

Abstract
In-situ X-ray absorption spectroscopy (XAS) at the oxygen K-edge was used to investigate the role of oxygen during the oxygen evolution reaction (OER) in an electrodeposited Ni-Fe(OxHy) electrocatalyst in alkaline pH. We show the rise of a pre-peak feature at 529 eV in the O K-edge spectra, correlated to the appearance of a shoulder at the Ni L3-edge and formation of oxidized Ni3+/4+-O. Then, for the first time, we track the spectral changes in a dynamic fashion in both the soft and hard X-ray regimes during cyclic voltammetry (in situ CV-XAS) to obtain a fine-tuned resolution of the potential-related changes. The pre-peak feature at the O K-edge likely signifies formation of an electron deficient oxygen site. The electrophilic oxygen species appears and disappears reversibly in correlation with the Ni2+ ↔ Ni3+/4+ process, and persists during OER catalysis as long the metal is oxidized. Our study provides new insight into OER electrocatalysis: Before onset of the O-O bond formation step, the catalytic oxyhydroxide has accumulated electron deficiencies by both, oxidation of transition metal ions and formation of partially oxidized oxygen sites.
Introduction
Reducing global carbon emissions will require efficient catalysts for use in solar-to-fuel conversion processes, where electrochemical water oxidation is the key to approach zero emissions1. For this, understanding the fundamental processes of the oxygen evolution reaction (OER, 4OH− → O2 + 2H2O + 4e−) from a mechanistic perspective is an important sub-goal for the design of highly efficient and functional electrocatalysts2,3. Ni-Fe oxyhydroxide (OxHy) electrocatalysts are currently the most active catalysts, and catalyze OER in alkaline media at low overpotentials with high O2 efficiency and turnover rates4–17. Combining Ni and Fe leads to an excessive increase in the catalytic activity, coincident with changes at the redox-active metal center, witnessing the electronic interaction between Ni and Fe sites7,18. The complexity of this interaction includes a modulation of the metal redox activity, which complicates the interpretation of the factors that scales with the OER activity. This have resulted in a controversy regarding the catalytically active structural and electronic state in mixed Ni-Fe catalysts7,14,18,19.
Friebel et al.14 presented in operando XAS investigations in combination with DFT + U calculations of mixed electrodeposited Ni-Fe catalysts with different compositions, which concluded that OER proceeds at lowest overpotential at Fe-sites embedded in the Ni(OOH) lattice. This study demonstrated in agreement with other studies that the active metal redox states can be described as Ni3+/4+-Fe3+ 10,20,21, however, according to work from us and other groups OER can also proceed on low-valent Ni2+ sites15,19. The same discussions have been held for the Fe-site where both Fe3+ 14,20 and Fe4+ sites19,21 have been observed. More recently, spectral fingerprints of Fe6+ sites were observed in a spectroscopic study in non-aqueous solvent by Hunter et al.22. This reveals an intriguing complexity of the catalytic system that may mask the true origin of the activity enhancement in binary Ni-Fe centers. Burke et al.23 recently presented a study with Fe-spiked KOH electrolyte that revealed two distinct catalytic sites, where the “highly-active” sites were proposed to be located at “edge” or “defect sites” since the activity was unrelated to long-term modulations of the bulk. The presence of “fast” and “slow” sites in Ni-Fe catalysts was also recently observed in a scanning electrochemical microscopy study by Ahn et al.24. The nature of the OER mechanism in alkaline electrolyte is not yet fully understood and has so far been rather simplified. The OER overpotential is assumed to be restricted by scaling relations between surface bound OER intermediates (O*, OH*, OOH*) that includes four proton-coupled electron transfer (PCET) steps resulting in high overpotential25. The presence of “active oxygen” species has recently been emphasized, where a non-concerted step has been proposed to occur in the mechanistic pathway by Koper and coworkers20,26,27. To entangle the role of the oxygen as a plausible redox-active site in Ni-Fe catalysts, further spectroscopic investigations would be required. X-ray spectroscopic investigations have so far principally been limited to the metal K-edges of the Ni and the Fe metal7,14,18, where studies involving in situ soft X-ray at the O K-edge of Ni-Fe catalysts is still lacking. This may provide useful information on the interaction and orbital mixing between the metal 3d and oxygen 2p sites28–30. In the only study known to us of a similar system, Yoshida et al. presented an in situ soft XAS study of an electrodeposited nickel-borate catalyst (Ni-Bi) in near neutral pH conditions, where changes in the O K-edge spectra were observed as a reversible peak near 529 eV, related to formation of Ni+3.6 in edge-sharing NiO6 octahedra31. Using in situ ambient pressure XPS, Ali-Löytty et al.32 investigated the O K-edge of an electrodeposited Ni-Fe catalyst, however corresponding changes around the O K pre-edge region was absent in this study.
In this study, we present the first in situ measurements in both the soft and the hard X-ray regimes under OER catalytic conditions at the O K-edge and the metal (Ni, Fe) L- and K- edges of an electrodeposited Ni-Fe(OxHy) oxygen evolution electrocatalyst in alkaline electrolyte. We discuss the involvement of the oxygen species in OER and we further correlate changes at the O K-edge in a dynamic fashion with changes occurring at the metal L- and K-edges during in situ CV cycling.
Experimental Section
Sample preparation
The Ni-Fe films were cathodically electrodeposited from aqueous solution using ultrapure Milli-Q water (>18.2 MΩ cm) containing 9 mM Ni(SO4) × 6H2O (Sigma-Aldrich), 9 mM Fe(SO4) × 7H2O (Sigma-Aldrich), and 25 mM (NH4)2SO4. The catalyst was electrodeposited on a 150 nm thick Si3N4 membrane (Silson) coated with a 1 nm Ti adhesion layer and a 20 nm Au conductive top layer employed as working electrode. A cathodic current density of −250 mA/cm2 was applied for 5 s on a limited area of 2 × 2 mm, which was considered as the geometric surface area of the working electrode. The metal loading was determined by total reflection X-ray fluorescence spectroscopy (TXRF), using a benchtop S2 Picofox spectrometer (Bruker) with a Mo Kα source at 40 kV and a Si-drift detector. The metal loading on the electrodes was determined to 510 nmol/cm2 of Ni and 275 nmol/cm2 of Fe (Ni:Fe = 65:35). The film thickness was estimated to 400 nm by assuming 0.125 µC/cm2 per 1 nm33.
Scanning Electron Microscopy (SEM)
The morphology of the catalyst film deposited on the Si3N4/Ti/Au was characterized using a LEO Gemini 1530 field emission scanning electron microscope (FESEM) at the Helmholtz Zentrum Berlin (HZB), of as-deposited catalysts and after the soft X-ray in situ OER characterization.
