

Abstract
Human papillomavirus (HPV) is recognised as the cause of precancerous and cancerous cervical lesions. Furthermore, in high-grade lesions, HPV is frequently integrated in the host cell genome and associated with the partial or complete loss of the E1 and E2 genes, which regulate the activity of viral oncoproteins E6 and E7. In this study, using a double-capture system followed by high-throughput sequencing, we determined the HPV integration status present in liquid-based cervical smears in an urban Gabonese population. The main inclusion criteria were based on cytological grade and the detection of the HPV16 genotype using molecular assays. The rate of HPV integration in the host genome varied with cytological grade: 85.7% (6/7), 71.4% (5/7), 66.7% (2/3) 60% (3/5) and 30.8% (4/13) for carcinomas, HSIL, ASCH, LSIL and ASCUS, respectively. For high cytological grades (carcinomas and HSIL), genotypes HPV16 and 18 represented 92.9% of the samples (13/14). The integrated form of HPV16 genotype was mainly found in high-grade lesions in 71.4% of samples regardless of cytological grade. Minority genotypes (HPV33, 51, 58 and 59) were found in LSIL samples, except HPV59, which was identified in one HSIL sample. Among all the HPV genotypes identified after double capture, 10 genotypes (HPV30, 35, 39, 44, 45, 53, 56, 59, 74 and 82) were detected only in episomal form. Our study revealed that the degree of HPV integration varies with cervical cytological grade. The integration event might be a potential clinical prognostic biomarker for the prediction of the progression of neoplastic lesions.
Introduction
Cervical cancer (CC) is the fourth most common cancer in women worldwide and is the leading cause of cancer deaths among women living in sub-Saharan Africa1,2. Among the 200 human papillomavirus (HPV) genotypes identified to date, only 50 genotypes that are capable of infecting the cervical epithelium are classified as “low-risk” (LR) or “high-risk” (HR). LR genotypes are associated with lesions that may regress spontaneously whereas only 16 HPV genotypes classified as HR (16, 18, 31, 33, 34, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68, 70) are explicitly associated with cervical cancer3,4.
HPV infection includes integration of the HPV genome into the host genome. This integration leads to the linearization of the HPV genome, usually somewhere the region of the E1 and E2 genes, but can also cause the partial or total deletion of these genes5,6. The loss of one or both of these genes leads to the overexpression of the E6 and E7 genes, a condition that contributes to oncogenesis and the progression of low-grade lesions to more severe lesions and ultimately carcinoma. Determining the physical state of the viral genome (integrated or episomal state) and the insertion site may provide a better understanding on how this integration mechanism promotes carcinogenesis7,8. HPV integration has been studied using various techniques. The very first approaches included Southern blots and fluorescent in situ hybridization9–11, but these methods require a large amount of fresh DNA. Various PCR-based approaches requiring less DNA were then developed. Given that the integration of the HPV genome into the host genome generally induces the partial or total deletion of the E1 or E2 genes, the identification of integration status relies on the failure to amplify these genes in their entirety6,12–14. More customized molecular techniques such as restriction site PCR (RS-PCR)15 and the detection of integrated papillomavirus sequences PCR (DIPS-PCR)16 have also been developed, but also cannot distinguish between integrated and episomal forms, or a mixture of these. To overcome this limitation, quantitative real-time PCR that can measure E2/E6 copy numbers have also been developed to determine the ratio of both forms present in a sample5. More recently, next-generation sequencing (NGS) has been successfully applied to detecting integration events which consisted in screening for viral-host chimeric junctions. The whole-genome sequencing coupled with read mapping for analysis from DNA extracted from HeLa cell line has been used to determine that the integration event occurred near the c-myc oncogene, demonstrating an example of host chromosomal alteration caused by a viral integration associated with an cancer17. Similarly, exome sequencing can characterize HPV integration: using the blood from a metastatic cervical carcinoma patient, 1.2 billion reads were generated to detect HPV integration by mapping reads on a reference human genome. This approach identified integrated HPV 18 even in the presence of the episomal form of the same HPV genotype. Another study, using the RNA-seq technique and based on discordant paired-end reads aligned to the viral and the host genomes18, revealed an integration rate of 82.3% in cervical squamous cell carcinoma. However, this method can only detect integration sites within coding regions. Finally, the highly sensitive and recent approach combining NGS with capture technology has been used on different types of samples such as snap-frozen19–21 or paraffin-embedded22,23 tissues from biopsies of carcinoma and adenocarcinoma cases. Still other studies are based on squamous cell carcinoma and cervical adenocarnicoma cell lines24–26. The different types of samples and bioinformatics approaches have revealed several integration sites across the human genome with various integration rates increasing with the severity of the lesions27,28. Regarding bioinformatics techniques, mapping approaches generally either rely on paired-end reads aligned with the viral and host genomes separately23,27 or aligned with an index of combined viral-host genomes20,29. Mapping algorithms have also been employed in conjunction with the Smith and Watterman algorithm to increase sensitivity30.
In this study, using a double-capture system followed by high-throughput sequencing, we determined the HPV genotype(s) present in liquid-based cervical (LBC) smears performed on an urban Gabonese population. The HPV integration status, as well as the possible integration site(s), were also explored. Our study revealed that the degree of HPV integration varies with the severity of the cytological cervical grade (from atypical to carcinoma cells). Moreover, this integration appeared to be associated with large deletions at the genomic insertion points. The specific integration of HPV samples suggests that HPV integration may be a potential early-stage biological and clinical prognostic biomarker for the prediction of the progression of neoplastic lesions.
