

Abstract
The cell cycle is strictly ordered to ensure faithful genome duplication and chromosome segregation. Underlying this order are control mechanisms that dictate when a cell transitions from one phase to the next. Much is known about control of the G1/S, G2/M, and metaphase/anaphase transitions, but there is no known control mechanism for the S/G2 transition. Here, we show cells transactivate the mitotic gene network as they exit S phase through a CDK1-directed FOXM1 phosphorylation switch. Intrinsic activation of the checkpoint kinase ATR by ETAA1 during DNA replication blocks this switch until the end of S phase. Loss of the ATR-FOXM1 pathway deregulates the S/G2 transition, leading to premature mitosis, under-replicated DNA and DNA damage. Thus, ATR couples DNA replication to mitosis by enforcing an S/G2 checkpoint, preserving genome integrity.
One Sentence Summary:
ATR represses FOXM1-dependent expression of a mitotic gene network until the S/G2 transition ensuring completion of DNA replication.
To avoid mitotic errors and loss of genetic information during cell division, cells must completely replicate their DNA before mitosis (1–3). How cells sense the end of S phase and coordinate these processes is a fundamental biological question. A checkpoint monitoring the end of DNA replication has been proposed (4, 5), but regulators and effectors of such an S/G2 checkpoint have yet to be identified. Extrinsic sources of DNA damage can activate the checkpoint kinase ATR (ATM and Rad3-related) delaying the cell cycle in S or G2 through post-translational modification of the CDC25-cyclin dependent kinase (CDK) pathway (6). Because ATR is essential for cell viability (7, 8) and prevents premature mitosis during normal proliferation (9, 10), we hypothesized it may also function intrinsically during normal S-phase to coordinate DNA replication and mitosis.
To test this hypothesis, we utilized quantitative image-based cytometry (QIBC) (11) to simultaneously distinguish G1, S, G2, and mitotic populations by plotting the fluorescence signals of EdU, DAPI, and histone H3-pS10 (fig. S1, A-E). Non-cancerous hTERT RPE-1 and MCF10A cells were studied to minimize the confounding cell cycle deregulation common to cancer cells. After pulse-labeling cells with EdU and nocodazole arrest (Fig. 1A and fig. S2A), we quantified the H3-pS10 signal as a function of time (fig. S2, B-D). S-phase cells treated with ATR inhibitors (ATRi) exhibited accelerated mitotic entry (Fig. 1B). Surprisingly, ATR inhibition in G2 cells did not change mitotic entry rates, whereas WEE1 inhibition in G2 phase triggered rapid mitotic entry (Fig. 1C). This suggests that ATR does not act during a normal G2 phase to regulate mitotic entry, unlike its response to DNA damage. This extends previous findings (9, 10) and suggests that it is in S-phase that ATR acts to control mitotic entry.
Figure 1. ATR represses transactivation of a mitotic gene network during S phase.
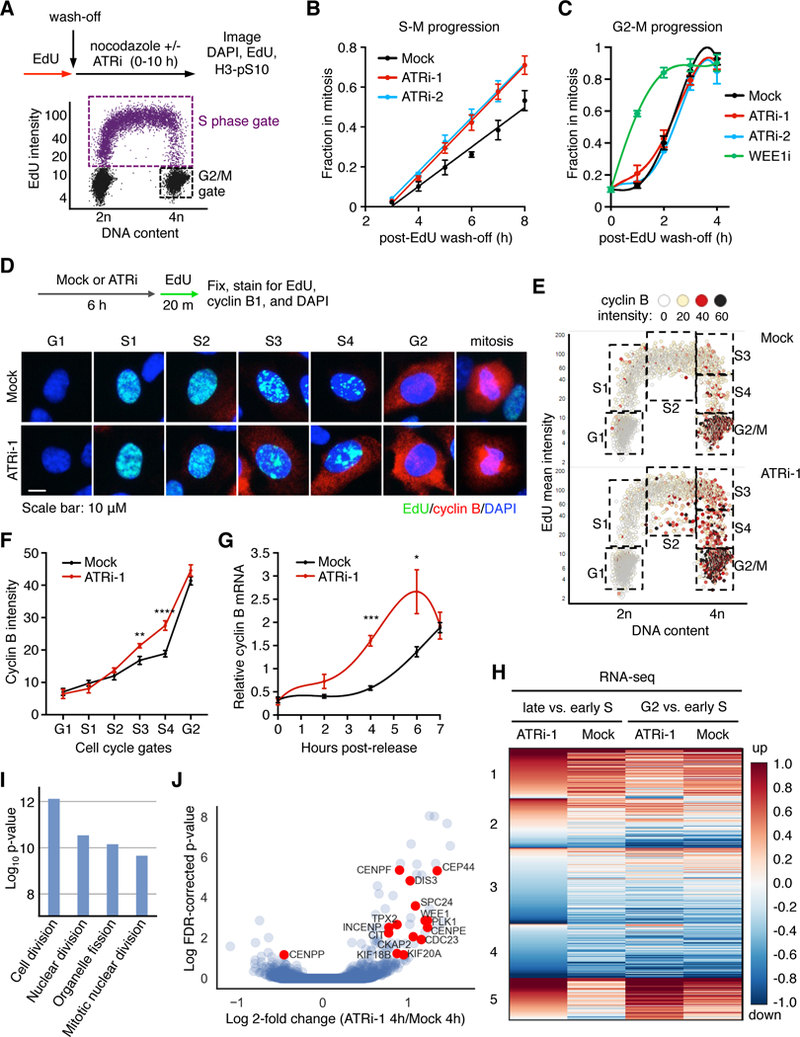
