

Fungicides are essential to control diseases in agriculture because many crops are highly susceptible to pathogens. However, many pathogens rapidly evolve resistance to fungicides. A large body of studies have described specific mutations conferring resistance and have often made inferences about the origins of resistance based on sequencing data from the target gene alone. Here, we show the de novo acquisition of resistance to the ubiquitously used azole fungicides in genetically isolated populations of the wheat pathogen Zymoseptoria tritici in Tasmania, Australia. We confirm evidence for parallel evolution through genome-scale analyses of representative worldwide populations. The emergence of complex resistance haplotypes following a well-documented recent introduction of azoles into Australian farming practices demonstrates how rapidly chemical resistance evolves in agricultural ecosystems.
KEYWORDS: azole, CYP51, Zymoseptoria tritici, fungicide resistance
ABSTRACT
Zymoseptoria tritici is a globally distributed fungal pathogen which causes Septoria tritici blotch on wheat. Management of the disease is attempted through the deployment of resistant wheat cultivars and the application of fungicides. However, fungicide resistance is commonly observed in Z. tritici populations, and continuous monitoring is required to detect breakdowns in fungicide efficacy. We recently reported azole-resistant isolates in Australia; however, it remained unknown whether resistance was brought into the continent through gene flow or whether resistance emerged independently. To address this question, we screened 43 isolates across five Australian locations for azole sensitivity and performed whole-genome sequencing on 58 isolates from seven locations to determine the genetic basis of resistance. Population genomic analyses showed extremely strong differentiation between the Australian population recovered after azoles began to be used and both Australian populations recovered before azoles began to be used and populations on different continents. The apparent absence of recent gene flow between Australia and other continents suggests that azole fungicide resistance has evolved de novo and subsequently spread within Tasmania. Despite the isolates being distinct at the whole-genome level, we observed combinations of nonsynonymous substitutions at the CYP51 locus identical to those observed elsewhere in the world. We observed nine previously reported nonsynonymous mutations as well as isolates that carried a combination of the previously reported L50S, S188N, A379G, I381V, Y459DEL, G460DEL, and N513K substitutions. Assays for the 50% effective concentration against a subset of isolates exposed to the tebuconazole and epoxiconazole fungicides showed high levels of azole resistance. The rapid, parallel evolution of a complex CYP51 haplotype that matches a dominant European haplotype demonstrates the enormous potential for de novo resistance emergence in pathogenic fungi.
IMPORTANCE Fungicides are essential to control diseases in agriculture because many crops are highly susceptible to pathogens. However, many pathogens rapidly evolve resistance to fungicides. A large body of studies have described specific mutations conferring resistance and have often made inferences about the origins of resistance based on sequencing data from the target gene alone. Here, we show the de novo acquisition of resistance to the ubiquitously used azole fungicides in genetically isolated populations of the wheat pathogen Zymoseptoria tritici in Tasmania, Australia. We confirm evidence for parallel evolution through genome-scale analyses of representative worldwide populations. The emergence of complex resistance haplotypes following a well-documented recent introduction of azoles into Australian farming practices demonstrates how rapidly chemical resistance evolves in agricultural ecosystems.
INTRODUCTION
Globally distributed fungal diseases are an increasing threat to both natural and agricultural ecosystems due to rapid anthropogenic spread and climate change (1, 2). Among these diseases caused by fungi, pathogens of wheat are of increasing concern in countries around the world due to their potential to cause total crop losses (3). Currently, there are two main methods used to limit the impact of these diseases on wheat yields: (i) deployment of resistant host cultivars and (ii) the application of chemical fungicides. Both methods, however, are susceptible to breakdowns in efficacy, due to the rapid evolution of pathogen populations (4, 5). The breakdown of host resistance is devastating because it can take many years for breeders to develop replacement cultivars. In the absence of effective host resistance, fungicides are heavily relied upon. However, intensive application of fungicides accelerates the evolution of resistant pathogen populations (6).
Azole fungicides, specifically, demethylation inhibitors, are one of three major classes of fungicides used to control fungal disease on cereal crops (7, 8). Azoles target the CYP51 (erg11) gene, which in fungi is part of the pathway responsible for the biosynthesis of sterols, a key component of the fungal cell membrane (7, 9). Azoles were introduced into commercial agriculture in the mid to late 1970s and now account for nearly 50% of all fungicides used in Europe (8).
The molecular mechanisms that drive increased resistance in plant pathogens are best studied in the wheat pathogen Zymoseptoria tritici (9, 10). Z. tritici is a globally distributed pathogen of wheat that is predominant in temperate, wet climates, such as northern Europe (8, 11). The pathogen causes the disease known as Septoria tritici blotch (STB) and undergoes regular cycles of sexual reproduction during the growing season (12, 13). The pathogen has also been shown to migrate long distances (14, 15). Splash-dispersed, asexual pycnidiospores drive the development of epidemics in the field, while new infections are initiated from sexual ascospores each growing season (16, 17). Azole resistance in Z. tritici has been attributed to three mechanisms: (i) nonsynonymous (NS) mutations in the coding region of the CYP51 gene that can alter the binding of the drug, (ii) mutations that lead to CYP51 overexpression, or (iii) overexpression of efflux pumps (9, 18).