Materials and Methods
Study population and cervical sample collection
The women participating in this study were recruited during a previous multi-centre cross-sectional study carried out at two hospitals in Libreville, Gabon31. The main inclusion criteria were based on cytology and the presence of the HPV16 genotype detected using molecular assays. The characteristics of this recruitment and molecular genotyping of HPV have been described previously31. Here, all specimens were divided into five groups according to cytological results: atypical squamous cells of undetermined significance (ASCUS, group 1, n = 13), low-grade squamous intraepithelial lesions (LSIL, group 2, n = 5), atypical squamous cells which do not rule out high-grade squamous intra-epithelial lesions (ASCH, group 3, n = 3), high-grade squamous intraepithelial lesions (HSIL, group 4, n = 7) and carcinoma (group 5, n = 7). This study was approved by the Medical Ethics Committee of Gabon (Consent Number PROT No. 0010/2013/SG/CNE), and was authorised by the Gabonese Ministry of Health (No. 00775/MS/CAB.M/SG/DGS) and the Scientific Committee of the Centre International de Recherches Médicales de Franceville (CIRMF). All experiments were performed in accordance with relevant guidelines and regulations and written informed consent was obtained for all included patients.
Double-capture preparation and sequencing
DNA was quantified using the Quant-iT assay (Invitrogen) and a fixed amount of DNA (500 ng) was fragmented using a Covaris M220 ultrasonicator according to the manufacturer’s instructions. The 450 bp DNA fragments were used to construct a genomic library with the SeqCap EZ Reagent Kit (Roche NimbleGen, Madison, USA) according to the manufacturer’s recommendations. All nucleic acid purification steps were carried out using Agencourt AMpure XP beads (Beckmann Coulter, the Netherlands). After the first steps of end repair, adapter ligation and first pre-capture PCR, a specific HPV capture was performed with SeqCap EZ probes with incubation at 40 °C over the night. After the various stages of washing and amplification of the fragments captured by PCR, a second capture is carried out in order to optimize the enrichment of the HPV fragments and to maximise the fraction of HPV reads in the bioinformatics analyses. A volume of 4 ul of the previous library obtained after the first capture is used as input for the second capture using the same protocol. The final library quality and size were assessed on an Agilent BioAnalyser 2100 (Agilent Technologies, USA) and Illumina Sequencing was conducted using a Miseq benchtop sequencer: 300 cycle runs were performed and 150 nucleotide paired-end reads were obtained.
Bioinformatics analysis and determination of viral genome integration sites in the human genome
The analysis pipeline is showed in Fig. 1. The quality of the generated reads was initially assessed and filtered using the CLC Workbench 10.0.1 quality control and trimming tool. All filtered paired-end reads were concatenated to obtain longer fragments, and then mapped against complete HPV genomes available in GenBank using CLC Workbench 10.0.1 with at least 40% of the read length aligned and 90% identity (L40/I90). The integration sites were defined as breakpoints with chimeric reads corresponding to human and viral sequences within a concatenated or single read. The process of finding the chimeric reads was based on several successive mappings against viral and human genomes (Fig. 1). A mapping approach instead of BLAST alignment was chosen to save computational time32. The chimeric reads have human and viral parts of variable size and, during the successive mappings against viral and human references, potential short sequences may match either reference in a nonspecific manner, leading to false positives. Since no assumptions could be made on the size of the viral and human sequences of chimeric reads, the mapping parameters had to be flexible enough to avoid missing reads with short chimeric fragments, but specific enough to ignore possible false positives due to those tiny chimeric fragments. Therefore, the cut-offs were set to 40% of the entire reads, which had to align with at least 90% identity (L40/I90). Since HPV genotypes are classified in different types based on at least a 10% divergence, the preliminary high-stringency (L90/I90) mapping against a large collection of GenBank’s complete HPV genomes allowed for a rapid distinction between the HPV genotypes involved in potential integration events. The reads were then grouped by HPV genotype. Detection of chimeric reads was then carried out by mapping all reads with the mapping parameters described above: for each identified HPV genotype, the mapped reads were recovered and then mapped against the Hg38 human genome assembly. All groups of mapped reads were individually recovered. For each group, an integration event was determined if (i) all the detected chimeric reads had the same breakpoint position and (ii) the human and viral chimeric fragments had variable lengths, avoiding as far as possible duplicate read issues. Each recovered group was mapped one last time against the sequence of the specific HPV genotype to confirm the presence of chimeric reads and to determine the breakpoint site within the viral genome. A table summarising the chromosomal localisation for each viral integration was generated. Similarly, the previously determined loci of the integration breakpoint were verified by the overlap between the UCSC cytogenetic bands and the chimeric read locations. For samples in which episomal and integrated forms were both observed (Fig. 2), there were two types of mixed profiles, depending on which form predominated based on coverage ratio. A form was determined as predominant if its coverage was at least 10 times higher than the other form’s coverage.
Figure 1.
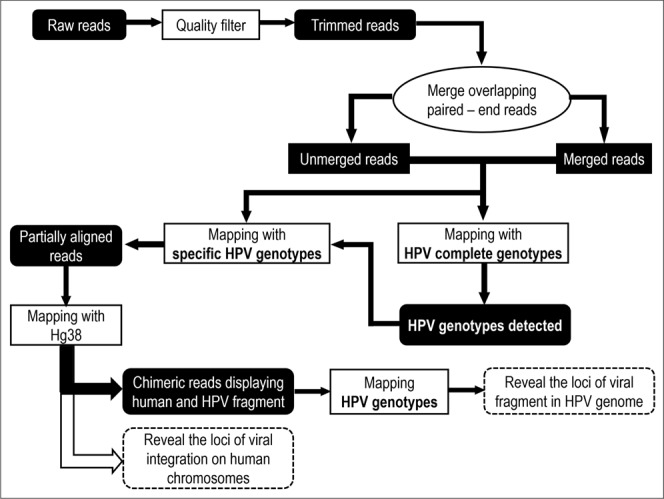
Analysis pipeline for determining integration breakpoint.
Figure 2.

Detection of putative episomal or integrated forms of the virus. An integration event was detected when the coverage of the genomic region between the unaligned part of the read mapped on viral reference was low.