(A) S-Mor G2-M progression assay showing the distribution of cells at T=0 and gating scheme. See fig. S2, A-D for full description of assay. (B and C) Fraction of S-phase gated cells (B) or G2/M-gated cells (C) in mitosis as a function of time. Error bars represent the SEM of 3 independent experiments. (D) Experimental design and representative images of cells in G1, early to late S phase (S1-S4), G2, or mitosis. (E) QIBC plots of total DAPI intensity vs. EdU mean intensity, and mean cyclin B cytoplasmic intensity on a color scale. Boxes indicate gated populations used for further analysis. (F) Mean cyclin B cytoplasmic intensities of gated populations shown in (E). Error bars represent the SEM of 4 independent experiments. **p < 0.05, ****p< 0.001. (G) RT-qPCR analysis of cyclin B1 mRNA isolated from synchronized cells described in fig. S6. Error bars represent the SEM from 4 independent experiments. ***p < 0.01. (H) Unsupervised clustering analysis from every pairwise comparison between mock or ATRi and early S, late S, or G2 synchronized cells (selected comparisons shown). Heatmap indicates the fold-change of normalized RNA-seq reads between indicated samples (n=3). (I) The top 4 GO terms for Group genes clustered in (H). (J) Volcano plot of Log10 FDR-corrected p-values and log 2-fold change in gene expression of ATRi/Mock of late S phase cells.
These observations are in principle consistent with ATR inhibition shortening S phase. Nevertheless, a pulse-chase-pulse assay (fig. S3A), showed that the effect of ATR inhibition on S phase is small compared to its shortening of S and G2 together (fig. S3, B-D). Using the EYFP-PCNA biosensor in live MCF10A cells to measure S-phase duration (12) (fig. S4, A-D), we found ATRi shortened S phase by 45 min, irrespective of when it was added (fig. S4, E-G). This is also less than the approximate 2–3 hour shortening of the combined S and G2 phase (Fig. 1B). We conclude that ATR activity in S phase controls the subsequent duration of G2.
At the end of S phase, mitotic factors begin to accumulate, including cyclin B (13). We hypothesized that ATR may control S-M progression by regulating the build-up of these pro-mitotic factors and tested this idea by monitoring cyclin B expression over the cell cycle. Notably, ATR inhibition led to premature cyclin B accumulation in S phase (Fig. 1, D-F). This effect was observed with multiple ATR inhibitors, in multiple cell lines, and was time-dependent (fig. S5). As cyclin B accumulation is controlled transcriptionally (14), we asked whether ATR inhibition temporally deregulates cyclin B transcription. We synchronized cells to early S phase (fig. S6, A-D) and measured cyclin B mRNA (CCNB1) as cells progressed into G2. In cells not treated with ATRi, cyclin B mRNA remained unchanged until late S phase, when it increased concomitant with the S to G2 transition (Fig. 1G and fig. S6D). By contrast, ATR inhibition prematurely increased cyclin B1 mRNA levels in early S phase cells (Fig. 1G) indicating ATR controls its transcription.