In situ X-ray absorption spectroscopy and electrochemical measurements
In situ electrochemical X-ray absorption measurements of the O K-edge and Ni, Fe L-edges were performed with a PEEK transmission cell in transmission mode34, at the LiXEdrom 2.0 end-station at the U56-2 PGM2 beamline at BESSY II (HZB). The layout of the liquid cell used for the in situ measurements is illustrated in Fig. 1. Similar to previous work35, the thin layer of electrolyte is confined between two assembled Si3N4 membranes (100 nm). One of them is coated with 1 nm Ti, 20 nm Au and the Ni-Fe catalyst, which serves as working electrode. A Pt-wire was used as counter electrode, and a leak-free Ag/AgCl (Ø = 1 mm) applied as reference electrode. The electrochemical measurements were controlled using a Bio-Logic SP-200 potentiostat. All electrode potentials are reported on the reversible hydrogen electrode scale (ERHE = 0.21 VAg/AgCl + 0.059 V × pH). The two He-chambers of the XAS cell (He-1 and He-2) were constantly purged during the measurements in order to maximize the X-ray transmission signal by removing air and water vapor. Applying He-pressure also allows to bend the two membranes so that the thickness of the electrolyte layer is reduced and thus the detrimental absorption of the soft X-ray at the O K-edge is decreased36. All measurements were carried out in 0.1 M KOH electrolyte pH 13. A syringe pump was used to keep a constant electrolyte flow of 4 µl/s. The resistance in the sXAS cell during the in situ measurements was estimated to ca 200 Ω. This pressure was varying with the He-pressure, whereas this was kept constant. The measurements are shown without iR-compensation.
Figure 1.
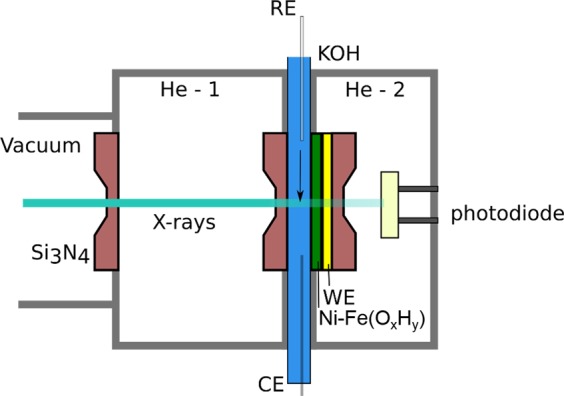
A schematic illustration of the soft X-ray transmission cell used in this study. The catalyst is deposited on the Au-coated silicon nitride membrane window closest to the photodiode. The electrolyte is confined between the two Si3N4 membranes. The GaAs photodiode measures the X-ray transmitted through the electrolyte and membranes.
The slit width was set to 50 µm to obtain a spectral resolution of 200 meV at an energy of 535 eV. The cell was located out of focus by 50 cm as described in our previous reports35. The He pressure inside the cell was kept constant at 6 kPa during the in situ measurements. Photons were collected using a GaAs photodiode (Hamamatsu G1127). The current was measured using a 6514 Keithley electrometer with 2 nA range sensitivity. Every spectrum was averaged on three consecutives scans and were normalized to the current produced by the beam hitting the refocusing mirror before the sample. At the O K-edge the spectra were calibrated using the pre-edge of water at 535 eV37–40 and normalized to the the NEXAFS region above 570 eV. In order to reveal the contribution of the different electronic states present in the sample, we decomposed the O K-edge spectrum in its spectral components using multi-peak fitting in Matlab, similar to the procedure followed by Guo et al.41. For more details, see Supporting Information. The Ni and Fe L-edges were calibrated using the L3 peaks of NiO at 852 eV42 and that of Fe2O3 at 708.5 eV43. A linear background was subtracted and the intensities were normalized to the maximum intensity of the L3 edge. To avoid radition damage, we significantly reduced the exposition of the catalyst to X-ray by selecting an experimental geometry where the incoming photons are attenuated by the electrolyte solution before hitting the catalyst sample. In order to ensure the reproducebility of the measurements, 3 repetitions were carried out for each configuration.
In situ XAS in the hard X-ray regime at the Ni and Fe K-edges were carried out at the KMC-3 beamline (BESSY II, HZB). A silicon (111) double-crystal monochromator was used for selecting a fixed X-ray excitation energy. The electrochemical cell was made of PTFE, and the Ni-Fe catalyst electrodeposited on Au-coated glassy carbon working electrodes (area 0.2 cm2). A Pt-mesh was used as counter electrode, and a leak free Ag/AgCl filled with 3 M KCl as reference electrode. The resistance in the cell was estimated to ca 30 Ω. The sample was placed 45 °C to the incoming X-ray beam using back-side illumination, and the fluoresence monitored using a scintillation detector. The amplified signal was recorded with a Bio-Logic SP-200 potentiostat. For more details, see reference44.
Cyclic Voltammetry during X-ray absorption spectroscopy
Cyclic voltammetry-X-ray absorption measurements (CV-XAS) was performed at the U56-2 PGM2 and KMC-3 beamlines for the sXAS (O K and Ni, Fe L) and for the hard XAS (Ni, Fe K), respectively. The energy of the beam was set to a constant value of 854.1 eV for the Ni L, 529 eV for O K, 842 eV for Ni K, and 726 eV, and for Fe K-edges. The scan-rate was set to 5 mV/s or 10 mV/s while the Keithley signal was recorded with an analog-to-digital (AD) converter using the provided EC-lab software from Bio-Logic. The photons were collected with a photodiode and the resulting current was read from the output of the Keithley. The shown CV curves is an average of at least 16 individual CV cycles between ~1–1.7 V vs RHE.
Results and Discussion
Electrochemical Properties and Surface Morphologies
An electrodeposited Ni-Fe oxyhydroxide (OxHy) catalyst was investigated for the oxygen evolution reaction using in situ X-ray absorption spectroscopy in the soft and hard X-ray regimes. SEM characterization showed that the Si3N4/Ti/Au electrode substrate was decorated with platelet-like sheets, with a structure similar to the ones reported for other electrodeposited Ni-Fe oxyhydroxide catalysts (see Supporting Information Figure S1a)19,45,46. The Ni-Fe samples examined after the XAS investigations (after OER characterization) showed similar structure as the freshly as-deposited catalyst (see Fig. 1b). Total reflection X-ray fluorescence spectroscopy (TXRF) confirmed that the catalyst had a Fe-content of 35 atomic %. Measurements of an empty electrode substrate in comparison to the substrate with the electrodeposited Ni65Fe35 catalyst confirmed a negligible OER activity of the bare glassy carbon substrate, confirming that the OER activity originates from the Ni65Fe35 electrocatalyst (see Figure S2).
In-situ X-ray absorption spectroscopy at the nickel and iron L- and K-edges
The Ni65Fe35 catalyst was investigated using soft X-ray (sXAS) at the Ni and Fe L-edges at several electrode potentials. The applied potential was increased by steps of 0.05 V into the OER region. For 3d transition metals, sXAS at the metal L-edge directly measures the unoccupied TM-3d states through dipole-allowed 2p-to-3d transitions.