While different azole compounds remain more or less effective to control STB in Europe, there is clear evidence, particularly from the United Kingdom, Ireland, and France, of a slow erosion of their effectiveness starting in the early 2000s (5, 19–24). The shift in azole sensitivity has been attributed to an accumulation of multiple NS substitutions in the CYP51 gene (25, 26). Many of these mutations have now been validated experimentally, whereby specific NS substitutions within CYP51 decrease azole sensitivity when heterologously expressed in yeast (21, 26). Despite being a single-site fungicide, there are a diverse set of azole compounds that target different amino acids within the CYP51 protein. These slight differences in target site seem to be important, as no single NS substitution or combinations of substitutions confer high levels of resistance to all azole compounds (18, 22, 27).
To date, there are over 35 NS mutations reported (reviewed by Cools and Fraaije [9] and Huf et al. [28]). In field populations of Z. tritici, there are sets of NS mutations within the CYP51 gene that are mutually exclusive, indicating that there are trade-offs that prevent certain combinations of mutations from occurring in a single isolate (22, 29, 30). Due to its importance in resistance, the CYP51 gene from multiple field populations of Z. tritici in Europe, Africa, North America, and Australia has been sequenced or genotyped (22, 29–32). Brunner et al. showed that sensitive CYP51 alleles have essentially disappeared from European populations sampled after widespread use of the fungicide (29). Frequent recombination likely brought together multiple different NS substitutions, which in combination confer higher levels of azole resistance (29). More recently, Estep et al. (2015) have reported the recent emergence of azole fungicide resistance in populations of Z. tritici from Oregon (30). Here, however, the authors found that the most recently sampled isolates carried no more than a single mutation associated with azole resistance, which they attributed to a very recent emergence. This is in sharp contrast to the most recently genotyped isolates from Europe, which carry complex and very diverse combinations of NS mutations (23, 24, 28, 33).
In Australia, azoles account for more than 98% of registered fungicides and were not widely used to control leaf diseases until they began to be used in 2002 to control the development of the wheat disease stripe rust (Puccinia striiformis f. sp. tritici, yellow rust) (34). The contribution of fungicides in controlling STB in Australia in 1998 was estimated to be 0%, but this swiftly rose to 19% in 2009 (35). This was during a period when Z. tritici was at historically low levels in wheat fields due to the millennial drought (2001 to 2009) (A. Milgate, personal observation). Starting in 2010, Z. tritici reemerged as a serious disease of wheat in the high-rainfall zones of southeastern continental Australia as well as Tasmania. Favorable environmental conditions in these regions allowed disease severity to reach thresholds capable of causing yield losses. In response, farmers began applying fungicides against Z. tritici for disease control, typically using two applications per crop in Victoria, compared to three in Tasmania. Azole resistance was detected in Victoria and Tasmania in 2011. This was only 9 years after the beginning of the widespread use of the fungicide on wheat and only 1 to 2 years since Z. tritici had been observed to be causing disease in these regions (36). Since the first detection of azole resistance, annual monitoring of field populations shows that azole resistance is now established in Tasmania (A. Milgate, personal observation) (36). This geographically isolated population provides a unique opportunity to investigate the emergence and spread of azole resistance. This work used whole-genome resequencing to place the newly resistant Tasmanian population in the context of the global pathogen diversity. We then examined the sequence diversity of the CYP51 gene and contrasted this to relatedness at the whole-genome scale. For a subset of isolates, we measured the levels of resistance against tebuconazole and epoxiconazole. Together these analyses comprehensively characterize the emergence of azole resistance in a genetically isolated plant pathogen population.
RESULTS
Population structure of azole-resistant isolates from Tasmania.
In order to understand the origins of azole resistance in Tasmania, we selected a set of 58 isolates sampled across seven locations in Tasmania in 2014 for Illumina whole-genome resequencing (see Table S1 in the supplemental material). There were insufficient sequencing reads from isolate WAI1820 for it to be used for further population analyses. Data from an additional 13 Australian Z. tritici genomes sampled from Tasmania, New South Wales (NSW), and Victoria were also included in all further analyses (37). Resequencing data from four global field populations were used to place these isolates in relation to isolates from the rest of the world (38). In total, 218 genomes were used for whole-genome comparisons and represented approximately 5 populations: Tasmania 2014 (Australian isolates recovered after azoles began to be used [Australian post-azole isolates]), New South Wales 2001 (Australian isolates recovered before azoles began to be used [Australian pre-azole isolates]), Oregon 1992 (pre-azole isolates), Israel 1990 (pre-azole isolates), and Switzerland 1999 (post-azole isolates). All quality-controlled short-read data were aligned to the sequence of reference isolate IPO323, and single nucleotide polymorphisms (SNPs) were called using the GATK tool. After filtering for low-quality variants and minor alleles that occurred in <5% of the isolates and some additional thinning, the final data set contained 44,845 SNPs spanning core Z. tritici chromosomes 1 to 13. We conducted a principal component analysis (PCA) based on the genome-wide SNP data to estimate the genetic similarities among the isolates (Fig. S1). Consistent with earlier population genetic analyses, the pre-azole Australian population (NSW, 2001) formed a genetically distinct group that was distant from the isolates from the rest of the world (Israel, Oregon, and Switzerland) (39). However, the post-azole Tasmanian isolates formed a second, distinct group that was distant from all other populations. The additional Australian isolates resequenced by McDonald et al. (37) and sampled prior to 2011 clustered tightly with the pre-azole Australian population (2001). These isolates spanned multiple different sampling locations and times, including two isolates, WAI332 and WAI320, sampled in 1979 and 1980, respectively. Only one isolate sampled prior to 2011, WAI327 (NSW 2002), clustered together with the Tasmanian group. WAI327 is the only exception among the 97 Australian isolates whose genomes were sequenced, including isolate WAI147, sampled at the same time and location. We have conservatively excluded WAI327 from further analyses as a potential sample mix-up error. Together, these data show that the two Australian populations (the pre- and post-azole populations) are distinct both from each other and from the populations in the rest of the world.
