

Abstract
Monogenic autoinflammatory disorders are a group of conditions defined by systemic or localized inflammation without identifiable causes, such as infection. In contrast to classical primary immunodeficiencies that manifest with impaired immune responses, these disorders are due to defects in genes that regulate innate immunity leading to constitutive activation of pro-inflammatory signaling. Through studying patients with rare autoinflammatory conditions, novel mechanisms of inflammation have been identified that bare on our understanding not only of basic signaling in inflammatory cells, but also of the pathogenesis of more common inflammatory diseases and have guided treatment modalities. Autoinflammation has further been implicated as an important component of cardiovascular, neurodegenerative, and metabolic syndromes. In this review, we will focus on a subset of inherited enzymatic deficiencies that lead to constitutive inflammation, and how these rare diseases have provided insights into diverse areas of cell biology not restricted to immune cells. In this way, Mendelian disorders of the innate immune system, and in particular loss of catalytic activity of enzymes in distinct pathways, have expanded our understanding of the interplay between many seemingly disparate cellular processes. We also explore the overlap between autoinflammation, autoimmunity, and immunodeficiency, which has been increasingly recognized in patients with dysregulated immune responses.
Keywords: autoinflammation, innate immunity, mutations, enzyme deficiency, metabolic sensors, ubiquitination, protein homeostasis
Introduction
Autoinflammatory disorders are characterized by recurrent or persistent systemic or organ specific inflammation without inciting event and classically present with elevated acute phase reactants (1, 2). Characterization of these disorders has primarily focused on familial forms of disease due to highly penetrant mutations as was the case with the identification of the MEFV gene responsible for Familial Mediterranean Fever (FMF) and mutations in the TNFRSF1A gene as the cause of dominantly inherited TRAPS (Tumor Necrosis Factor Receptor Associated Periodic Syndrome). As genetic sequencing technology and analysis have improved and cost has decreased, there have now been nearly 30 genes identified as causative for autoinflammatory disorders (3). Many of the earliest identified monogenic autoinflammatory diseases were directly related to constitutive inflammasome activation and include FMF and cryopyrinopathies, or loss of a critical inhibitory mechanism as in deficiency of IL-1 (DIRA) or IL-36 (DITRA) receptor antagonist leading to imbalanced cytokine receptor signaling (4–9). Examples such as these have led to classification systems focused on the primary molecular pathways that are altered and thus diseases have been denoted as inflammasomopathies, interferonopathies, and NF-kB related autoinflammatory disorders (10–13). These classifications have helped identify shared mechanisms of disease pathogenesis and principles of treatment. Generally, autoinflammatory disorders are due to gene dysregulation restricted to hematopoietic lineages, whereas involvement of non-inflammatory cells is limited. Although most monogenic autoinflammatory disorders can be placed into this paradigm, many newly identified disorders seem to defy this classification and they have revealed a role for pathways not previously linked to immune function.
Here we will focus on classifying a subset of disorders by the specific biochemical deficiency as opposed to the clinical manifestations or immune mechanism that is disrupted (Table 1). These disorders will be organized by the affected cellular function to highlight the unexpected links between specific biochemical processes and immune dysregulation. We will review disorders that are due to loss of a enzymatic activity and how these diseases may reveal important aspects not only of immunology but of basic cellular signaling. Enzymatic deficiencies offer unique potential treatment strategies based on either accumulation of toxic substrates or loss of catalytic products and can theoretically be treated with enzyme replacement therapy.
Table 1.
Summary of diseases, genes, and inheritance for autoinflammatory disorders discussed.
| Disease | Acronym | MIM disease | Inheritance | Gene/protein | Transcript ID |
|---|---|---|---|---|---|
| Sideroblastic anemia, B-cell immunodeficiency, developmental delay and periodic fevers | SIFD | 616084 | AR | TRNT1/TRNT1 | NM_001302946 |
| Majeed syndrome | – | 609628 | AR | LPIN2/LPIN2 | NM_014646.2 |
| PLCγ2-associated antibody deficiency, and immune dysregulation | PLAID | 614468 | AD | PLCG2/PLCγ2 | NM_002661.3 |
| Autoinflammation, PLCγ2-associated antibody deficiency, and immune dysregulation | APLAID | 614878 | AD | PLCG2/PLCγ2 | NM_002661.3 |
| Monogenic systemic JIA/IBD | – | – | AR | C13ORF31/LACC1/FAMIN | NM_153218 |
| Mevalonate kinase deficiency | MKD/HIDS | 251170/260920 | AR | MVK/MVK | NM_000431.3 |
| Haploinsufficiency A20 | HA20 | 616744 | AD/De novo | TNFAIP3/A20 | NM_006290.3 |
| Otulipenia/Otulin-related autoinflammtory syndrome | ORAS | 615712 | AR | OTULIN/OTULIN | NM_138348.5 |
| RIPK1 deficiency | – | 618108 | AR | RIPK1/RIPK1 | NM_003804 |
| Deficiency of adenosine deaminase 2 | DADA2 | 615688 | AR | CECR1/ADA2/ADA2 | NM_001282225.1 |
Disorders Due to Disruption of Protein Translation and Homeostasis
tRNA Nucleotidyltransferase, CCA-Adding, 1 (TRNT1) Deficiency
The TRNT1 gene encodes a ubiquitously expressed tRNA nucleotidyltransferase, CCA-Adding, 1 (TRNT1) that is essential for protein synthesis. TRNT1 adds and repairs the conserved CCA sequence at the 3′ end of all precursor cytosolic and mitochondrial transfer ribonucleic acids (tRNAs), a step necessary for the attachment of conjugate amino acids (14). TRNT1 also regulates RNA stability through tRNA decay mechanisms and may play an important role in reducing levels of non-coding RNAs (15). TRNT1 is localized to the mitochondria via a 41 amino acid transit peptide and is expressed in all tissues. The crystal structure of human TRNT1 (PDB ID:1Ou5) shows that the protein functions as a homodimer via intermolecular disulfide bond (16). Complete deficiency of Trnt1 in mice is embryonic lethal further highlighting the essential function of this gene.
Bi-allelic loss of function mutations in TRNT1 lead to a recessively inherited syndrome named SIFD for sideroblastic anemia, B-cell immunodeficiency, developmental delay, and periodic fevers (Figure 1A) (17, 18). Given the ubiquitous expression of TRNT1, it is not surprising that reduced function of the enzyme leads to a complex phenotype. To date, more than 30 patients have been reported with significant clinical and immunologic heterogeneity (17, 19–22). At the severe end of the spectrum are patients with neonatal-onset severe anemia and prominent extramedullary erythropoiesis, profound immunodeficiency, metabolic and neurological abnormalities (17). In this first published cohort of 12 patients, median survival was 48 months and seven patients died due to cardiac or multiorgan failure. Recurrent fever has been reported in most but not all patients with SIFD. Immunodeficiency in SIFD is primarily due to defects in B cells differentiation and can manifest early in life or can be progressive and present later (23). T and NK cell numbers are in the low-normal range and some patients also carry a diagnosis of combined variable immunodeficiency but without serious bacterial or viral infections. At the milder end of the spectrum are patients with non-syndromic retinitis pigmentosa and subtle hematological features (24, 25).
Figure 1.

Autoinflammatory diseases due to dysregulation in proteostasis. SIFD (A) and CANDLE (B) syndromes.
SIFD-associated variants are loss-of-function and they include missense, non-sense, frameshift, and splice site mutations. TRNT1 pathogenic variants are either novel or have a low frequency in the general population. As is the case with many other recessively inherited diseases, a subset of patients were identified in founder populations with homozygous pathogenic variants. Hypomorphic mutations in TRNT1 reduce protein expression, affect protein stability, or alter its catalytic efficiency (18, 26). So far, no patients have been identified with biallelic non-sense or frameshift mutation, further emphasizing that residual TRNT1 protein is essential for development. In TRNT1-deficient cells, maturation of nuclear and mitochondrial tRNAs is impaired, which leads to a defect in global protein synthesis (21, 27). As result, these cells are unable to maintain protein homeostasis under stress conditions. Protein degradation pathways, which are essential for clearance of unprocessed/misfolded proteins, eventually become insufficient to remove an excessive load of misfolded proteins in mutant cells. Accumulation of misfolded proteins can then result in cell death and release of various proinflammatory cytokines through the unfolded protein response (UPR). Although ER stress and activation of the UPR has not been experimentally demonstrated in TRNT1-deficient cells, there are examples of other similar systemic inflammatory diseases of dysregulated protein homeostasis, such as Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) and POMP-related autoinflammation and immune dysregulation disease (PRAID). Both CANDLE and PRAID syndrome result from defects in proteasome assembly leading to the accumulation of ubiquinated proteins triggering cellular stress and the type 1 interferon (IFN) response (Figure 1B) (28, 29). In addition to the UPR stress-mediated inflammatory response, reactive oxygen species (ROS) accumulation has been observed in cultured SIFD patients' fibroblasts, and elevated ROS levels can activate other inflammatory pathways such as the NLRP3 inflammasome and type I interferon response (21, 30). Furthermore, some clinical manifestations of SIFD resemble patients with mitochondrial disorders and patient-derived fibroblasts showed decrease in cellular respiration and oxidative phosphorylation (31).
Thus, far, it has not been investigated why hypomorphic mutations in TRNT1 cause severe defects in differentiation of hematopoietic cells, but it may have to do with tissue-specific regulatory functions or the specific metabolic demands of immune cells. Similar tissue specific clinical manifestations have been identified in genetic syndromes with loss of tRNA synthetases, enzymes responsible for tRNA conjugation to specific amino acids, despite their essential function across all tissues (32).