The in situ sXAS spectra at the Ni L-edges are shown in Fig. 2a. The electrochemical activity during the measurement is shown in the Supporting Information Figure S3. The overall spectral line shape consists of features in two regions, L3-edge around 852 eV (features A and B) and L2–edge around 870 eV (features C and D), resulting from the core- hole spin-orbital-coupling split. The fine splitting in both L3 and L2 is due to the crystal field effect from local ligand environment47,48. Those features result from dipole transitions from the 2p core level of the metal to empty states taking atomic multiplet effects into account which are sensitive to the electronic and oxidation state of the metal, and to the local geometry47. Upon increasing the potential from 0.98 V to 1.53 V vs. RHE, the amplitude of feature B is intensified. When the potential is reversed the amplitude of feature B drops down to its initial value. The intensity ratio of the double-peak features in the Ni L3 region (at 852 and 854.1 eV) fingerprints the oxidation state of the Ni atoms in the catalyst49,50. Our measurements do not support formation of Ni4+ due to the absence of a shift in the position of the enhanced feature B, i.e. the 2nd shoulder of the L3 doublet feature. However, the rise of feature B during OER conditions reveals the presence of Ni species in oxidation state +3, showing that the Ni65Fe25 catalyst oxidized from Ni2+ → Ni3+ 51. This observation is in accordance with previous reports of oxidation states obtained from Ni K-edge measurements of mixed Ni-Fe catalysts during OER catalytic potential7,14,50.
Figure 2.

In Situ XAS of electrodeposited Ni-Fe catalyst at (a) the Ni L-edge and (b) Fe L-edge. Potential values are in V vs RHE, where “rev” means “reverse” direction when stepping from higher to lower electrode potentials, and “OCP” stands for open circuit potential. The measurements were carried out in 0.1 M KOH, and the potentials are reported on the RHE scale.
In Fig. 2b, the Fe L-edges are shown, with the L3-edge peak around 707 eV (features A’ and B’) and the L2-edge peak around 720 eV (features C’ and D’). In contrast to the Ni L-edge, there is no observed potential dependence of the spectral features on the Fe L-edge, hence our measurements are compatible with Fe+3 both during non-catalytic and OER catalytic potential. This observation is in agreement with previous studies at the metal K-edge of Ni-Fe catalysts7,14. Oxidation states of Fe4+ or higher have on the other hand been reported during OER18,20,21. We cannot rule out that a small fraction of surface related Fe atoms may enter a higher oxidation state in our catalysts, however if the Fe atoms that changes oxidation state are a minority in number (for example appear at edge or defect sites) they may not be possible to resolve in these measurements. It is also possible that the Fe species formed during OER are too reactive to be observed under the current reaction conditions, in line with previous discussions regarding the reactivity and equilibrium states of intermediates occurring in the catalytic cycle7,22.
To support our observations at the metal L-edges, we performed in situ XAS measurements in the hard X-ray regime at the Ni and Fe K-edges, where the spectral fingerprints are well known and oxidation states can be extracted from the edge positions. These measurements showed that the Ni oxidation process begin at 1.48 V vs. RHE, seen as an edge shift to higher energies (see Figure S4a). The Fe K-edge on the other hand did not undergo a pronounced edge shift, however a small visible change in the white line intensity occurred at electrode potentials above the threshold of Ni oxidation, in accordance with other work on Ni-Fe catalysts during in situ conditions (see Figure S4b)7,9,14,21. It has been discussed whether this corresponds to an oxidation state change at the Fe site or a change in the geometry as a response to the oxidation state increase at the Ni site during OER9,14,21. In accordance with these studies, the changes we observe in the white line intensity may indicate that a small fraction of Fe atoms enters a higher oxidation state in our mixed Ni-Fe catalyst.
The Ni K-edge showed an edge shift of +1.2 eV at the highest measured potential, which corresponds to an oxidation state increase of 0.7 units, determined using calibration curves with known reference compounds7. This is in agreement with the transition from Ni2+ to Ni3+, hence confirming the observations at the Ni L-edge. From the Ni K pre-edge, contributions from a metallic phase was evident in our Ni65Fe35 catalyst, which was also reported in a recent study of a similar electrodeposited Ni-Fe catalyst9. Using linear combination of reference compounds, we could correct for this metallic contribution (see Figure S5a,b). The oxidation state increase of the pure oxide phase (after subtraction of the metallic phase) was estimated to 1.1 units, so slightly higher oxidation state than without corrections for the metallic contribution. This is still compatible with Ni3+, however we cannot exclude that a fraction of the Ni atoms enters an oxidation state higher than +3. Therefore, we conclude that the Ni atoms in our Ni65Fe35 catalyst do not reach “full” oxidation with Ni in oxidation state between +3 and +47,14. We also noticed that on the reverse scan a portion of the Ni atoms needed a longer time to relax back to the initial reduced Ni2+ ground state, also observed in previous studies20,52.
In situ soft X-ray absorption spectroscopy at the O K-edge
X-ray absorption spectroscopy at the O K-edge was used to probe the local bonding and symmetry properties of the X-ray excited oxygen atom. The O1s spectra of the electrodeposited Ni-Fe catalyst under applied potential are shown in Fig. 3a. The O K-edge spectra can be divided into three distinct regions. The high-energy region above 540 eV corresponding to the extended X-ray absorption fine structure. The spectra presented here were normalized to the intensity in this region.
Figure 3.

In situ XAS O k-edge spectra of electrodeposited Ni-Fe catalyst measured at various applied potentials. Potential values are given vs RHE., where “rev” means the reverse direction from higher to lower potentials. (b) O K-edge prefeature region at 1.18 V and (c) at 1.78 V vs RHE. Blue dots represent the experimental points, colored dashed lines the fit components of the multi-peak fitting and solid color lines the fit data model. The peak at 529 eV is the O1s-O(2p)/Ni(t2g) transition and appears at OER potential due to the increase in oxidation state of Ni. The measurements were carried out in 0.1 M KOH. (d) Schematic illustration showing the correlation between the changes at the O K-edge at 529 eV and the Ni L-edge at 854.1 eV; a partial donation of the higher electron density on the oxygen site (Oδ−) to the Ni metal site (O → Ni charge transfer).
The low energy region between 525 eV and 534 eV is the region of main interest in this work and a detailed view is presented in Fig. 3b,c. This low energy region typically originates from electronic transitions from the O1s shell to transition metal 3d orbitals hybridized with oxygen 2p orbitals28,48,53–55. Four peaks could be distinguished at 529 eV, 529.9 eV, 531.2 eV and 532.5 eV. We assign these to the transitions from O(1 s) to O(2p) orbitals mixed with Ni(3d)t2g, Fe(3d)t2g, O(*) of O2 gas, and Fe(3d)eg, respectively28,56,57. The peak positions were determined with Gaussian multi-peak fitting (see Figure S6a–f, Tables S1–S2 for fit parameters, and Table S3 for literature peak assignment). A reference spectrum of the O K-edge for O2 gas is shown in Figure S7. Spectral intensities between 534 eV and 540 eV result from near edge transitions of the water molecule, specifically at 535 eV, and at the main- and post-edge at 537.5 eV and 542 eV, respectively. Note, that main- and post-edge features are saturated. The observed intensity variations of main- and post-edge do not correlate with the applied potential but increase with measurement time (see Figure S6e,f). These changes may also be influenced by slow variations in the He pressure of the sXAS transmission cell, which can cause changes in the electrolyte layer thickness between the two assembled Si3N4 membranes. Similar changes of the pre-peak to main-peak ratio were observed for a transmission cell by Schreck et al.40, who correlated this to inhomogeneity of the liquid layer.
