tissues and organs.[1] In addition, the absence of desmosomes in skeletal muscle, different non-sense-mediated RNA decay mechanisms, gene modifiers effects, and very limited regenerative capacity of the adult heart may also have a role in different phenotypes.[9,37]
In conclusion, we have reported a novel phenotype with desminopathy and performed functional identification of a potentially pathogenic splice-site mutation (c.735+1G>T) in DES. It has provided additional evidence supporting the genetic pathogenic mechanism associated with desminopathy. These findings are important because the earlier patients with desminopathy are identified and treated, the greater the likelihood of preventing sudden cardiac arrest, and other complications. This study extends our knowledge of the spectrum of phenotype and their geographic distribution in desminopathy.
Funding
This work was supported by grants from CAMS Innovation Fund for Medical Sciences (No. 2016-I2M-1-002), the National Key Research and Development Program of China (No. 2016YFC1300100), National Natural Science Foundation of China (No. 81600305, No. 81400187), Beijing Municipal Science and Technology Commission (No. Z151100003915078), and PUMC Graduate Innovation Fund (2018-1002-01-14).
Conflicts of interest
None.
Supplementary Material
Footnotes
How to cite this article: Fan P, Lu CX, Dong XQ, Zhu D, Yang KQ, Liu KQ, Zhang D, Zhang Y, Meng X, Tan HQ, Yu LT, Dou KF, Liu YX, Zhang X, Zhou XL. A novel phenotype with splicing mutation identified in a Chinese family with desminopathy. Chin Med J 2019;00:00–00. doi: 10.1097/CM9.0000000000000001
Peng Fan and Chao-Xia Lu contributed equally to this work.
References
- 1.Clemen CS, Herrmann H, Strelkov SV, Schroder R. Desminopathies: pathology and mechanisms. Acta Neuropathol 2013;125:47–75. doi: 10.1007/s00401-012-1057-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med 2000;342:770–780. doi: 10.1056/nejm200003163421104. [DOI] [PubMed] [Google Scholar]
- 3.Gudkova A, Kostareva A, Sjoberg G, Smolina N, Turalchuk M, Kuznetsova I, et al. Diagnostic challenge in desmin cardiomyopathy with transformation of clinical phenotypes. Pediatr Cardiol 2013;2:467–470. doi: 10.1007/s00246-012-0312-x. [DOI] [PubMed] [Google Scholar]
- 4.Hong D, Wang Z, Zhang W, Xi J, Lu J, Luan X, et al. A series of Chinese patients with desminopathy associated with six novel and one reported mutations in the desmin gene. Neuropathol Appl Neurobiol 2011;37:257–270. doi: 10.1111/j.1365-2990.2010.01112.x. [DOI] [PubMed] [Google Scholar]
- 5.van Spaendonck-Zwarts KY, van Hessem L, Jongbloed JD, de Walle HE, Capetanaki Y, van der Kooi AJ, et al. Desmin-related myopathy. Clin Genet 2011;80:354–366. doi: 10.1111/j.1399-0004.2010.01512.x. [DOI] [PubMed] [Google Scholar]
- 6.Wahbi K, Behin A, Charron P, Dunand M, Richard P, Meune C, et al. High cardiovascular morbidity and mortality in myofibrillar myopathies due to DES gene mutations: a 10-year longitudinal study. Neuromuscul Disord 2012;22:211–218. doi: 10.1016/j.nmd.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 7.Weihl CC, Iyadurai S, Baloh RH, Pittman SK, Schmidt RE, Lopate G, et al. Autophagic vacuolar pathology in desminopathies. Neuromuscul Disord 2015;25:199–206. doi: 10.1016/j.nmd.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Claeys KG, Fardeau M, Schroder R, Suominen T, Tolksdorf K, Behin A, et al. Electron microscopy in myofibrillar myopathies reveals clues to the mutated gene. Neuromuscul Disord 2008;18:656–666. doi: 10.1016/j.nmd.2008.06.367. [DOI] [PubMed] [Google Scholar]
- 9.Azzimato V, Genneback N, Tabish AM, Buyandelger B, Knoll R. Desmin, desminopathy and the complexity of genetics. J Mol Cell Cardiol 2016;92:93–95. doi: 10.1016/j.yjmcc.2016.01.017. [DOI] [PubMed] [Google Scholar]
- 10.Lazarides E. Intermediate filaments as mechanical integrators of cellular space. Nature 1980;283:249–256. doi: org/10.1038/283249a0. [DOI] [PubMed] [Google Scholar]
- 11.Tian C, Fuller C, Miles L, Jefferies J, Ryan T, Sawnani H, et al. A novel homozygous desmin nonsense mutation causes pediatric onset autosomal recessive desminopathy with severe cardiomyopathy. Neuromuscular Disorders 2016;S114–S115. doi: 10.1016/j.nmd.2016.06.107. [Google Scholar]
- 12.Brodehl A, Dieding M, Biere N, Unger A, Klauke B, Walhorn V, et al. Functional characterization of the novel DES mutation p.L136P associated with dilated cardiomyopathy reveals a dominant filament assembly defect. J Mol Cell Cardiol 2016;91:207–214. doi: 10.1016/j.yjmcc.2015.12.015. [DOI] [PubMed] [Google Scholar]
- 13.Ojrzynska N, Bilinska ZT, Franaszczyk M, Ploski R, Grzybowski J. Restrictive cardiomyopathy due to novel desmin gene mutation. Kardiol Pol 2017;7:723 doi: 10.5603/KP.2017.0129. [DOI] [PubMed] [Google Scholar]
- 14.Goldfarb LG, Park KY, Cervenakova L, Gorokhova S, Lee HS, Vasconcelos O, et al. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet 1998;19:402–403. doi: 10.1038/1300. [DOI] [PubMed] [Google Scholar]