To address whether ATR controls the transcription of additional genes, we performed RNA-seq in mock-and ATR-inhibited cells synchronized to early S phase, late S phase, or early G2 phase (fig. S6D). Unsupervised clustering analysis revealed a group (group 5) of genes that were prematurely upregulated with ATRi in S phase (Fig. 1H, fig. S6E, and Table S1). Consistent with their normal G2 expression, GO-term analysis of group 5 showed enrichment of genes involved in the G2/M transition and mitosis (Fig. 1, I and J). These findings suggest that a number of genes needed for G2/M are poised for transcriptional activation in early S phase, but are repressed by ATR until the end of S phase.
We next asked how ATR controls transactivation of this G2/M gene network. To this end, we analyzed publically available ChIP-seq data for transcription factor enrichment at the promoters of group 5 genes. Both B-MYB and FOXM1 were highly enriched at these sites (fig. S7A). B-MYB and FOXM1 are phosphorylated and activated by a cyclin A-dependent CDK, allowing B-MYB to recruit FOXM1 to promoters and execute a mitotic transcriptional program (15–21). We asked whether knockdown of either protein would prevent premature cyclin B accumulation. B-MYB knockdown prevented cells from entering S phase, suggesting an independent role for B-MYB in early cell cycle progression (fig. S7, B-D). Importantly, however, FOXM1 knockdown, which did not block S phase entry, prevented cyclin B accumulation in S phase cells following ATR inhibition (fig. S7, B and E), confirming that FOXM1 activity drives premature cyclin B expression.
To determine whether ATR regulates B-MYB and FOXM1, we monitored their phosphorylation following ATR inhibition in synchronized cells. In mock-treated cells, B-MYB and FOXM1 underwent minimal S-phase phosphorylation; however, both transcription factors exhibited a profound mobility shift, indicative of rapid hyper-phosphorylation, at or near the S/G2 transition. ATR inhibition allowed immediate B-MYB and FOXM1 hyper-phosphorylation (Fig. 2, A and B, and fig. S7F). In asynchronous cells, single-cell analysis of FOXM1 phosphorylation at T600 (fig. S8A), a CDK1/2-dependent modification promoting FOXM1 activation (16, 22), revealed that FOXM1 phosphorylation rises in early S phase, remains steady throughout S phase, and then rises again in G2. Strikingly, ATR inhibition caused premature phosphorylation of FOXM1 in early and late S phase cells, raising S phase pFOXM1 levels to G2 levels (Fig. 2, C-F, and fig. S8B). Premature phosphorylation in S phase was observed within 10 minutes of ATR inhibition (fig. S8, C and D), and when replication was fully blocked with thymidine (fig. S9A). Moreover, mRNA FISH analysis of PLK1, a FOXM1 target, revealed the premature expression of PLK1 after ATR inhibition in cells with or without thymidine (fig. S9, B and C). These findings clearly demonstrate that ATR suppresses premature FOXM1 phosphorylation and downstream transcription in all S phase cells, irrespective of S phase progression.
Figure 2. ATR controls an S/G2 FOXM1 phosphorylation switch.