To better understand the placement of the post-azole Tasmanian population in the overall genetic structure of the species, we analyzed the relatedness among all resequenced isolates by use of the FineStructure algorithm. FineStructure estimates the relatedness between individuals by dividing each individual genome into short haplotype blocks that span the reference assembly. This is followed by a full pairwise comparison between each isolate that estimates the number of shared genomic haplotype blocks. Isolates that are more closely related share more haplotype blocks with each other than isolates that are more distantly related. The final results are displayed as a heat map (similarity matrix) that shows the number of shared haplotype blocks between all pairs of individuals (Fig. 1). This heat map was also used to draw the dendrogram at the top of Fig. 1, which shows the relatedness between larger groups of individuals (i.e., between populations). Individual isolate names can be viewed alongside the dendrogram in Fig. S2A. This analysis clearly defines the five global populations, with some subpopulations being found within them (Fig. 1). Consistent with the preliminary PCA, the post-azole Tasmanian population was distinct from the isolates in the rest of the world and shared few to no haplotype regions with populations from outside Australia (Fig. 1). This analysis also suggests that the pre- and post-azole populations from Australia are more closely related to each other than to populations in the rest of the world. Within Tasmania there is evidence for at least four distinct subpopulations, with strong support values on each of the branches within the dendrogram (>97% posterior support; Fig. 1). These four groups did not correlate with the sampling locations (Table S1), indicating that there is population subdivision and gene flow within Tasmania.
FIG 1.

Fine structure analysis of the resistant Tasmanian population in comparison to population in the rest of the world. This heat map shows the coancestry matrix of 218 Z. tritici individuals. The coloring of the heat map represents the number of shared genomic regions between different Z. tritici individuals. A strong population structure is visible where large blocks appear on the diagonal, whereby groups of related individuals share more of their genome with each other than with other individuals from other populations. The dendrogram shown at the top of the heat map shows which groups of isolates (populations) are more closely related to each other. Posterior support values are drawn only on branches where they were less than 1. For ease of viewing, the major clusters are colored according to the population of origin. The subgroups within the Tasmanian population (G1 to G4) are also colored.
Global distribution of CYP51 nucleotide haplotypes.
To investigate sequence variation at the CYP51 locus, all resequenced isolates’ genomes were assembled de novo and their CYP51 sequences were extracted using the BLASTn program. In addition, all available CYP51 sequences deposited in NCBI were downloaded and included in the alignment (NCBI was last accessed on 8 March 2018). To include as many of the NCBI sequences as possible, the full-length CYP51 coding sequence was trimmed to a final alignment length of 1,298 bp, including indels (bases 409 to 1705 of the coding region from reference isolate IPO323). This alignment, which includes the NCBI accession numbers, was used to generate a nucleotide haplotype network (Fig. 2A; all sequences are available in Table S2). This network shows which isolates share an identical CYP51 nucleotide sequence. Figure 2A shows that the post-azole Tasmanian isolates share their partial CYP51 nucleotide sequences with isolates from other parts of the world, mainly, isolates recovered in Europe after 2000. This network is constructed with ∼68% of the total coding region of CYP51. While this is only a partial alignment of the gene, it is a clear indication that the post-azole Tasmanian isolates share many of the same CYP51 mutations (both synonymous and NS) with isolates from the rest of the world.
FIG 2.

Distribution of mutations in the CYP51 gene in the resequenced Australian genomes and publicly available sequences. Each colored pie chart represents a unique CYP51 nucleotide sequence (haplotype); the size of the pie chart is proportional to the number of individuals with that sequence. Individuals are colored according to their population of origin, given in the legend. Single point mutations are indicated by small black dashes. Small black circles are inferred but unsampled haplotypes. The length of the branches is not proportional to the distance between haplotypes. Potential recombination events are shown, where loops connect different haplotypes. (A) Haplotype network constructed with de novo-assembled genomes and all available CYP51 sequences from NCBI. The network was constructed with a partial sequence of CYP51 from bases 409 to 1705 (inclusive), based on the predicted coding start site in the reference isolate, IPO323. (B) Haplotype network constructed only with CYP51 sequences from isolates with whole-genome sequences. The network was built using the complete genomic DNA sequence of CYP51 from the start to the stop codon. Each unique protein is outlined in a black dashed box. Amino acid substitutions that arose only once in the network are highlighted in black bold text. Amino acid substitutions that appear in multiple distinct nucleotide backgrounds are in red. The left and right halves of the network are indicated by the red asterisk flanked by black arrows.
