

Abstract
Background:
Osteogenesis imperfecta (OI), a heritable bone fragility disorder, is mainly caused by mutations in COL1A1 gene encoding α1 chain of type I collagen. This study aimed to investigate the COL1A1 mutation spectrum and quantitatively assess the genotype-phenotype relationship in a large cohort of Chinese patients with OI.
Methods:
A total of 161 patients who were diagnosed as OI in Department of Endocrinology of Peking Union Medical College Hospital from January 2010 to December 2017 were included in the study. The COL1A1 mutation spectrum was identified by next generation sequencing and confirmed by Sanger sequencing. A new clinical scoring system was developed to quantitatively assess the clinical severity of OI and the genotype-phenotype relationship was analyzed. The independent sample t-test, analysis of variance, Mann-Whitney U-test, Chi-squared test, Pearson correlation, and multiple linear regression were applied for statistical analyses.
Results:
Among 161 patients with OI, 32.9% missense mutations, 16.8% non-sense mutations, 24.2% splice-site mutations, 24.8% frameshift mutations, and 1.2% whole-gene deletions were identified, of which 38 variations were novel. These mutations led to 53 patients carrying qualitative defects and 67 patients carrying quantitative defects in type I collagen. Compared to patients with quantitative mutations, patients with qualitative mutations had lower alkaline phosphatase level (296 [132, 346] U/L vs. 218 [136, 284] U/L, P = 0.009) and higher clinical score (12.2 ± 5.3 vs. 7.4 ± 2.4, P < 0.001), denoting more severe phenotypes including shorter stature, lower bone mineral density, higher fracture frequency, more bone deformity, vertebral compressive fractures, limited movement, and dentinogenesis imperfecta (DI). Patients would not present with DI if the glycine substitutions happened before the 79th amino acid in triple helix of α1 chains.
Conclusions:
This presented distinctive COL1A1 mutation spectrum in a large cohort of Chinese patients with OI. This new quantitative analysis of genotype-phenotype correlation would be helpful to predict the prognosis of OI and genetic counseling.
Keywords: Osteogenesis imperfecta, COL1A1, Clinical scoring system, Genotype, Phenotype
Introduction
Osteogenesis imperfecta (OI) is a rare heritable bone disorder with incidence of 1/15,000 to 20,000 births, which is characterized by decreased bone mineral density (BMD), impaired bone strength, recurrent bone fractures, and resulting in progressive bone deformity (BD).[1] Moreover, OI can also cause a variety of complex extra-skeletal manifestations including dentinogenesis imperfecta (DI), ligamentous laxity, blue sclera (BS), hearing impairment, and so on.[2] OI is traditionally classified as type I, type II, type III, and type IV, which indicate the mild type, perinatal lethal type, most severe type of survival and moderate type, respectively.[2] However, as the high heterogeneity of patients with OI, the traditional classification could not express the degree of clinical severity of OI quantitatively.
As we know, the pathogenesis of OI is closely correlated to the abnormal structure or quantity of type I collagen. As the most abundant bone matrix protein, type I collagen plays crucial roles in maintaining the integrity of bone structure and bone strength. It is an ordered heterotrimer that consisted of 2 α1 chains and 1 α2 chain, which are encoded by COL1A1 and COL1A2 genes, respectively.[3] Qualitative or quantitative defects of type I collagen induced by gene mutations would lead to OI.[4] Generally, in the random selection of abnormal chains, 75% of mutant heterotrimers contain 1 or more defective α1 chains.[5] Recently, collagen alterations induced by COL1A1 mutations are divided into 2 categories, including mutations causing a defect to synthesize the products and leading to quantitative defects of type I collagen, or mutations leading to produce structural abnormal collagen molecules and causing the substitution of a glycine within the Gly-X-Y triplet domain of the triple helix.[6]
Although a lot of COL1A1 mutations had been reported, the genotype-phenotype relationship of patients with OI have not been fully illuminated,[6-11] and results of such studies were elusive in China because of the limited sample size of OI.[12-14] However, OI not only damages the life quality of the patients, but also brings heavy social and economic burden on their families. Therefore, it is critically important to assess clinical severity and reveal the genotype-phenotype relationship of patients OI, which will be helpful to genetic counseling and predicting the prognosis of OI. This study investigated the COL1A1 mutation spectrum and evaluated clinical severity of OI quantitatively by a clinical scoring system in a large Chinese cohort of OI. In addition, this study analyzed the genotype-phenotype relationship in these patients with OI.
Methods
Ethical approval
The study was conducted in accordance with the Declaration of Helsinki and was approved by the Scientific Ethics Committee of Peking Union Medical College Hospital. All participants or their parents provided written informed consents before they participated in this study.
Subjects
A total of 161 patients from 146 unrelated families who were diagnosed as OI in Department of Endocrinology in Peking Union Medical College Hospital from January 2010 to December 2017 were included in the study. There were 107 males and 54 females with median visiting age of 10.7 years (range: 0–64 years).