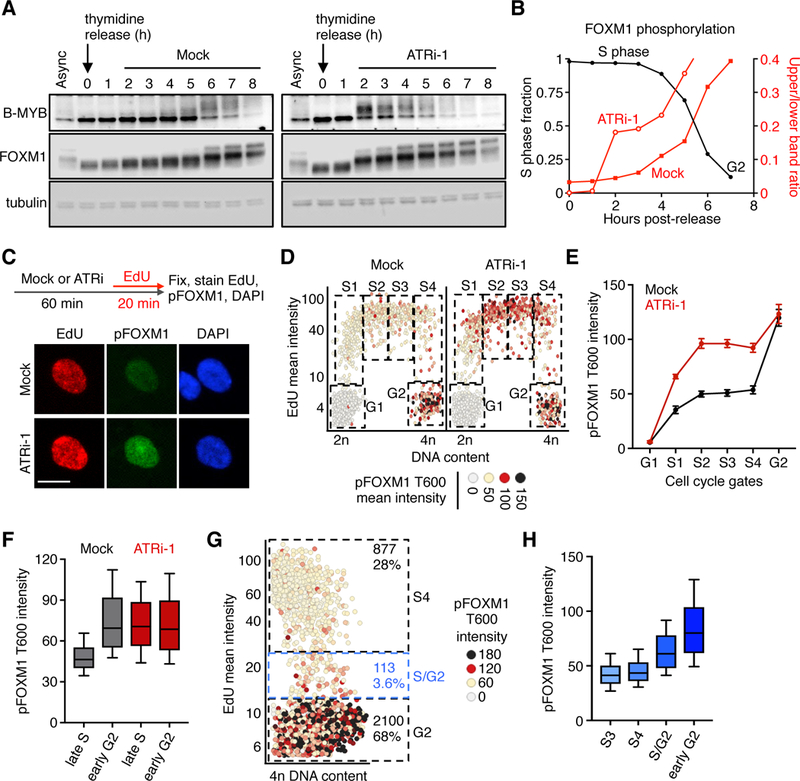
(A) Western blots of cells synchronized as in fig S6A and mock- or ATRi-treated. (B) The ratio of upper to lower FOXM1 band intensities from blots shown in (A) are plotted as red lines on the right y-axis. Black lines (left y-axis) represent the fraction of S phase synchronized cells. S phase fraction was calculated as described in fig. S6, B-D. (C) Experimental design and representative images of S phase hTERT RPE-1 cells. (D) QIBC plots of total DAPI intensity vs. EdU mean intensity, and mean pFOXM1 T600 intensity on a color scale. Boxes indicate gated populations used for further analysis. (E) Median pFOXM1 intensities of gated populations shown in (D). Error bars represent the SEM of 4 independent experiments. (F) pFOXM1 T600 mean intensity in late S or early G2 cells treated as described in (C). (G) QIBC plot of EdU-labeled cells with 4n DNA content. Boxes indicate gated populations used for analysis in (H). Numbers indicate number of cells in each gated population. The S/G2 population was determined as described in fig. S10. (H) pFOXM1 T600 mean intensity of late S phase cells (S3 and S4), S/G2 cells, or early G2 cells. In (F) and (G), whiskers indicate the 10th and 90th percentiles. Boxes indicate the 25th-75th percentile and median value.
The marked difference in S and G2 phase pFOXM1 levels suggests that there might be a unique regulatory event that defines the S/G2 transition, akin to the inactivation of APC/CCdh1 at the G1/S transition or the activation of nuclear cyclin B/CDK1 at the G2/M transition (23, 24). Thus, we measured pFOXM1 levels during late S (S3 and S4), the S/G2 transition (fig. S10), and early G2. pFOXM1 levels rose abruptly in the S/G2 population. As this population comprises less than 4% of the late S and early G2 cells, phosphorylation occurs within a narrow time window (Fig. 2, G and H). Live-cell imaging with the EYFP-PCNA biosensor followed by fixation and pFOXM1 imaging confirmed that FOXM1 phosphorylation occurs rapidly at the S/G2 transition (fig. S11, A and B). Thus, the S/G2 transition is tightly regulated by ATR ensuring FOXM1 phosphorylation occurs in a switch-like manner upon the completion of S phase, rather than gradually throughout S phase.
Paradoxically, a cyclin A-dependent CDK phosphorylates FOXM1, but cyclin A-CDK activity gradually increases throughout S phase (25), inconsistent with the observed switch-like behavior. As cyclin A can bind to either CDK2 or CDK1, we hypothesized that different cyclin A-CDK complexes are active in S phase and at the S/G2 transition. Therefore, we measured pFOXMl levels following either CDK2 or CDK1 inhibition. Two different CDK2 inhibitors had little effect on FOXM1 phosphorylation at a range of doses (Fig. 3, A and B and fig. S12A) although both decreased EdU incorporation (fig. S12B). In striking contrast, both rapid FOXM1 phosphorylation at the S/G2 transition and premature phosphorylation in ATR-inhibited cells were CDK1 dependent (Fig. 3, A and C, and fig. S12). Furthermore, inhibition of an analogsensitive CDK1 (CDK1-as) (26) in RPE-1 cells reduced FOXM1 phosphorylation, and CDK2 inhibition had no additional effect (fig. S13). These results confirm the specific role of CDK1 in mediating FOXM1 phosphorylation at the S/G2 transition.