Most patients are treated symptomatically with blood transfusions, IgG replacement therapy, and corticosteroids, while the disease morbidity and mortality remains high. Necessity for blood transfusions for anemia is often increased during fevers. Anti-TNF therapy suppresses inflammation reducing the need for blood transfusions, and improving growth, although it is not clear if non-immune functions improve under these therapies (21). Molecular diagnosis in early life is crucial to prevent some of the severe disease consequences. Hematopoietic stem cell transplantation (HSCT) has been attempted and has helped with hematological features, however one patient died of transplant-related complications (17).
Second Messenger Mediated Diseases
LPIN2-Deficiency
LPIN2 is a member of the lipin family of enzyme, which function in glycerolipid biosynthesis (33, 34). There are 2 other lipin family member enzymes, LPIN1, and LPIN3. LPIN2 is a phosphatidate phosphatase (PAP) that catalyzes the conversion of phosphatidic acid to diacylglycerol (DAG), a critical byproduct necessary for the production of triacylgycerol, phophatidylcholine, and phosphatidylethanolamine (35). DAG works as a secondary lipid messenger by activating protein kinases and RAS signaling pathways among others (36). In addition to the PAP activity, LPIN1 and 2 may function as a transcriptional co-activator with peroxisome proliferator-activated receptors (PPARα) (37). LPIN1 and LPIN2 are expressed in macrophages and seem to have opposite roles in regulating immune responses (38, 39). While LPIN1 acts as a proinflammatory mediator during Toll-like receptor (TLR) signaling, LPIN2 downregulates proinflammatory signaling induced by saturated fatty acids (40). In vitro depletion of LPIN2 causes increased expression of IL-6 and TNF in macrophages. In vivo studies of LPIN2 function have also demonstrated its role as a negative regulator of the NLRP3 inflammasome and TLR4 signaling (41). LPIN2-deficient mouse bone marrow derived macrophages (BMDM) display low cholesterol levels, an increase in ATP-promoted potassium flux, and increased activity of the PTX7 receptor. Thus, reduced function of LPIN2 results in upregulation of the inflammatory pathways and leads to overproduction of IL-1β, IL-18, and TNF cytokines. Similar results were obtained in primary patient-derived macrophages. These studies established a critical link between lipid biosynthesis and inflammation.
LPIN2 loss of function in humans leads to Majeed Syndrome, a rare, recessively inherited disorder that is characterized by the triad of early-onset chronic recurrent multifocal osteomyelitis (CRMO), dyserythropoietic anemia (typically microcytic), and neutrophilic skin lesions (Figure 2A) (42, 43). While CRMO and skin inflammation can be explained by a loss of LPIN2 anti-inflammatory function, the molecular mechanism of anemia is less clear. Interestingly, LPIN1 is highly expressed in muscle, and loss of enzymatic activity leads to a recessively inherited muscle disease (44). Given the ubiquitous expression of LPIN1 and LPIN2 and the widespread importance of glycerolipids in diverse cellular functions, it is intriguing why deficiency of these enzymes would cause these very distinct, tissue-specific phenotypes. To date, 14 cases of Majeed syndrome have been reported, all in consanguineous families of Middle Eastern ancestry. As all patients carry a private homozygous mutation there is a limited number of identified pathogenic variants (Infevers). Causal variants include mostly non-sense and splice site mutations that lead to reduced protein expression. A single pathogenic missense variant, p.Ser734Leu, was shown in vitro to abolish the PAP activity of LPIN2, suggesting that this activity is important for the pathogenesis of the disease (45).
Figure 2.

Autoinflammatory diseases due to dysregulation in lipid-mediated signaling. LPIN2 (A), PLAID (B), and APLIAD (C) syndromes.
IL-1 blockade results in dramatic improvement in clinical and laboratory parameters of inflammation in Majeed syndrome, confirming that loss of LPIN2 in these patients leads to autoinflammation via enhanced IL-1β production (46).
PLCG2-Associated Diseases
Phospholipase C (PLC) enzymes have a key role in the regulation of variety of cellular functions (47). The PLC family of enzymes include PLCγ1 and PLCγ2, encoded by PLCG1 and PLCG2 genes, respectively. These enzymes catalyze the hydrolysis of phospholipids and generate secondary messengers that provide a signal between many cellular receptors with downstream intracellular pathways. While PLCG1 is ubiquitously expressed, PLCG2 is highly expressed in hematopoietic cells. PLCγ2 is activated by immune receptors such as B cell and Fc receptors and has an important role in mediating innate immune responses (48). In response to receptor stimulation, PLCγ2 catalyzes the formation of the second messengers inositol triphosphate (IP3) and DAG from phosphatidylinositol 4,5-bisphosphate (PIP2). IP3 binds to IP3 receptors on the endoplasmic reticulum, resulting in calcium release and subsequent activation of various signaling pathways including mitogen activated protein kinases (MAPK). DAG remains bound to the cell membrane where it can activate protein kinase C (PKC) and other kinases. Pathogenic mutations in PLCG2 lead to two distinct phenotypes: PLCγ2-associated antibody deficiency and immune dysregulation (PLAID) and autoinflammation, antibody deficiency, and immune dysregulation (APLAID) (Figures 2B,C).
PLAID is a dominantly inherited disease characterized by cold-induced urticarial or blistering rash with onset in infancy, variable degrees of immunodeficiency and autoimmunity, and skin granuloma formation (49). In some patients, the skin lesions get worse over time leading to tissue destruction. APLAID is also dominantly inherited and presents early in life with fever, blistering, or erythematous relapsing skin lesions often triggered by heat and sweating, arthralgia, ocular inflammation, enterocolitis, and progressive interstitial lung disease (50). To date over thirty patients with PLAID and only two patients with APLAID have been reported (51). The main distinguishing features between PLAID and APLAID are that PLAID patients have cold-induced urticaria and prominent autoimmune features as compared to APLAID patients. Features of autoimmunity were found in about 25% of patients with PLAID. B cell immunodeficiency is in the spectrum of both diseases, and manifests with recurrent bacterial respiratory and GI infections. Common laboratory findings include antibody deficiency (low serum IgG and IgM) and decreased levels of circulating CD19+ and class-switched memory B cells.
The disease pathophysiology is due to a complex combination of temperature-sensitive, cell-specific gain and loss-of-function variants in the PLCγ2 signaling pathway (52). PLAID families were found to have in-frame genomic deletions spanning the cSH2 autoinhibitory domain of PLCγ2. This domain blocks the active site of the enzyme and deletion of this region results in diminished PLCγ2-mediated signaling at physiologic temperature but enhanced signaling at sub-physiologic temperatures. Primary B and NK cells from PLAID patients have an anergic phenotype while mast cells spontaneously activate when exposed to lower temperature due to loss of the autoinhibitory domain. On the other hand, APLAID patients, who carry a gain-of-function missense mutation instead of a deletion in the same cSH2 autoinhibitory domain of PLCγ2, have very different cellular and clinical phenotypes. The gain-of-function mutation identified in APLAID patients, p.Ser707Tyr, causes temperature-independent production of IP3, intracellular Ca2+ release and ERK phosphorylation in patient-derived PBMCs (50). In addition, there is evidence for Ca-dependent activation of the NLRP3 inflammasome in primary cells (53). Collectively, these data suggest constitutive hyper activation of myeloid cells in APLAID.
PLAID and APLAID patients with noticeable immunodeficiency require immunoglobulin replacement therapy, while patients with a severe inflammatory phenotype have proven difficult to treat. Although the NLRP3 inflammasome was shown to have a role in mediating inflammation in APLAID, IL-1 inhibitors have been mostly ineffective. Similarly, other cytokine inhibitors have been trialed but were ineffective in suppressing disease activity. This observation suggests function for other yet unknown pathways in the disease pathogenesis.
Metabolic Sensors Implicated in Autoinflammatory Diseases
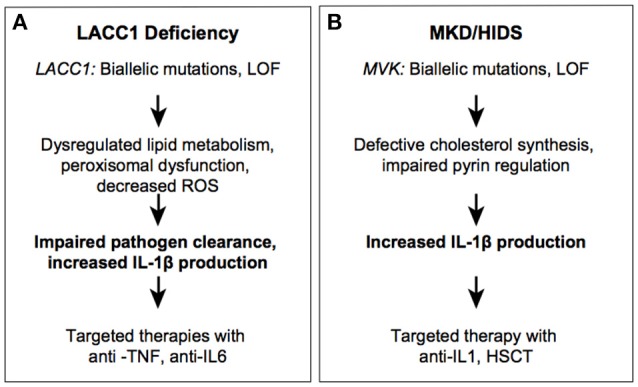
LACC1 Deficiency
LACC1 encodes for the protein FAMIN (Fatty Acid Metabolism and Immunity Nexus), which has homology to bacterial multicopper oxidoreductase enzymes (54, 55). FAMIN interacts with fatty acid synthase (FASN) and localizes to the peroxisome. FAMIN is predominantly expressed in macrophages, where it regulates fatty acid oxidation and lipogenesis to maximize metabolism (55). The exact mechanism of how FAMIN controls lipid balance is not clear but patient monocytes with different LACC1 alleles have altered laccase (phenol-oxidoreductase) suggesting loss of this enzymatic activity may be the mechanism of disease (56, 57).