Results
Description of the HPV genotypes detected after sequencing the captured fragments
The double-capture system was designed to detect 17 high-risk (HR) HPV genotypes (Table S1). In this study, 35 samples were tested; independently of the cytological grade of the cervical smears tested, 15 HPV genotypes were detected, of which 10 and 5 were HR and low-risk (LR) genotypes, respectively (Tables 1 and S2). Of these 15 HPV genotypes, 6 genotypes (HPV51 + 5 LR) did not belong to the expected detection spectrum of the designed probes and appeared to be captured by homology with the targeted genotypes. The HPV16 genotype was found in 100% (35/35) of samples. However, the integrated status was showed in only 15.4% (2/13), 33.3% (1/3), 57.1% (4/7) and 71.4% (5/7) of ASCUS, ASCH, HSIL and carcinoma samples, respectively. The HPV18 genotype was detected in only 25.7% of samples (9/35) and found integrated in 55.6% of samples (5/9). The HPV33 genotype was found in 65.7% of samples (23/35), but only once in integrated form in an LSIL sample. The other HR-HPV genotypes, most often detected in episomal form, were types HPV56 and HPV59 at 31.4% (11/35) and HPV58 at 37.1% (13/35). However, HPV56 and 58 were found in 81.8% (9/11) and 84.6% (11/13) of neoplasia and carcinoma samples (i.e. LSIL, HSIL, ASCH and carcinomas), whereas HPV59 was mainly found in ASCUS samples (63.6% (7/11)). The most frequent LR-HPV genotype was HPV82 with a rate of 34.2% (12/35 samples), with no particularly predominant cytological grade. The other HR and LR genotypes (HPV30, 39, 44, 45, 53, and 74) were detected in only one or two samples. HPV30, 45 and 74 were found in two samples (HSIL and LSIL) with significantly longer sequence lengths compared with the other samples (Tables 1 and S2).
Table 1.
Summary of HPV detected for each sample.
| Cytological grade | Sample | HPV genotypes |
|---|---|---|
| Carcinoma | A9 | HPV 16, 33 |
| B3 | HPV 16, 33, 56, 58, 82 | |
| B8 | HPV 16, 18, 56, 58 | |
| C6 | HPV 16, 18, 58 | |
| C8 | HPV 16, 18 | |
| C11 | HPV 16, 33 | |
| C12 | HPV 16, 18, 39, 58, 82 | |
| HSIL | A8 | HPV 16, 33, 35, 51, 59 |
| B1 | HPV 16, 33, 56, 58 | |
| B2 | HPV 16, 33, 56, 58 | |
| B5 | HPV 16, 33, 35, 39, 56, 58, 82 | |
| B12 | HPV 16, 30, 33, 39, 45, 56, 58, 74, 82 | |
| C5 | HPV 16, 18 | |
| C9 | HPV 16, 51 | |
| ASCH | A7 | HPV 16, 33, 35, 74 |
| B7 | HPV 16, 33, 35, 56, 58, 82 | |
| B9 | HPV 16, 18, 33, 56, 58, 82 | |
| LSIL | A2 | HPV 16, 33, 35, 59 |
| A11 | HPV 16, 33, 35, 45, 51, 59 | |
| B6 | HPV 16, 44, 56, 58, 82 | |
| B10 | HPV 16, 33, 56, 58, 82 | |
| C7 | HPV 16, 51 | |
| ASCUS | A1 | HPV 16, 33, 35, 59 |
| A3 | HPV 16, 33, 59 | |
| A4 | HPV 16, 33, 35, 59, 82 | |
| A5 | HPV 16, 33, 35, 59, 82 | |
| A6 | HPV 16, 33, 59 | |
| A10 | HPV 16, 33, 35, 59 | |
| B4 | HPV 16, 33, 56, 58, 82 | |
| B11 | HPV 16, 18, 33, 35, 56, 58, 59, 82 | |
| C1 | HPV 16, 18 | |
| C2 | HPV 16, 53 | |
| C3 | HPV 16 | |
| C4 | HPV 16 | |
| C10 | HPV 16, 18, 33, 51 |
Integration of the HPV genome in the human genome
The rate of HPV integration in the host genome varied with cytological grade: 30.8% (4/13), 60% (3/5), 66.7% (2/3), 71.4% (5/7) and 85.7% (6/7) for ASCUS, LSIL, ASCH, HSIL, and carcinomas, respectively (Table 2). The main integrated HPV genotypes, regardless of the cytological grade, were HPV16, 18, 33, 51, 58 and 59. For high cytological grades (carcinomas, HSIL and ASCH), genotypes HPV16 and 18 represented a 92.9% (12/13) of samples. The HPV16 genotype was mainly found in high-grade lesions in 71.4% of samples regardless of the cytological grade. Minority genotypes (HPV33, 51, 58 and 59) were found in LSIL samples, except HPV59 which was identified in one HSIL sample.
Table 2.
Summary of 21 HPV integration statuses.