At increased electrode potential we observe a rise of a new pre-peak feature at 529 eV, which appears at 1.48 V and disappears reversibly at reductive potentials (see Figure 3a and S6a). We note that this peak is coincident with the oxidation of Ni2+ → Ni3+ as seen in both the Ni L and K edges. In accordance with previous studies, we correlate the 529 eV feature to the hybridization between O(2p) and Ni(3d)t2g orbitals, t2g being the 3d orbital with lowest energy in the frame of molecular orbital theory for an octahedral complex28,41,48,54,56. The other new feature located right above this peak, more specifically at 529.9 eV, is assigned to interactions between the O(2p) and hybridized Fe(3d) orbitals. This peak showed a much smaller change with the applied electrode potential in comparison to the 529 eV peak. This implies that Fe undergoes a smaller modulation, which supports the observations at the Fe K- edge where only a small change was observed in the white line intensity. This shows that a small modification at the Fe site is likely, however much smaller in comparison to the changes at the Ni site.
A similar pre-peak at 529 eV was reported in a soft XAS study of a Ni-Bi catalyst in borate buffer (near neutral pH) by Yoshida et al.31, and was assigned to bridging oxygen atoms coordinated to oxidized Ni+3/+4. This was explained by the oxide oxygens (O) connected to oxidized Ni(OOH) which gives rise to a O K-edge peak at lower energies in comparison to hydroxide oxygens (-OH)31. This pre-peak feature is indeed present in the powder reference compound LiNiO2, where Ni is in oxidation state +3 (see Figure S8). In work from other groups on metal oxides, the rise of a pre-peak at such low energies has been given alternative explanations. In work by Suntivich et al.28 a pre-peak in the O K-edge spectra was observed in perovskite OER catalysts. This was explained as an increase in hybridization between 3(d) and O(2p) states, which could cause injection/extraction of electrons from oxygen to the metal site (Ni3+-O2− → Ni(3-δ)+ − O(2-δ)−), which was correlated to a more efficient OER activity, see Fig. 3d28. In work by Cho et al.58 Ni vacancies in NiO increased the formal charge on nearby atoms, which was proposed to favor charge transfer from O → Ni to stabilize a Ni2+-O− complex56,58. It is therefore possible that a charge redistribution occurs in concomitance with the oxidation of Ni2+ to Ni3+. In previous in situ XAS studies by us we have shown that Fe-dopants in mixed Ni-Fe catalysts lowers the average oxidation state of Ni atoms during OER15. This was correlated to a significant decrease in structural order based on the FT-EXAFS amplitudes, which could indicate a higher number of defects. If such vacancies facilitate O → Ni charge-transfer, it may explain the observed stabilization of low-valent Ni2+ sites in mixed Ni-Fe catalysts.
We also note the similarity between our pre-peak and a peak reported by Pfeifer et al.59–61 in a series of in situ O K-edge and combined DFT studies of IrO2. They assigned a pre-peak at 529 eV to formation of oxygen in formal oxidation state −1. The nature of these O1− species was experimentally confirmed to be highly electrophilic and hence likely to facilitate nucleophilic attack61. According to their DFT theoretical O K-edge spectra of IrO2, neither stable peroxide (O-O) nor superoxo (O-O−) species are likely to be represent the pre-peak at such low excitation energies as 529 eV60. The nature of the oxygens species related to the pre-peak was therefore thought to be singly adsorbed oxygen (O-CUS) at undercoordinated sites (µ1-O or µ2-O), which is only feasible at edge or defect sites in Ni-Fe oxyhydroxides. In a recent experimental study by Burke Stevens et al.1 of Ni-Fe catalysts prepared by spiking the electrolyte with Fe3+ ions, observations support that edge or defect sites are more reactive towards OER. Koper and coworkers discussed formation of “active oxygen” of superoxo character (Ni-OO−) during OER in NiOOH catalysts seen as a band between 950–1150 cm−1 in surface enhanced Raman spectroscopy, which was rising in correlation to the nickel oxidation to Ni3+-O(H)26,27. Based on this, a non-concerted proton-coupled electron transfer (PCET) step was included in the OER mechanism. In previous studies by us we also noticed a deviation from ideal Nernstian behavior of the nickel oxido redox couple in Ni-Fe catalysts, which we explained as a two proton-one electron transfer step22. This is in contradiction to the DFT study presented earlier by Friebel et al.14, where Fe was found to have the lowest overpotential and thus proposed as the active site, however, radicals were not discussed in the context.
Whether our observed oxygen species are formed at Ni or Fe sites is not possible to discriminate, however the largest change in metal oxidation state is observed at Ni sites. Formation of oxygen of radical character on the other hand would not force a change in the metal valency, whereby we cannot conclusively verify the site of formation. In addition, if species are formed at undercoordinated sites such as edge and defect sites it would be a minority of the total number of sites in our Ni65Fe35 catalyst.
In situ cyclic-voltammetry - X-ray absorption at O K-edge Ni L-edge
In order to fine-tune the changes observed in the XAS spectra with the OER activity in a dynamic fashion in our electrodeposited Ni65Fe35 catalyst, the XAS transmission signal was monitored during CV cycling at the O K, Ni L, and Ni K edges at a fixed beam energy. The Keithley signal was recorded via an analog-to-digital converter read directly by the Bio-Logic potentiostat software (see Fig. 4a). In the soft XAS regime, the energy was fixed either at the O K pre-peak energy at 529 eV or at 854.1 eV for the L-edge feature, since both these peaks were seen to change reversibly as a response to applied electrode potential. These were assigned to the hybridization of O(2p)-Ni(3d)eg at the O K-edge and the Ni L3 shoulder reflecting formation of Ni3+, respectively. At the Ni K-edge, the energy was fixed at the rising main edge (8346 eV) and at the Fe K-edge slightly above the main edge at the white-line intensity (7130 eV) where the largest change were visible.
Figure 4.

In situ XAS measurements of the Ni65Fe35(OxHy) catalyst in 0.1 M KOH (a) Soft XAS absorption intensities at the Ni L- and O K-edge at fixed beam energy while CV cycling between 0.98–1.68 V vs. RHE at a scan-rate of 5 mV/s. The X-ray absorption for the O K-edges was measured at 529 eV (red curve), and for the Ni L-edge at 854.1 eV (blue curve). The arrows indicate the scan direction. (b) The Ni K-edge shift at a fixed beam energy (8346 eV for Ni K and 7140 eV for Fe K, right axis) while CV cycling at 5 mV/s (left axis). The edge shift is reported as delta-eV where the baseline level was the edge position at 1.0 V vs. RHE. (c) Steady-state measurements showing the extracted peak areas under the corresponding O K peak (red) and Ni L peaks (blue, orange), and the edge shift at the K-edge (delta eV, green). The current density is shown on the left axis. The closed red, blue and black dots represent the anodic scan, the respective open dots the cathodic scan. For the Ni K-edge the cathodic scan runs above the anodic one and both are shown in green dots, and the open green dot is after an additional waiting time of 15 min at the end of the measurement. The Ni L peak was scaled to the O K peak for simplicity.
