

Abstract
Our goal was to examine whether in utero exposure to alcohol impaired reactivity of cerebral arterioles during development. We fed Sprague–Dawley dams a liquid diet with or without alcohol (3% ethanol) for the duration of pregnancy (21–23 days). Around 4–6 weeks after birth, we examined reactivity of cerebral arterioles to eNOS- (ADP) and nNOS-dependent (NMDA) agonists in the offspring. We found that in utero exposure to alcohol attenuated responses of cerebral arterioles to ADP and NMDA, but not to nitroglycerin in rats exposed to alcohol in utero. L-NMMA reduced responses to agonists in control rats, but not in rats exposed to alcohol in utero. Treatment of dams with apocynin for the duration of pregnancy rescued the impairment in reactivity to ADP and NMDA in the offspring. Protein expression of NOX-2 and NOX-4 was increased in alcohol rats compared to control rats. We also found an increase in superoxide levels in the cortex of rats exposed to alcohol in utero. Our findings suggest that in utero exposure to alcohol impairs eNOS and nNOS reactivity of cerebral arterioles via a chronic increase in oxidative stress.
Keywords: Cerebral circulation, fetal alcohol spectrum disorders, NOS, oxidative stress, superoxide
Introduction
Alcohol is one of the most commonly used chemical substances and exposure to alcohol in utero in humans causes fetal developmental abnormalities of the brain.1,2 These negative effects are defined as fetal alcohol spectrum disorders (FASD). These disorders comprise several alcohol-related abnormalities including fetal alcohol syndrome (FAS), partial fetal alcohol syndrome (pFAS), alcohol-related birth defects (ARBD) and alcohol-related neurodevelopmental disorder (ARND)1. While the prevalence of FASD is difficult to estimate, it is thought that 1% of children in the USA have some form of FASD.1 This is concerning since abnormalities caused by FASD last a lifetime and no treatment is available for these deficits.
In the brain, deficits produced by in utero exposure to alcohol are either structural or functional, and lead to cognitive decline, hyperactivity, and an increased incidence of epilepsy.2 In the past, the focus of FASD research has been on neuronal development, hippocampal morphology, and inflammation.3–5 In contrast, the cerebral microcirculation and its contribution to FASD have received very little attention. Although some studies have shown that in utero exposure to alcohol in the second trimester can impair changes in cerebral blood flow in response to hypoxia in fetal sheep,6,7 no studies have examined cellular mechanisms that account for this impairment. Additionally, no studies have examined whether in utero exposure to alcohol can impair reactivity of cerebral resistance arterioles (i.e. vessels that directly control cerebral blood flow) to agonists of eNOS and nNOS. Thus, our first goal was to determine the effect of in utero exposure to alcohol on reactivity of cerebral arterioles during development.
Oxidative stress has been shown to increase neuronal damage and death in several brain structures in FASD. Investigators have reported that exposure to alcohol in utero increases markers of oxidative stress, including damage to lipids, proteins, DNA, and alterations of endogenous antioxidants in hippocampus, striatum, hypothalamus, cerebellum, and cerebral cortex of mice and rats.8–11 Oxidative stress has also been shown to increase damage and loss of Purkinje cells and cerebellar granule neurons after exposure to alcohol in utero.11,12 It is then reasonable to suggest that oxidative stress may be linked to brain damage in FASD. Additionally, chronic oxidative stress may impair the cerebral vasculature and exacerbate damage linked to in utero exposure to alcohol. Thus, our second goal was to examine the role of oxidative stress in altered reactivity of cerebral arterioles by in utero exposure to alcohol.
Materials and methods
Experimental diets
All experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee at Louisiana State University Health Sciences Center-Shreveport and are in accordance with the National Institutes of Health guidelines. The manuscript was written in compliance with the ARRIVE guidelines. We used virgin adult male (16 weeks old, 300 g) and female (16 weeks old, 250 g) Sprague–Dawley rats. A pair of male and female rats was allowed to mate overnight. Females were then housed singly and assigned randomly to one of four groups: control liquid diet, 3% alcohol liquid diet, control+apocynin liquid diet, or 3% alcohol+apocynin liquid diet. Apocynin was added to the liquid diet in order to examine a role for oxidative stress in impaired responses of cerebral arterioles in rats exposed to alcohol in utero. Apocynin is an inhibitor of NADPH oxidase, preventing the assembly of NADPH oxidase by blocking the translocation of p47-phox and p67-phox to the membrane.16,17 Pregnant dams were fed one of these diets for the entire gestation period (21–23 days). Liquid diets were prepared daily. The control dams were fed a liquid diet that contains 1.0 kcal/ml of which 35% are derived from fat, 47% from carbohydrates, and 18% from protein. For the 3% alcohol groups, we fed the dams a liquid diet that contains 1.0 kcal/ml of which 35% are derived from fat, 18% from protein, 29% from carbohydrate, and 18% from alcohol. The total daily volume of diet fed to the control animals was based upon the daily consumption of diet by the alcohol animals. The alcohol diet resulted in a blood alcohol concentration of 13.6 ± 1 mM in dams. This is equivalent to a BAC of 0.07% in humans. The liquid diets fed to the dams were replaced by a normal chow/water diet after the birth of the pups. Rats were weaned at three weeks of age and placed in cages with rats of the same sex. Both male and female rats were used for the experiments described. For the control+apocynin and alcohol+apocynin groups, we added apocynin (10 mg/kg/day) to the liquid diets fed to the dams for the duration of pregnancy (21–23 days).