Phenotype assessment
We obtained a detailed medical history of the patients and performed a physical examination on them, including BD, BS, DI, and ligamentous laxity. Patients’ height and weight were measured by Harpenden stadiometer (Seritex Inc., East Rutherford, NJ, USA) and RGZ-120 weighing scale (Xiheng, Wuxi, China). The age- and gender-specific Z-scores of height and weight were calculated on the basis of reference data from the Chinese National Centers for Disease Control and Prevention.[15] BMD at lumbar spine 2–4 and proximal hip were measured by Dual energy X-ray absorptiometry (DXA, Lunar Prodigy Advance; GE Healthcare, Chicago, IL, USA), and the BMD Z-scores were calculated based on BMD data from age- and gender-matched normal Chinese children.[16,17] Information of bone fractures were evaluated including time of the initiation, frequency and site. Features of bone were assessed by X-ray films. Then patients were diagnosed as OI type I, III, or IV according to the Sillence classification based on clinical and radiological characteristics. As type II OI was neonatal lethal, no patient with this type OI was included.
To quantitatively evaluate the phenotypes, we used clinical scores to assess the severity of OI.[18,19] Clinical score = number of fractures (1–4) + fracture frequency (1–4) + vertebral compressive fracture (VCF) (0–1) + scoliosis (0–1) + bowing (0–8) + height Z-score (1–3) + BMD Z-score (1–4) + limited movement (0–1). Number of fractures scoring: 1, number of fractures < 10; 2, 10 ≤ number of fractures < 20; 3, 20 ≤ number of fractures < 30; 4, number of fractures ≥ 30. Frequency of fracture scoring: 1, frequency of fractures (per year) < 1; 2, 1 ≤ frequency of fractures (per year) < 2; 3, 2 ≤ frequency of fractures (per year) < 3; 4, frequency of fractures (per year) ≥ 3. VCF scoring: 0, non VCF; 1, with VCF. Scoliosis scoring: 0, non scoliosis; 1, with scoliosis. In the scoring system, bowing of the tibia, femur, humerus or forearm accounts for 1 point each. Height Z-score scoring: 1, height Z-score ≥ –2.0; 2, –3.0 ≤ height Z-score < –2.0; 3, height Z-score < –3.0. BMD Z-score scoring: 1, BMD Z-score ≥ –2.0; 2, –3.0 ≤ BMD Z-score < –2.0; 3, –4.0 ≤ BMD Z-score < –3.0; 4, BMD Z-score < –4.0. Limited movement scoring: 0, autonomic activity; 1, limited movement. A higher score meant severer clinical phenotype of OI.
Serum levels of alanine aminotransferase (ALT), creatinine (Cr), calcium (Ca), phosphate (P), alkaline phosphatase (ALP), and a bone formation marker were measured by automated analyzers. An automated electrochemiluminescence system (E170; Roche Diagnostics, Basel, Switzerland) was used to detect serum levels of beta cross-linked carboxy-terminal telopeptide of type I collagen (β-CTX, a bone resorption marker), 25-hydroxyvitamin D (25OHD), and intact parathyroid hormone (PTH). All these serum biochemical parameters were measured in the central clinical laboratory of Peking Union Medical College Hospital.
Detection of COL1A1 mutation
The patients’ genomic DNA was isolated from peripheral leukocytes under the standard procedures (QIAamp DNA Mini Kit (50); Qiagen, Duesseldorf, Germany). Genetic detection was performed through paired-end sequencing using a panel for next generation sequencing (NGS), which covered more than 700 genes associated with genetic bone disorders, including 19 candidate genes of OI (COL1A1, COL1A2, IFITM5, SERPINF1, CRTAP, P3H1, PPIB, SERPINH1, FKBP10, BMP1, PLOD2, SP7, TMEM38B, WNT1, CREB3H1, SPARC, PLS3, P4HB, SEC24D). The experimental procedures of the present research followed a protocol as described previously. The genomic DNA from patients were fragmented and ligated with end-repaired adaptors oligonucleotides. Then DNA fragments with adaptor molecules were purified and enriched by polymerase chain reaction (PCR). Each barcoded library product was pooled and subjected to enrichment of targeted genomic sequences, and sequenced on one lane of the IlluminaHiSeq 2000 (Illumina Inc., San Diego, CA, USA) with paired-end 100 bases read length. Illumina pipeline (version 1.3.4) was used to perform bioinformatic analysis processes of reads mapping, variants detection (including polymorphisms, single-nucleotide variants and small indels), and annotation.[20]
All the sequencing results were analyzed by Nucleotide BLAST software (http://blast.ncbi.nlm.nih.gov/Blast.cgi) to compare with the standard sequences in the database. We only further analyzed those mutations expected to produce damaged protein, including non-sense mutations, missense mutations, frameshift mutations, splice-site mutations, and whole-gene deletion. Prediction of missense mutations was performed by Polyphen2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT software (http://sift.jcvi.org/). Frameshift mutations were predicted using Genome Analysis Toolkit (https://software.broadinstitute.org/gatk/). Potential splice-site mutations were predicted with Berkeley Drosophila Genome Project (http://www.fruitfly.org/). The mutations identified by NGS were further confirmed by PCR using primers (Supplementary Table 1) and Sanger sequencing and co-segregation analysis in each OI family.