During the CV cycling, the absorption both at the O K (529 eV) and the Ni L (851.1 eV) peaks in the soft XAS regime were strongly correlated and followed nearly identical behavior (see Fig. 4a). Both peaks started to rise at the same potential (~1.40 V vs. RHE), which is at pre-catalytic potentials. Both traces steadily grow on the anodic scan and into the OER region, and continue to grow as the potential sweep is reversed until the potential reached the onset of the metal reduction wave. The O K and Ni L3 peaks then decline in correlation with the metal reduction towards the end of the CV scan. When the potential is reversed to the initial value of 0.98 V, the two signals are almost decayed back to the background levels. Evident from these measurements is that the pre-edge feature at the O K-edge is closely related to the changes at the Ni L3-edge peak. Hence, the Ni2+ → Ni3+ oxidation and the appearance of the O K pre-edge peak at 529 eV appear as two entangled processes and occur at electrode potentials prior to the onset of OER and hence prior to O-O bond coupling. This was further confirmed in the corresponding CV-XAS measurements at the Ni and Fe K-edges, where the oxidation state change of Ni and the modulation in white-line intensity of Fe were followed in a similar fashion (see Figs 4b and S9). These measurements confirmed that the oxidation state changes at Ni K edge and the changes in white line intensity at the Fe K edge are as well two closely related processes. Hence, the O K pre-edge peak at 529 eV, the oxidation of Ni2+ → Ni3+/4+, and the changes at the Fe K-edge are all correlated, and occurs at pre-catalytic potentials, which was also confirmed in steady-state measurements (see Fig. 4c). The correlation between the oxidation at the Ni K and Fe K edge was recently demonstrated in a study by González-Flores et al.9, where it was clear that these two processes set off prior to the onset of OER. We also take notice of the “hysteresis” with respect to the oxidized nickel seen by Yoshida et al.31, which reflects the peak separation between the oxidation/reduction peak visible in the CVs, where the reduction is more difficult than the oxidation. In our steady-state measurements it was evident that the reduction of Ni centers was slow, however if an additional waiting time of 15 min was added at the end of the measurements at 0.98 V significantly more Ni centers had relaxed (see Fig. 4c). Differences between the hard and soft X-ray regimes are therefore certain influenced by the time held at each potential.
In the previous soft XAS study by Yoshida et al.31 a Ni-Bi catalyst in 0.1 M potassium borate solution, the O K- pre-peak at 528.7 eV appeared at 0.7 V vs. Ag/AgCl (estimated by us to ~1.45 V vs. RHE assuming pH 9.2), coincident with the onset of the catalytic current. The corresponding Ni L- spectrum was not recorded at this potential, however, detection of Ni4+ was confirmed at 1.0 V vs. Ag/AgCl (~1.75 V vs. RHE) from measurements at the Ni K-edge31. Therefore, it is not possible to draw conclusions regarding the correlation between the metal oxidation and the onset of the O K edge pre-peak in their measurements. Interestingly, in the studies of IrO2 by Pfeifer et al.59–61, the theoretical simulations of the spectra concluded that the pre-peak at 529 eV assigned to O1− existed only at coordinatively unsaturated sites such as µ1-O and µ2-O sites, however protonated forms of these (µ1-OH and µ2-OH) or in-plane µ3-O in rutile type IrO2 were not compatible with a pre-peak at such low energies60. In our electrodeposited Ni65Fe35(OxHy) catalyst, the “bulk” of the oxyhydroxide layers is mainly composed of µ3-O(H) sites, whereas “edge” or “defect” sites would contain such µ1-O and µ2-O in the oxidized form. With regard to recent studies showing that edge or defect sites may be more reactive towards OER23,24, we speculate that the 529 eV peak reflects a more reactive site located at such undercoordinated sites. This would also require that the oxidation of Ni2+ → Ni3+/4+ happen prior to the onset of OER, which we observe. This would be in line with the picture presented by Yoshida et al.31 from the O K-edge measurements, where it was proposed that only surface species in NiOOH participate in OER.
What can be concluded from our soft XAS measurements –that adds new information for interpretation of the reaction intermediates occurring in the OER catalytic cycle - is that these O species appear and disappear in concomitance with the metal oxidation and reduction (Ni2+ ↔ Ni3+/4). Supported by work from other groups, this may be a more reactive O site that is electron deficient (partially oxidized) and of more radical character (denoted as O−2+δ), see proposed mechanistic scheme in Fig. 5.
Figure 5.
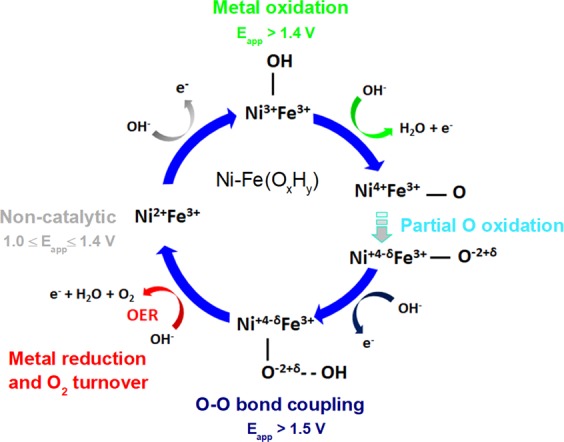
Mechanistic scheme summarizing the metal oxidation observed in our in situ XAS measurements of the Ni65Fe35(OxHy) catalyst. The resting state (non-catalytic potential) is observed between 1.0–1.4 V vs. RHE. Above this potential (>1.4 V) the oxidation of Ni2+ to Ni3+/4+ occurs. An oxidation state lower than +4 is usually observed both in Ni(OOH) and in mixed Ni-Fe(OxHy) catalysts. In the 1st metal oxidation step (shown in green), a discharge of an OH− group takes place, which adsorbs to a vacant site and increases the oxidation state by one unit. In this step a radical species may instead be formed (-OH∙) which would not result in a metal oxdation state increase (this is not shown in our cycle however may happen at both Fe and Ni sites). In the 2nd metal oxidation step, Ni is further oxidized to Ni4+. However, if there is a consecuetive step where charge re-distribution occurs from O 2p → Ni 3d, the average metal oxidation state is lowered (Ni+4-δ), and as a consequence the oxygen oxidation state is increased (O−2+δ). This step according to our O K-edge data is a stable state and is strongly correlated to the metal oxidation step, and occurs prior to O-O bond formation.
We therefore consider this as an additional step in the mechanistic cycle that occurs at potentials prior to the onset of OER. In the first metal oxidation step in the OER cycle, coordination of a hydroxyl group (-OH) is shown. There is a possibility that a species of radical character is formed already in this step (-OH∙)2,62,63, which would not require an increase in formal oxidation state of the metal. Since the O species we observe are likely to be in its deprotonated form60, the formation of this species is more probable to follow after the 2nd metal oxidation step which includes a deprotonation. This step occurs prior to the O-O bond coupling. We cannot distinguish whether the observed O species are located at Fe or Ni sites, however we have reasons to believe that they are closely associated with Ni sites owing to the large modulation seen at those sites according to our XAS data. Whether our observed O K species are distinct to the “active oxygen” species detected in Raman spectroscopic studies by Koper and coworkers26,27 or to the “negatively charged oxygen” ligands generated at Fe-centers, as proposed by us in previous work based on a non-Nernstian behavior of the redox couple is not entirely clear but not excluded20. Those changes as well all occurred in concomitance with the Ni2+ → Ni3+/4+ redox transition. This work hence demonstrates that all spectroscopic changes are well correlated with the metal oxidation state changes in the investigated Ni-Fe(OxHy) catalyst and occurs at pre-catalytic potentials. These all represents an equilibrium state well observed at room temperature conditions. It should be kept in mind that Fe ions may impact on the rate constants in the catalytic cycle of Ni-Fe catalysts. This may in turn introduce changes in the observed equilibrium states7. It was recently demonstrated by Goddard and coworkers in a theoretical study where Fe substitution in Ni(OOH) drastically changed the rate determining step, and thereby shifted the Ni4+ resting state in Ni(OOH) to a more reduced Ni3+ resting state in Ni-Fe(OOH). In recent work by Gray and coworkers it was shown that nonaqueous electrolyte may allow for spectroscopic detection of a broader range of metal oxidation states, where Fe atoms were observed to transit via a highly oxidized Fe6+ state in the OER catalytic cycle22.