Preparation of animals
The rats (offspring) were prepared for studies at 4–6 weeks of age. The experimenter was blinded to the groups. On the day of the experiment, the rats were anesthetized with thiobutabarbital sodium (Inactin, 100 mg/kg body weight, intraperitoneally), and a tracheotomy was performed. The rats were ventilated mechanically with room air and supplemental oxygen. A catheter was placed into a femoral vein for injection of supplemental anesthesia (10–30 mg/kg) as necessary, and a femoral artery was cannulated to measure arterial blood pressure and to obtain a blood sample for blood gases.
Cerebral arteriolar diameter
A craniotomy was prepared over the left parietal cortex to visualize the microcirculation of the cerebrum. The cranial window was suffused with artificial cerebrospinal fluid (2 ml/min) that was bubbled continuously with 95% nitrogen and 5% carbon dioxide. Artificial cerebrospinal fluid contained (in mmol/l): 124 NaCl, 2.5 KCl, 2.0 MgSO4, 1.25 KH2PO4, 26 NaHCO3, 10 glucose, 2.5 CaCl2. Temperature of the suffusate was maintained at 37 ± 1 ℃. The cranial window was connected via a three-way valve to an infusion pump, which allowed for infusion of agonists and antagonists into the suffusate. This method maintained a constant temperature, pH, PCO2, and PO2 of the suffusate during infusion of drugs. Arterial blood gases were monitored and maintained within normal limits throughout the experiment period. Diameter of cerebral arterioles was measured using a video image-shearing device. The cranial window was superfused with artificial cerebral spinal fluid for 30 min prior to testing responses of cerebral arterioles to the agonists.
Responses of cerebral arterioles were examined during superfusion of agonists that presumably produce dilation dependent on the release of nitric oxide via activation of eNOS: ADP (10 and 100 μM); or nNOS: NMDA (30 and 100 μM). We also examined responses of cerebral arterioles to nitroglycerin (1.0 and 10 μM), which dilates cerebral arterioles independent of NOS. Diameter of arterioles was measured before, and at 1-min intervals for 5 min during application of agonists. Baseline diameter of cerebral arterioles returned to control levels (before application of agonists) within 2–3 min after application of agonists was stopped. To determine the role of nitric oxide in ADP and NMDA-induced dilation of cerebral arterioles, we suffused the cranial window with NG-monomethyl-L-arginine (L-NMMA; 10 μM). Thus, after initially measuring responses of cerebral arterioles to the agonists, we started a continuous suffusion with L-NMMA. One hour after starting suffusion with L-NMMA, we retested responses of cerebral arterioles to the agonists.
Superoxide levels
We measured superoxide anion levels using lucigenin-enhanced chemiluminescence. After transcardially perfusion of the heart with cold normal saline, the brain was removed and immersed in a modified Krebs-HEPES buffer containing (in mmol/l): 118 NaCl, 4.7 KCl, 1.3 CaCl2, 1.2 MgCl2, 1.2 KH2PO2, 25 NaHCO3, 10 HEPES, and 5 glucose (pH 7.4). Tissue samples were weighed from the parietal cortex and were placed in polypropylene tubes containing 5 µmol/l lucigenin, and then they were read in a Berthold Sirius luminometer, which reports relative light units emitted integrated over 30-s intervals for 5 min. Data were corrected for background activity and normalized to tissue weight. In these studies, we measured superoxide anion levels under basal conditions and during exposure to NADPH (10 and 100 μM) for 5 min. Thus, we examined baseline superoxide levels and levels of superoxide during exposure to a substrate of NADPH oxidase.
Western blot analysis
Brains were extracted and cleaned with PBS. Parietal cortex or cerebral microvessels from both hemispheres were isolated and stored at −80 ℃. Frozen tissue samples (parietal cortex or cerebral microvessels isolated from brain tissue) were homogenized in 20% (weight/volume) ice-cold buffer that contained 10 mM Tris-HCl (pH 7.4),1% sodium dodecyl sulfate,1 mM sodium vanadate, 10 µg/ml aprotinin, 10 µg/ml eupeptin, and 1 mM phenylmethylsulfonylfluoride. Next, samples were centrifuged at 12,000 × g for 20 min at 4 ℃, and protein concentration in the supernatant was determined by the Bradford method (Bio-Rad, Richmond, CA) with bovine serum albumin as the standard. SDS-PAGE was performed on a 10% gel on which 5 µg of total protein/well was loaded. After SDS-PAGE, the protein was transferred onto a polyvinylidenedifluoride membrane. Immunoblotting analysis was performed using goat anti-gp91-phox (NOX-2) (Santa Cruz, CA), mouse anti-eNOS (BD Transduction Laboratories), and mouse anti-nNOS (Santa Cruz, CA), rabbit anti-SOD-1 (Santa Cruz, CA), goat anti-SOD-2 (Santa Cruz, CA), and goat anti NOX-4 (Santa Cruz, CA). Goat anti-mouse immunoglobulin G, donkey anti-goat immunoglobin G, and goat anti-rabbit immunoglobin G were used as the secondary antibodies. An enhanced chemiluminescence substrate (Pierce) was applied to the membrane, followed by an exposure within an EpiChemi II Darkroom (UVP BioImaging, Upland, CA, USA) for visualization with the Worklab digital imaging system. Kodak 1D software was used to quantify the signal. The expression of eNOS, nNOS, SOD-1, SOD-2, NOX-2, and NOX-4 was calculated as the ratio of intensity of the band of the protein of interest relative to the intensity of the GAPDH band detected on the same blots.