The haplotype network drawn in Fig. 2B is constructed with the full-length CYP51 coding region (1,907 bp) from 217 de novo-assembled genomes (Table S3). Each unique protein haplotype within this nucleotide network is labeled to highlight NS substitutions that appear in multiple haplotypes. The network can roughly be divided into two halves (left and right). In the larger data set that includes sequences from NCBI, multiple complex reticulations link different nucleotide haplotypes across these two halves (Fig. 2A). As mentioned above, however, this network represents only 68% of the coding region. Using a smaller number of individuals but the complete gene sequence, there is less evidence of recombination across the entirety of the gene, based on the lack of reticulation (loops) between the two halves (Fig. 2B). There is evidence of recombination restricted to the right half of the network (Fig. 2B). Several NS mutations appear multiple times in distinct nucleotide haplotypes on either extreme of the network, for example, the L50S mutation, present in ST99CH_28 (left) versus ST99CH_3A4 (right) separated by over 45 single nucleotide polymorphisms. Similarly, the combination of L50S and Y461S appears in very distinct nucleotide haplotypes on either side of the red asterisk in the network, with no reticulations connecting these haplotype sequences. This indicates that these NS mutations have arisen multiple times in different genetic backgrounds. Other NS mutations are restricted to either side of the network. For example, the Y137F mutation appears in only a single haplotype restricted to the right side of the network. Similarly, the G460/Y461 deletion and the S188N NS substitutions are found only in nucleotide haplotypes forming the left side of the network.
The network shown in Fig. 2B reveals two dominant nucleotide haplotypes in the pre-azole Australian population, separated by 38 point mutations. On the right side of the network is the CYP51 sequence of the reference isolate, IPO323, which is sensitive to azoles and referred to as the wild type (WT). The most frequent pre-azole Australian haplotype on the right side of the network (AUS_1A5) is unique at the nucleotide level. The amino acid sequence is, however, identical to that of the IPO323 WT allele. On the left side of the network, the most frequent pre-azole haplotype (AUS_1A4) contains three NS substitutions that differ from the WT: L50S, S188N, and N513K. This haplotype is shared with a few isolates from the Swiss population. No haplotype sampled in the pre-azole population was found in the post-azole population, indicating a strong population shift over time at this locus.
There are three most-frequent haplotypes in post-azole Australia which appear at distinct tips of the network shown in Fig. 2B (WAI1820, to the left of the red asterisk; ST99CH_3A9, to the right of the red asterisk; and WAI321, to the right of the red asterisk). These post-azole haplotypes contain NS substitutions that were all previously observed in azole-resistant isolates from around the world (Fig. 2A) (9, 28). On the right side of the network, two distinct azole-resistant haplotypes instead of WT alleles were sampled. One haplotype carries a single NS substitution known to confer azole resistance, the Y137F mutation. The second carries both the L50S and Y461S mutations. On the left extremity of the network, the most frequent haplotype contains a combination of seven NS mutations that differ from the WT sequence. This haplotype is shared with other global isolates in Fig. 2A, though this is based on only a partial alignment of the whole coding sequence. Isolates carrying this combination of NS substitutions have previously been reported to be highly resistant to azoles (9, 28).
NS mutations in CYP51 and their effect on resistance in Australian isolates.
A summary of all of the NS substitutions previously associated with resistance and found within Australia to date is provided in Table 1 (isolate-specific details are provided in Table S4). This includes information for additional isolates that were screened for known CYP51 mutations via partial Sanger sequencing of the CYP51 gene (Table S4). In Australian isolates sampled prior to 2002, four protein isoforms were identified. The most common isoforms identified in the pre-2002 (historical) isolates were the wild-type (IPO323) protein haplotype, followed by combinations made up of the L50S, S188N, and N513K site mutations. This specific combination of mutations has been found in European isolates, but no azole resistance assays have been conducted to see if they increase azole resistance (9). In the post-2010 samples, nine isoforms were identified, and the WT was not present among these isolates. A significant evolutionary shift was the emergence of isoform 11, carrying seven NS mutations present exclusively in Tasmania (Fig. 2B, bottom left). The CYP51 nomenclature proposed very recently by Huf et al. (28) is also included in Table 1, and new haplotypes following this naming convention are listed in Table S5.
TABLE 1.
Distribution of CYP51 isoforms within Australiaa
| Unified code nameb | Australian CYP51 isoform no.c | Mutation(s) | No. of isolates | Presentd: |
|
|---|---|---|---|---|---|
| Before 2002 | After 2010 | ||||
| WT | 1 | Wild type | 23 | Yes | |
| A2 | 2 | L50S | 3 | Yes | |
| A13 | 3 | N513K | 1 | Yes | |
| B7 | 4 | L50S, S188N | 3 | Yes | Yes |
| C18 | 5 | L50S, S188N, N513K | 22 | Yes | Yes |
| C22 | 6 | L50S, S188N, Y461S | 1 | Yes | |
| A12 | 7 | Y461S | 1 | Yes | |
| B8 | 8 | L50S, Y461S | 34 | Yes | |
| B14 | 9 | Y137F, Y461S | 5 | Yes | |
| A3 | 10 | Y137F | 39 | Yes | |
| G1 | 11 | L50S, S188N, A379G, I381V, 459DEL,e 460DEL, N513K | 30 | Yes | |
| Total | 162 | ||||
The table summarizes all combinations of NS mutations found in Australia to date and includes some isolates which were not part of the whole-genome resequencing analyses.