The first link between LACC1 and disease was identified through an association with leprosy and Crohn's disease (CD) by genome wide association studies (GWAS) (58–61). Interestingly leprosy and Crohn's disease are both granulomatous disorders, and share the same LACC1 risk allele, the specific missense p.Ile254Val variant (rs3764147; MAF = 0.27). Later studies have demonstrated an association between a SNP (rs2121033; MAF = 0.27) downstream of LACC1 with Behcet's Disease (Figure 3A) (62, 63). It is possible that these two susceptibility alleles are in linkage disequilibrium and therefore inherited on the same haplotype. LACC1 was subsequently identified as a cause of recessively inherited monogenic CD, based upon a single consanguineous Saudi Arabian family with multiple members presenting with early-onset disease (64). The novel p.Cys284Arg variant segregated with the disease phenotype in this large family with four affected and seven unaffected members. This mutation is predicted to be detrimental for protein stability and activity, although this has not been confirmed experimentally. LACC1 was further implicated in monogenic autoinflammatory disease through identification of families with recessively inherited systemic juvenile idiopathic arthritis (SoJIA) (65). The same mutation, p.Cys284Arg, identified in the family with CD, was identified in 13 children affected with juvenile idiopathic arthritis from five consanguineous Saudi Arabian families. All patients had symmetrical polyarthritis, the characteristic quotidian fevers, evanescent rash and were homozygous for the missense mutation p.Cys284Arg (65). Interestingly, none of the patients with inflammatory arthritis had CD or ulcerative colitis. Thus, the same population-specific variant has been linked to distinct inflammatory phenotypes. Two recent studies reported 19 additional patients with primarily juvenile idiopathic arthritis, all from Middle Eastern consanguineous families, with distinct genotypes including p.M1I, p.R414*, p.I330del, p.I254V, p.Cy370Tyrfs*6, and p. T276fs*2 in LACC1 (66, 67). Some of these mutations have been shown to result in decreased protein expression, confirming that the disease is caused by loss of function of FAMIN.
Figure 3.
Autoinflammatory diseases due to impaired metabolic sensors. LACC1 (A) and MVK (B) syndromes.
Effective treatment modalities reported include TNF blockade with adalimumab or IL-6 inhibition with tocilizumab (66). Given the fact that LACC1/FAMIN controls reactive oxygen species production in macrophages, loss of this enzyme was predicted to result in decreased IL-1β production. However, knockout and knock-in disease mutation murine models show increased IL-1β, likely due to compromised pathogen clearance (55). Similar findings were made in patient-derived cells with a p.I254V homozygous variant, which revealed decreased ROS production and decreased NOD2-dependent bacterial clearance (56). The central function of LACC1 in granulomatous disease and pathogen clearance is similar to that of NOD2, a key pathogen recognition receptor mutated in a Mendelian granulomatous disease, Blau Syndrome, and polygenic CD. Because the same LACC1 mutations, p.Ile254Val and p.Cys284Arg, have been associated with multiple phenotypes including, leprosy, BD, SoJIA and early-onset CD, it is likely that environmental factors, such as microbiota, play an important role in disease expressivity and manifestations. The study of phenotypes associated with mutations in NOD2 and LACC1 implies that environmental factors may be important in the pathogenesis of more common granulomatous diseases including sarcoidosis (68). The critical position of FAMIN in cellular energy homeostasis reveals unique regulatory functions that exist in phagocytes to ensure a balance of energy sources available for clearance of pathogens.
Mevalonate Kinase-Associated Diseases
Another example of dysregulated immunometabolism is mevalonate kinase deficiency/hyper IgD syndrome (MKD/HIDS). Mevalonate kinase (MVK) is a key enzyme in the biosynthesis of cholesterol and isoprenoids, catalyzing the conversion of mevalonic acid to mevalonate-5-phosphate. Through this role in cholesterol synthesis, MVK regulates levels of geranylgeranyl pyrophosphate, which is important for prenylation and regulation of the small GTPases. RhoGTPases inhibit the pyrin inflammasome by activating protein kinase N enzymes (PKNs), which suppress pyrin function (9). Protein geranylgeranylation is also critical for TLR-induced activation of phosphatidylinositol-3-OH kinase (PI(3)K) through the interaction between the small GTPase KRAS and the PI(3)K catalytic subunit p110delta. Compromised (PI(3)K) activity results in constitutive activation of pyrin (69). Thus, loss of MVK activity leads to reduced prenylation of GTPases, which in turn results in increased activity of the pyrin inflammasome and excessive production of IL-1β.
Recessive loss of function mutations in MVK can result in a spectrum of disease ranging from severe multisystem disease (mevalonic aciduria; MKD) to a milder autoinflammatory disease (HIDS) (Figure 3B) (70). Patients with MKD manifest dysmorphic features, psychomotor retardation, progressive cerebellar ataxia, and systemic inflammation. Patients with HIDS have episodes of high fever, rash, abdominal pain, aphthous ulcers, pharyngitis, swollen lymph nodes, and arthralgia/arthritis. These symptoms are often triggered by immunization or other stresses. Cells from patients with HIDS still have residual MVK enzymatic activity (about 1–10% of the activity found in healthy control cells), whereas patients with MKD have complete deficiency in enzyme activity (71, 72). Most severe MKD-associated mutations create truncated proteins, whereas HIDS-associated mutations are missense substitutions and are thought to impair MVK stability (73, 74). The inflammatory phenotype in patients with HIDS is ameliorated with anti-IL-1 therapy consistent with the finding that MVK-deficient PBMCs secrete higher levels of IL-1β (75–77).
Post-translation Modifying Enzymes in Autoinflammatory Diseases
Several recently described autoinflammatory disorders highlight the role of post-translational modifications (PTMs), in the regulation of TNF and IL-1β signaling pathways and NF-κB activation (10, 78). Ubiquitination is the covalent attachment of an evolutionarily conserved 76-amino acid ubiquitin (Ub) protein to target substrates in the form of a monomer or polymers (ubiquitin chains; Ub chains). Ubiquitin chains can be conjugated at different lysine residues (K6, K11, K29, K33, K48, K63) along with the amino terminal methionine (M1), which can determine the fate of the modified protein. Proteins conjugated with Lys48 (K48) Ub chains are targeted for degradation via the ubiquitin-proteasome system (UPS), while Lys63 (K63) linked and linear Ub chains have essential roles in promoting signaling cascades. Ubiquitination can be reversed by a class of enzymes known as deubiqutylases or deubiquitinases (DUBs). There are more than 100 known DUBs expressed in various cells and with different degrees of specificity for Ub chains. Several DUBs are highly expressed in hematopoietic cells where they function as negative regulators of NF-kB signaling (A20, OTULIN, CYLD, and Cezanne). Mutations in genes encoding some of these DUB enzymes, along with other PTM enzymes such as kinases, have been recently implicated in autoinflammatory disorders.
Haploinsufficiency of TNFAIP3/A20 (HA20)
TNFAIP3/A20 has two enzymatic activities that synergize to restrict inflammatory responses: it has deubiquitinase activity by which it causes hydrolysis of K63 Ub linkages in receptor signaling complexes, and it has E3 Ub ligase activity through which substrates are modified with K48 Ub chains to target them for proteasomal degradation (79). TNFAIP3/A20 is a 790-residue protein that consists of an amino-terminal ovarian tumor domain (OTU) followed by 7 zinc finger domains (ZFs). The ZnF4 domain is essential for A20 E3 ligase activity and dimerization. The protein has a critical role as negative regulator of canonical NF-κB signaling. A20 null mice exhibit multi-organ inflammation, cachexia, and early lethality, while conditional A20 knockout in B cells, T cells and/or epithelial cells alone do not lead to spontaneous inflammation suggesting that the inflammatory phenotype is specific to myeloid cells (80).
Heterozygous loss-of-function mutations in TNFAIP3/A20 are associated with an autoinflammatory disease named haploinsufficiency of A20 (HA20) (Figure 4A) (81). Most patients with HA20 present with an early-onset autoimmune/autoinflammatory phenotype, with a variety of organ-specific features that are analogous to diseases such as Behcet's disease, systemic lupus erythematosus, Hashimoto's thyroiditis, autoimmune lymphoproliferative syndrome, and Crohn's disease. Many patients present with oral and genital ulcers, which are uncommon symptoms in other monogenic autoinflammatory diseases (82–86). There is substantial variability in expressivity of disease even among patients with the same genotype. Recently, large deletions on chromosome 6 encompassing up to 55 genes including TNFAIP3, were identified in patients with systemic inflammation, psychomotor and growth delay, and situs inversus (87, 88). The heterotaxy (abnormal organ arrangement) was likely the consequence of the deleted CITED2 gene (89, 90). Thus, anti-inflammatory therapy should be considered in patients with a complex phenotype and heterozygous genomic deletions on Chr. 6q23-q24.
Figure 4.

Autoinflammatory diseases due to dysregulation in post-translational modification. HA20 (A), Otulipenia (B), and RIPK1 deficiency (C) syndromes.
HA20-associated mutations are mostly located within the OTU domain and lead to truncated proteins of different length, with some partial protein products stably expressed in patient cells (91). Whether these truncated protein affect signaling remains unclear, although a dominant negative mode of action for these byproducts could mechanistically explain the phenotype. In addition to germline mutations in TNFAIP3, a low-frequency somatic mutation was reported in two unrelated Japanese families, which is of particular concern for genetic counseling and disease reoccurrence risk stratification (84). Irrespective of mutation type and position, patients with active disease have high serum levels of many proinflammatory cytokines such as IL-1β, IL-6, IL-9, IL-17, IL-18, TNF, suggesting constitutive activation of canonical NF-κB and NLRP3 inflammasome pathways (81, 84). Therapies with cytokine inhibitors, such as anti-TNF, anti-IL-1, and anti-IL-6 have been successfully employed in controlling disease activity. HSCT was curative in one reported patient (92).
Otulipenia/Otulin-Related Autoinflammatory Syndrome (ORAS)
OTULIN is a highly conserved deubiquitinase that hydrolyzes Met1-linked (linear) Ub chains from conjugated substrates. OTULIN has a critical role in regulation of angiogenesis and is required for craniofacial and neuronal development by regulating canonical Wnt signaling (93). OTULIN also functions as a negative regulator of the canonical NF-kB pathway by deubiquitinating the linear ubiquitination chain assembly complex (LUBAC) (94). LUBAC, which consists of HOIP, HOIL1, and SHARPIN, catalyzes linear ubiquitination and is essential for NF-κB signaling (95). Conditional knockout of Otulin in immune lineages results in a viable but severe inflammatory phenotype in mice with a more severe phenotype in the KO myeloid lineage than lymphoid lineage (96).