Statistical analysis
Two-way ANOVA was used to compare findings between groups of rats. Bonferroni correction was used to test for significance between groups of rats. No difference was observed between male and female rats, and thus all data were pooled. Values are mean ± SEM. A p-value of 0.05 or less was considered to be significant.
Results
Responses to agonists
Baseline diameter of cerebral arterioles, mean arterial pressure, and body weight were similar in all groups at 4–6 weeks of age (Table 1).
Table 1.
Baseline parameters of 4–6-week-old rats.
| Control (8) | 3% EtOH (7) | Control + Apocynin (8) | 3% EtOH + Apocynin (7) | |
|---|---|---|---|---|
| Baseline Diameter (Microns) | 42 ± 1 | 42 ± 1 | 36 ± 3 | 43 ± 4 |
| Mean Arterial Pressure (mmHg) | 95 ± 5 | 102 ± 5 | 105 ± 7 | 101 ± 5 |
| Body Weight (g) | 149 ± 11 | 157 ± 6 | 166 ± 6 | 153 ± 21 |
Note: Values are means ± SE. N for each group is given in parentheses.
Application of ADP, NMDA, and nitroglycerin produced dose-related dilation of cerebral arterioles in 4–6-week-old rats (Figure 1(a) to (c)). However, the magnitude of vasodilation in response to ADP and NMDA, but not to nitroglycerin, was reduced in rats exposed to alcohol in utero.
Figure 1.
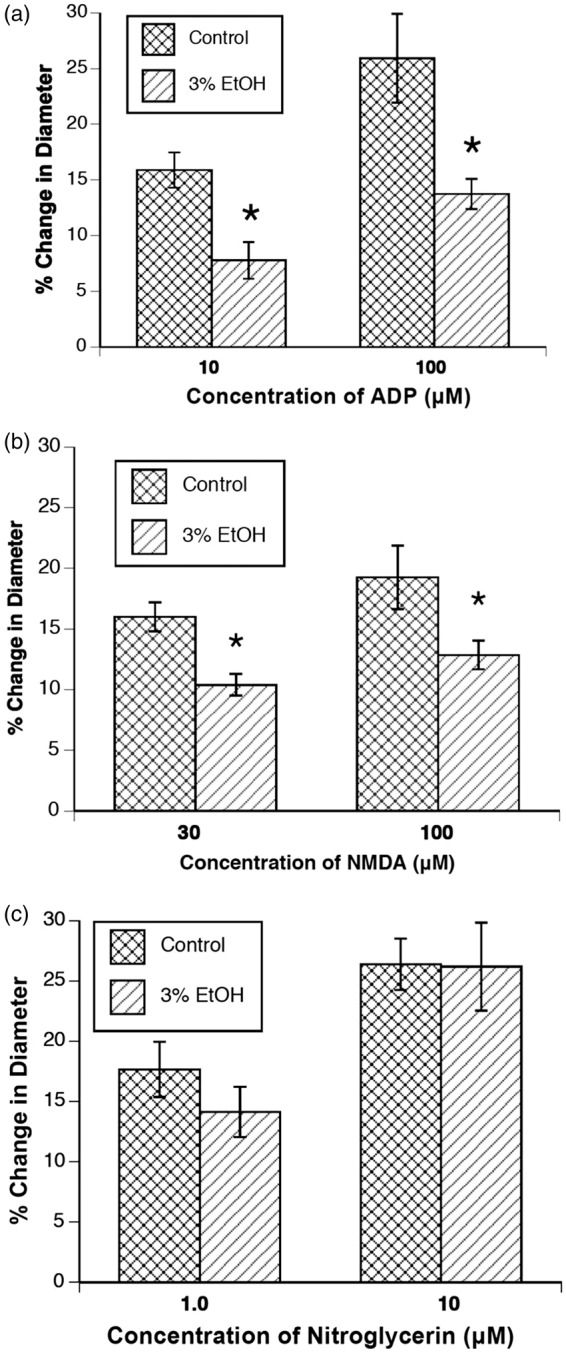
Responses of cerebral arterioles to ADP (a), NMDA (b), and nitroglycerin (c) in 4–6-week-old control rats (n = 8) and rats exposed to alcohol in utero (n = 7). Values are means ± SE. *p < 0.05 versus responses in control rats.
Effect of apocynin
First, treatment of dams with apocynin had no effect on the responses of control rats. Second, treatment of the dams with apocynin for the entire duration of pregnancy rescued impaired responses of cerebral arterioles to ADP and NMDA (Figure 2). Treatment with apocynin did not influence responses to nitroglycerin (Figure 2).
Figure 2.