| Cytological grade | Sample | HPV genotype | Status of integration | Main integration | Supplemental integrations | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromosomal position of integration sites | Size of deleted area (bp) | Chromosomal localisation | Nature of region | Gene or other disrupted sequence | Genes nearby (within 500 kb) | Chromosomal position of integration sites | Chromosomal localisation | ||||
| Carcinoma | C8 | HPV16 | Majority | 11:4141137.0.4141147 | 10 | 11p15.4 | non-coding region | RRM1, STIM1 | |||
| B8 | HPV18 | Majority | 13:73970531.0.20:52677001*** | N.A. | 13q22.1 & 20q13.2 | 13: intron 20: non-coding region | 13: KLF12 20: RP4-715N11.2 (lincRNA) | ||||
| A9 | HPV16 | Majority | 8 :42662221.0.42662224 | 3 | 8p11.21 | non-coding region | |||||
| B3 | HPV16 | Minority | 3 :167913519 | 0 | 3q26.1 | non-coding region | LINC01330 (transcribed pseudogene) | GOLIM4, SERPINI1 | |||
| C11 | HPV16 | Minority | 2 :16078561.0.16078617 | 226 | 2p24.3 | non-coding region | GACAT3 (lincRNA) | MYCN | |||
| C6 | HPV16 | Minority | 10 :4650152 | 0 | 10p15.1 | non-coding region | |||||
| HSIL | A8 | HPV59 | Majority | N.D. | N.D. | N.D. | |||||
| B1 | HPV16 | Majority | 11:62609491.0.62678847 | 69356 | 11q12.3 | intron | GANAB | SCGB1A1, AHNAK, EEF1G, TUT1, RP11-864I4.1, MTA2, ROM1, EML3, B3GAT3, INTS5, GANAB, METTL12, BSCL2, HNRNPUL2, ZBTB3, RP11-831H9.11, LRRN4CL, TTC9C, POLR2G, C11orf98, HNRNPUL2-BSCL2, TAF6L, NXF1, RP11-727F15.14, LBHD, GNG3, TMEM223, STX5, TMEM179B, WDR74, UBXN1, UQCC3, SLC3A2 | |||
| C5 | HPV18 | Majority | 17:62864111.0.62866626 | 2515 | 17q24.2 | non-coding region | TANC2, MARCH10, MRC2 | 44398322; 44616177; 45120465 | 17q21.32; 17q21.33 | ||
| C9 | HPV16 | Minority | 14:53825501.0.55712832 | 1887331 | 14q22.2 & 14q22.3 | non-coding region non-coding region | AL162759.1 (lincRNA) | KTN1, FBXO34, WDHD1, SAMD4A, GCH1, CGRRF1, CDKN3, BMP4* FERMT2, DDHD1, STYX, PSMC6, GNPNAT1, TXNDC16, PTGDR, PELI2, TMEM260** | |||
| B2 | HPV16 | Minority | 6:126248596 | 0 | 6q22.32 | non-coding region | CENPW, TRMT11 | ||||
| B5 | HPV16 | Minority | 6:107221398.0.107226375 | 4977 | 6q21.1 | intron | PDSS2 | BEND3, C6orf203 | |||
| ASCHL | B9 | HPV18 | Majority | 20:5751892 | 0 | 20p12.3 | intron region | C20orf196 | CHGB, TRMT6, MCM8, GPCPD1 | ||
| A7 | HPV16 | Minority | 13:73626724 | 0 | 13q22.1 | non-coding region | LINC00393 (lincRNA) | KLF12 | |||
| SIL | B6 | HPV58 | Minority | 2:1608294 | 0 | 2p25.3 | non-coding region | AC144450.1 (antisense) | PXDN, MYT1L, SNTG2, TPO | ||
| C7 | HPV51 | Minority | 2:133183995.0.133185219 | 1224 | 2q21.2 | intron | NCKAP5 | Chr2:3338144; Chr5:5744895 | 2p25.3; 5p15.32 | ||
| B10 | HPV33 | Minority | 10:74594325 | 0 | 10q22.1 | intron | ADK | KAT6B | |||
| ASCUS | A10 | HPV16 | Majority | 8:24011443.0.24011455 | 12 | 8p21.2 | non-coding region | STC1 | |||
| B11 | HPV18 | Majority | 21:43348764.0.43348768 | 4 | 21q22.3 | non-coding region | SIK1, HSF2BP, H2BFS, CRYAA, U2AF1 | ||||
| C10 | HPV18 | Majority | 22:41151127.0.41152240 | 1113 | 22q13.2 | intron | EP300 | L3MBTL2, RANGAP1, ZC3H7B, TEF, RBX1, XPNPEP3 | |||
| B4 | HPV16 | Minority | 5:147283983.0.147285227 | 1244 | 5q33.1 | intron | STK32A | DPYSL3, PPP2R2B | |||
N.D. Not determined; *deleted genes between breakpoint of HPV genome; **genes nearby; lncRNA: long non-coding RNA; ***chromosomal translocation.
Among all the HPV genotypes identified after double capture, 10 genotypes (HPV30, 35, 39, 44, 45, 53, 56, 59, 74 and 82) were detected only in episomal form; no virus-human chimeric sequences were detected after mapping them on reference sequences. For samples in which episomal and integrated forms were both observed, there were two types of mixed profiles, depending on which form predominated based on coverage ratio. One mixed profile Epi/Int showed a predominance of the episomal form compared with the integrated form, whereas for the other mixed profile Int/Epi, the integrated form predominated the episomal form. The mixed Int/Epi HPV profile was found in 47.6% (10/21) of samples, whereas the mixed Epi/Int profile was observed in 52.8% (11/21) of samples. In high-grade lesions (carcinomas, HSIL and ASCH), both profiles were found in equivalent proportions, whereas in all LSIL samples, only the Epi/Int profile was observed. HPV16 and 18 were respectively found in 40% and 50% of Int/Epi samples. HPV16 was involved in 75% of Epi/Int samples, whereas HPV18 was not detected in any Epi/Int samples.
Chromosomal localisation of viral genome integration
For all cytological grades, viral integration in the host genome was sometimes followed by deletion of either part of the viral genome or part of the host genome at the integration site. The breakpoint sites in the viral genome, before integration, were located in one of four viral genes (E1, E2, L1, and L2) independently of the cytological grade. A breakpoint site in the E2 gene represented about 40% (12/30) of integrations followed by E1, L1 and L2 with 26.7% (8/30), 23.3% (7/30) and 10% (3/30), respectively. The partial or total deletion of the E2 gene was found in 84.2% of cases (16/19), with the first breakpoint site being located upstream or downstream from the gene and the second site being located either within or further downstream from the gene. The E2 deletion was found in all carcinomas, ASCH and LSIL samples and in 75% and 80% of ASCUS and HSIL samples, respectively. The breakpoint sites were found outside the E2 gene in only two samples; namely either within the E1 gene or between the L1 and L2 genes for ASCUS and HSIL samples.