Recessively inherited, loss of function mutations in the OTU domain of OTULIN are associated with the early-onset severe inflammatory disease (Figure 4B) (96, 97). Patients present with failure to thrive, recurrent fevers, rash, joint swelling, and gastrointestinal inflammation. The cutaneous manifestations include painful erythematous rashes, subcutaneous skin nodules, and lipodystrophy. In one highly inflamed patient, skin biopsy showed neutrophilic dermatitis and panniculitis. To date, three mutations (2 missense and 1 frameshift) in five individuals from Middle Eastern, consanguineous families, have been reported. The phenotypic characteristics of OTULIN deficiency will expand with increased awareness and identification of new mutations. Pathogenic mutations affect the catalytic activity of OTULIN thereby impairing removal of linear ubiquitination; mutant cells accumulate linear Ub chains on various substrates such as IKKγ/NEMO, RIPK1, and ASC (97). Increased linear ubiquitination of IKKγ and RIPK1 results in constitutive activation of the canonical NF-kB pathway. Patient-derived immune cells produce higher levels of many proinflammatory cytokines that are secreted both by myeloid cells and T cells, such as IL-17 and IFNγ. Therapy with TNF inhibitors has been very effective in suppressing systemic inflammation and in improving growth and development. Hematopoietic stem cell transplantation (HSCT) may also hypothetically rescue the hematological phenotype although it has not been attempted due to the favorable response to treatment with TNF-inhibitors.
Receptor Interacting Protein Kinase 1 (RIPK1)-Associated Immunodeficiency and Autoinflammation
Receptor Interacting Protein Kinase 1 (RIPK1) is a widely expressed serine/threonine kinase that regulates TNFR (TNF receptor) signaling and cell death pathways (98). RIPK1 plays an important role in the regulation of immune responses through its dual functions either in activating NF-κB signaling or initiating cell death programs. RIPK1 serves as a scaffold in plasma membrane-associated protein complexes that transmit signals from TNFR1, TLR2, and TLR4 cell surface receptors. Stimulation of these receptors activates the canonical NF-κB pathway, MAPK, and pro-survival genes. Cytoplasmic RIPK1 kinase activity is important for initiation of cell death signaling pathways, apoptosis and necroptosis (99, 100). RIPK1 kinase activity acts primarily in initiating necroptosis, a caspase-independent form of cell death, through auto-phosphorylation, which triggers a protein interaction cascade. During necroptosis, the plasma membrane permeabilizes, releasing damage associated molecular patterns (DAMPs) that can in turn lead to a prolonged immune response. RIPK1 has been extensively studied in murine models, with knockout mice displaying postnatal lethality due to systemic inflammation and increased cell death (101, 102). Inhibition of RIPK1 kinase activity using necroptosis inhibitors, necrostatin-1 and its analogs, can ameliorate a variety of mouse models of disease, including septic shock, myocardial infarction, and amyotrophic lateral sclerosis, along with inflammatory phenotypes such as rheumatoid arthritis and ulcerative colitis implicating necroptosis as a major contributor in disease pathogenesis (103).
Recently, four patients with severe immunodeficiency, gut inflammation, and progressive polyarthritis were identified with RIPK1 homozygous loss of function alleles (Figure 4C) (104). The disease causing variants are small nucleotide deletions in the N-terminal kinase domain that lead to frameshift and loss of a substantial portion of the protein. The autoinflammatory phenotypic features consists of a GI inflammation with variable onset and severity, which suggests that environmental factors may modulate the disease expressivity. The four described patients suffered from recurrent infections, mostly viral but some bacterial and fungal infection. Immunophenotyping revealed lymphopenia, most prominently of CD4+ T cells, with one patient demonstrating low antibody titers and another with low serum immunoglobulins (IgG, IgM, IgA). RIPK1-deficient patient fibroblasts stimulated with TNF exhibit impaired MAPK activation, reduced cytokine production and decreased cell viability consistent with the immunodeficient phenotype. Similar to fibroblasts, LPS-stimulated patient monocytes showed decreased production of proinflammatory cytokines including IL-6, IL-12, and TNF however, they secreted high levels of IL-1β. Cell death in cultured fibroblasts was predominantly mediated by necroptosis as apoptosis inhibitors were unable to reduce cell death. Although the mechanism of inflammation in humans, like mice, is due to increased cell death via necroptosis, the absence of immunodeficiency in Ripk1 −/− mice and the viability of null alleles in humans, hints at a unique role for necroptosis specific to human immune cells.
This disease phenotype is fairly severe and with high mortality. As IL-1β likely contributes to both IBD and arthritis in these patients, anti-IL-1 therapy may be considered to suppress the inflammatory component of the disease although none of the reported patients received targeted IL-1 inhibitors. Three of the patients with RIPK1 deficiency underwent HSCT, two died shortly after transplant due to infectious causes.
Nucleic Acid Regulation in Autoinflammatory Disease
Adenosine Deaminase 2 Gene (ADA2) Deficiency
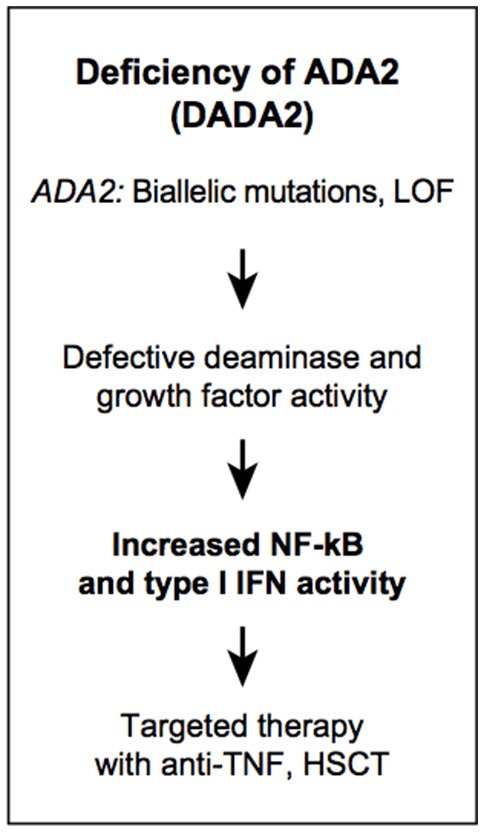
Adenosine deaminase (ADA) is an aminohydrolase that regulates purine metabolism and adenosine homeostasis by catalyzing the deamination of adenosine (Ado) and 2′-deoxyadenosine (dAdo) into inosine and deoxyinosine, respectively. Extracellular levels of adenosine increase during hypoxia and tissue injury and can lead to persistent inflammation through activation of adenosine receptors (AdoR) (105). In humans, there are two isoenzymes encoded by different genes, ADA1 (also known as ADO) and ADA2 (also known as CECR1) that have different enzymatic properties. ADA2 has a 100-fold higher Michaelis-Menton constant for adenosine (Km = 2 mM) than ADA1 and is not essential for intracellular deaminase activity. ADA2 is a secreted protein and under physiological conditions is present in low levels in plasma. In addition, ADA2 shares homology with adenosine deaminase growth factors (ADGFs) that have been shown to play a role in development (106). ADA1 and 2 are both highly expressed in immune cells and are critical for the development of the immune system. ADA1 is predominantly expressed in lymphocytes, while ADA2 is secreted by activated myeloid cells (107).
The first human disease linked to a defect in ADA1 function was severe combined immunodeficiency. A complete deficiency of ADA1 is fatal early in life, while patients with partial ADA1 deficiency present with milder clinical symptoms, later in childhood. In the absence of ADA1, deoxyadenosine nucleotides accumulate in lymphocytes and are toxic resulting in T-B-NK- SCID (108). Deficiency of adenosine deaminase type 2 (DADA2) is a recessively inherited disease manifesting with fevers, vasculitis, livedo racemosa, early-onset ischemic stroke, liver disease, and mild immunodeficiency (Figure 5) (109, 110). The vasculopathy/vasculitis associated with DADA2 predominantly affects small- and medium-sized arteries with histologic findings consistent with necrotizing ANCA-negative vasculitis or polyarteritis nodosa (109, 111). Livedo racemosa is the most common skin manifestation, and it can be complicated by skin ulcerations, distal arterial occlusion, and sometimes digital necrosis (112). Neurological manifestations result from lacunar ischemic infarcts in the deep-brain nuclei, midbrain, and/or brainstem. Some patients were reported with intracerebral hemorrhage as the first disease manifestation (111, 113). Hematological manifestations were not appreciated initially, however it has become evident that some patients present with hypocellular bone marrow (114–116). Their clinical presentations include aplastic anemia, pure red cell aplasia, lymphopenia, neutropenia, thrombocytopenia, and hypogammaglobilinemia, in particular low IgG and IgM levels. Immunodeficiency is generally milder than in patients with ADA1 deficiency, and resembles common variable immunodeficiency (115). Lymphoproliferative features are also in the spectrum of DADA2 hematopoietic manifestations (117, 118). Autoimmune findings in DADA2 have been found in a small subset of patients, and manifest as autoimmune cytopenias, transiently positive lupus anticoagulant, and systemic lupus. This is consistent with increased type 1 interferon (IFN) gene expression signature in blood samples of patients with DADA2 (119, 120).
Figure 5.
Autoinflammatory diseases due to dysregulation in purine metabolism. DADA2.