- 15.Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009;37:e67 doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 17.Pertea M, Lin X, Salzberg SL. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res 2001;29:1185–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaildrat P, Killian A, Martins A, Tournier I, Frebourg T, Tosi M. Use of splicing reporter minigene assay to evaluate the effect on splicing of unclassified genetic variants. Methods Mol Biol 2010;653:249–257. doi: 10.1007/978-1-60761-759-4_15. [DOI] [PubMed] [Google Scholar]
- 19.Cooper TA. Use of minigene systems to dissect alternative splicing elements. Methods 2005;37:331–340. doi: 10.1016/j.ymeth.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 20.Goldfarb LG, Vicart P, Goebel HH, Dalakas MC. Desmin myopathy. Brain 2004;127:723–734. doi: 10.1093/brain/awh033. [DOI] [PubMed] [Google Scholar]
- 21.Park KY, Dalakas MC, Goebel HH, Ferrans VJ, Semino-Mora C, Litvak S, et al. Desmin splice variants causing cardiac and skeletal myopathy. J Med Genet 2000;37:851–857. doi: 10.1136/jmg.37.11.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakai K, Sakamoto H. Construction of a novel database containing aberrant splicing mutations of mammalian genes. Gene 1994;141:171–177. doi: org/10.1016/0378-1119(94)90567-3. [DOI] [PubMed] [Google Scholar]
- 23.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet 1992;90:41–54. [DOI] [PubMed] [Google Scholar]
- 24.Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015;347:1254806 doi: 10.1126/science.1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dagvadorj A, Olive M, Urtizberea JA, Halle M, Shatunov A, Bonnemann C, et al. A series of West European patients with severe cardiac and skeletal myopathy associated with a de novo R406W mutation in desmin. J Neurol 2004;251:143–149. doi: 10.1007/s00415-004-0289-3. [DOI] [PubMed] [Google Scholar]
- 26.Cetin N, Balci-Hayta B, Gundesli H, Korkusuz P, Purali N, Talim B, et al. A novel desmin mutation leading to autosomal recessive limb-girdle muscular dystrophy: distinct histopathological outcomes compared with desminopathies. J Med Genet 2013;50:437–443. doi: 10.1136/jmedgenet-2012-101487. [DOI] [PubMed] [Google Scholar]
- 27.Dunand M, Lobrinus JA, Jeannet PY, Behin A, Claeys KG, Selcen D, et al. Confirmation that abnormal desmin accumulation and migration are due to a desmin gene mutation in a familial cardiomyopathy and distal myopathy. Neuromuscul Disord 2009;19:802 doi: 10.1016/j.nmd.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 28.Kostareva A, Gudkova A, Sjoberg G, Kiselev I, Moiseeva O, Karelkina E, et al. Desmin mutations in a St. Petersburg cohort of cardiomyopathies. Acta Myol 2006;25:109–115. [PubMed] [Google Scholar]
- 29.Nalini A, Gayathri N, Richard P, Cobo AM, Urtizberea JA. New mutation of the desmin gene identified in an extended Indian pedigree presenting with distal myopathy and cardiac disease. Neurol India 2013;61:622–626. doi: 10.4103/0028-3886.125269. [DOI] [PubMed] [Google Scholar]
- 30.McDonald KK, Stajich J, Blach C, Ashley-Koch AE, Hauser MA. Exome analysis of two limb-girdle muscular dystrophy families: mutations identified and challenges encountered. PLoS One 2012;7:e48864 doi: 10.1371/journal.pone.0048864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med 2014;16:601–608. doi: 10.1038/gim.2013.204. [DOI] [PubMed] [Google Scholar]
- 32.Goldfarb LG, Olivé M, Vicart P, Goebel HH. Intermediate filament diseases: desminopathy. Adv Exp Med Biol 2008;131–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ripoll-Vera T, Zorio E, Gamez JM, Molina P, Govea N, Cremer D. Phenotypic patterns of cardiomyopathy caused by mutations in the desmin gene. A clinical and genetic study in two inherited heart disease units. Rev Esp Cardiol (Engl Ed) 2015;68:1027–1029. doi: 10.1016/j.rec.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 34.Sharma S, Conover GM, Elliott JL, Der Perng M, Herrmann H, Quinlan RA. alphaB-crystallin is a sensor for assembly intermediates and for the subunit topology of desmin intermediate filaments. Cell Stress Chaperones 2017;22:613–626. doi: 10.1007/s12192-017-0788-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su H, Wang X. The ubiquitin-proteasome system in cardiac proteinopathy: a quality control perspective. Cardiovasc Res 2010;2:253–262. doi:10.1093/cvr/cvp287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ravikumar B, Sarkar S, Rubinsztein DC. Clearance of mutant aggregate-prone proteins by autophagy. Methods Mol Biol 2008;445:195–211. doi: 10.1007/978-1-59745-157-4_13. [DOI] [PubMed] [Google Scholar]
- 37.Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, et al. Dynamics of cell generation and turnover in the human heart. Cell 2015;161:1566–1575. doi: 10.1016/j.cell.2015.05.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.