Deletion of host genome sequences was observed at the integration sites in 57.9% of samples (11/19) (Table 2). The size of the deletion varied from 3 to 1.88 × 106 nucleotides for carcinoma (A9) and HSIL (C9) samples. In the other cases (8/19), the viral genome integrated without any modifications to the host genome (Table 2). As shown in previous studies33, we did not detect any preferential integration sites, whether in a chromosome or at a particular locus. Of the 21 samples, 28.6% (6/21) of the integration sites were found in a unique chromosome, whereas the other integration sites were found at least twice in a single chromosome but in different loci (Table 2). Chromosome 2 showed the most integrations, with three integrations located at different loci, in two low-grade lesions (LSIL - 2p25.3 and 2q21.2) as well as in a carcinoma (2p24.3) (Table 2). Only one case of two integration sites was found in a HSIL sample, and the sites were located in two different loci (C9 - 14q22.2 and 14q22.3), with the deletion of 1.8 × 106 nucleotides between these two sites. Moreover, as shown in previous studies34,35, only one case of chromosomal translocation between chromosomes 13 and 20 (B8 - 13q22.1 & 20q13.2) of an HPV18 integration was observed in a carcinoma. Finally, we observed multiple integration events with the same genotype in only two samples, LSIL and HSIL: either on the same chromosome but in different loci (C5 - 17q24.2 versus 17q21.32 to 17q21.33), or in a different locus on the same chromosome or on a different chromosome (C7 - 2q21.2 versus 2p25.3 and 5p15.32).
The detailed analysis of the localisation of HPV integration in each locus previously identified showed that in 36.4% of samples (8/22), HPV integration occurred in an intron whereas in 40.8% of samples (9/22) it occurred in a non-coding region. In parallel, in 22.8% of cases (5/22), the integration occurred either in a long non-coding RNA (3/22) or in an antisense RNA or a putative transcript of a pseudo-gene (2/22). Except for two carcinoma samples (B8 and C7), there was always a group of genes found in close proximity to the HPV integration site (+/−500 kb), among which some may be involved in cell division or are oncogenes such as MYCN (C11) or RRM1 (C8)36,37. Similarly, HPV integration appeared to have occurred in the intron of a gene involved in cellular processes such as the Kruppel-like factor 12 (KLF12) or the coding gene for the E1A binding protein p300 in a carcinoma (B8) or an ASCUS (C10), respectively. Finally, in this study, only two HSIL samples were associated with a large loss of a chromosomal region (60 and 1800 kb respectively for cases B1 and C9). In each sample, the loss was accompanied by the deletion of several entire genes (introns and exons) or of eight exons for the UBXN1 gene for example (B1) (Table 2).
Discussion
Here, we used a double-capture system to investigate HPV integration sites in the human genome on cervical smears with precancerous and cancerous lesions (LSIL and HSIL) or undetermined atypical cells (ASCUS and ASCH). Previous studies based on cervical biopsies have demonstrated viral genome integration in the early stages of lesion development21,27. For example, a study of high-throughput viral integration detection (HIVID) identified 3667 integration breakpoints of HPV with integration rates comprised between 44.4% and 71.4% according to the clinicopathological stage considered (CIN1 to CIN3)21. Furthermore, these integration rates increased significantly with the severity of neoplastic lesions. However, 87% of these integration points (reported by Hu et al.) may, in fact, be experimental or computational artefacts38. The various studies on integration sites based on a double-capture system followed by high-throughput sequencing of the captured fragments also report variable integration rates in precancerous lesions as well as invasive carcinomas23,27. Nevertheless, although integration rates may vary among studies, they are significantly higher in CIN3 than in CIN1 and CIN2. It is difficult to compare these data with those obtained in our study, because we did not use cervical biopsies, but LBC smears on which only cytological analyses were performed. However, although it is not possible to attain perfect correspondence between histological and cytological data, HSIL and ASCH cases are usually indicative of high-grade lesions and are comparable to the CIN3 stage, and LSIL cases to the CIN1 stage. Based on our cytological data, we observed the same pattern of increased integration with increased lesion severity (60%, 85.7% and 85.7% respectively for LSIL, HSIL and carcinomas). Unlike previous studies, we also analysed smears with undetermined atypical cells (ASCUS). Despite the limited number of these samples, the integration rate found in ASCUS was even lower than in the LSIL samples, with respectively 30.7% and 60%. Altogether, our results tend to confirm that the viral genome integration process is an event that occurs very early in the development of neoplastic lesions39. However, of four ASCUS patients in whom integration was revealed, two of them died less than four years after the first cervical cancer investigations and one developed precancerous lesions without dying. The last patient has not been located, but it is very probable that she did not develop cervical cancer because she does not appear in the pathology monitoring program that is currently being conducted in Gabon40. The other located patients with ASCUS status in which no integration was found have not died or developed high-grade precancerous lesions. As described in literature, these data appear to suggest that some cytological analyses did not reveal high-grade (HSIL) precancerous lesions in patients after performing the tests41. Although the number of LSIL cases in this study was small compared with the other cytological grades, the integrated HPV genotypes were neither HPV16 nor 18, two genotypes that were involved in many high-grade lesion cases in our cohort. In addition, the genotypes identified in our LSIL (HPV33, 51, and 58) cases have been associated with carcinomas in other studies, albeit not the most frequently found genotypes. All these data suggest that the integration of the HPV genome is not a rare event and that it already occurs at a low-grade lesion stage. However, given that HPV33, 51 and 58 genotypes were never found integrated in more severe grades, it is possible that the size of our high-grade and carcinoma cohort is too small to reveal them. Other studies led in Gabon have confirmed that genotypes HPV16 and 18 are the two main genotypes found in carcinomas, followed by genotypes HPV33 and 58, but in much lower proportions42,43. These studies also demonstrate that the minor genotypes such as HPV33 and 58 can also be found in co-detection with HPV16 or 18, and therefore may not necessarily be responsible for the cancerous lesion Indeed, given that cervical smears can collect a large number of cells from the cervix, and not only from the area of the cancerous lesion, it is not surprising to find multi-infections of HPV42,43, unlike a biopsy which mainly collect cells infected by a single genotype from the lesion. In this study, there was no evidence that these minor genotypes associated with HPV16 and/or 18, particularly in high-grade lesions, were integrated into human genome. In any case, whatever the cytological grade considered, no simultaneous integration of different genotypes has been found.