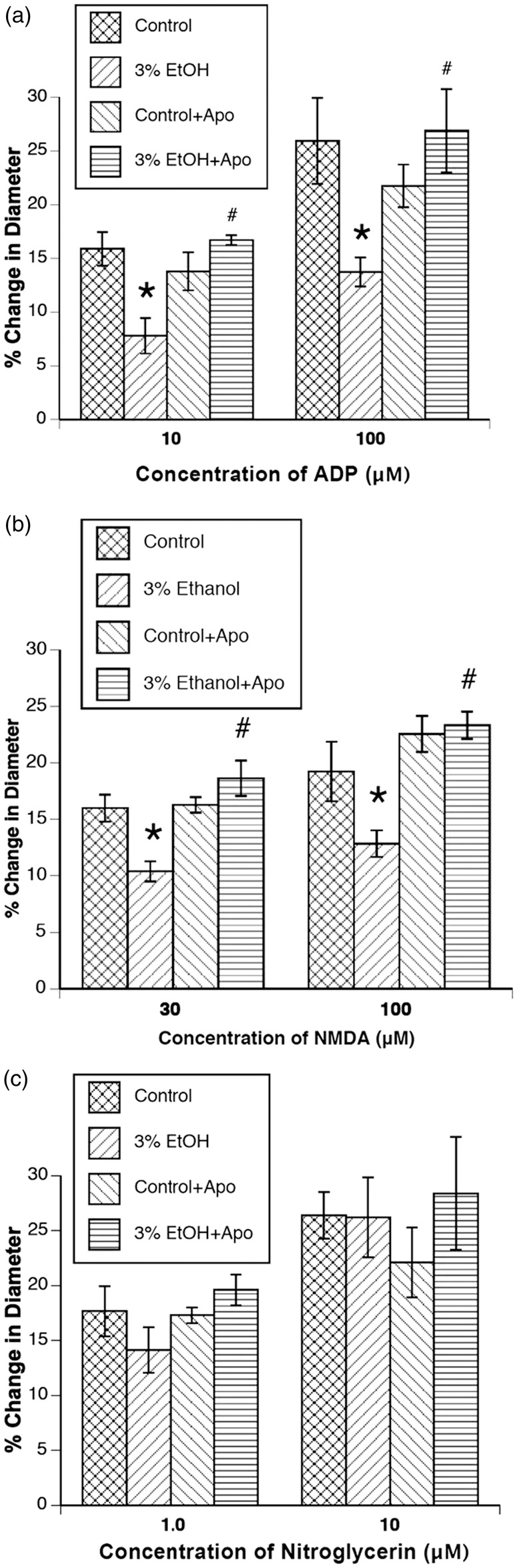
Responses of cerebral arterioles to ADP (a), NMDA (b), and nitroglycerin (c) in control rats (n = 8), rats exposed to alcohol in utero (n = 7), control rats+apocynin (n = 8), and rats exposed to alcohol in utero + apocynin (n = 7). Values are means ± SE. *p < 0.05 versus responses in control rats. #p < 0.05 versus responses in 3% EtOH rats.
Effect of L-NMMA
Baseline diameter of cerebral arterioles before and after L-NMMA application is found in Table 2. L-NMMA reduced baseline diameter in control rats, control rats + apocynin, and in rats exposed to alcohol in utero + apocynin, but had no effect on baseline diameter of rats exposed to alcohol in utero at 4–6 weeks of age (Table 2). We found that L-NMMA reduced responses to ADP and NMDA in control rats, control rats + apocynin, and rats exposed to alcohol in utero + apocynin (Figure 3). Additionally, L-NMMA reduced responses of cerebral arterioles to 100 µM ADP in alcohol animals. L-NMMA had no effect on responses of cerebral arterioles to nitroglycerin.
Table 2.
Baseline diameters of cerebral arterioles before and after application of L-NMMA.
| Control (8) | 3% EtOH (7) | Control + Apocynin (7) | 3% EtOH + Apocynin (4) | |
|---|---|---|---|---|
| Diameter Before L-NMMA (Microns) | 43 ± 2 | 45 ± 3 | 45 ± 3 | 59 ± 6 |
| Diameter After L-NMMA (Microns) | 30 ± 4* | 46 ± 6 | 34 ± 2* | 44 ± 3* |
Note: Values are means ± SE.N for each group is given in parentheses.
p < 0.05 versus baseline diameter before L-NMMA application.
Figure 3.

Responses of cerebral arterioles to ADP (a), NMDA (b), and nitroglycerin (c) in control rats (n = 8), rats exposed to alcohol in utero (n = 7), control rats + apocynin (n = 7) and rats exposed to alcohol in utero + apocynin (n = 4) before and during suffusion of L-NMMA (10 μM). Values are means ± SE. *p < 0.05 versus response before L-NMMA.
Superoxide
We found that superoxide levels from cortex tissue were increased in rats exposed to alcohol in utero under basal conditions (Figure 4). After stimulation with NADPH, we found a significant increase in the ability to produce superoxide in animals exposed to alcohol in utero (Figure 4).
Figure 4.

Superoxide levels at baseline and during stimulation with NADPH (10 and 100 μM) in control rats (n = 24), rats exposed to alcohol in utero (n = 24), control rats+apocynin (n = 24), and rats exposed to alcohol+apocynin (n = 24). Values are means ± SE.*p < 0.05 versus responses in control rats. #p < 0.05 versus responses in rats exposed to alcohol in utero.
Expression of eNOS, nNOS, NOX-2, NOX-4, SOD-1, and SOD-2
We did not observe a significant difference in the expression of eNOS in microvessels or nNOS in the parietal cortex tissue after exposure to alcohol in utero in 4–6-week-old rats (Figure 5(a) and (b)). However, we found that in utero exposure to alcohol significantly increased the expression of NOX-2 in cerebral microvessels (Figure 5(c)). Additionally, we found an increase in NOX-4 expression in cortex tissue (Figure 5(d)). SOD-1 and SOD-2 expression levels were unchanged in cortex tissue (Figure 5(e) and (f)).