To date, there are more than 170 patients diagnosed with DADA2 described from all over the world (121–123). Many DADA2-associated mutations are present at a low frequency in the general population and it is possible that the disease is still underdiagnosed. In addition to standard genetic testing by sequencing, DADA2 can be diagnosed by measuring plasma or serum ADA2 activity. ADA2 activity testing is recommended to confirm the pathogenicity of novel variants identified by sequencing. The molecular mechanisms by which mutations in ADA2 lead to disease are still largely unknown. Proinflammatory cytokines have been identified in skin biopsies and blood samples of DADA2 patients however, the function of ADA2 in differentiation of hematopoietic and endothelial cells has not been comprehensively investigated. Deficiency of ADA2 is associated with monocyte-macrophage polarization toward the M1 subset, and M1 macrophages are known to promote inflammation, although it is not clear if this is directly dependent on adenosine, aminohydrolase catalytic activity on a novel substrate, growth factor function, or another as yet unidentified mechanism.
Primary treatment of DADA2 relies on anti-TNF agents in patients with a mostly inflammatory phenotype, while patients with bone marrow failure may require HSCT (124, 125). Identification of patients with ADA1-SCID and DADA2 has helped identify unexpected roles for adenosine as a key molecule in the regulation of immune physiology.
Interferonopathies
Interferonopathies are a group of systemic inflammatory diseases with continuum of features of autoinflammation and autoimmunity. The underlying mechanism of inflammation in monogenic interferonopathies is a defect in sensing and degradation of nucleic acids leading to constitutive activation of innate immune responses (126, 127). These disorders show a striking resemblance to congenital viral infections although occur in the absence of any identified pathogen.
Two of the best studied diseases are Aicardi-Goutières syndrome (AGS) and stimulator of interferon genes (STING)-associated vasculopathy with onset in infancy (SAVI). AGS is a heterogenous early-onset disorder manifesting with basal ganglia calcifications, encephalopathy, neurological impairments and other features of autoimmunity. Disease-associated mutations have been identified in genes that either impact nucleic acid degradation (TREX1, SAMHD1, RNASEH2A, RNASEH2B, RNASEH2C, DNASE2), or sensing of nucleic acids (IFIH1/MDA5) or RNA editing (ADAR) and result in activation of the type I interferon pathway. Patients with SAVI have gain-of-function mutations in TMEM173, which encodes the STING protein, and they present with prominent skin lesions, small vessel vasculitis, peripheral amputations, and interstitial lung disease. STING is an endoplasmic transmembrane protein that functions as an indirect sensor of endogenous or pathogen-derived cytosolic dsDNA (128). Upon binding to cGAMP, which is generated by the sensing receptor cGAS, STING activates IRF3, a transcription factor for type 1 interferon and related genes. Production of type 1 interferon cytokines results in an amplification loop that can be blocked by JAK inhibitors providing further insights into the mechanism of pathogenesis (129). Type I interferonopathies mechanisms, disease manifestations, and treatment have been reviewed in great detail elsewhere (127, 130).
Conclusions
The disorders discussed in this review exemplify how unbiased identification of mutated genes in patients with rare autoinflammatory diseases has uncovered novel mechanisms governing innate immune responses. Collectively, these findings point to previously unrecognized functions for these genes and their associated pathways. These atypical autoinflammatory genes not only help identify unique pathways regulating inflammatory pathways, but also illuminate shared features with other disorders based on treatment response and clinical manifestations (Figure 6). It is unclear whether these genes have a specific function in immune system regulation or if the relatively specific immune phenotypes are due to particular sensitivity of the immune system to disruption in basic cell machinery and metabolism. Furthermore, unlike disruption of canonical autoinflammatory pathways, mutations in many of these genes can result in concomitant overactivity of the innate immune system (autoinflammation) and of the adaptive immune system (autoimmunity) and be accompanied with ineffective immune responses (immunodeficiency). This provides evidence for cell-specific function of these enzymes in the regulation of metabolic and immune signaling pathways. Besides targeted anti-cytokine therapies, hematopoietic stem cell transplantation has been attempted in some patients refractory to conventional treatments, and can cure hematological and immunological manifestations. Like with other metabolic disorders, early diagnosis and treatment is critical for preventing disease complications. Unbiased genetic sequencing of patients with early-onset immune dysregulation disorders will continue to identify novel disease-causing genes and further delineate the molecular mechanisms governing inflammation in humans.
Figure 6.

Overlapping and unique features of autoinflammatory diseases. Outer pentagon shows pathways altered in autoinflammatory disease, central pentagon highlights genes in the corresponding node found mutated in monogenic disease (dark blue), and inner pentagon showing shared clinical features and treatment (light blue).
Web Link
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs. Hirotsugu Oda and Kalpana Manthiram for critical reading and suggestions in the preparation of the manuscript.
References
- 1.De Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol. (2015) 33:823–74. 10.1146/annurev-immunol-032414-112227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol. (2017) 18:832–42. 10.1038/ni.3777 [DOI] [PubMed] [Google Scholar]
- 3.Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. (2018) 38:129–43. 10.1007/s10875-017-0465-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, Van Royen-Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. (2009) 360:2426–37. 10.1056/NEJMoa0807865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei XY, Fraitag S, et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med. (2011) 365:620–8. 10.1056/NEJMoa1013068 [DOI] [PubMed] [Google Scholar]
- 6.Cowen EW, Goldbach-Mansky R. DIRA, DITRA, and new insights into pathways of skin inflammation: what's in a name? Arch Dermatol. (2012) 148:381–4. 10.1001/archdermatol.2011.3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffman HM, Broderick L. The role of the inflammasome in patients with autoinflammatory diseases. J Allergy Clin Immunol. (2016) 138:3–14. 10.1016/j.jaci.2016.05.001 [DOI] [PubMed] [Google Scholar]
- 8.Masters SL, Lagou V, Jeru I, Baker PJ, Van Eyck L, Parry DA, et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci Transl Med. (2016) 8:332ra345. 10.1126/scitranslmed.aaf1471 [DOI] [PubMed] [Google Scholar]
- 9.Park YH, Wood G, Kastner DL, Chae JJ. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol. (2016) 17:914–21. 10.1038/ni.3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aksentijevich I, Zhou Q. NF-kappaB pathway in autoinflammatory diseases: dysregulation of protein modifications by ubiquitin defines a new category of autoinflammatory diseases. Front Immunol. (2017) 8:399. 10.3389/fimmu.2017.00399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oda H, Kastner DL. Genomics, biology, and human illness: advances in the monogenic autoinflammatory diseases. Rheum Dis Clin North Am. (2017) 43:327–45. 10.1016/j.rdc.2017.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davidson S, Steiner A, Harapas CR, Masters SL. An update on autoinflammatory diseases: interferonopathies. Curr Rheumatol Rep. (2018) 20:38. 10.1007/s11926-018-0748-y [DOI] [PubMed] [Google Scholar]
- 13.Harapas CR, Steiner A, Davidson S, Masters SL. An update on autoinflammatory diseases: inflammasomopathies. Curr Rheumatol Rep. (2018) 20:40. 10.1007/s11926-018-0750-4 [DOI] [PubMed] [Google Scholar]
- 14.Wellner K, Betat H, Morl M. A tRNA's fate is decided at its 3' end: collaborative actions of CCA-adding enzyme and RNases involved in tRNA processing and degradation. Biochim Biophys Acta Gene Regul Mech. (2018) 1861:433–41. 10.1016/j.bbagrm.2018.01.012 [DOI] [PubMed] [Google Scholar]
- 15.Wilusz JE, Whipple JM, Phizicky EM, Sharp PA. tRNAs marked with CCACCA are targeted for degradation. Science (2011) 334:817–21. 10.1126/science.1213671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Augustin MA, Reichert AS, Betat H, Huber R, Morl M, Steegborn C. Crystal structure of the human CCA-adding enzyme: insights into template-independent polymerization. J Mol Biol. (2003) 328:985–94. 10.1016/S0022-2836(03)00381-4 [DOI] [PubMed] [Google Scholar]