In this study, the detection of integration sites was based on successive read mappings against viral references and then host genome (Fig. 3). This method successfully identified the integration position within the host genome and which part of the viral genome was involved. However, issues on the specificity and sensitivity of this method must be addressed. Other studies on HPV integration in the human genome are also based either on mapping tools or sequence alignment tools such as BLASTN or BLAT29,44. Alignment-based methods seem prima facie unbiased regarding chimera composition because no assumptions, concerning the size of the host and viral sequences in chimeric reads, are made. Therefore, the risk of missing host/viral chimeric reads resulting from integration sites should be low, regardless of the size of human and viral sequences of these chimera. Nevertheless, querying a database comprising only one organism introduces a bias. For instance, BLASTN is designed to detect similarities and is likely to find distant homology between unrelated sub-sequences, thereby ‘forcing’ the alignment to some extent. The same “artificial” human reads generated from Hg38 (Table 3) were also used for an alignment using BLAST (evalue = 0.0001) and HPV references from Table S1. This alignment resulted in 136,245/58,333,246 (2.3%) false positives. On the other hand, mapping methods may be questionable when it comes to specificity and working with genome sizes with different orders of magnitude. In fact, regarding the mapping parameters, especially the identity percentage and the minimum length of a read required to consider the latter as mapped, the reads may be assigned to the wrong genome23. Looking for chimeric reads by mapping in local alignment may inadvertently lead to finding a short sub-sequence that maps to both organisms. Our analysis showed that the decrease in the minimum length in either local or global mode for mapping parameters (CLC genomics), expressed in percentage or with a fixed number of bases, led to more viral reads being mapped on the Hg 38 genome (Table 3), increasing the rate of false positives. Consequently, although rare, cases mistaking a viral sub-sequence for a human one (or vice-versa) may lead to confusion regarding integration events. Therefore, the very stringent local mapping parameters used in this study were likely to limit the risk of false positives (Table 3). By choosing carefully the parameters, our mapping approach, more than saving computational time, allowed for keeping low the potential rate of false positives that can be returned by alignment methods. However, these stringent parameters can either be the reason for a lower integration rate than in other studies or may mask other events, such as multiple integration, by eliminating chimeric reads that are weakly represented. For instance, we observed only two cases of multiple integrations with the same genotype on the same chromosome. Lastly, as described in the literature21,23, micro-homologies were systematically observed in regions adjacent to integration sites of the viral genome in the host’s genome whatever the mapping parameters used (data not shown).
Figure 3.

Chimeric reads from mapping against viral reference are next mapped again human reference. The reads that aligned are in solid colour and the unaligned part is faded. If those chimeric reads mapped concordantly on the same human chromosome, then an integration event was called for this chromosome. Therefore the detection of integration events is based on chineric reads, mapped concordantly on viral reference, associated with low coverage and mapping on a single human chromosome.
Table 3.
Mapping parameters used for finding chimeric reads with CLC genomics workbench 10. The reads used for these comparisons were produced using a sequencing read generator tool (ART_ILLUMINA v2.5.8).
| Tool parameters | CLC mapping | human reads-HPV references | HPV reads-human reference | |
|---|---|---|---|---|
| •match score = 1 | Gobal alignment | 90% identity | 4,572,369 (7.8%) | 1,448,604 (99.9%) |
| •mismatch cost = 2 | 40% length | |||
| •Linear gap cost of insertion/deletion | 90% identity | 4,576,999 (7.8%) | 1,448,607 (99.9%) | |
| •Insertion cost = 3 | 30% length | |||
| •Deletion cost = 3 | Local alignment | 90% identity | 10,226 (0.01%) | 6,365 (0.4%) |
| 40% length | ||||
| 90% identity | 49,806 (0.08%) | 17,688 (1.2%) | ||
| 30% length | ||||
| Total reads | 58,333,246 (human) | 1,448,670 (HPV) | ||
Finally, owing to the greater sensitivity in the detection of integration phenomena, the analysis of our data regarding integration shows that they can be classified into two types of profiles: one in which the integrated form predominates a residual episomal form, and those for which the episomal form dominates the integrated form. This residual episomal- predominant form (Epi/Int) can be due to the presence of several cells infected by the same HPV genotype, but at a different stage. Additionally, the Epi/Int form could be the result of smaller number of cells which harbours a higher number of copies compared with the cells with an integrated form where there would be only one copy per cell. On the other hand, when the integrated form of the genotype is predominant (Int/Epi), the cell is probably already in the clonal multiplication phase due to the disruption in cellular functions linked to the expression of genes E6 and E7. The development of cervical cancer is a process that requires several stages in which many genetic alterations intervene, activating many cellular oncogenes or inactivating tumour suppressor genes. In either case, the expression of the HPV viral oncogenes E6 and E7 is required during all stages of tumour progression. The integration of the viral genome often leads to the partial deletion of viral genes such as E1, E2, L1, and L2. In our study, all observed integrations were accompanied by the loss of viral sequences, with deletion of the E2 ORF in 84.2% of the cases. As suggested in other studies, the loss of this region may induce the deregulation of the expression of oncogenes E7 and E645. However, the analysis of these integration sites in the human genome cannot explain the link between viral integration and the cellular modifications that we observed, except in some samples in which integration occurred close to genes known to be involved either in the cellular division process (RRM1) or to act as proto-oncogenes (c-myc)33. Transcriptomic analyses to confirm these modifications in expression would be difficult to perform because our initial samples are a mix of healthy and transformed cells obtained from a cervical smear.
To conclude, the integration rate of HPV genomes in the host genome varies according to the cytological grade considered, as observed on biopsies in other studies. The possibility of detecting these integration events from smears performed in liquid-based environments has many advantages for developing countries or for those whose at-risk populations have little or limited access to a health system. Smears are easy to perform and do not require any particular conditions during their transport to the analysis laboratory. In addition to other molecular investigations, the early screening for HPV integration in low-grade or atypical lesions can be used as a clinical prognostic biomarker for better prediction of the progression of these neoplastic lesions.
Supplementary information
Acknowledgements
We acknowledge Heïdi Lançon and Dr Carolyn Engel-Gautier for editing the English in the manuscript. The authors wish to thank all the Gabonese women who participated in this study and the staff of ICL and CHUL for their wonderful devotion and care to the patients included in this study. This study was supported by INCa (France) and the CIRMF, which is supported by the Government of Gabon, Total-Fina-Elf Gabon, and the Ministère de la Coopération Française. The funders had no role in study design, data analysis or preparation of the manuscript.