Unified code names for CYP51 protein isoforms proposed by Huf et al. (28).
Isoform number reported in public reports to growers in Australia.
When the isolate was sampled.
DEL, a 3-nucleotide deletion of the amino acid.
Azole resistance was measured in a subset of isolates and reported previously (36). We expanded this analysis with a subset of isolates from Tasmania, Victoria, NSW, and South Australia, including some of our resequenced isolates, and measured the 50% effective concentration (EC50) values for two azoles, tebuconazole and epoxiconazole (Fig. 3; Table S6). We observed three levels of resistance. The first was the baseline sensitive response of the wild type, with EC50 values ranging from 0.027 to 0.69 (mg/liter), and the second level included isoforms 4, 8, and 10 containing the site mutations Y461S and Y137F, with EC50 values ranging from 0.067 to 4.597 mg/liter for tebuconazole. The third level of resistance was the highest associated with isoform 11, which contained 7 mutations, with EC50 values ranging from 4.481 to 8.181 mg/liter for tebuconazole. The isoforms were consistently ranked in terms of resistance to both fungicides. However, epoxiconazole consistently showed lower EC50 values than tebuconazole.
FIG 3.
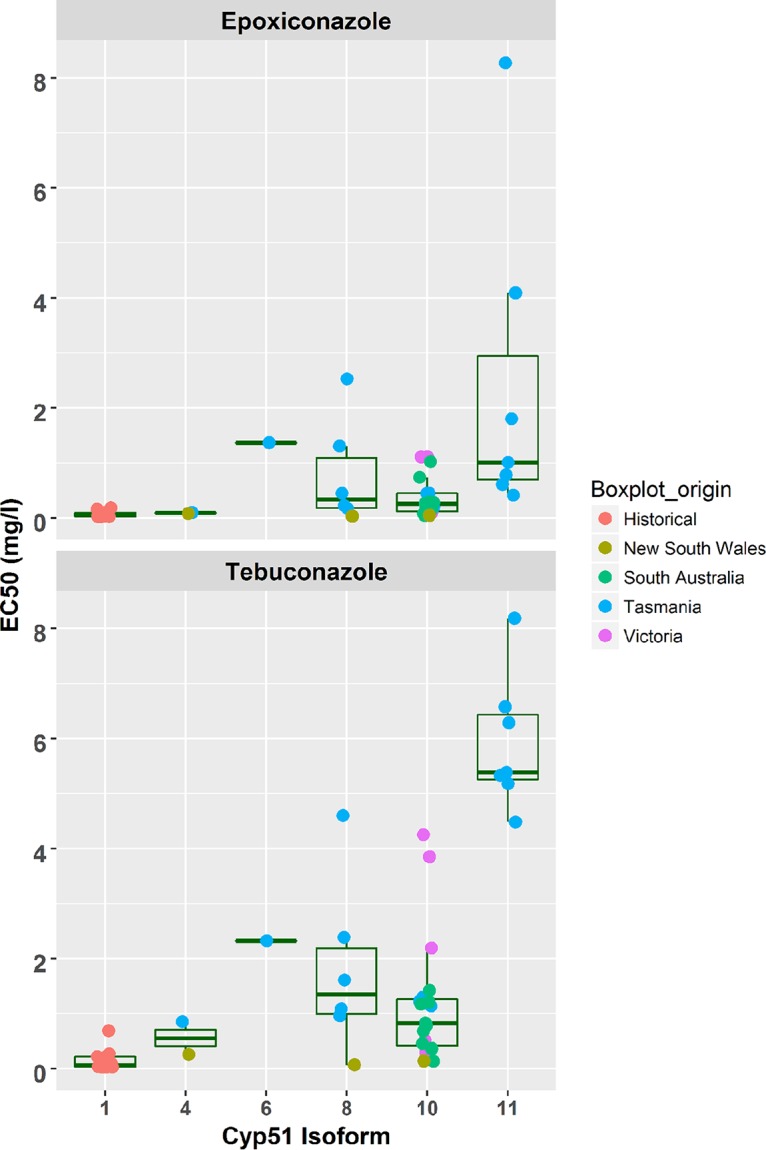
Measured EC50 values of tebuconazole and epoxiconazole for known CYP51 amino acid haplotypes coded to show their origin. The number of isolates tested was as follows: isoform 1 (wild type), n = 11; isoform 4 (L50S, S188N), n = 2; isoform 6 (L50S, S188N, Y461S), n = 1; isoform 8 (L50S, Y461S), n = 6; isoform 10 (Y137F), n = 16; and isoform 11 (L50S, S188N, A379G, I381V, 459DEL, 460DEL, N513K [where DEL indicates deletion of the amino acid]), n = 7.
Australian isolates do not carry the cytB G143A mutation conferring resistance to strobilurins.
To investigate whether the azole-resistant Tasmanian isolates were also resistant to strobilurins, we aligned the sequence of the mitochondrial cytochrome b gene extracted from the de novo assemblies. This gene is the main target of strobilurins, and a single point mutation, G143A, confers full resistance. We found that all Australian isolates carried the WT G143 allele and were sensitive to strobilurins (NCBI GenBank accession numbers MH699681 to MH699754).