The absence from the Osteogenesis Imperfecta Variant Database (https:// oi.gene.le.ac.uk/home.php) was the criterion to identify the novel mutations. Mutations in COL1A1 that led to stop codons or frameshifts were classified as quantitative mutations. Mutations that induced amino acid substitution in α1 chains in type I collagen were regarded as qualitative mutations.
Statistical analysis
Data with normal distribution were expressed as mean ± standard deviation and analyzed by independent sample t-test or analysis of variance, while those with abnormal distribution were presented as median (Q1, Q3) and analyzed by Mann-Whitney U-test. Covariance analysis was used to compare serum levels of biochemical parameters in different groups after adjusted by age and gender. The Chi-squared test was used to analyze categorical variables. Relationships between the positions of helical glycine substitutions in α1 chains of type I collagen and phenotypic features of patients with OI were analyzed using Pearson correlation. Multiple linear regression analysis was applied to examine the associations between bone turnover biomarker level and clinical scores. The SPSS software version 23.0 (SPSS Inc., Chicago, IL, USA) was used to perform all statistical analyses. A two-tailed P < 0.05 was considered statistical significance.
Results
Phenotypes of patients with OI carrying COL1A1 mutations
According to Sillence classification, this study included 90 (55.9%) patients with type I, 36 (22.4%) patients with type III, and 35 (21.7%) patients with type IV OI. There was no significant difference in gender among these OI types (P = 0.980). Patients with type III OI were the earliest onset with median age of 0.5 years (U = 26.35, P < 0.001), but their median visit age (12.3 years) was the oldest (U = 9.43, P = 0.009). The 69.4% patients with type I OI, 34.3% with type III and 51.4% with type IV presented positive family history (χ2 = 13.18, P = 0.001). The clinical scores were the lowest in patients with type I OI (6.6 ± 2.0), and the highest in patients with type III OI (16.0 ± 3.9, F = 178.79, P < 0.001). Compared with patients with type I OI, type III had shorter height and lower weight, severer skeletal lesions, including lower BMD Z-score at lumbar spine and proximal hip, higher fracture number and frequency, more proportion of BD, VCFs, limited movement, wormian bones and DI, and patients with type IV OI were in moderate severity of phenotypes. The phenotype difference between OI types was consistent with the clinical score. BS (94.4–97.7%) and ligamentous laxity (61.3–80.0%) were the common manifestations among different OI types, while hearing impairment was infrequent (2.9–5.7%). There was no significant difference in serum levels of Ca, P, β-CTX, 25OHD, PTH, ALT, and Cr among different OI types, but serum ALP level was lowest in patients with type III OI (U = 8.21, P = 0.016) [Table 1]. In addition, as shown in Supplementary Figure 1, there was an independent negative correlation between ALP level and clinical score, which indicated that lower ALP level maybe predicted severer clinical manifestation.
Table 1.
Demographic and clinical characteristics of patients with OI with COL1A1 mutations in this study
COL1A1 mutation spectrum of Chinese patients with OI
In 161 patients with OI, 53 (32.9%) missense mutations, 27 (16.8%) non-sense mutations, 39 (24.2%) splice-site mutations, 40 (24.8%) frameshift mutations, and 2 (1.2%) whole-gene deletions in COL1A1 were found, 38 of which were novel variants, including 10 missense mutations, 3 non-sense mutations, 18 frameshift mutations, and 7 splice mutations [Table 2]. Sanger sequencing results for a part of these mutations are shown in Figure 1. The details of COL1A1 mutation spectrum are shown in Figure 2. In patients with type I OI, frameshift mutation was the most common mutation and in patients with type III/IV OI, missense mutation was the most common mutation. There were 14 mutations found in more than 2 unrelated families. Among the 53 missense mutations, 42 (79.2%) led to of a glycine substitution in the Gly-X-Y triplet domain of the triple helix. Among the 42 glycine mutations, substitutions by serine were the most common (n = 24, 57.1%), followed by arginine (n = 8, 19.0%), alanine (n = 3, 7.1%), asparaginic acid (n = 2, 4.8%), valine (n = 2, 4.8%), cysteine (n = 1, 2.4%), glutamic acid (n = 1, 2.4%), and lysine (n = 1, 2.4%) substitutions.
Table 2.