Figure 3. ATR is active until G2, preventing CDKl-dependent FOXM1 phosphorylation.
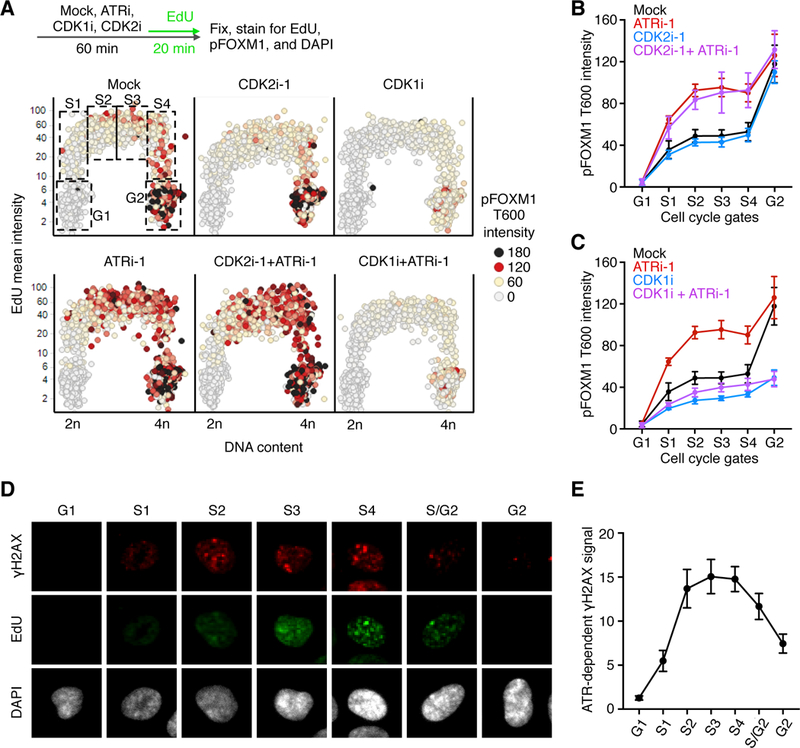
(A) Experimental design and QIBC plots of total DAPI intensity vs. EdU mean intensity, and mean pFOXM1 T600 intensity on a color scale. Boxes indicate gated populations used for further analysis. (B and C) Median pFOXM1 intensities of gated populations shown in (A). Error bars represent the SEM of 3 independent experiments. (D) Representative images of G1, early-to-late S (S1-S/G2), or G2 hTERT RPE-1 cells. (E) ATR activity during an unperturbed cell cycle as described in fig. S14. Error bars represent the SEM of 3 independent experiments
As the data suggest that CDK1 is rapidly activated at the S/G2 transition to phosphorylate FOXM1, we hypothesized that ATR activity may abruptly decline as S phase ends relieving an intrinsic checkpoint. Thus, we measured ATR activity by quantifying the effect of ATR inhibition on phosphorylation of the histone variant H2AX, at Ser139 (γH2AX). The ATR-dependent γH2AX signal increased as cells entered S phase, peaked in mid-and late-S phase, and rapidly decreased as cells completed DNA replication (Fig. 3, D and E, and fig. S14, A-D). Thus, ATR is active throughout S phase, but its activity drops at the S/G2 transition allowing rapid CDK1-dependent FOXM1 phosphorylation and transactivation of the G2/M gene network.