DISCUSSION
In this study, we identified a genetically distinct Tasmanian Z. tritici population that independently acquired a complex haplotype of resistance to azole fungicides. Whole-genome comparisons with the genomes of a global collection of isolates showed very low genome-wide relatedness between this population and other Z. tritici populations, ruling out gene flow as the source of the resistance haplotype. The post-azole Australian Z. tritici population has acquired the same set of NS substitutions in the CYP51 gene that have been observed in Europe and North America (25). These NS substitutions and their combinations conferred levels of azole resistance that were broadly consistent with previously described results (21, 22, 25–27). CYP51 isoforms similar to Tasmanian isoform 11 have been sampled in the United Kingdom and France (9, 22, 40). Unlike in Europe, the Tasmanian population remained sensitive to the second most commonly used fungicide class, the strobilurins. This is an important result, as strobilurin resistance is present in Europe at frequencies as high as 98% and often precedes the development of azole resistance (33, 41). This means that most isolates found in Europe are resistant to both fungicide classes. Therefore, if European or North American isolates were the source of azole resistance now found in Tasmania via migration or gene flow, the allele conferring strobilurin resistance would also have been introduced into these isolates. We conclude that azole resistance has emerged independently in Tasmania.
Analyses using genome-wide SNP markers clearly showed that the post-azole population from Tasmania is a population genetically distinct from the populations in the rest of the world. Based on this analysis, the most closely related population is the pre-azole Australian population; however, isolates from either population are clearly distinguishable from each other. An additional 12 Australian isolates, which were sequenced in a previous study, grouped tightly with either the post-azole or the pre-azole population (37). The grouping of these isolates into either Australian population is important because they were sequenced independently of both field populations and serve as a quality control for sequencing batch effects, indicating that the relationships that we observed in this data set are true. Within Tasmania there is evidence for at least four distinct subpopulations; however, there was no correlation between these genetic groups and the sampling location. Further, there was no correlation between these groups and CYP51 alleles. This indicates that there is gene flow within Tasmania, which has led to the widespread distribution of the resistant CYP51 haplotypes. Further sequencing would be required to understand the origins of the four subdivided populations that currently exist in Tasmania.
Here we used population-level data to show two dominant but distinct CYP51 haplotypes in the pre-azole Australian population. We also show in the pre-azole population that these two genetically distinct susceptible alleles coexist in one population. We observed a dramatic shift in the frequency of CYP51 nucleotide haplotypes in the post-azole populations, whereby the most frequent haplotypes in the pre-azole populations disappeared after azole application. The replacement of susceptible haplotypes with resistant ones is commonly observed in other populations around the world (29, 30, 33, 42). This replacement is a strong indication of the selective pressure that azoles exert on susceptible populations.
Despite being highly distinct at the whole-genome level, the post-azole Australian isolates’ partial CYP51 nucleotide sequences were found to be identical to sequences from other populations around the world (18, 28, 29, 40). This includes both NS and synonymous nucleotide substitutions. Of the reported NS mutations that increased EC50 values, the L50S and Y461S mutations were the most commonly observed substitutions in post-azole Australian isolates. These two mutations appeared in distinct nucleotide haplotypes located at both extremes of the haplotype network and likely arose independently. Of these two mutations, the L50S substitution was present in the pre-azole Australian population; however, previous studies have conclusively shown that this mutation alone does not confer azole resistance (9). This is a dramatic example of parallel evolution, whereby the exact same complex combinations of NS and synonymous mutations found elsewhere in the world are now found in the genetically distinct populations in Tasmania. These observations strongly suggest that there are combinatorial constraints of mutations at this locus (i.e., epistasis). These constraints are driven by the differential effects of individual mutations on resistance to different azole compounds (18, 40).
In Europe, declines in the efficacy of azoles have been recorded as occurring over decades (21 years for epoxiconazole) (17, 19). In Australia, regular foliar applications of fungicides started in approximately 2002 (35). Tebuconazole and propiconazole were widely applied on wheat after 2002 as a foliar spray to primarily control stripe rust (34). Epoxiconazole has not been used as frequently in Australia for the control of diseases in wheat until more recently (APVMA; https://portal.apvma.gov.au/pubcris). This well-known history of fungicide usage puts the maximum time for the development of azole resistance in Z. tritici to ∼9 years (2002 to 2011) (36). Isoform 11, carrying the complex combination of seven NS substitutions, was detected in Tasmania in 2014. In the United Kingdom, the same combination of site mutations was not detected until almost 30 years after the introduction of azole fungicides (9, 22). Additional factors, such as when azoles were used and which specific azoles were used in Europe versus Australia, could also strongly influence the intensity of selection for resistance. However, there appears to have been a more rapid evolution of complex resistant CYP51 haplotypes, especially isoform 11, in Australia than in Europe.