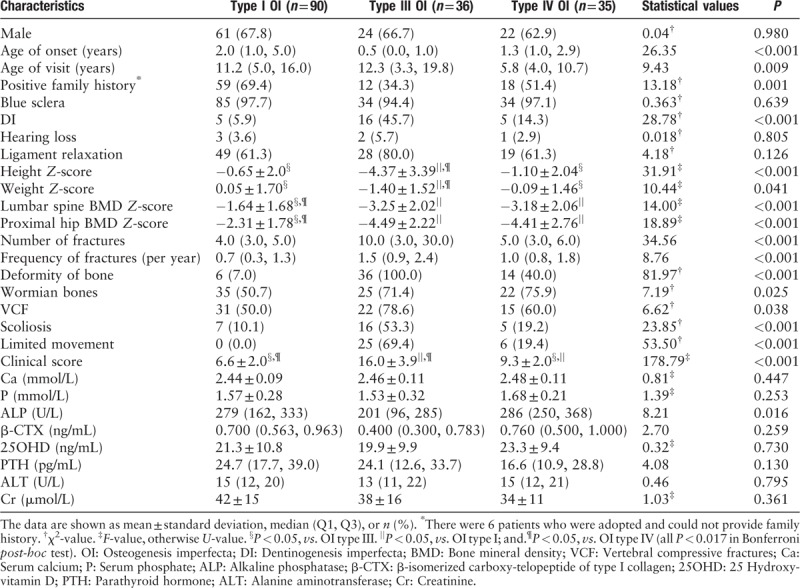
Novel variants of COL1A1 detected by next generation sequencing in this study
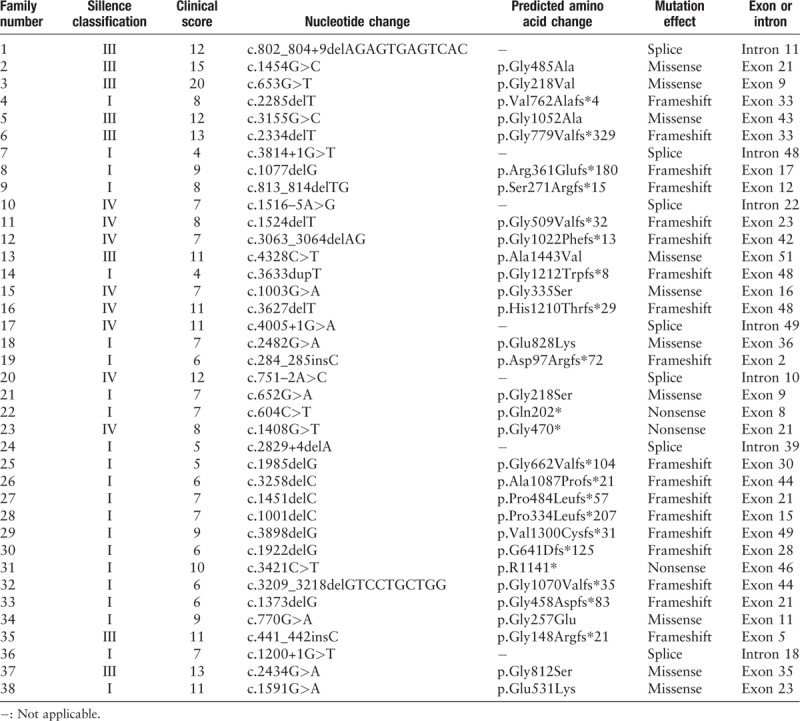
Figure 1.
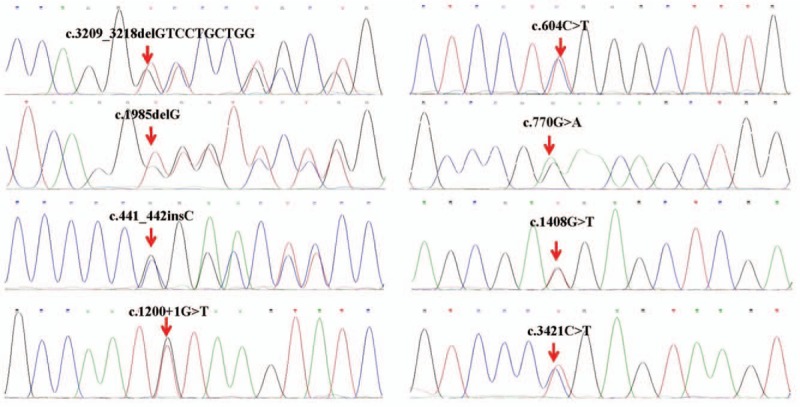
Sanger sequencing for a part of COL1A1 mutations.
Figure 2.
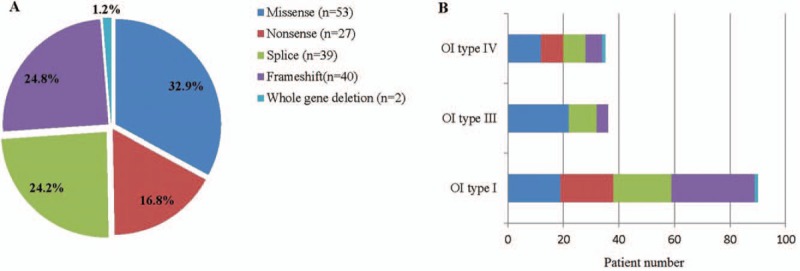
Mutation spectrum of COL1A1 mutations in patients with OI. (A) Mutation types of COL1A1 in patients with OI. (B) COL1A1 mutation spectrum in patients with different Sillence classification. OI: Osteogenesis imperfecta.
Genotype-phenotype correlation
The OI phenotypes were found to be correlated with genotypes of COL1A1. As shown in Table 3, the median onset age of patients with qualitative mutations was 1.0 year, which was earlier than patients with quantitative mutations (2.0 years, P = 0.005). Compared with quantitative mutations in COL1A1 gene, patients with qualitative mutations had significantly higher clinical score (7.4 ± 2.4 for quantitative and 12.2 ± 5.3 for qualitative, t = –6.61, P < 0.001), which denoted severer phenotype.