Figure 5.

The effect of in utero exposure to alcohol on the expression of eNOS (a), nNOS (b), NOX-2 (c), NOX-4 (d), SOD-1 (e), and SOD-2 (f). (Top) Representative Western blot in parietal cortex or cerebral arterioles (Cont=control, n = 4; EtOH = rats exposed to alcohol in utero, n = 4, Cont+A = control+apocynin, n = 4; EtOH+A = rats exposed to alcohol in utero + apocynin, n = 4). (Bottom) Quantified data from the Western blots. Values are mean ± SE. *p < 0.05 versus responses in control rats. #p < 0.05 versus responses in 3% EtOH rats.
Discussion
There are several new findings from this study. First, exposure to alcohol in utero impairs dilation of cerebral arterioles in response to agonists that act via eNOS and nNOS in young (4–6-week-old) rats. Second, treatment of the dams with apocynin rescued the impaired reactivity linked to exposure to alcohol in utero. Third, L-NMMA reduced responses of cerebral arterioles to ADP and NMDA in all groups except rats exposed to alcohol in utero. Fourth, superoxide levels are increased at baseline and after stimulation with NADPH in rats exposed to alcohol in utero. Fifth, in utero exposure to alcohol upregulates the expression of NOX-2 and NOX-4 in microvessels and tissue from the parietal cortex, respectively, but in utero exposure does not alter the expression of eNOS, nNOS, SOD-1, or SOD-2. Based on these findings, we suggest that impaired NOS-dependent dilation of cerebral arterioles in animals exposed to alcohol in utero is related to increased levels of superoxide, presumably via NOX-2- and NOX-4-dependent mechanisms.
Consideration of methods
We used ADP as our eNOS-dependent agonist, NMDA as our nNOS-dependent agonist, and nitroglycerin as our NOS-independent agonist. ADP binds to the P2Y12 G protein-coupled receptor, NMDA opens NMDA receptors to allow the inward passage of Ca2+ ions, while nitroglycerin releases free nitric oxide following enzymatic action by dehydrogenases.13–15 ADP appears to dilate cerebral arterioles through a pathway involving eNOS.18–21 While some22 have suggested that dilation of cerebral arterioles in mice in response to ADP may be related to the release of endothelium-derived hyperpolarizing factor (EDHF) through activation of potassium channels, others23–27 have suggested that potassium channel activation does not have a significant role in dilation of cerebral arterioles to ADP in rats and rabbits. In the case of NMDA, it has been shown that NMDA dilates cerebral arterioles through a pathway involving nNOS.25,28–31 Thus, it appears that ADP and NMDA are appropriate agonists to examine eNOS- and nNOS-dependent dilation in cerebral arterioles.
We used L-NMMA to determine a role for NO in responses to ADP and NMDA. L-NMMA is a nonspecific inhibitor of all isoforms of NOS. We used L-NMMA to examine if the responses to ADP and NMDA in our groups were specifically NOS-dependent. We and several others have used L-NMMA extensively to test specificity of NOS-dependent agonists.32–36 We found decreased responses to ADP and NMDA in control rats, control+apocynin rats, and rats exposed to alcohol in utero + apocynin following application of L-NMMA. Rats exposed to alcohol in utero did not display a similar attenuation in responses to ADP and NMDA following L-NMMA, except for 100 µM ADP. This is likely due to already reduced responses to agonists in this group. Additionally, we found that L-NMMA reduced cerebral arteriolar baseline diameter in control rats, control+apocynin rats, and rats exposed to alcohol in utero + apocynin. We did not observe this effect in rats exposed to alcohol in utero. Because eNOS- and nNOS-dependent responses are impaired in rats exposed to alcohol in utero, an inhibitor of NOS like L-NMMA likely has very little effect on reducing baseline diameter of cerebral arterioles.
We used apocynin to determine a role of NADPH oxidase in impaired responses of cerebral arterioles in rats exposed to alcohol in utero. Apocynin is a nonspecific inhibitor of NADPH oxidase, preventing the assembly of NADPH oxidase by blocking the translocation of p47-phox and p67-phox to the membrane.16,17 Treatment of dams with apocynin in utero reduced the production of superoxide in the parietal cortex of rats exposed to alcohol. This suggests that the increase in oxidative stress observed in rats exposed to alcohol in utero may be linked to activation of NADPH oxidase. To further explore the effect of alcohol exposure and superoxide, we examined whether apocynin would reduce the impairment observed in rats exposed to alcohol in utero to ADP and NMDA. We found that apocynin alleviated impaired eNOS- and nNOS-dependent reactivity of cerebral arterioles in rats exposed to alcohol in utero. It appears that apocynin is a fitting inhibitor of NADPH oxidase and improves responses to eNOS and nNOS agonists, in addition to reducing superoxide levels in animals exposed to alcohol in utero. Alternatively, there is some evidence by Heumuller et al.37 that apocynin may act as an antioxidant rather than an inhibitor of NADPH oxidase in endothelial and smooth muscle cells. It has been suggested that apocynin is a prodrug that is activated by myeloperoxidase and forms the dimer diapocynin in leukocytes. Endothelial and smooth muscle cells fail to form diapocynin and fail to activate the less effective monomer apocynin. Instead, in vascular smooth muscle, redox-sensitive p38-mitogen-activated kinase (Akt) and extracellular signal-regulated kinase 1/2 is inhibited by apocynin, suggesting an alternative antioxidant pathway catalyzed by apocynin that does not involve NADPH oxidase.37
We measured the levels of superoxide using lucigenin-enhanced chemiluminescence. We found that in utero exposure to alcohol increased basal levels of superoxide in parietal cortex tissue in 4–6-week-old rats and after NADPH, which was used to stimulate NADPH oxidase activity. This suggests an increased production of superoxide in these animals that may contribute to increased oxidative stress in rats exposed to alcohol in utero.