Author Contributions
N.B. conceived and designed the study. P.B.M. performed all the molecular biology assays for HPV genotyping and the generation of all libraries whereas I.L. performed the sequencing. A.A.N.M. and N.B. performed bioinformatics analysis. A.A.N.M., P.M.B., I.L., I.H.K., E.J., S.D.D., X.S.G., E.M.L., E.B. and N.B. authors analysed the data. A.A.N.M., P.B.M. and N.B. wrote the manuscript. I.H.K. and I.L. participated in the critical reading of the manuscript. A.A.N.M., P.M.B., I.L., I.H.K., E.J., S.D.D., X.S.G., E.M.L., E.B. and N.B. authors read and approved the final manuscript.
Data Availability
All raw data produced during this study are available Under Accession Number: SUB4880803.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Andriniaina Andy Nkili-Meyong and Pamela Moussavou-Boundzanga contributed equally.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-37871-2.
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer burden: Globocan 2000. International journal of cancer. 2001;94:153–156. doi: 10.1002/ijc.1440. [DOI] [PubMed] [Google Scholar]
- 2.Soerjomataram I, et al. Global burden of cancer in 2008: a systematic analysis of disability-adjusted life-years in 12 world regions. Lancet (London, England) 2012;380:1840–1850. doi: 10.1016/s0140-6736(12)60919-2. [DOI] [PubMed] [Google Scholar]
- 3.Bosch FX, Munoz N. The viral etiology of cervical cancer. Virus research. 2002;89:183–190. doi: 10.1016/S0168-1702(02)00187-9. [DOI] [PubMed] [Google Scholar]
- 4.Walboomers JM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. The Journal of pathology. 1999;189:12–19. doi: 10.1002/(sici)1096-9896(199909)189:112::aid-path4313.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 5.Arias-Pulido H, Peyton CL, Joste NE, Vargas H, Wheeler CM. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. Journal of clinical microbiology. 2006;44:1755–1762. doi: 10.1128/jcm.44.5.1755-1762.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Das BC, Sharma JK, Gopalakrishna V, Luthra UK. Analysis by polymerase chain reaction of the physical state of human papillomavirus type 16 DNA in cervical preneoplastic and neoplastic lesions. The Journal of general virology. 1992;73(Pt 9):2327–2336. doi: 10.1099/0022-1317-73-9-2327. [DOI] [PubMed] [Google Scholar]
- 7.Carow, K. et al. Viral-Cellular DNA Junctions as Molecular Markers for Assessing Intra-Tumor Heterogeneity in Cervical Cancer and for the Detection of Circulating Tumor DNA. International journal of molecular sciences18, 10.3390/ijms18102032 (2017). [DOI] [PMC free article] [PubMed]
- 8.Han L, et al. Comparative study of HPV16 integration in cervical lesions between ethnicities with high and low rates of infection with high-risk HPV and the correlation between integration rate and cervical neoplasia. Experimental and therapeutic medicine. 2015;10:2169–2174. doi: 10.3892/etm.2015.2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choo KB, Pan CC, Han SH. Integration of human papillomavirus type 16 into cellular DNA of cervical carcinoma: preferential deletion of the E2 gene and invariable retention of the long control region and the E6/E7 open reading frames. Virology. 1987;161:259–261. doi: 10.1016/0042-6822(87)90195-4. [DOI] [PubMed] [Google Scholar]
- 10.Cooper K, Herrington CS, Stickland JE, Evans MF, McGee JO. Episomal and integrated human papillomavirus in cervical neoplasia shown by non-isotopic in situ hybridisation. Journal of clinical pathology. 1991;44:990–996. doi: 10.1136/jcp.44.12.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snijders PJ, et al. Human papillomavirus (HPV) type 16 and 33 E6/E7 region transcripts in tonsillar carcinomas can originate from integrated and episomal HPV DNA. The Journal of general virology. 1992;73(Pt 8):2059–2066. doi: 10.1099/0022-1317-73-8-2059. [DOI] [PubMed] [Google Scholar]
- 12.Gudleviciene Z, et al. Integration of human papillomavirus type 16 in cervical cancer cells. Open medicine (Warsaw, Poland) 2015;10:1–7. doi: 10.1515/med-2015-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kahla S, Kochbati L, Chanoufi MB, Maalej M, Oueslati R. HPV-16 E2 physical status and molecular evolution in vivo in cervical carcinomas. The International journal of biological markers. 2014;29:e78–85. doi: 10.5301/jbm.5000051. [DOI] [PubMed] [Google Scholar]
- 14.Vernon SD, Unger ER, Miller DL, Lee DR, Reeves WC. Association of human papillomavirus type 16 integration in the E2 gene with poor disease-free survival from cervical cancer. International journal of cancer. 1997;74:50–56. doi: 10.1002/(SICI)1097-0215(19970220)74:1<50::AID-IJC9>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 15.Thorland EC, et al. Human papillomavirus type 16 integrations in cervical tumors frequently occur in common fragile sites. Cancer research. 2000;60:5916–5921. [PubMed] [Google Scholar]
- 16.Luft F, et al. Detection of integrated papillomavirus sequences by ligation-mediated PCR (DIPS-PCR) and molecular characterization in cervical cancer cells. International journal of cancer. 2001;92:9–17. doi: 10.1002/1097-0215(200102)9999:9999<::AID-IJC1144>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 17.Liang WS, et al. Simultaneous characterization of somatic events and HPV-18 integration in a metastatic cervical carcinoma patient using DNA and RNA sequencing. International journal of gynecological cancer: official journal of the International Gynecological Cancer Society. 2014;24:329–338. doi: 10.1097/igc.0000000000000049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ojesina AI, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506:371–375. doi: 10.1038/nature12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Integrated genomic and molecular characterization of cervical cancer. Nature543, 378–384, 10.1038/nature21386 (2017). [DOI] [PMC free article] [PubMed]
- 20.Chung TK, et al. Genomic aberrations in cervical adenocarcinomas in Hong Kong Chinese women. International journal of cancer. 2015;137:776–783. doi: 10.1002/ijc.29456. [DOI] [PubMed] [Google Scholar]