- 17.Wiseman DH, May A, Jolles S, Connor P, Powell C, Heeney MM, et al. A novel syndrome of congenital sideroblastic anemia, B-cell immunodeficiency, periodic fevers, and developmental delay (SIFD). Blood (2013) 122:112–23. 10.1182/blood-2012-08-439083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakraborty PK, Schmitz-Abe K, Kennedy EK, Mamady H, Naas T, Durie D, et al. Mutations in TRNT1 cause congenital sideroblastic anemia with immunodeficiency, fevers, and developmental delay (SIFD). Blood (2014) 124:2867–71. 10.1182/blood-2014-08-591370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wedatilake Y, Niazi R, Fassone E, Powell CA, Pearce S, Plagnol V, et al. TRNT1 deficiency: clinical, biochemical and molecular genetic features. Orphanet J Rare Dis. (2016) 11:90. 10.1186/s13023-016-0477-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barton C, Kausar S, Kerr D, Bitetti S, Wynn R. SIFD as a novel cause of severe fetal hydrops and neonatal anaemia with iron loading and marked extramedullary haemopoiesis. J Clin Pathol. (2018) 71:275–8. 10.1136/jclinpath-2017-204698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giannelou A, Wang H, Zhou Q, Park YH, Abu-Asab MS, Ylaya K, et al. Aberrant tRNA processing causes an autoinflammatory syndrome responsive to TNF inhibitors. Ann Rheum Dis. (2018) 77:612–9. 10.1136/annrheumdis-2017-212401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lougaris V, Chou J, Baronio M, Gazzurelli L, Lorenzini T, Soresina A, et al. Novel biallelic TRNT1 mutations resulting in sideroblastic anemia, combined B and T cell defects, hypogammaglobulinemia, recurrent infections, hypertrophic cardiomyopathy and developmental delay. Clin Immunol. (2018) 188:20–2. 10.1016/j.clim.2017.11.008 [DOI] [PubMed] [Google Scholar]
- 23.Frans G, Moens L, Schaballie H, Wuyts G, Liston A, Poesen K, et al. Homozygous N-terminal missense mutation in TRNT1 leads to progressive B-cell immunodeficiency in adulthood. J Allergy Clin Immunol. (2017) 139:360–3 e366. 10.1016/j.jaci.2016.06.050 [DOI] [PubMed] [Google Scholar]
- 24.Deluca AP, Whitmore SS, Barnes J, Sharma TP, Westfall TA, Scott CA, et al. Hypomorphic mutations in TRNT1 cause retinitis pigmentosa with erythrocytic microcytosis. Hum Mol Genet. (2016) 25:44–56. 10.1093/hmg/ddv446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hull S, Malik AN, Arno G, Mackay DS, Plagnol V, Michaelides M, et al. Expanding the phenotype of TRNT1-related immunodeficiency to include childhood cataract and inner retinal dysfunction. JAMA Ophthalmol. (2016) 134:1049–53. 10.1001/jamaophthalmol.2015.5833 [DOI] [PubMed] [Google Scholar]
- 26.Leibovitch M, Hanic-Joyce PJ, Joyce PBM. In vitro studies of disease-linked variants of human tRNA nucleotidyltransferase reveal decreased thermal stability and altered catalytic activity. Biochim Biophys Acta Proteins Proteom. (2018) 1866:527–40. 10.1016/j.bbapap.2018.02.002 [DOI] [PubMed] [Google Scholar]
- 27.Sasarman F, Thiffault I, Weraarpachai W, Salomon S, Maftei C, Gauthier J, et al. The 3' addition of CCA to mitochondrial tRNASer(AGY) is specifically impaired in patients with mutations in the tRNA nucleotidyl transferase TRNT1. Hum Mol Genet. (2015) 24:2841–7. 10.1093/hmg/ddv044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Torrelo A. CANDLE syndrome as a paradigm of proteasome-related autoinflammation. Front Immunol. (2017) 8:927. 10.3389/fimmu.2017.00927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poli MC, Ebstein F, Nicholas SK, De Guzman MM, Forbes LR, Chinn IK, et al. Heterozygous truncating variants in POMP escape nonsense-mediated decay and cause a unique immune dysregulatory syndrome. Am J Hum Genet. (2018) 102:1126–42. 10.1016/j.ajhg.2018.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fremond ML, Melki I, Kracker S, Bondet V, Duffy D, Rice GI, et al. Comment on: ‘Aberrant tRNA processing causes an autoinflammatory syndrome responsive to TNF inhibitors’ by Giannelou et al: mutations in TRNT1 result in a constitutive activation of type I interferon signalling. Ann Rheum Dis. (2018). 10.1136/annrheumdis-2018-213745. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 31.Liwak-Muir U, Mamady H, Naas T, Wylie Q, Mcbride S, Lines M, et al. Impaired activity of CCA-adding enzyme TRNT1 impacts OXPHOS complexes and cellular respiration in SIFD patient-derived fibroblasts. Orphanet J Rare Dis. (2016) 11:79. 10.1186/s13023-016-0466-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyer-Schuman R, Antonellis A. Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease. Hum Mol Genet. (2017) 26:R114–27. 10.1093/hmg/ddx231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peterfy M, Phan J, Xu P, Reue K. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat Genet. (2001) 27:121–4. 10.1038/83685 [DOI] [PubMed] [Google Scholar]
- 34.Reue K. The lipin family: mutations and metabolism. Curr Opin Lipidol. (2009) 20:165–70. 10.1097/MOL.0b013e32832adee5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donkor J, Sariahmetoglu M, Dewald J, Brindley DN, Reue K. Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. J Biol Chem. (2007) 282:3450–7. 10.1074/jbc.M610745200 [DOI] [PubMed] [Google Scholar]
- 36.Carrasco S, Merida I. Diacylglycerol, when simplicity becomes complex. Trends Biochem Sci. (2007) 32:27–36. 10.1016/j.tibs.2006.11.004 [DOI] [PubMed] [Google Scholar]
- 37.Zhang P, Reue K. Lipin proteins and glycerolipid metabolism: roles at the ER membrane and beyond. Biochim Biophys Acta Biomembr. (2017) 1859(9 Pt B):1583–95. 10.1016/j.bbamem.2017.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valdearcos M, Esquinas E, Meana C, Gil-De-Gomez L, Guijas C, Balsinde J, et al. Subcellular localization and role of lipin-1 in human macrophages. J Immunol. (2011) 186:6004–13. 10.4049/jimmunol.1003279 [DOI] [PubMed] [Google Scholar]
- 39.Meana C, Pena L, Lorden G, Esquinas E, Guijas C, Valdearcos M, et al. Lipin-1 integrates lipid synthesis with proinflammatory responses during TLR activation in macrophages. J Immunol. (2014) 193:4614–22. 10.4049/jimmunol.1400238 [DOI] [PubMed] [Google Scholar]
- 40.Valdearcos M, Esquinas E, Meana C, Pena L, Gil-De-Gomez L, Balsinde J, et al. Lipin-2 reduces proinflammatory signaling induced by saturated fatty acids in macrophages. J Biol Chem. (2012) 287:10894–904. 10.1074/jbc.M112.342915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lorden G, Sanjuan-Garcia I, De Pablo N, Meana C, Alvarez-Miguel I, Perez-Garcia MT, et al. Lipin-2 regulates NLRP3 inflammasome by affecting P2X7 receptor activation. J Exp Med. (2017) 214:511–28. 10.1084/jem.20161452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferguson PJ, Chen S, Tayeh MK, Ochoa L, Leal SM, Pelet A, et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). J Med Genet. (2005) 42:551–7. 10.1136/jmg.2005.030759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cox AJ, Ferguson PJ. Update on the genetics of nonbacterial osteomyelitis in humans. Curr Opin Rheumatol. (2018) 30:521–5. 10.1097/BOR.0000000000000530 [DOI] [PubMed] [Google Scholar]
- 44.Zeharia A, Shaag A, Houtkooper RH, Hindi T, De Lonlay P, Erez G, et al. Mutations in LPIN1 cause recurrent acute myoglobinuria in childhood. Am J Hum Genet. (2008) 83:489–94. 10.1016/j.ajhg.2008.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Donkor J, Zhang P, Wong S, O'loughlin L, Dewald J, Kok BP, et al. A conserved serine residue is required for the phosphatidate phosphatase activity but not the transcriptional coactivator functions of lipin-1 and lipin-2. J Biol Chem. (2009) 284:29968–78. 10.1074/jbc.M109.023663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herlin T, Fiirgaard B, Bjerre M, Kerndrup G, Hasle H, Bing X, et al. Efficacy of anti-IL-1 treatment in Majeed syndrome. Ann Rheum Dis. (2013) 72:410–3. 10.1136/annrheumdis-2012-201818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koss H, Bunney TD, Behjati S, Katan M. Dysfunction of phospholipase Cgamma in immune disorders and cancer. Trends Biochem Sci. (2014) 39:603–11. 10.1016/j.tibs.2014.09.004 [DOI] [PubMed] [Google Scholar]
- 48.Gorjestani S, Yu M, Tang B, Zhang D, Wang D, Lin X. Phospholipase Cgamma2 (PLCgamma2) is key component in Dectin-2 signaling pathway, mediating anti-fungal innate immune responses. J Biol Chem. (2011) 286:43651–9. 10.1074/jbc.M111.307389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ombrello MJ, Remmers EF, Sun G, Freeman AF, Datta S, Torabi-Parizi P, et al. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med. (2012) 366:330–8. 10.1056/NEJMoa1102140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou Q, Lee GS, Brady J, Datta S, Katan M, Sheikh A, et al. A hypermorphic missense mutation in PLCG2, encoding phospholipase Cgamma2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet. (2012) 91:713–20. 10.1016/j.ajhg.2012.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aderibigbe OM, Priel DL, Lee CC, Ombrello MJ, Prajapati VH, Liang MG, et al. Distinct cutaneous manifestations and cold-induced leukocyte activation associated with PLCG2 mutations. JAMA Dermatol. (2015) 151:627–34. 10.1001/jamadermatol.2014.5641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Milner JD. PLAID: a syndrome of complex patterns of disease and unique phenotypes. J Clin Immunol. (2015) 35:527–30. 10.1007/s10875-015-0177-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chae JJ, Park YH, Park C, Hwang IY, Hoffmann P, Kehrl JH, et al. Connecting two pathways through Ca 2+ signaling: NLRP3 inflammasome activation induced by a hypermorphic PLCG2 mutation. Arthritis Rheumatol. (2015) 67:563–7. 10.1002/art.38961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lisova ZA, Lisov AV, Leontievsky AA. Two laccase isoforms of the basidiomycete Cerrena unicolor VKMF-3196. Induction, isolation and properties. J Basic Microbiol. (2010) 50:72–82. 10.1002/jobm.200900382 [DOI] [PubMed] [Google Scholar]