We used both male and female rats in our studies. In the past we have shown that 3-month-old rats that were chronically fed alcohol with a liquid diet showed sex-related differences in responses of cerebral arterioles to acetylcholine, ADP, kainate, and NMDA.38 In the present study, we did not observe sex-related differences in eNOS or nNOS-dependent responses of cerebral arterioles in control rats or in rats exposed to alcohol in utero. Nevertheless, we did not expect any differences at 4–6 weeks of age due to rats not reaching puberty until 6–8 weeks of age.39
Consideration of previous studies
Very few studies have examined the effect of in utero exposure to alcohol on vascular function. One study used thoracic aortas harvested from 25-week-old rats exposed to alcohol in utero.40 They observed that responses to carbamylcholine chloride, an endothelium-dependent agonist, were impaired in adult rats that had been exposed to alcohol in utero, while relaxation to an NO donor (sodium nitroprusside) was similar between control rats and rats exposed to alcohol in utero. Our studies expand this concept by exploring the effect of agonists of eNOS and nNOS on cerebral arterioles of rats exposed to alcohol in utero and mechanisms that may involve the action of superoxide and NADPH oxidase.
Other studies have examined the effect of in utero exposure to alcohol on large cerebral arteries in sheep. One study by Mayock et al.41 found that in utero exposure to alcohol impaired the increase in cerebral blood flow in response to hypoxia. Another study from this group42 using the same model found that responses of cerebral arterioles to adenosine were not affected. Additionally, they found no changes in cerebral microvessel density in this model.41 In the present study, we found impaired responses of cerebral arterioles to agonist of eNOS and nNOS in rats exposed to alcohol in utero. Our studies extend beyond those of Mayock et al. by examining proteins involved in vasodilation and inhibition of those responses through mechanisms involving the effect of oxidative stress.
Investigators have examined the influence of in utero exposure to alcohol on oxidative stress in brain. Previous studies have found increased expression of p22phox, p27phox, NOX activator 1 (NOXA1), and NOX organizer 1 (NOXO1) in mouse embryos following exposure to alcohol.43 In the present study, we found increased expression of NOX-2 and NOX-4 in rats exposed to alcohol in utero. Other studies found that use of antioxidants, including vitamin C, E, and beta-carotene, protected against alcohol induced cell loss,44 reduced ROS production and NF-kappa B activation,45 and protected against ethanol combined with ischemia in embryonic rat hippocampal cultures.46 Additionally, protein expression of NOX-2 and NOX-4 remained elevated in rats exposed to alcohol and apocynin in utero. It is possible that alcohol exposure in utero would induce overexpression of proteins that have an important role in the production of superoxide, but the presence of apocynin, which prevents the assembly of the NADPH oxidase complex, would reduce the production of superoxide and rescue the impairment in reactivity observed in cerebral arterioles in response to agonists of NOS. Nonetheless, SOD-1 and SOD-2 protein expression were not altered, suggesting that antioxidant pathways are not affected in cerebral arterioles.
In summary, we examined the effects of in utero exposure to alcohol on eNOS- and nNOS-dependent reactivity of cerebral arterioles, superoxide levels, and expression of key enzymes responsible for oxidative stress. We found that in utero exposure to alcohol impaired responses of cerebral arterioles to eNOS and nNOS-dependent agonists in 4–6-week-old rats. In addition, we found expression of proteins involved in endothelial vasodilation and oxidative stress to be altered by in utero exposure alcohol. Further, we found that treatment of dams with apocynin prevented impaired reactivity in cerebral arterioles in rats exposed to alcohol in utero. Based on these findings, we suggest that in utero exposure to alcohol impairs eNOS- and nNOS-dependent pathways by mechanisms that result in an increase in oxidative stress. We speculate that impaired responses of cerebral arterioles to activation of essential vasodilator pathways following in utero exposure to alcohol may have an important role in the pathogenesis of cognitive and structural brain aberrancies. The loss of vasodilator capacity would impair changes in cerebral blood flow after an increase in nutritional demand and result in a scenario of poor brain oxygenation and nutrition. This may produce an ischemic cerebral environment that may exacerbate brain dysfunction.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
All authors contributed extensively to the work presented in this paper. WGM conceived the study, SGC and WGM designed the experiments, SGC performed the experiments, and SGC and WGM analyzed the data and wrote the manuscript.