In mammalian cells, ATR is activated through a RAD9A-TOPBP1 or ETAA1-dependent pathway (6, 27–29). Knockdown of ETAA1 reduced ATR activity in an unperturbed S phase whereas RAD9A knockdown did not, even though it reduced γH2AX levels following hydroxyurea-induced replication stress (Fig. 4A, and fig. S14E). Moreover, deletion of the ETAA1 ATR-activation domain in HCT116 cells greatly reduced ATR activity in an unperturbed S phase but not in response to replication stress, while auxin-mediated degradation of a TOPBPl-mAID fusion protein had the opposite effect (Fig. 4, B and C and fig. S15). Thus, ETAA1 acts during an unperturbed S phase to activate ATR and enforce the intrinsic S/G2 checkpoint, whereas TOPBP1-RAD9A activates ATR to enforce the replication stress response.
Figure 4. The ETAA1-ATR checkpoint couples S phase and mitosis.
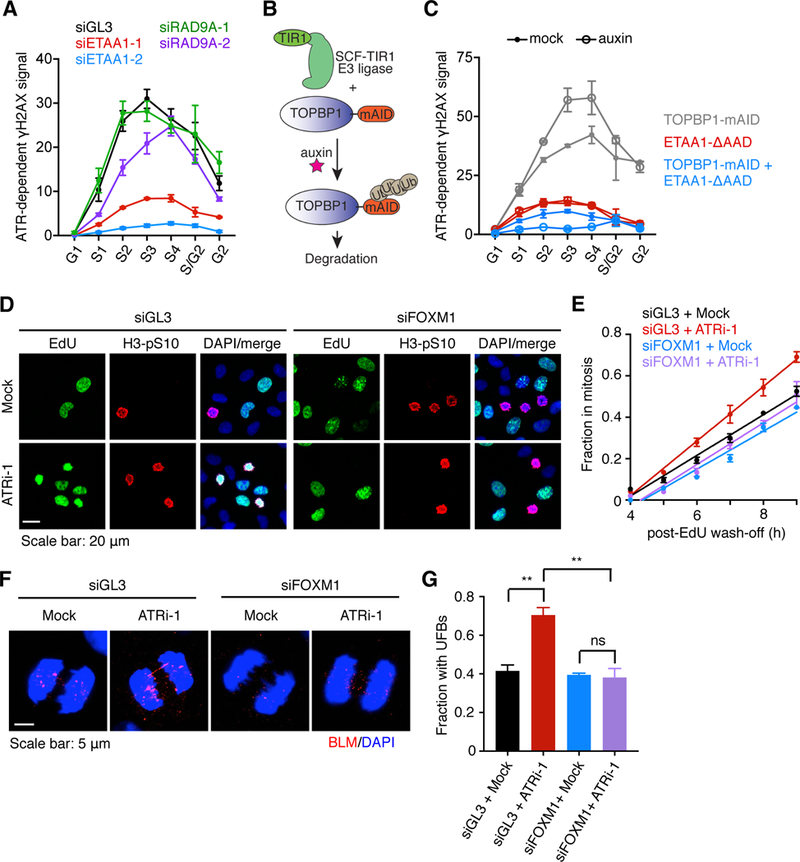
(A) ATR activity in siRNA-transfected cells during an unperturbed cell cycle as described in fig. S14. Error bars represent the SEM of 3 independent experiments (B) Illustration of the mAID degron system fused to TOPBP1. (C) ATR activity in HCT116 cell lines mock-treated or auxin-treated for 2 h as described in fig. S15A. Error bars represent the SEM of 3 independent experiments (D and E) S-M progression assay (see Fig. 1, A and B) in hTERT RPE-1 cells 40 h after siRNA transfection. Representative images 6 h post EdU-wash-off. (E) Fraction of S phase-gated cells in mitosis as a function of time post-EdU wash-off. Error bars represent the SEM of 3 independent experiments. (F) Representative images of anaphase cells 40 h after siRNA transfection and then mock- or ATRi-treated for 8 h. (G) Fraction of cells with UFBs. Error bars represent the SEM of three independent experiments.
Once activated, ATR enforces its checkpoint functions in part by phosphorylating and activating CHK1, an effector kinase that ensures CDK inhibition by regulating CDC25 (6). CHK1 inhibition also triggered premature phosphorylation of FOXM1 and S phase accumulation of cyclin B (fig. S16, A-C). Thus, the S/G2 transition is controlled by the repressive activity of the ETAA1-ATR-CHK1 pathway on the cyclin A-CDK1-FOXM1 axis until the end of S phase (fig. S16D).