As conclusion, the signature at ~529 eV in the O K spectra may indicate formation of partly electron deficient oxygen sites (O−2+δ) related to the formation of oxidized Ni3+/4+, and persist throughout OER catalytic as long as oxidized metal species prevail. This means that prior to the O-O bond formation step, the Ni-Fe(OxHy) catalyst has accumulated electron deficiencies. The true nature and role of these electropositive oxygen sites in the OER mechanism would need further in-depth investigations.
Conclusions
In summary, we have demonstrated that in situ XAS in the soft and hard X-ray regimes can deliver in-depth information on the role of the oxygen in an electrodeposited Ni65Fe35(OxHy) catalyst during oxygen evolution conditions. By scrutinizing a feature seen at the O K-edge, we conclude that the appearance of a pre-peak at 529 eV occurs concomitant with changes at Ni L-edge peak 854.1 eV and Ni K and Fe K-edges, corresponding to the process formation of oxidized Ni3+/4+. By combining CV cycling and in situ sXAS, we further retrieve a more fine-tuned potential dependence of these entangled processes. We confirm that the O K pre-peak and the Ni oxidation occurs at pre-catalytic potentials prioir to O-O bond formation, and persists during OER catalysis as long as there is oxidized nickel. The pre-peak feature implies increased hybridization between O(2p) and Ni3(d) states, which has been proposed to induce charge transfer from O → Ni. This gives rise to an electron deficient oxygen site, which fits with previous assignment of the pre-peak at 529 eV to O1−. These sites are likely to arise from undercoordinated µ1-O and µ2-O sites, which are found at “edge” or “defect” sites of the oxyhydroxide layers. These sites may play an important role in the OER activity.
Supplementary information
Acknowledgements
We gratefully acknowledge financial support by the Helmholtz Association (VH-NG-1140), the Bundesministerium für Bildung und Forschung (IN-SITU-XAS, 05K16KE2) and the Deutsche Forschungsgemeinschaft (Cluster of Excellence UniCat, EXC 314-2). We acknowledge the Helmholtz-Zentrum Berlin (HZB) for providing access to the beamlines U56-2 PGM2 and KMC-3 at the Berlin synchrotron radiation source (BESSYII, Berlin-Adlershof) and we thank the BESSYII/HZB staff members, in particular Dr. Ivo Zizak, for important support.
Author Contributions
D.D. performed and analyzed XAS measurements. M.G. prepared samples, performed XAS measurements, analyzed electrochemistry data, measured TXRF. P.C. prepared samples, performed XAS measurements, measured TXRF. L.X. supported the XAS measurements, collected SEM data. K.M.L. and H.D. designed and supervised the experiments. All authors wrote the manuscript.
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Dorian Drevon and Mikaela Görlin contributed equally.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-37307-x.
References
- 1.Lewis NS, Nocera DG. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. 2006;103:15729–15735. doi: 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dau H, et al. The Mechanism of Water Oxidation: From Electrolysis via Homogeneous to Biological Catalysis. ChemCatChem. 2010;2:724–761. doi: 10.1002/cctc.201000126. [DOI] [Google Scholar]
- 3.Hong WT, et al. Toward the rational design of non-precious transition metal oxides for oxygen electrocatalysis. Energy Environ. Sci. 2015;8:1404–1427. doi: 10.1039/C4EE03869J. [DOI] [Google Scholar]
- 4.Kleiman-Shwarsctein A, Hu Y-S, Stucky GD, McFarland EW. NiFe-oxide electrocatalysts for the oxygen evolution reaction on Ti doped hematite photoelectrodes. Electrochem. commun. 2009;11:1150–1153. doi: 10.1016/j.elecom.2009.03.034. [DOI] [Google Scholar]
- 5.Gong M, et al. An Advanced Ni–Fe Layered Double Hydroxide Electrocatalyst for Water Oxidation. J. Am. Chem. Soc. 2013;135:8452–8455. doi: 10.1021/ja4027715. [DOI] [PubMed] [Google Scholar]
- 6.Corrigan DA. The Catalysis of the Oxygen Evolution Reaction by Iron Impurities in Thin Film Nickel Oxide Electrodes. J. Electrochem. Soc. 1987;134:377. doi: 10.1149/1.2100463. [DOI] [Google Scholar]
- 7.Görlin M, et al. Oxygen evolution reaction dynamics, faradaic charge efficiency, and the active metal redox states of Ni-Fe oxide water splitting electrocatalysts. J. Am. Chem. Soc. 2016;138:5603–5614. doi: 10.1021/jacs.6b00332. [DOI] [PubMed] [Google Scholar]
- 8.Dionigi F, Strasser P. NiFe-Based (Oxy)hydroxide Catalysts for Oxygen Evolution Reaction in Non-Acidic Electrolytes. Adv. Energy Mater. 2016;6:1600621. doi: 10.1002/aenm.201600621. [DOI] [Google Scholar]
- 9.Gonzáles-Flores, D. et al. Nickel-iron catalysts for electrochemical water oxidation – redox synergism investigated by in-situ X-ray spectroscopy with millisecond time resolution. Sustain. Energy Fuels, 10.1039/C8SE00114F (2018).