The rapid emergence of azole resistance in Tasmania is even more significant when placed in the context of the recommended application rates of fungicides in Australia compared to the rest of the world. In Australia the maximum label dose applied for triazoles is half that for equivalent triazole active ingredients applied in the United Kingdom. For example, as a single application, epoxiconazole is available formulated in products such as Opus 125 (BASF) in Australia for wheat at a maximum dose of 62.5 g of active ingredient applied per ha, while in the United Kingdom, epoxiconazole is available as Opus (BASF) and has a maximum dose of 125 g of active ingredient applied per ha (19). What is apparent from this study is that the evolution toward resistance in Australia has occurred at a much lower azole dose than in the United Kingdom. This finding suggests that further research is required to clarify the impact that dose has on the selection of resistant mutations. In contrast to the rapid evolution of azole resistance, to date, there has been no detection of the G143A mutation in the cytB gene conveying resistance to strobilurins, despite these chemicals being available for use in Australia since 2004. Use of these chemicals, however, has been negligible due in part to price and strict maximum residue levels for strobilurins imposed by some export markets. This highlights Australia as a unique case study whereby regulatory limits have influenced a chemical’s usage and, thereby, the speed of the evolution of resistance to different chemistries in pathogen populations.
Sequencing of the CYP51 alleles from mainland Australia has shown that azole-resistant haplotypes are now present in these populations. Further whole-genome sequencing, under way, is required to determine if these haplotypes are representative of these populations and whether these azole-resistant haplotypes are migrants from Tasmania. Together, these data have captured the emergence of azole resistance in a genetically distinct fungal pathogen population. Here, we show data that demonstrate that complex CYP51 haplotypes have emerged in less than 10 years after widespread azole use. More recent studies from azole-resistant populations in Europe show that other NS substitutions, not yet observed in Australia, are now the most common alleles in these populations (23). As farmers respond to the current situation by choosing more efficacious products or increasing the number of fungicide sprays per crop, it remains to be seen whether these more recent mutations observed in Europe will also be selected for in the Australian populations. Monitoring of the Australian populations will allow a rare chance to measure the changes in these allelic frequencies during the establishment phase of fungicide resistance.
MATERIALS AND METHODS
Fungal growth and storage.
Isolates were grown on yeast sucrose agar (YSA; 10 g/liter yeast extract, 10 g/liter sucrose, 1.5% agar) at 22°C for 4 days. Yeast-like growth was scraped from the surface of the plate and used for DNA extraction. Isolates were stored at −80°C in 30% glycerol. DNA was extracted with a Qiagen DNeasy plant minikit (Qiagen, Victoria, Australia).
Growth assays and EC50 measurements.
The fungicide resistance screening was modified from the methods of Stammler et al. (43) and Cools et al. (25). A 100-μl aliquot of yeast extract-malt extract (YMS) liquid medium (4 g/liter yeast extract, 10 g/liter malt extract, 4 g/liter glucose; Amyl Media Pty Ltd., Dandenong South, Victoria, Australia) and 25 mg/liter gentamicin (Merck Millipore, Bayswater, Victoria, Australia) amended with decreasing concentrations of epoxiconazole (Soprano 125SC; Adama Australia Pty Ltd., NSW, Australia) and tebuconazole (Orius 430SC; Adama Australia Pty Ltd., NSW, Australia) (100, 30, 10, 3, 1, 0.3, 0.1, 0.03, 0.01, 0.003, and 0.001 liter−1) was added to the wells of 96-well flat-bottomed microtiter plates (Thermo Scientific Nunc, catalog number NUN 167008; Thermo Fisher Scientific Australia Pty Ltd.). After 7 days at 18°C on YMS agar (4 g/liter yeast extract, 4 g/liter malt extract, 4 g/liter sucrose, 15 g/liter agar, 25 mg/liter gentamicin), spore suspensions at a concentration of 1 × 106 spores ml−1 were prepared for each isolate in YMS liquid medium. Aliquots of a 100-μl spore suspension were added to each well, and then the plate was sealed with adhesive plate sealer (catalog number AB-0580; Thermo Scientific, UK) to avoid evaporation. Each isolate was screened in a minimum of 2 experiments, and each experiment contained 4 replicates of each isolate. Average values for each isolate were used to calculate the EC50 values, described below. Four replicate wells were also used per fungicide concentration as blanks (fungicide solution plus YMS liquid medium). The plates were incubated at 18°C in the dark with continuous agitation on an orbital shaker at 120 rpm (Ratek Instruments Pty Ltd., Boronia, Victoria, Australia). Growth was measured by determination of the absorbance at 405 nm using a Metertech AccuReader microplate reader (Fisher Biotec, Wembley, WA, Australia) and was measured at 0, 2, 5, and 7 days postinoculation.
Statistical methods.
Fungicide sensitivities were determined at the 50% effective concentrations (EC50) for each isolate. For each fungicide, two microtiter plates constituted a complete set of treatments with the 11 fungicide concentrations (100, 30, 10, 3, 1, 0.3, 0.1, 0.03, 0.01, 0.003, and 0.001 mg liter−1) plus a zero control allocated to the 12th column. Cubic smoothing splines fitted as linear mixed models in ASReml (version 3.0) (44) according to the methods of Verbyla et al. (45) were used to model curvilinear trends of the standardized absorbance readings as a function of the fungicide concentration. Predicted isolate growth curves were modeled at three time points, 2, 5, and 7 days after inoculation, for all concentrations. A mean spline was then fitted to these slopes, and the EC50 that was predicted from this model occurs at 50% of the maximum slope. In addition, the R package drc (57) was also used to fit the 4-parameter log-logistic function to calculate the EC50 and standard errors (see Table S6 in the supplemental material).