- 21.Hu Z, et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nature genetics. 2015;47:158–163. doi: 10.1038/ng.3178. [DOI] [PubMed] [Google Scholar]
- 22.Duncavage EJ, et al. Hybrid capture and next-generation sequencing identify viral integration sites from formalin-fixed, paraffin-embedded tissue. The Journal of molecular diagnostics: JMD. 2011;13:325–333. doi: 10.1016/j.jmoldx.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmes A, et al. Mechanistic signatures of HPV insertions in cervical carcinomas. Npj Genomic Medicine. 2016;1:16004. doi: 10.1038/npjgenmed.2016.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adey A, et al. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature. 2013;500:207–211. doi: 10.1038/nature12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akagi K, et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome research. 2014;24:185–199. doi: 10.1101/gr.164806.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Landry JJ, et al. The genomic and transcriptomic landscape of a HeLa cell line. G3 (Bethesda, Md.) 2013;3:1213–1224. doi: 10.1534/g3.113.005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, et al. Genome-wide profiling of the human papillomavirus DNA integration in cervical intraepithelial neoplasia and normal cervical epithelium by HPV capture technology. Sci Rep. 2016;6:35427. doi: 10.1038/srep35427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Dai SZ, Chu HJ, Cui HF, Xu XY. Integration sites and genotype distributions of human papillomavirus in cervical intraepithelial neoplasia. Asian Pacific journal of cancer prevention: APJCP. 2013;14:3837–3841. doi: 10.7314/APJCP.2013.14.6.3837. [DOI] [PubMed] [Google Scholar]
- 29.Chandrani P, et al. NGS-based approach to determine the presence of HPV and their sites of integration in human cancer genome. British journal of cancer. 2015;112:1958–1965. doi: 10.1038/bjc.2015.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackson R, et al. Functional variants of human papillomavirus type 16 demonstrate host genome integration and transcriptional alterations corresponding to their unique cancer epidemiology. BMC genomics. 2016;17:851. doi: 10.1186/s12864-016-3203-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moussavou PB, et al. Molecular analysis of human Papillomavirus detected among women positive for cervical lesions by visual inspection with acetic acid/Lugol’s iodine (VIA/VILI) in Libreville, Gabon. Infect Agent Cancer. 2016;11:50. doi: 10.1186/s13027-016-0098-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scholz MB, Lo CC, Chain PS. Next generation sequencing and bioinformatic bottlenecks: the current state of metagenomic data analysis. Current opinion in biotechnology. 2012;23:9–15. doi: 10.1016/j.copbio.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 33.Wentzensen N, Vinokurova S, von Knebel Doeberitz M. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer research. 2004;64:3878–3884. doi: 10.1158/0008-5472.CAN-04-000964/11/3878. [DOI] [PubMed] [Google Scholar]
- 34.Koopman LA, et al. Recurrent integration of human papillomaviruses 16, 45, and 67 near translocation breakpoints in new cervical cancer cell lines. Cancer research. 1999;59:5615–5624. [PubMed] [Google Scholar]
- 35.Zhao JW, et al. HPV16 integration probably contributes to cervical oncogenesis through interrupting tumor suppressor genes and inducing chromosome instability. Journal of experimental & clinical cancer research: CR. 2016;35:180. doi: 10.1186/s13046-016-0454-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su C, et al. ERCC1, RRM1 and BRCA1 mRNA expression levels and clinical outcome of advanced non-small cell lung cancer. Medical oncology (Northwood, London, England) 2011;28:1411–1417. doi: 10.1007/s12032-010-9553-9. [DOI] [PubMed] [Google Scholar]
- 37.Wang N, et al. Increased expression of RRM2 by human papillomavirus E7 oncoprotein promotes angiogenesis in cervical cancer. British journal of cancer. 2014;110:1034–1044. doi: 10.1038/bjc.2013.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dyer N, Young L, Ott S. Artifacts in the data of Hu et al. Nature genetics. 2016;48:2–4. doi: 10.1038/ng.3392. [DOI] [PubMed] [Google Scholar]
- 39.Hopman AH, et al. Transition of high-grade cervical intraepithelial neoplasia to micro-invasive carcinoma is characterized by integration of HPV 16/18 and numerical chromosome abnormalities. The Journal of pathology. 2004;202:23–33. doi: 10.1002/path.1490. [DOI] [PubMed] [Google Scholar]
- 40.Berthet N, et al. COFAC-Col: A Cervical Cancer Control Networking Initiative in Five French-Speaking African Countries. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2016;25:1004–1005. doi: 10.1158/1055-9965.EPI-15-1248. [DOI] [PubMed] [Google Scholar]
- 41.Arbyn, M. et al. Human papillomavirus testing versus repeat cytology for triage of minor cytological cervical lesions. The Cochrane database of systematic reviews, Cd008054, 10.1002/14651858.CD008054.pub2 (2013). [DOI] [PMC free article] [PubMed]
- 42.Zoa Assoumou S, et al. Human papillomavirus genotypes distribution among Gabonese women with normal cytology and cervical abnormalities. Infect Agent Cancer. 2016;11:2. doi: 10.1186/s13027-016-0046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zoa-Assoumou S, et al. Human papillomavirus genotypes distribution in cervical cancer cases in Gabon. Infectious agents and cancer. 2016;11:42. doi: 10.1186/s13027-016-0091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katz JP, Pipas JM. SummonChimera infers integrated viral genomes with nucleotide precision from NGS data. BMC bioinformatics. 2014;15:348. doi: 10.1186/s12859-014-0348-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peter M, et al. Frequent genomic structural alterations at HPV insertion sites in cervical carcinoma. The Journal of pathology. 2010;221:320–330. doi: 10.1002/path.2713. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data produced during this study are available Under Accession Number: SUB4880803.