References
- 1.Riley EP, Infante MA, Warren KR. Fetal alcohol spectrum disorders: an overview. Neuropsychol Rev 2011; 21: 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones KL. The effects of alcohol on fetal development. Birth Defects Res C Embryo Today 2011; 93: 3–11. [DOI] [PubMed] [Google Scholar]
- 3.Ullah I, Ullah N, Naseer MI, et al. Neuroprotection with metformin and thymoquinone against ethanol-induced apoptotic neurodegeneration in prenatal rat cortical neurons. BMC Neurosci 2012; 13: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gil-Mohapel J, Boehme F, Patten A, et al. Altered adult hippocampal neuronal maturation in a rat model of fetal alcohol syndrome. Brain Res 2011; 1384: 29–41. [DOI] [PubMed] [Google Scholar]
- 5.Sozo F, O'Day L, Maritz G, et al. Repeated ethanol exposure during late gestation alters the maturation and innate immune status of the ovine fetal lung. Am J Physiol – Lung Cell Mol Physiol 2009; 296: L510–L518. [DOI] [PubMed] [Google Scholar]
- 6.Gleason CA, Iida H, Hotchkiss KJ, et al. Newborn cerebrovascular responses after first trimester moderate maternal ethanol exposure in sheep. Pediatr Res 1997; 42: 39–45. [DOI] [PubMed] [Google Scholar]
- 7.Ngai AC, Mondares RL, Mayock DE, et al. Fetal alcohol exposure alters cerebrovascular reactivity to vasoactive intestinal peptide in adult sheep. Neonatology 2008; 93: 45–51. [DOI] [PubMed] [Google Scholar]
- 8.Petkov VV, Stoianovski D, Petkov VD, et al. Lipid peroxidation changes in the brain in fetal alcohol syndrome. Biull Eksp Biol Med 1992; 113: 500–502. [PubMed] [Google Scholar]
- 9.Shirpoor A, Nemati S, Ansari MH, et al. The protective effect of vitamin E against prenatal and early postnatal ethanol treatment-induced heart abnormality in rats: a 3-month follow-up study. Int Immunopharmacol 2015; 26: 72–79. [DOI] [PubMed] [Google Scholar]
- 10.Ramachandran V, Perez A, Chen J, et al. In utero ethanol exposure causes mitochondrial dysfunction, which can result in apoptotic cell death in fetal brain: a potential role for 4-hydroxynonenal. Alcohol Clin Exp Res 2001; 25: 862–871. [PubMed] [Google Scholar]
- 11.Heaton MB, Paiva M, Madorsky I, et al. Ethanol effects on neonatal rat cortex: comparative analyses of neurotrophic factors, apoptosis-related proteins, and oxidative processes during vulnerable and resistant periods. Brain Res Dev Brain Res 2003; 145: 249–262. [DOI] [PubMed] [Google Scholar]
- 12.Light KE, Belcher SM, Pierce DR. Time course and manner of Purkinje neuron death following a single ethanol exposure on postnatal day 4 in the developing rat. Neuroscience 2002; 114: 327–337. [DOI] [PubMed] [Google Scholar]
- 13.Lewis CJ, Ennion SJ, Evans RJ. P2 purinoceptor-mediated control of rat cerebral (pial) microvasculature; contribution of P2X and P2Y receptors. J Physiol (Lond) 2000; 527: 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawand NB, Reddig WJ, Cashin AE, et al. NMDA receptors and associated signaling pathways: a role in knee joint blood flow regulation. Eur J Pharmacol 2004; 499: 155–161. [DOI] [PubMed] [Google Scholar]
- 15.Ranadive SM, Eugene AR, Dillon GA, et al. Comparison of the vasodilatory effects of sodium nitroprusside versus nitroglycerin. J Appl Physiol 2017; 123: 402–406. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stolk J, Hiltermann TJ, Dijkman JH, et al. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol 1994; 11: 95–102. [DOI] [PubMed] [Google Scholar]
- 17.Johnson DK, Schillinger KJ, Kwait DM, et al. Inhibition of NADPH oxidase activation in endothelial cells by ortho-methoxy-substituted catechols. Endothelium 2002; 9: 191–203. [DOI] [PubMed] [Google Scholar]
- 18.Faraci FM. Role of endothelium-derived relaxing factor in cerebral circulation: large arteries vs. microcirculation. Am J Physiol 1991; 261: H1038–H1042. [DOI] [PubMed] [Google Scholar]
- 19.Mayhan WG. Endothelium-dependent responses of cerebral arterioles to adenosine 5'-diphosphate. J Vasc Res 1992; 29: 353–358. [DOI] [PubMed] [Google Scholar]
- 20.Ayajiki K, Okamura T, Toda N. Involvement of nitric oxide in endothelium-dependent, phasic relaxation caused by histamine in monkey cerebral arteries. Jpn J Pharmacol 1992; 60: 357–362. [DOI] [PubMed] [Google Scholar]
- 21.You J, Johnson TD, Childres WF, et al. Endothelial-mediated dilations of rat middle cerebral arteries by ATP and ADP. Am J Physiol 1997; 273: H1472–H1477. [DOI] [PubMed] [Google Scholar]
- 22.Faraci FM, Lynch C, Lamping KG. Responses of cerebral arterioles to ADP: eNOS-dependent and eNOS-independent mechanisms. Am J Physiol Heart Circ Physiol 2004; 287: H2871–H2876. [DOI] [PubMed] [Google Scholar]
- 23.Brayden JE. Hyperpolarization and relaxation of resistance arteries in response to adenosine diphosphate. Distribution and mechanism of action. Circ Res 1991; 69: 1415–1420. [DOI] [PubMed] [Google Scholar]