- 55.Cader MZ, Boroviak K, Zhang Q, Assadi G, Kempster SL, Sewell GW, et al. C13orf31 (FAMIN) is a central regulator of immunometabolic function. Nat Immunol. (2016) 17:1046–56. 10.1038/ni.3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lahiri A, Hedl M, Yan J, Abraham C. Human LACC1 increases innate receptor-induced responses and a LACC1 disease-risk variant modulates these outcomes. Nat Commun. (2017) 8:15614. 10.1038/ncomms15614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Assadi G, Vesterlund L, Bonfiglio F, Mazzurana L, Cordeddu L, Schepis D, et al. Functional analyses of the Crohn's disease risk gene LACC1. PLoS ONE (2016) 11:e0168276. 10.1371/journal.pone.0168276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jostins L, Ripke S, Weersma RK, Duerr RH, Mcgovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491:119–24. 10.1038/nature11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu H, Irwanto A, Fu X, Yu G, Yu Y, Sun Y, et al. Discovery of six new susceptibility loci and analysis of pleiotropic effects in leprosy. Nat Genet. (2015) 47:267–71. 10.1038/ng.3212 [DOI] [PubMed] [Google Scholar]
- 60.Assadi G, Saleh R, Hadizadeh F, Vesterlund L, Bonfiglio F, Halfvarson J, et al. LACC1 polymorphisms in inflammatory bowel disease and juvenile idiopathic arthritis. Genes Immun. (2016) 17:261–4. 10.1038/gene.2016.17 [DOI] [PubMed] [Google Scholar]
- 61.Wang D, Fan Y, Malhi M, Bi R, Wu Y, Xu M, et al. Missense variants in HIF1A and LACC1 contribute to leprosy risk in Han Chinese. Am J Hum Genet. (2018) 102:794–805. 10.1016/j.ajhg.2018.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takeuchi M, Mizuki N, Meguro A, Ombrello MJ, Kirino Y, Satorius C, et al. Dense genotyping of immune-related loci implicates host responses to microbial exposure in Behcet's disease susceptibility. Nat Genet. (2017) 49:438–43. 10.1038/ng.3786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu P, Du L, Hou S, Su G, Yang L, Hu J, et al. Association of LACC1, CEBPB-PTPN1, RIPK2 and ADO-EGR2 with ocular Behcet's disease in a Chinese Han population. Br J Ophthalmol. (2018) 102:1308–14. 10.1136/bjophthalmol-2017-311753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patel N, El Mouzan MI, Al-Mayouf SM, Adly N, Mohamed JY, Al Mofarreh MA, et al. Study of Mendelian forms of Crohn's disease in Saudi Arabia reveals novel risk loci and alleles. Gut (2014) 63:1831–2. 10.1136/gutjnl-2014-307859 [DOI] [PubMed] [Google Scholar]
- 65.Wakil SM, Monies DM, Abouelhoda M, Al-Tassan N, Al-Dusery H, Naim EA, et al. Association of a mutation in LACC1 with a monogenic form of systemic juvenile idiopathic arthritis. Arthritis Rheumatol. (2015) 67:288–95. 10.1002/art.38877 [DOI] [PubMed] [Google Scholar]
- 66.Kallinich T, Thorwarth A, Von Stuckrad SL, Rosen-Wolff A, Luksch H, Hundsdoerfer P, et al. Juvenile arthritis caused by a novel FAMIN (LACC1) mutation in two children with systemic and extended oligoarticular course. Pediatr Rheumatol Online J. (2016) 14:63 10.1186/s12969-016-0124-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Karacan I, Ugurlu S, Sahin S, Everest E, Kasapcopur O, Tolun A, et al. LACC1 gene defects in familial form of juvenile arthritis. J Rheumatol. (2018) 45:726–8. 10.3899/jrheum.170834 [DOI] [PubMed] [Google Scholar]
- 68.Szymanski AM, Ombrello MJ. Using genes to triangulate the pathophysiology of granulomatous autoinflammatory disease: NOD2, PLCG2 and LACC1. Int Immunol. (2018) 30:205–13. 10.1093/intimm/dxy021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Akula MK, Shi M, Jiang Z, Foster CE, Miao D, Li AS, et al. Control of the innate immune response by the mevalonate pathway. Nat Immunol. (2016) 17:922–9. 10.1038/ni.3487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Van Der Burgh R, Ter Haar NM, Boes ML, Frenkel J. Mevalonate kinase deficiency, a metabolic autoinflammatory disease. Clin Immunol. (2013) 147:197–206. 10.1016/j.clim.2012.09.011 [DOI] [PubMed] [Google Scholar]
- 71.Stoffels M, Simon A. Hyper-IgD syndrome or mevalonate kinase deficiency. Curr Opin Rheumatol. (2011) 23:419–23. 10.1097/BOR.0b013e328349c3b1 [DOI] [PubMed] [Google Scholar]
- 72.Ter Haar NM, Jeyaratnam J, Lachmann HJ, Simon A, Brogan PA, Doglio M, et al. The phenotype and genotype of mevalonate kinase deficiency: a series of 114 cases from the eurofever registry. Arthritis Rheumatol. (2016) 68:2795–805. 10.1002/art.39763 [DOI] [PubMed] [Google Scholar]
- 73.Houten SM, Frenkel J, Rijkers GT, Wanders RJ, Kuis W, Waterham HR. Temperature dependence of mutant mevalonate kinase activity as a pathogenic factor in hyper-IgD and periodic fever syndrome. Hum Mol Genet. (2002) 11:3115–24. 10.1093/hmg/11.25.3115 [DOI] [PubMed] [Google Scholar]
- 74.Mandey SH, Schneiders MS, Koster J, Waterham HR. Mutational spectrum and genotype-phenotype correlations in mevalonate kinase deficiency. Hum Mutat. (2006) 27:796–802. 10.1002/humu.20361 [DOI] [PubMed] [Google Scholar]
- 75.Frenkel J, Rijkers GT, Mandey SH, Buurman SW, Houten SM, Wanders RJ, et al. Lack of isoprenoid products raises ex vivo interleukin-1beta secretion in hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis Rheum. (2002) 46:2794–803. 10.1002/art.10550 [DOI] [PubMed] [Google Scholar]
- 76.Galeotti C, Meinzer U, Quartier P, Rossi-Semerano L, Bader-Meunier B, Pillet P, et al. Efficacy of interleukin-1-targeting drugs in mevalonate kinase deficiency. Rheumatology (2012) 51:1855–9. 10.1093/rheumatology/kes097 [DOI] [PubMed] [Google Scholar]
- 77.De Benedetti F, Gattorno M, Anton J, Ben-Chetrit E, Frenkel J, Hoffman HM, et al. Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N Engl J Med. (2018) 378:1908–19. 10.1056/NEJMoa1706314 [DOI] [PubMed] [Google Scholar]
- 78.Steiner A, Harapas CR, Masters SL, Davidson S. An update on autoinflammatory diseases: relopathies. Curr Rheumatol Rep. (2018) 20:39. 10.1007/s11926-018-0749-x [DOI] [PubMed] [Google Scholar]
- 79.Wertz IE, O'rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature (2004) 430:694–9. 10.1038/nature02794 [DOI] [PubMed] [Google Scholar]
- 80.Das T, Chen Z, Hendriks RW, Kool M. A20/tumor necrosis factor alpha-induced protein 3 in immune cells controls development of autoinflammation and autoimmunity: lessons from mouse models. Front Immunol. (2018) 9:104. 10.3389/fimmu.2018.00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet. (2016) 48:67–73. 10.1038/ng.3459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aeschlimann FA, Batu ED, Canna SW, Go E, Gul A, Hoffmann P, et al. A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann Rheum Dis. (2018) 77:728–35. 10.1136/annrheumdis-2017-212403 [DOI] [PubMed] [Google Scholar]
- 83.Berteau F, Rouviere B, Nau A, Le Berre R, Sarrabay G, Touitou I, et al. Response to: ‘A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease’. Ann Rheum Dis. (2018). 10.1136/annrheumdis-2018-213347. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 84.Kadowaki T, Ohnishi H, Kawamoto N, Hori T, Nishimura K, Kobayashi C, et al. Haploinsufficiency of A20 causes autoinflammatory and autoimmune disorders. J Allergy Clin Immunol. (2018) 141:1485–8 e1411. 10.1016/j.jaci.2017.10.039 [DOI] [PubMed] [Google Scholar]
- 85.Lawless D, Pathak S, Scambler TE, Ouboussad L, Anwar R, Savic S. A case of adult-onset still's disease caused by a novel splicing mutation in TNFAIP3 successfully treated with tocilizumab. Front Immunol. (2018) 9:1527. 10.3389/fimmu.2018.01527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zheng C, Huang Y, Ye Z, Wang Y, Tang Z, Lu J, et al. Infantile Onset Intractable Inflammatory Bowel Disease Due to Novel Heterozygous Mutations in TNFAIP3 (A20). Inflamm Bowel Dis. (2018) 24:2613–20. 10.1093/ibd/izy165 [DOI] [PubMed] [Google Scholar]
- 87.Franco-Jarava C, Wang H, Martin-Nalda A, Alvarez SD, Garcia-Prat M, Bodet D, et al. TNFAIP3 haploinsufficiency is the cause of autoinflammatory manifestations in a patient with a deletion of 13Mb on chromosome 6. Clin Immunol. (2018) 191:44–51. 10.1016/j.clim.2018.03.009 [DOI] [PubMed] [Google Scholar]
- 88.Viel S, Cheyssac E, Pescarmona R, Besson L, Till M, Viremouneix L, et al. Large deletion in 6q associated to A20 haploinsufficiency and thoracoabdominal heterotaxy. Ann Rheum Dis. (2018) 77:1697–8. 10.1136/annrheumdis-2018-213300 [DOI] [PubMed] [Google Scholar]