Many genes within the FOXM1 network promote mitotic entry (30), and FOXM1 overexpression is sufficient to accelerate mitotic entry (20). Therefore, we asked if the effect of ATR inhibition on S-M progression was due to FOXM1 deregulation or whether it resulted from another function of ATR. To do so, we knocked down FOXM1 to a level that still allows cells to enter mitosis yet also suppresses premature cyclin B expression (fig. S7E). Under these conditions, we found that FOXM1 knockdown largely rescued the accelerated S-M progression in ATR-inhibited cells (Fig. 4, D and E). These findings indicate that ATR slows the rate of mitotic entry primarily through its control of FOXM1.
The deregulated S/G2 transition induced by ATR inhibition may lead to incomplete DNA replication. Perturbations that allow cells to enter mitosis with under-replicated DNA, including ATR inhibition, induce ultrafine anaphase bridges (UFBs) coated by BLM helicase (9, 31). Importantly, partial FOXM1 knockdown fully negated UFB formation induced by ATR inhibition, suggesting that premature transactivation of the mitotic gene network prevents completion of DNA replication (Fig. 4, F and G). If UFBs are unresolved in mitosis, DNA breakage may occur, leading to 53BP1 body formation (32). We found that ATR inhibition increased the formation of 53BP1 bodies in the daughter G1 cells, and, partial FOXM1 knockdown reduced their formation (fig. S17). Thus, proper control of the S/G2 phosphorylation switch ensures timely transactivation of the mitotic gene network and is critical to prevent premature mitotic entry and subsequent DNA damage resulting from under-replicated DNA.
Our data unveil an S/G2 phosphorylation switch, controlled by ATR, that initiates the G2/M transcription program. By repressing CDK1, ATR blocks this switch until the end of S phase to ensure the temporal order of later cell cycle processes. Although the nature of the signal activating ATR in an unperturbed S phase is still unclear, the role of ETAA1 suggests that ssDNA generated by ongoing replication, rather than unreplicated DNA itself, is a component of the signal. Transiently-formed RPA-coated ssDNA could serve as a platform for colocalizing both ETAA1 and ATR, sustaining ATR activity throughout S phase. The decline in ATR activity that occurs once replication is complete would signal a clear S/G2 transition, executed via a CDK1-pFOXM1 switch (fig. S18).
We propose this intrinsic function of ATR in normal cells may effectively serve as the checkpoint long proposed to monitor the completion of replication (4, 5). Loss of this function causes an identity crisis allowing G2 phase to overlap with S phase, leading to incomplete replication, an early mitosis, and subsequent DNA damage. Because the ATR-FOXM1 pathway ensures that G2 events are dependent on the completion of S phase, we refer to this as an intrinsic S/G2 checkpoint. Given the frequent overexpression of FOXM1 in cancer (33), deregulation of this fundamental cell cycle transition could be a common event in many cancers that contributes to their genome instability.
Supplementary Material
Acknowledgments:
We thank Fena Ochs, Jim Ferrell, and members of the Cimprich laboratory for helpful discussions and feedback on the manuscript. This work was supported by grants from the NIH to K.A.C. (ES016486) and D C. (CA102729) and Wellcome grants (107022 and 203149) to W.C.E. J.C.S was supported by a Postdoctoral Fellowship, PF-15–165-01-DMC from the American Cancer Society and holds a Postdoctoral Enrichment Program Award from the Burroughs Wellcome Fund. S.H. was supported by a fellowship from the German Research Foundation DFG (HA 6996/1–1). F.C.-S. was supported by a CONACYT fellowship from the Mexican Government. RNA-seq data are available in the GEO repository (accession number GSE116131).