Table 3.
Comparison of phenotypes between quantitative and qualitative mutations in COL1A1
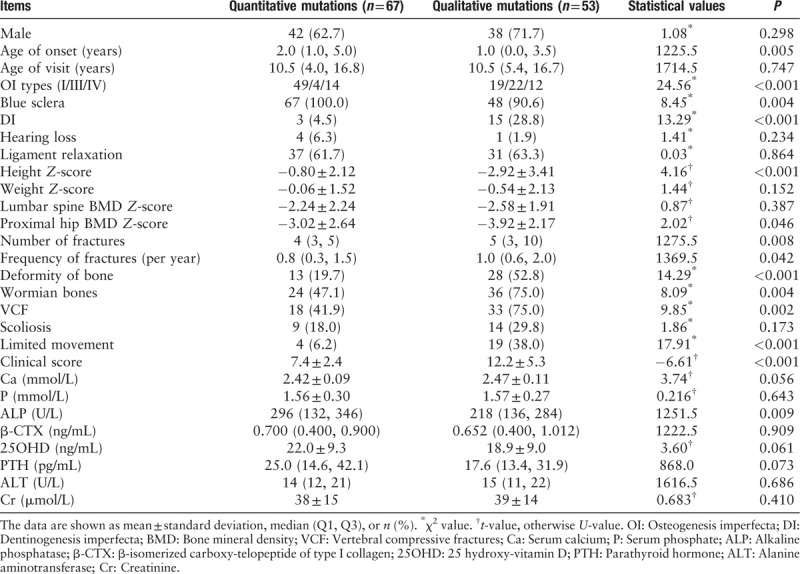
Patients with qualitative mutations had lower BMD at proximal hip (Z-score, –3.92 ± 2.17) than patients with quantitative mutations (Z-score, –3.02 ± 2.64, t = 2.02, P = 0.046). And patients with qualitative mutations had higher median fracture number (5 vs. 4, U = 1275.5, P = 0.008) and frequency (1.0 vs. 0.8, U = 1369.5, P = 0.042) and more proportion of BD (52.8% vs. 19.7%, χ2 = 14.29, P = 0.042), wormian bones (75.0% vs. 47.1%, χ2 = 8.09, P = 0.004), VCF (75.0% vs. 41.9%, χ2 = 9.85, P = 0.002), and limited movement (38.0% vs. 6.2%, χ2 = 17.91, P < 0.001), compared with patients with quantitative mutations in COL1A1. Patients with qualitative mutations had lower ALP level than patients with quantitative mutations in type I collagen. The height of patients with qualitative mutations in COL1A1 was significantly shorter than patients with quantitative defects (–0.80 ± 2.12 for quantitative and –2.92 ± 3.41 for qualitative, t = 4.16, P < 0.001). All patients with quantitative changes of type I collagen had blue sclera, which was 90.6% in qualitative collagen defect group. Patients with qualitative mutations were more prone to suffer from DI (28.8%) than quantitative mutations (4.5%, χ2 = 14.29, P < 0.001). Moreover, the patients would not present DI when the glycine substitutions happened before 79th amino acid in triple helix of collagen type I α1 chain [Figure 3]. However, there was no association between phenotypes and positions or types of glycine substitution (Supplementary Table 2).
Figure 3.
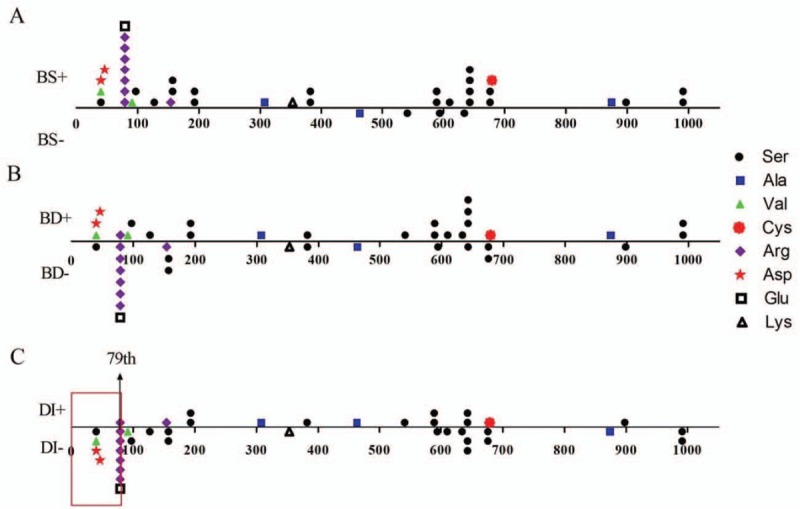
Relationships between helical glycine substitutions positions in collagen type I α1 chains and phenotypes. (A) Relationship between the positions of glycine substitutions and BS. (B) Relationship between the positions of glycine substitutions and BD. (C) Relationship between the positions of glycine substitutions and DI. The red frame indicated that patients would not present DI if the glycine substitutions happened before the 79th amino acid in triple helix of collagen type I α1 chains. BS: Blue sclera; BD: Bone deformity; DI: Dentinogenesis imperfecta.