- 89.Sperling S, Grimm CH, Dunkel I, Mebus S, Sperling HP, Ebner A, et al. Identification and functional analysis of CITED2 mutations in patients with congenital heart defects. Hum Mutat. (2005) 26:575–82. 10.1002/humu.20262 [DOI] [PubMed] [Google Scholar]
- 90.Lopes Floro K, Artap ST, Preis JI, Fatkin D, Chapman G, Furtado MB, et al. Loss of Cited2 causes congenital heart disease by perturbing left-right patterning of the body axis. Hum Mol Genet. (2011) 20:1097–110. 10.1093/hmg/ddq554 [DOI] [PubMed] [Google Scholar]
- 91.Takagi M, Ogata S, Ueno H, Yoshida K, Yeh T, Hoshino A, et al. Haploinsufficiency of TNFAIP3 (A20) by germline mutation is involved in autoimmune lymphoproliferative syndrome. J Allergy Clin Immunol. (2017) 139:1914–22. 10.1016/j.jaci.2016.09.038 [DOI] [PubMed] [Google Scholar]
- 92.Duncan CJA, Dinnigan E, Theobald R, Grainger A, Skelton AJ, Hussain R, et al. Early-onset autoimmune disease due to a heterozygous loss-of-function mutation in TNFAIP3 (A20). Ann Rheum Dis. (2018) 77:783–6. 10.1136/annrheumdis-2016-210944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rivkin E, Almeida SM, Ceccarelli DF, Juang YC, Maclean TA, Srikumar T, et al. The linear ubiquitin-specific deubiquitinase gumby regulates angiogenesis. Nature (2013) 498:318–24. 10.1038/nature12296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Heger K, Wickliffe KE, Ndoja A, Zhang J, Murthy A, Dugger DL, et al. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature (2018) 559:120–4. 10.1038/s41586-018-0256-2 [DOI] [PubMed] [Google Scholar]
- 95.Shimizu Y, Taraborrelli L, Walczak H. Linear ubiquitination in immunity. Immunol Rev. (2015) 266:190–207. 10.1111/imr.12309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Damgaard RB, Walker JA, Marco-Casanova P, Morgan NV, Titheradge HL, Elliott PR, et al. The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell (2016) 166:1215–30 e1220. 10.1016/j.cell.2016.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhou Q, Yu X, Demirkaya E, Deuitch N, Stone D, Tsai WL, et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc Natl Acad Sci USA. (2016) 113:10127–32. 10.1073/pnas.1612594113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.He S, Wang X. RIP kinases as modulators of inflammation and immunity. Nat Immunol. (2018) 19:912–22. 10.1038/s41590-018-0188-x [DOI] [PubMed] [Google Scholar]
- 99.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature (2015) 517:311–20. 10.1038/nature14191 [DOI] [PubMed] [Google Scholar]
- 100.Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. (2017) 18:127–36. 10.1038/nrm.2016.149 [DOI] [PubMed] [Google Scholar]
- 101.Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature (2014) 513:90–4. 10.1038/nature13608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rickard JA, O'donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell (2014) 157:1175–88. 10.1016/j.cell.2014.04.019 [DOI] [PubMed] [Google Scholar]
- 103.Newton K, Manning G. Necroptosis and inflammation. Annu Rev Biochem. (2016) 85:743–63. 10.1146/annurev-biochem-060815-014830 [DOI] [PubMed] [Google Scholar]
- 104.Cuchet-Lourenco D, Eletto D, Wu C, Plagnol V, Papapietro O, Curtis J, et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science (2018) 361:810–3. 10.1126/science.aar2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hershfield MS. New insights into adenosine-receptor-mediated immunosuppression and the role of adenosine in causing the immunodeficiency associated with adenosine deaminase deficiency. Eur J Immunol. (2005) 35:25–30. 10.1002/eji.200425738 [DOI] [PubMed] [Google Scholar]
- 106.Zavialov AV, Yu X, Spillmann D, Lauvau G, Zavialov AV. Structural basis for the growth factor activity of human adenosine deaminase ADA2. J Biol Chem. (2010) 285:12367–77. 10.1074/jbc.M109.083527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Meyts I, Aksentijevich I. Deficiency of adenosine deaminase 2 (DADA2): updates on the phenotype, genetics, pathogenesis, and treatment. J Clin Immunol. (2018) 38:569–78. 10.1007/s10875-018-0525-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mccusker C, Upton J, Warrington R. Primary immunodeficiency. Allergy Asthma Clin Immunol. (2018) 14:61. 10.1186/s13223-018-0290-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. (2014) 370:921–31. 10.1056/NEJMoa1307362 [DOI] [PubMed] [Google Scholar]
- 110.Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. (2014) 370:911–20. 10.1056/NEJMoa1307361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Garg N, Kasapcopur O, Foster J, II, Barut K, Tekin A, Kizilkilic O, et al. Novel adenosine deaminase 2 mutations in a child with a fatal vasculopathy. Eur J Pediatr. (2014) 173:827–30. 10.1007/s00431-014-2320-8 [DOI] [PubMed] [Google Scholar]
- 112.Gonzalez Santiago TM, Zavialov A, Saarela J, Seppanen M, Reed AM, Abraham RS, et al. Dermatologic features of ADA2 deficiency in cutaneous polyarteritis nodosa. JAMA Dermatol. (2015) 151:1230–4. 10.1001/jamadermatol.2015.1635 [DOI] [PubMed] [Google Scholar]
- 113.Lee PY, Huang Y, Zhou Q, Schnappauf O, Hershfield MS, Li Y, et al. Disrupted N-linked glycosylation as a disease mechanism in deficiency of ADA2. J Allergy Clin Immunol. (2018) 142:1363–5.e8. 10.1016/j.jaci.2018.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ben-Ami T, Revel-Vilk S, Brooks R, Shaag A, Hershfield MS, Kelly SJ, et al. Extending the clinical phenotype of adenosine deaminase 2 deficiency. J Pediatr. (2016) 177:316–20. 10.1016/j.jpeds.2016.06.058 [DOI] [PubMed] [Google Scholar]
- 115.Schepp J, Proietti M, Frede N, Buchta M, Hubscher K, Rojas Restrepo J, et al. Screening of 181 patients with antibody deficiency for deficiency of adenosine deaminase 2 sheds new light on the disease in adulthood. Arthritis Rheumatol. (2017) 69:1689–700. 10.1002/art.40147 [DOI] [PubMed] [Google Scholar]
- 116.Cipe FE, Aydogmus C, Serwas NK, Keskindemirci G, Boztug K. Novel mutation in CECR1 leads to deficiency of ADA2 with associated neutropenia. J Clin Immunol. (2018) 38:273–7. 10.1007/s10875-018-0487-x [DOI] [PubMed] [Google Scholar]
- 117.Alsultan A, Basher E, Alqanatish J, Mohammed R, Alfadhel M. Deficiency of ADA2 mimicking autoimmune lymphoproliferative syndrome in the absence of livedo reticularis and vasculitis. Pediatr Blood Cancer (2018) 65:e26912. 10.1002/pbc.26912 [DOI] [PubMed] [Google Scholar]
- 118.Trotta L, Martelius T, Siitonen T, Hautala T, Hamalainen S, Juntti H, et al. ADA2 deficiency: clonal lymphoproliferation in a subset of patients. J Allergy Clin Immunol. (2018) 141:1534–7 e1538. 10.1016/j.jaci.2018.01.012 [DOI] [PubMed] [Google Scholar]
- 119.Uettwiller F, Sarrabay G, Rodero MP, Rice GI, Lagrue E, Marot Y, et al. ADA2 deficiency: case report of a new phenotype and novel mutation in two sisters. RMD Open (2016) 2:e000236. 10.1136/rmdopen-2015-000236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Skrabl-Baumgartner A, Plecko B, Schmidt WM, Konig N, Hershfield M, Gruber-Sedlmayr U, et al. Autoimmune phenotype with type I interferon signature in two brothers with ADA2 deficiency carrying a novel CECR1 mutation. Pediatr Rheumatol Online J. (2017) 15:67. 10.1186/s12969-017-0193-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Van Montfrans JM, Hartman EA, Braun KP, Hennekam EA, Hak EA, Nederkoorn PJ, et al. Phenotypic variability in patients with ADA2 deficiency due to identical homozygous R169Q mutations. Rheumatology (2016) 55:902–10. 10.1093/rheumatology/kev439 [DOI] [PubMed] [Google Scholar]
- 122.Caorsi R, Penco F, Grossi A, Insalaco A, Omenetti A, Alessio M, et al. ADA2 deficiency (DADA2) as an unrecognised cause of early onset polyarteritis nodosa and stroke: a multicentre national study. Ann Rheum Dis. (2017) 76:1648–56. 10.1136/annrheumdis-2016-210802 [DOI] [PubMed] [Google Scholar]
- 123.Rama M, Duflos C, Melki I, Bessis D, Bonhomme A, Martin H, et al. A decision tree for the genetic diagnosis of deficiency of adenosine deaminase 2 (DADA2): a French reference centres experience. Eur J Hum Genet. (2018) 26:960–71. 10.1038/s41431-018-0130-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Van Eyck L, Liston A, Meyts I. Mutant ADA2 in vasculopathies. N Engl J Med. (2014) 371:478–9. 10.1056/NEJMc1405506 [DOI] [PubMed] [Google Scholar]
- 125.Hashem H, Kumar AR, Muller I, Babor F, Bredius R, Dalal J, et al. Hematopoietic stem cell transplantation rescues the hematological, immunological, and vascular phenotype in DADA2. Blood (2017) 130:2682–8. 10.1182/blood-2017-07-798660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Crow YJ, Manel N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol. (2015) 15:429–40. 10.1038/nri3850 [DOI] [PubMed] [Google Scholar]
- 127.Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J Exp Med. (2016) 213:2527–38. 10.1084/jem.20161596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. (2014) 371:507–18. 10.1056/NEJMoa1312625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest. (2018) 128:3041–52. 10.1172/JCI98814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Martinez-Quiles N, Goldbach-Mansky R. Updates on autoinflammatory diseases. Curr Opin Immunol. (2018) 55:97–105. 10.1016/j.coi.2018.09.014 [DOI] [PubMed] [Google Scholar]