References and Notes:
- 1.Morgan DO, The cell cycle : principles of control. Primers in biology (New Science Press Ltd in association with Oxford University Press, London, 2007), pp.xxvii, 297 p. [Google Scholar]
- 2.Rao PN, Johnson RT, Mammalian cell fusion: studies on the regulation of DNA synthesis and mitosis. Nature 225, 159–164 (1970). [DOI] [PubMed] [Google Scholar]
- 3.Johnson RT, Rao PN, Mammalian cell fusion: induction of premature chromosome condensation in interphase nuclei. Nature 226, 717–722 (1970). [DOI] [PubMed] [Google Scholar]
- 4.Hartwell LH, Weinert TA, Checkpoints: controls that ensure the order of cell cycle events. Science 246, 629–634 (1989). [DOI] [PubMed] [Google Scholar]
- 5.Elledge SJ, Cell cycle checkpoints: preventing an identity crisis. Science 274, 1664–1672 (1996). [DOI] [PubMed] [Google Scholar]
- 6.Saldivar JC, Cortez D, Cimprich KA, The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown EJ, Baltimore D, ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 14, 397–402 (2000). [PMC free article] [PubMed] [Google Scholar]
- 8.de Klein A et al. , Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol 10, 479–482 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Eykelenboom JK et al. , ATR activates the S-M checkpoint during unperturbed growth to ensure sufficient replication prior to mitotic onset. Cell Rep 5, 1095–1107 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Ruiz S et al. , A Genome-wide CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to ATR Inhibitors. Mol Cell 62, 307–313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toledo LI et al. , ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Hahn AT, Jones JT, Meyer T, Quantitative analysis of cell cycle phase durations and PC12 differentiation using fluorescent biosensors. Cell Cycle 8, 1044–1052 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akopyan K et al. , Assessing kinetics from fixed cells reveals activation of the mitotic entry network at the S/G2 transition. Mol Cell 53, 843–853 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Fung TK, Poon RY, A roller coaster ride with the mitotic cyclins. Semin Cell Dev Biol 16, 335–342 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Chen YJ et al. , A conserved phosphorylation site within the forkhead domain of FoxM1B is required for its activation by cyclin-CDK1. J Biol Chem 284, 30695–30707 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laoukili J et al. , Activation of FoxM1 during G2 requires cyclin A/Cdk-dependent relief of autorepression by the FoxM1 N-terminal domain. Mol Cell Biol 28, 3076–3087 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson C et al. , Cell-cycle regulation of B-Myb protein expression: specific phosphorylation during the S phase of the cell cycle. Oncogene 12, 1855–1864 (1996). [PubMed] [Google Scholar]
- 18.Lane S, Farlie P, Watson R, B-Myb function can be markedly enhanced by cyclin A-dependent kinase and protein truncation. Oncogene 14, 2445–2453 (1997). [DOI] [PubMed] [Google Scholar]
- 19.Sala A et al. , Activation of human B-MYB by cyclins. Proc Natl Acad Sci 94, 532–536 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laoukili J et al. , FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol 7, 126–136 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Sadasivam S, Duan S, DeCaprio JA, The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev 26, 474–489 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luscher-Firzlaff JM, Lilischkis R, Luscher B, Regulation of the transcription factor FOXM1c by Cyclin E/CDK2. FEBS Lett 580, 1716–1722 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Cappell SD, Chung M, Jaimovich A, Spencer SL, Meyer T, Irreversible APC(Cdhl) Inactivation Underlies the Point of No Return for Cell-Cycle Entry. Cell 166, 167–180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santos SD, Wollman R, Meyer T, Ferrell JE Jr., Spatial positive feedback at the onset of mitosis. Cell 149, 1500–1513 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spencer etal SL., The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 155, 369–383 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hochegger H et al. , An essential role for Cdk1 in S phase control is revealed via chemical genetics in vertebrate cells. J Cell Biol 178, 257–268 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee YC, Zhou Q, Chen J, Yuan J, RPA-Binding Protein ETAA1 Is an ATR Activator Involved in DNA Replication Stress Response. Curr Biol 26, 3257–3268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haahr P et al. , Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat Cell Biol 18, 1196–1207 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Bass TE et al. , ETAA1 acts at stalled replication forks to maintain genome integrity. Nat Cell Biol 18, 1185–1195 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindqvist A, Rodriguez-Bravo V, Medema RH, The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol 185, 193–202 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan KL, Palmai-Pallag T, Ying S, Hickson ID, Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 11, 753–760 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Lukas C et al. , 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 13, 243–253 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Laoukili J, Stahl M, Medema RH, FoxM1: at the crossroads of ageing and cancer. Biochim Biophys Acta 1775, 92–102 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.