Discussion
This study with the large sample size investigated the genotype-phenotype relationship in patients with OI carrying COL1A1 mutations. The results not only outlined an abundant and significant COL1A1 mutation spectrum in Chinese patients with OI, but also quantitatively indicated an important genotype-phenotype correlation using a clinical scoring system.
This scoring system included 8 major criteria,[18,19] including number of fractures, fracture frequency, VCF, scoliosis, bowing, height Z-score, BMD Z-score, and limited movement. The higher the score, the more severe the condition was. The clinical score of patients with type I was the lowest while the patients with type III OI was the highest, which confirmed that the clinical scoring system could quantitatively reflect clinical severity of OI. Patients with type III OI were early onset and lag in diagnosis, which led to not in time of the treatment and aggravated the condition of OI.[21] In aspect of extra-skeletal manifestation, BS was a common feature of patients with OI.[14,22] Consistent with the previous studies,[21,23] patients with types III and IV OI had higher proportion of DI than type I patients, which may be interpreted as dentin and bone were similar in the composition of the extracellular matrix.[24] The progressive hearing loss was reported in 50% to 60% of adult patients with OI, but it was found in 5.0% to 26.7% of pediatric patients with OI,[14,25] which was similar in the patients of this study. This phenomenon may be related to the short course of OI in children.
In the genotype analysis, missense mutation in COL1A1 was dominant in this cohort, but the proportion was lower than reports in Canada (47.3%),[6] Sweden (60.9%),[21] Vietnam (67.6%),[26] and India (85.7%).[27]We further found that missense mutation in COL1A1 could explain 21.1% mild OI, 34.3% moderate, and 61.1% severe OI. Similar to previous studies, glycine substituted by serine was the most common.[14,22,28] Moreover, this study found 38 novel variations, which contributed to extended the gene mutant spectrum of OI and reveal its pathogenesis. In addition, c.769G>A (p.Gly257Arg), c.1678G>A (p.Gly560Ser), c.2299G>A (p.Gly767Ser), c.2461G>A (p.Gly821Ser), c.3505G>A (p.Gly1169Ser), c.1081C>T (p.Arg361∗), c.1792C>T (p.Arg598∗), c.2644C>T (p.Arg882∗), c.3076C>T (p.Arg1026∗), c.1155+1G>A (Splicing variant), and c.1821+1G>A (Splicing variant) could be hotpot mutations for human OI, which had been reported for 6 to 37 times previously (https://oi.gene.le.ac.uk/home.php).
As for the analysis of the relationship between genotype and phenotype, this study confirmed that the clinical phenotype of patients with collagen qualitative defects was severer than patients with collagen quantitative defects, which was similar to previous reports.[6,22] BMDs of patients with collagen qualitative defects were lower than the patients with collagen quantitative defects, especially at proximal hip. In consistence with the results of this study, previous studies had revealed that COL1A1 quantitative mutations led to milder bone fragility than COL1A1 qualitative mutations.[6,11,22] Lower than Caucasian,[29] this study observed that 18.0% and 29.8% of patients with quantitative and qualitative mutations had scoliosis, respectively. Similarly, this study also found that the height of patients with collagen quantitative defects was taller than the patients with triple helical mutations.[6] In addition, this study also found wormian bone was more common in patients with qualitative mutations than patients with quantitative mutations. Two studies revealed patients with serine substitutions presented more severe bone phenotypes,[11,30] but this study did not observe this phenomenon. In this study, the position and type of glycine substitutions within the α1 (I) triple helical domain were not found to have association with bone lesions of OI. Rauch et al[28] observed that OI patients with mutations close to carboxyl-terminal end of type I collagen may not present BS, but this study did not found this phenomenon. This study did not find relationship between hearing loss and genotypes in COL1A1, which was similar to previous studies.[25,31] In Europe studies, patients with glycine substitutions before the 127th amino acids of the α1 (I) triple helical domain would have no DI,[21,28] but this study found the point was the 79th amino acids.
The mechanism of genotype-phenotype correlation in OI could relate to that COL1A1 quantitative mutations resulted in the amount of type I procollagen synthesis reduced by half of the amount and causing mild bone fragility. However, qualitative mutations in COL1A1 gene led to the structural abnormalities in collagen molecules, which could result in more severe bone lesions. Another finding in this study was that patients with qualitative mutations had lower serum ALP level than patients with quantitative mutations, and ALP level was negatively correlated to clinical severity of OI. ALP, as a bone formation marker, had important roles in promoting bone mineralization.[32,33] Therefore, this study conjectured that patients with OI with COL1A1 qualitative mutations may have been more severely damaged of osteoblasts function, which aggravated bone lesions.
Although this research expanded the spectrum of mutations in COL1A1, and revealed meaningful correlation between genotype and phenotype of OI, there were some limitations. Firstly, it was a cross-sectional study, which could not reveal the relationship of genotype and efficacy of bisphosphonates in patients with OI. Secondly, this study did not perform functional experiment to verify the protein variation of splice-site mutations. Thirdly, this study did not intensively evaluate the influence of gene mutations in COL1A1 on bone microstructure and bone biomechanical properties.