- 10.Landon J, et al. Spectroscopic Characterization of Mixed Fe–Ni Oxide Electrocatalysts for the Oxygen Evolution Reaction in Alkaline Electrolytes. ACS Catal. 2012;2:1793–1801. doi: 10.1021/cs3002644. [DOI] [Google Scholar]
- 11.Oliver-Tolentino MA, et al. An Approach to Understanding the Electrocatalytic Activity Enhancement by Superexchange Interaction toward OER in Alkaline Media of Ni–Fe LDH. J. Phys. Chem. C. 2014;118:22432–22438. doi: 10.1021/jp506946b. [DOI] [Google Scholar]
- 12.Görlin M, et al. Dynamical changes of a Ni-Fe oxide water splitting catalyst investigated at different pH. Catal. Today. 2016;262:65–73. doi: 10.1016/j.cattod.2015.10.018. [DOI] [Google Scholar]
- 13.Louie MW, Bell AT. An Investigation of Thin-Film Ni–Fe Oxide Catalysts for the Electrochemical Evolution of Oxygen. J. Am. Chem. Soc. 2013;135:12329–12337. doi: 10.1021/ja405351s. [DOI] [PubMed] [Google Scholar]
- 14.Friebel D, et al. Identification of Highly Active Fe Sites in (Ni, Fe)OOH for Electrocatalytic Water Splitting. J. Am. Chem. Soc. 2015;137:1305–1313. doi: 10.1021/ja511559d. [DOI] [PubMed] [Google Scholar]
- 15.Klaus S, Cai Y, Louie MW, Trotochaud L, Bell AT. Effects of Fe Electrolyte Impurities on Ni(OH)2/NiOOH Structure and Oxygen Evolution Activity. J. Phys. Chem. C. 2015;119:7243–7254. doi: 10.1021/acs.jpcc.5b00105. [DOI] [Google Scholar]
- 16.Corrigan DA. Effect of Coprecipitated Metal Ions on the Electrochemistry of Nickel Hydroxide Thin Films: Cyclic Voltammetry in 1 M KOH. J. Electrochem. Soc. 1989;136:723. doi: 10.1149/1.2096717. [DOI] [Google Scholar]
- 17.Qiu Y, Xin L, Li W. Electrocatalytic Oxygen Evolution over Supported Small Amorphous Ni–Fe Nanoparticles in Alkaline Electrolyte. Langmuir. 2014;30:7893–7901. doi: 10.1021/la501246e. [DOI] [PubMed] [Google Scholar]
- 18.Bates MK, Jia Q, Doan H, Liang W, Mukerjee S. Charge-Transfer E ff ects in Ni − Fe and Ni − Fe − Co Mixed-Metal Oxides for the Alkaline Oxygen Evolution Reaction. Acs Catal. 2016;6:155–161. doi: 10.1021/acscatal.5b01481. [DOI] [Google Scholar]
- 19.Chen JYC, et al. Operando Analysis of NiFe and Fe Oxyhydroxide Electrocatalysts for Water Oxidation: Detection of Fe 4+ by Mössbauer Spectroscopy. J. Am. Chem. Soc. 2015;137:15090–15093. doi: 10.1021/jacs.5b10699. [DOI] [PubMed] [Google Scholar]
- 20.Görlin M, et al. Tracking catalyst redox states and reaction dynamics in Ni-Fe oxyhydroxide oxygen evolution reaction (OER) electrocatalysts: The role of catalyst support and electrolyte pH. J. Am. Chem. Soc. 2017;139:jacs.6b12250. doi: 10.1021/jacs.6b12250. [DOI] [PubMed] [Google Scholar]
- 21.Wang D, et al. In Situ X-ray Absorption Near-Edge Structure Study of Advanced NiFe(OH) x Electrocatalyst on Carbon Paper for Water Oxidation. J. Phys. Chem. C. 2015;119:19573–19583. doi: 10.1021/acs.jpcc.5b02685. [DOI] [Google Scholar]
- 22.Hunter, B. M. et al. Trapping an Iron(VI) Water-Splitting Intermediate in Nonaqueous Media. Joule 2–17, 10.1016/j.joule.2018.01.008 (2018).
- 23.Stevens MB, Trang CDM, Enman LJ, Deng J, Boettcher SW. Reactive Fe-Sites in Ni/Fe (Oxy)hydroxide Are Responsible for Exceptional Oxygen Electrocatalysis Activity. J. Am. Chem. Soc. 2017;139:11361–11364. doi: 10.1021/jacs.7b07117. [DOI] [PubMed] [Google Scholar]
- 24.Ahn HS, Bard AJ. Surface Interrogation Scanning Electrochemical Microscopy of Ni1-xFexOOH (0 < x < 0.27) Oxygen Evolving Catalyst: Kinetics of the ‘fast’ Iron Sites. J. Am. Chem. Soc. 2016;138:313–318. doi: 10.1021/jacs.5b10977. [DOI] [PubMed] [Google Scholar]
- 25.Man IC, et al. Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces. ChemCatChem. 2011;3:1159–1165. doi: 10.1002/cctc.201000397. [DOI] [Google Scholar]
- 26.Trześniewski BJ, et al. In-situ observation of active oxygen species in Fe-containing Ni-based oxygen evolution catalysts: the effect of pH on electrochemical activity. J. Am. Chem. Soc. 2015;137:15112–15121. doi: 10.1021/jacs.5b06814. [DOI] [PubMed] [Google Scholar]
- 27.Diaz-Morales O, Ferrus-Suspedra D, Koper MTM. The importance of nickel oxyhydroxide deprotonation on its activity towards electrochemical water oxidation. Chem. Sci. 2016;7:2639–2645. doi: 10.1039/C5SC04486C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suntivich J, et al. Estimating Hybridization of Transition Metal and Oxygen States in Perovskites from OK-edge X-ray Absorption Spectroscopy. J. Phys. Chem. C. 2014;118:1856–1863. doi: 10.1021/jp410644j. [DOI] [Google Scholar]
- 29.Minasian SG, et al. Covalency in metal-oxygen multiple bonds evaluated using oxygen K-edge spectroscopy and electronic structure theory. J Am Chem Soc. 2013;135:1864–1871. doi: 10.1021/ja310223b. [DOI] [PubMed] [Google Scholar]
- 30.Liu J, Liu J, Song W, Wang F, Song Y. The role of electronic interaction in the use of Ag and Mn 3 O 4 hybrid nanocrystals covalently coupled with carbon as advanced oxygen reduction electrocatalysts. J. Mater. Chem. A. 2014;2:17477–17488. doi: 10.1039/C4TA03937H. [DOI] [Google Scholar]
- 31.Yoshida M, et al. Direct Observation of Active Nickel Oxide Cluster in Nickel–Borate Electrocatalyst for Water Oxidation by In Situ O K-Edge X-ray Absorption Spectroscopy. J. Phys. Chem. C. 2015;119:19279–19286. doi: 10.1021/acs.jpcc.5b06102. [DOI] [Google Scholar]
- 32.Ali-Löytty H, et al. Ambient-Pressure XPS Study of a Ni–Fe Electrocatalyst for the Oxygen Evolution Reaction. J. Phys. Chem. C. 2016;120:2247–2253. doi: 10.1021/acs.jpcc.5b10931. [DOI] [Google Scholar]
- 33.Bediako DK, et al. Structure–Activity Correlations in a Nickel–Borate Oxygen Evolution Catalyst. J. Am. Chem. Soc. 2012;134:6801–6809. doi: 10.1021/ja301018q. [DOI] [PubMed] [Google Scholar]
- 34.Schwanke C, Xi L, Lange KM. A soft XAS transmission cell for operando studies. J. Synchrotron Radiat. 2016;23:1390–1394. doi: 10.1107/S1600577516014697. [DOI] [PubMed] [Google Scholar]
- 35.Xi L, et al. In Situ L-Edge XAS Study of a Manganese Oxide Water Oxidation Catalyst. J. Phys. Chem. C. 2017;121:12003–12009. doi: 10.1021/acs.jpcc.7b02331. [DOI] [Google Scholar]