- 24.Chrissobolis S, Ziogas J, Chu Y, et al. Role of inwardly rectifying K(+) channels in K(+)-induced cerebral vasodilatation in vivo. Am J Physiol Heart Circ Physiol 2000; 279: H2704–H2712. [DOI] [PubMed] [Google Scholar]
- 25.Faraci FM, Breese KR. Nitric oxide mediates vasodilatation in response to activation of N-methyl-D-aspartate receptors in brain. Circ Res 1993; 72: 476–480. [DOI] [PubMed] [Google Scholar]
- 26.Faraci FM, Heistad DD. Role of ATP-sensitive potassium channels in the basilar artery. Am J Physiol 1993; 264: H8–H13. [DOI] [PubMed] [Google Scholar]
- 27.Taguchi H, Heistad DD, Kitazono T, et al. Dilatation of cerebral arterioles in response to activation of adenylate cyclase is dependent on activation of Ca(2+)-dependent K + channels. Circ Res 1995; 76: 1057–1062. [DOI] [PubMed] [Google Scholar]
- 28.Busija DW, Leffler CW. Dilator effects of amino acid neurotransmitters on piglet pial arterioles. Am J Physiol 1989; 257: H1200–H1203. [DOI] [PubMed] [Google Scholar]
- 29.Faraci FM, Breese KR, Heistad DD. Responses of cerebral arterioles to kainate. Stroke 1994; 25: 2080–2083. discussion 2084. [DOI] [PubMed] [Google Scholar]
- 30.Faraci FM, Brian JE. 7-Nitroindazole inhibits brain nitric oxide synthase and cerebral vasodilatation in response to N-methyl-D-aspartate. Stroke 1995; 26: 2172–2175. discussion 2176. [DOI] [PubMed] [Google Scholar]
- 31.Fergus A, Lee KS. Regulation of cerebral microvessels by glutamatergic mechanisms. Brain Res 1997; 754: 35–45. [DOI] [PubMed] [Google Scholar]
- 32.Arrick DM, Li C, Mayhan WG. Sex-related differences in reactivity of cerebral arterioles during moderate exercise training. Microcirculation 2016; 23: 549–557. [DOI] [PubMed] [Google Scholar]
- 33.Kang LS, Masilamani S, Boegehold MA. Juvenile growth reduces the influence of epithelial sodium channels on myogenic tone in skeletal muscle arterioles. Clin Exp Pharmacol Physiol 2016; 43: 1199–1207. [DOI] [PubMed] [Google Scholar]
- 34.Petersen L, Bek T. The diameter response of retinal arterioles in diabetic maculopathy is reduced during hypoxia and is unaffected by the inhibition of cyclo-oxygenase and nitric oxide synthesis. Graefes Arch Clin Exp Ophthalmol 2016; 254: 2339–2346. [DOI] [PubMed] [Google Scholar]
- 35.Mayhan WG, Scott JP, Arrick DM. Influence of type 1 diabetes on basal and agonist-induced permeability of the blood-brain barrier. Physiol Rep 2015; 3: e12653, . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yen W, Cai B, Yang J, et al. Endothelial surface glycocalyx can regulate flow-induced nitric oxide production in microvessels in vivo. PLoS ONE 2015; 10: e0117133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heumüller S, Wind S, Barbosa-Sicard E, et al. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 2008; 51: 211–217. [DOI] [PubMed] [Google Scholar]
- 38.Sun H, Mayhan WG. Sex difference in nitric oxide synthase-dependent dilatation of cerebral arterioles during long-term alcohol consumption. Alcohol Clin Exp Res 2005; 29: 430–436. [DOI] [PubMed] [Google Scholar]
- 39.Leibowitz SF, Akabayashi A, Alexander J, et al. Puberty onset in female rats: relationship with fat intake, ovarian steroids and the peptides, galanin and enkephalin, in the paraventricular and medial preoptic nuclei. J Neuroendocrinol 2009; 21: 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turcotte LA, Aberle NS, Norby FL, et al. Influence of prenatal ethanol exposure on vascular contractile response in rat thoracic aorta. Alcohol 2002; 26: 75–81. [DOI] [PubMed] [Google Scholar]
- 41.Mayock D, Ness D, Mondares R, et al. Binge alcohol exposure in the second trimester attenuates fetal cerebral blood flow response to hypoxia. J Appl Physiol 2007; 102: 972–977. [DOI] [PubMed] [Google Scholar]
- 42.Mayock DE, Ngai AC, Mondares RL, et al. Effects of binge alcohol exposure in the second trimester on intracerebral arteriolar function in third trimester fetal sheep. Brain Res 2008; 1226: 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong J, Sulik KK, Chen SY. The role of NOX enzymes in ethanol-induced oxidative stress and apoptosis in mouse embryos. Toxicol Lett 2010; 193: 94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marino MD, Aksenov MY, Kelly SJ. Vitamin E protects against alcohol-induced cell loss and oxidative stress in the neonatal rat hippocampus. Int J Dev Neurosci 2004; 22: 363–377. [DOI] [PubMed] [Google Scholar]
- 45.Peng Y, Kwok KHH, Yang P-HH, et al. Ascorbic acid inhibits ROS production, NF-kappa B activation and prevents ethanol-induced growth retardation and microencephaly. Neuropharmacology 2005; 48: 426–434. [DOI] [PubMed] [Google Scholar]
- 46.Mitchell JJ, Paiva M, Heaton MB. Vitamin E and beta-carotene protect against ethanol combined with ischemia in an embryonic rat hippocampal culture model of fetal alcohol syndrome. Neurosci Lett 1999; 263: 189–192. [DOI] [PubMed] [Google Scholar]