In conclusion, this study presented the distinctive COL1A1 mutation spectrum in the large Chinese patients with OI. In particular, this study identified 38 novel mutations, which expanded the known pathogenic spectrum of COL1A1 gene. Furthermore, this new quantitative analysis of genotype-phenotype correlation would be helpful to predict the prognosis of OI and genetic counseling.
Acknowledgements
The authors appreciate all of the patients and their families for their participation.
Funding
This study is supported by grants from the National Natural Science Foundation of China (No. 81570802), Chinese Academy of Medical Sciences Innovative Fund for Medical Sciences (CIFMS; No. 2016-I2M-3-003), and The National Key Research and Development Program of China (No. 2016YFC0901501).
Conflicts of interest
None.
Supplementary Material
Footnotes
How to cite this article: Li LJ, Lyu F, Song YW, Wang O, Jiang Y, Xia WB, Xing XP, Li M. Genotype-phenotype relationship in a large cohort of osteogenesis imperfecta patients with COL1A1 mutations revealed by a new scoring system. Chin Med J 2019;00:00–00. doi: 10.1097/CM9.0000000000000013
References
- 1.Forlino A, Marini JC. Osteogenesis imperfecta. Lancet 2016;387:1657–1671. doi: 10.1016/S0140-6736(15)00728-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Dijk FS, Sillence DO. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A 2014;164A:1470–1481. doi: 10.1002/ajmg.a.36545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forlino A, Cabral WA, Barnes AM, Marini JC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol 2011;7:540–557. doi: 10.1038/nrendo.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Y, Asan, Ma D, Lv F, Xu X, Wang J, et al. Gene mutation spectrum and genotype-phenotype correlation in a cohort of Chinese osteogenesis imperfecta patients revealed by targeted next generation sequencing. Osteoporos Int 2017;28:2985–2995. doi: 10.1007/s00198-017-4143-8. [DOI] [PubMed] [Google Scholar]
- 5.Bateman JF, Boot-Handford RP, Lamande SR. Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat Rev Genet 2009;10:173–183. doi: 10.1038/nrg2520. [DOI] [PubMed] [Google Scholar]
- 6.Rauch F, Lalic L, Roughley P, Glorieux FH. Relationship between genotype and skeletal phenotype in children and adolescents with osteogenesis imperfecta. J Bone Miner Res 2010;25:1367–1374. doi: 10.1359/jbmr.091109. [DOI] [PubMed] [Google Scholar]
- 7.Benusiene E, Kucinskas V. COL1A1 mutation analysis in Lithuanian patients with osteogenesis imperfecta. J Appl Genet 2003;44:95–102. [PubMed] [Google Scholar]
- 8.Hartikka H, Kuurila K, Korkko J, Kaitila I, Grenman R, Pynnonen S, et al. Lack of correlation between the type of COL1A1 or COL1A2 mutation and hearing loss in osteogenesis imperfecta patients. Hum Mutat 2004;24:147–154. doi: 10.1002/humu.20071. [DOI] [PubMed] [Google Scholar]
- 9.Ries-Levavi L, Ish-Shalom T, Frydman M, Lev D, Cohen S, Barkai G, et al. Genetic and biochemical analyses of Israeli osteogenesis imperfecta patients. Hum Mutat 2004;23:399–400. doi: 10.1002/humu.9230. [DOI] [PubMed] [Google Scholar]
- 10.Venturi G, Tedeschi E, Mottes M, Valli M, Camilot M, Viglio S, et al. Osteogenesis imperfecta: clinical, biochemical and molecular findings. Clin Genet 2006;70:131–139. doi: 10.1111/j.1399-0004.2006.00646.x. [DOI] [PubMed] [Google Scholar]
- 11.Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat 2007;28:209–221. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee KS, Song HR, Cho TJ, Kim HJ, Lee TM, Jin HS, et al. Mutational spectrum of type I collagen genes in Korean patients with osteogenesis imperfecta. Hum Mutat 2006;27:599 doi: 10.1002/humu.9423. [DOI] [PubMed] [Google Scholar]
- 13.Kataoka K, Ogura E, Hasegawa K, Inoue M, Seino Y, Morishima T, et al. Mutations in type I collagen genes in Japanese osteogenesis imperfecta patients. Pediatr Int 2007;49:564–569. doi: 10.1111/j.1442-200X.2007.02422.x. [DOI] [PubMed] [Google Scholar]
- 14.Zhang H, Yue H, Wang C, Hu W, Gu J, He J, et al. Clinical characteristics and the identification of novel mutations of COL1A1 and COL1A2 in 61 Chinese patients with osteogenesis imperfecta. Mol Med Rep 2016;14:4918–4926. doi: 10.3892/mmr.2016.5835. [DOI] [PubMed] [Google Scholar]
- 15.Li H, JiCY ZongXN. Height and weight standardized growth charts for Chinese children and adolescents aged 0 to 18 years (in Chinese). Chin J Pediatr 2009;7:487–492. doi: 10.3760/cma.j.issrr0578-1310.2009.07.003. [PubMed] [Google Scholar]