- 36.Nagasaka M, Hatsui T, Horigome T, Hamamura Y, Kosugi N. Development of a liquid flow cell to measure soft X-ray absorption in transmission mode: A test for liquid water. J. Electron Spectros. Relat. Phenomena. 2010;177:130–134. doi: 10.1016/j.elspec.2009.11.001. [DOI] [Google Scholar]
- 37.Pan G, et al. Nanobubbles at Hydrophilic Particle–Water Interfaces. Langmuir. 2016;32:11133–11137. doi: 10.1021/acs.langmuir.6b01483. [DOI] [PubMed] [Google Scholar]
- 38.Wernet P. The Structure of the First Coordination Shell in Liquid Water. Science (80-.). 2004;304:995–999. doi: 10.1126/science.1096205. [DOI] [PubMed] [Google Scholar]
- 39.Myneni S, et al. Spectroscopic probing of local hydrogen-bonding structures in liquid water. J. Phys. Condens. Matter. 2002;14:L213–L219. doi: 10.1088/0953-8984/14/8/106. [DOI] [Google Scholar]
- 40.Schreck S, Gavrila G, Weniger C, Wernet P. A sample holder for soft x-ray absorption spectroscopy of liquids in transmission mode. Rev. Sci. Instrum. 2011;82:103101. doi: 10.1063/1.3644192. [DOI] [PubMed] [Google Scholar]
- 41.Guo J, et al. Direct Observation of Two Electron Holes in a Hematite Photoanode during Photoelectrochemical Water Splitting. J. Phys. Chem. C. 2012;116:16870–16875. doi: 10.1021/jp304254k. [DOI] [Google Scholar]
- 42.Aziz EF, et al. Direct Contact versus Solvent-Shared Ion Pairs in NiCl 2 Electrolytes Monitored by Multiplet Effects at Ni(II) L Edge X-ray Absorption. J. Phys. Chem. B. 2007;111:4440–4445. doi: 10.1021/jp070085r. [DOI] [PubMed] [Google Scholar]
- 43.Hocking RK, et al. Fe L-Edge X-ray Absorption Spectroscopy Determination of Differential Orbital Covalency of Siderophore Model Compounds: Electronic Structure Contributions to High Stability Constants. J. Am. Chem. Soc. 2010;132:4006–4015. doi: 10.1021/ja9090098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zaharieva I, et al. Water oxidation catalysis – role of redox and structural dynamics in biological photosynthesis and inorganic manganese oxides. Energy Environ. Sci. 2016;9:2433–2443. doi: 10.1039/C6EE01222A. [DOI] [Google Scholar]
- 45.Trotochaud L, Young SL, Ranney JK, Boettcher SW. Nickel–Iron Oxyhydroxide Oxygen-Evolution Electrocatalysts: The Role of Intentional and Incidental Iron Incorporation. J. Am. Chem. Soc. 2014;136:6744–6753. doi: 10.1021/ja502379c. [DOI] [PubMed] [Google Scholar]
- 46.Browne MP, et al. The goldilocks electrolyte: examining the performance of iron/nickel oxide thin films as catalysts for electrochemical water splitting in various aqueous NaOH solutions. J. Mater. Chem. A. 2016;4:11397–11407. doi: 10.1039/C6TA03903K. [DOI] [Google Scholar]
- 47.De Groot, F. M. F. & Kotani, A. Core Level Spectroscopy of Solids (2008).
- 48.Lafuerza S, et al. Origin of the pre-peak features in the oxygen K-edge x-ray absorption spectra of LaFeO 3 and LaMnO 3 studied by Ga substitution of the transition metal ion. J. Phys. Condens. Matter. 2011;23:325601. doi: 10.1088/0953-8984/23/32/325601. [DOI] [PubMed] [Google Scholar]
- 49.Wang H, et al. L-edge X-ray absorption spectroscopy of some Ni enzymes: Probe of Ni electronic structure. J. Electron Spectros. Relat. Phenomena. 2001;114–116:855–863. doi: 10.1016/S0368-2048(00)00370-4. [DOI] [Google Scholar]
- 50.Zheng X, et al. Theory-driven design of high-valence metal sites for water oxidation confirmed using in situ soft X-ray absorption. Nat. Chem. 2017;10:149–154. doi: 10.1038/nchem.2886. [DOI] [PubMed] [Google Scholar]
- 51.Lin F, et al. Origin of electrochromism in high-performing nanocomposite nickel oxide. ACS Appl. Mater. Interfaces. 2013;5:3643–3649. doi: 10.1021/am400105y. [DOI] [PubMed] [Google Scholar]
- 52.Batchellor AS, Boettcher SW. Pulse-Electrodeposited Ni–Fe (Oxy)hydroxide Oxygen Evolution Electrocatalysts with High Geometric and Intrinsic Activities at Large Mass Loadings. ACS Catal. 2015;5:6680–6689. doi: 10.1021/acscatal.5b01551. [DOI] [Google Scholar]
- 53.Suntivich J, et al. Design principles for oxygen-reduction activity on perovskite oxide catalysts for fuel cells and metal-air batteries. Nat. Chem. 2011;3:546–550. doi: 10.1038/nchem.1069. [DOI] [PubMed] [Google Scholar]
- 54.Hayes JR, Grosvenor AP. An x-ray absorption spectroscopic study of the electronic structure and bonding of rare-earth orthoferrites. J. Phys. Condens. Matter. 2011;23:465502. doi: 10.1088/0953-8984/23/46/465502. [DOI] [PubMed] [Google Scholar]
- 55.Li N, et al. Influence of iron doping on tetravalent nickel content in catalytic oxygen evolving films. Proc. Natl. Acad. Sci. 2017;114:1486–1491. doi: 10.1073/pnas.1620787114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.J. Winter, M. Chemistry of the d-block elements (1994).
- 57.Wasinger EC, de Groot FMF, Hedman B, Hodgson KO, Solomon EI. L-edge X-ray Absorption Spectroscopy of Non-Heme Iron Sites: Experimental Determination of Differential Orbital Covalency. J. Am. Chem. Soc. 2003;125:12894–12906. doi: 10.1021/ja034634s. [DOI] [PubMed] [Google Scholar]
- 58.Cho D-Y, et al. Spectroscopic investigation of the hole states in Ni-deficient NiO films. J. Mater. Chem. C. 2013;1:4334. doi: 10.1039/c3tc30687a. [DOI] [Google Scholar]
- 59.Pfeifer V, et al. Reactive oxygen species in iridium-based OER catalysts. Chem. Sci. 2016;7:6791–6795. doi: 10.1039/C6SC01860B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pfeifer V, et al. In situ observation of reactive oxygen species forming on oxygen-evolving iridium surfaces. Chem. Sci. 2017;8:2143–2149. doi: 10.1039/C6SC04622C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pfeifer V, et al. The electronic structure of iridium oxide electrodes active in water splitting. Phys. Chem. Chem. Phys. 2016;18:2292–2296. doi: 10.1039/C5CP06997A. [DOI] [PubMed] [Google Scholar]
- 62.Xiao, H., Shin, H. & Goddard, W. A. Synergy between Fe and Ni in the optimal performance of (Ni, Fe)OOH catalysts for the oxygen evolution reaction. Proc. Natl. Acad. Sci. 201722034, 10.1073/pnas.1722034115 (2018). [DOI] [PMC free article] [PubMed]
- 63.Burke Stevens M, Trang CDM, Enman LJ, Deng J, Boettcher SW. J. Am. Chem. Soc. 2017;139:11361–11364. doi: 10.1021/jacs.7b07117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.