- 16.Xu H, Zhao Z, Wang H, Ding M, Zhou A, Wang X, et al. Bone mineral density of the spine in 11,898 Chinese infants and young children: a cross-sectional study. PLoS One 2013;8:e82098 doi: 10.1371/journal.pone.0082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan LJ, Lei SF, Chen XD, Liu MY, Guo YF, Xu H, et al. Establishment of peak bone mineral density in Southern Chinese males and its comparisons with other males from different regions of China. J Bone Miner Metab 2007;25:114–121. doi: 10.1007/s00774-006-0737-5. [DOI] [PubMed] [Google Scholar]
- 18.Mrosk J, Bhavani GS, Shah H, Hecht J, Krüger U, Shukla A, et al. Diagnostic strategies and genotype-phenotype correlation in a large Indian cohort of osteogenesis imperfecta. Bone 2018;110:368–377. doi: 10.1016/j.bone.2018.02.029. [DOI] [PubMed] [Google Scholar]
- 19.Aglan MS, Hosny L, El-Houssini R, Abdelhadi S, Salem F, Elbanna RA, et al. A scoring system for the assessment of clinical severity in osteogenesis imperfecta. J Child Orthop 2012;6:29–35. doi: 10.1007/s11832-012-0385-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang JY, Liu Y, Song LJ, Lv F, Xu XJ, San A, et al. Novel mutations in SERPINF1 result in rare osteogenesis imperfecta type VI. Calcif Tissue Int 2017;100:55–66. doi: 10.1007/s00223-016-0201-z. [DOI] [PubMed] [Google Scholar]
- 21.Lindahl K, Astrom E, Rubin CJ, Grigelioniene G, Malmgren B, Ljunggren O, et al. Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta. Eur J Hum Genet 2015;23:1042–1050. doi: 10.1038/ejhg.2015.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang ZL, Zhang H, Ke YH, Yue H, Xiao WJ, Yu JB, et al. The identification of novel mutations in COL1A1, COL1A2, and LEPRE1 genes in Chinese patients with osteogenesis imperfecta. J Bone Miner Metab 2012;30:69–77. doi: 10.1007/s00774-011-0284-6. [DOI] [PubMed] [Google Scholar]
- 23.Lin HY, Chuang CK, Su YN, Chen MR, Chiu HC, Niu DM, et al. Genotype and phenotype analysis of Taiwanese patients with osteogenesis imperfecta. Orphanet J Rare Dis 2015;10:152 doi: 10.1186/s13023-015-0370-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Opsahl VS, Gaucher C, Bardet C, Rowe PS, George A, Linglart A, et al. Tooth dentin defects reflect genetic disorders affecting bone mineralization. Bone 2012;50:989–997. doi: 10.1016/j.bone.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swinnen FK, Coucke PJ, De Paepe AM, Symoens S, Malfait F, Gentile FV, et al. Osteogenesis Imperfecta: the audiological phenotype lacks correlation with the genotype. Orphanet J Rare Dis 2011;6:88 doi: 10.1186/1750-1172-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ho DB, Zhytnik L, Maasalu K, Kändla I, Prans E, Reimann E, et al. Mutation analysis of the COL1A1 and COL1A2 genes in Vietnamese patients with osteogenesis imperfecta. Hum Genomics 2016;10:27 doi: 10.1186/s40246-016-0083-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stephen J, Shukla A, Dalal A, Girisha KM, Shah H, Gupta N, et al. Mutation spectrum of COL1A1 and COL1A2 genes in Indian patients with osteogenesis imperfecta. Am J Med Genet A 2014;164A:1482–1489. doi: 10.1002/ajmg.a.36481. [DOI] [PubMed] [Google Scholar]
- 28.Rauch F, Lalic L, Roughley P, Glorieux FH. Genotype-phenotype correlations in nonlethal osteogenesis imperfecta caused by mutations in the helical domain of collagen type I. Eur J Hum Genet 2010;18:642–647. doi: 10.1038/ejhg.2009.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato A, Ouellet J, Muneta T, Glorieux FH, Rauch F. Scoliosis in osteogenesis imperfecta caused by COL1A1/COL1A2 mutations - genotype-phenotype correlations and effect of bisphosphonate treatment. Bone 2016;86:53–57. doi: 10.1016/j.bone.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 30.Bodian DL, Chan TF, Poon A, Schwarze U, Yang K, Byers PH, et al. Mutation and polymorphism spectrum in osteogenesis imperfecta type II: implications for genotype-phenotype relationships. Hum Mol Genet 2009;18:463–471. doi: 10.1093/hmg/ddn374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pillion JP, Vernick D, Shapiro J. Hearing loss in osteogenesis imperfecta: characteristics and treatment considerations. Genet Res Int 2011;983942 doi: 10.4061/2011/983942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whyte MP. Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann N Y Acad Sci 2010;1192:190–200. doi: 10.1111/j.1749-6632.2010.05387.x. [DOI] [PubMed] [Google Scholar]
- 33.Lim EK, Keem JO, Yun HS, Jung J, Chung BH. Smart nanoprobes for the detection of alkaline phosphatase activity during osteoblast differentiation. Chem Commun 2015;51:3270–3272. doi: 10.1039/c4cc09620g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.