PCR screening of known CYP51 alleles.
Nucleotide substitutions at sites of the CYP51 gene known to be associated with azole resistance were determined by sequencing of the complete gene after PCR amplification. The gene was amplified in two sections and sequenced in both directions for each isolate using the primers CYP1L and CYP2L, CYP51 Fwd2 and CYP51 Fwd3, and CYP51 Rev1 and CYP51 Rev3, described previously (22, 36). Sequencing was carried out at the Australian Genome Research Facility (AGRF), Melbourne, Australia. For each isolate, the sequence reads were visually checked and trimmed manually. Consensus sequences were compared to the wild-type sequence using the BioEdit sequence alignment editor (46).
Whole-genome resequencing and single nucleotide polymorphism genotyping.
Genomic DNA was submitted to the Australian Genome Research Facility (AGRF) for whole-genome resequencing on an Illumina HiSeq 2000 sequencer. DNA was indexed and then pooled into a single library using an Illumina Nextera kit and submitted for 100-bp paired-end sequencing. Raw sequence data were trimmed with Trimmomatic (v0.27) with the following settings: ILLUMINACLIP:NexteraPE-PE.fa:2:30:10 LEADING:5 TRAILING:5 SLIDINGWINDOW:4:15 MINLEN:45. European resequenced isolates as well as the Australian population from 2001 were sequenced, and quality was assessed as described previously (NCBI BioProject accession numbers PRJNA178194 and PRJNA327615) (39, 47).
Reads were mapped to the sequence of the IPO323 reference genome (http://genome.jgi.doe.gov/Mycgr3/Mycgr3.home.html) using the bowtie2 (v2.2.3) program. The following settings were used for read mapping: --very-sensitive-local --local -p 12 -X 1000. The resulting .sam files were converted to .bam files and sorted using samtools (v0.1.19). The Piccard (v1.87) program was used to remove PCR duplicates from the .bam files for SNP calling (java -jar piccard.jar -tool MarkDuplicates) (https://broadinstitute.github.io/picard/). SNP calling was performed with GATK (v3.7) (https://software.broadinstitute.org/gatk/). The haplotype caller was run on each .bam file separately, and the final genotypes were called on the individual gvcfs with UnifiedGenotyper. Low-quality SNPs and genotype calls were flagged in the final variant call format (VCF) file by GATK’s Variant Filtration tool with the following settings: AN < 150, QD < 5.0, MQ < 20.0, ReadPosRankSum_lower < -2.0, ReadPosRankSum_upper > 2.0, MQRankSum_lower < -2.0, MQRankSum_upper > 2.0, BaseQRankSum_lower < -2.0, and BaseQRankSum_upper > 2.0. SNPs that did not meet these criteria were subsequently removed from the data set using VCFtools option --remove-filtered-all (48). Trimmed reads were also assembled de novo for the Tasmanian isolates using the SPAdes (v3.9) program (49).
Sequence analyses.
The de novo assemblies were queried for the CYP51 locus from isolate IPO323 using a locally installed BLASTn database (50). The CYP51 loci were extracted from these de novo assemblies using a previously published Python script (51). These extracted sequences along with all available CYP51 sequences from Z. tritici available in NCBI were aligned with the ClustalW program implemented in Geneious (v9.1.8) software (52). This alignment was manually trimmed so that all sequences were the same total length. The minimum-spanning network was estimated with the TCS program implemented in PopArt (v1.7) software (53, 54). After quality control, the reads from each sample were mapped to the genome of the reference strain, IPO323, and single nucleotide polymorphisms were called for each isolate with GATK and output as a VCF, as described above. This VCF data set was further filtered to remove isolates that had more than 25% missing data points across the whole genome and was further filtered to include only biallelic SNPs sampled every 500 bp using VCFtools. We also restricted our analysis to core chromosomes 1 to 13 of isolate IPO323. This stringently filtered VCF file was split into a separate file for each chromosome with VCFtools: --chr. Each chromosome VCF was converted to PLINK .ped format with plink (v1.90b5.2): --vcf input.vcf --vcf-half-cal h --chr $i.chr --recode 12 --const-fid 0 --allow-extra-chr (55). These .ped formatted files were finally converted to chromopainter input with plink2chromopainter.pl (https://people.maths.bris.ac.uk/~madjl/finestructure/toolsummary.html). Note that this outputs diploid-formatted .phase files, which were manually edited to make haploid-formatted files by removing the second genotype line for each individual using bash sed. A uniform recombination file was generated for each haploid phase file with makeuniformrecfile.pl. FineStructure was run under several different assumptions but achieved the best convergence assuming that there was genetic linkage between SNP markers and a starting effective population size (Ne) of 500 (ChromopainterLinked model [CPL]). The highest posterior state is shown in Fig. S2 in the supplemental material. The order of isolates shown in Fig. S2 is the same as that shown in Fig. 1. FineStructure (v2) was run using both the unlinked (fs ZymoFungicide_CPU.cp -v -import CPU_settings.txt -ploidy 1 -go) and the linked (fs ZymoFungicide_Ne10000.cp -n -v -import CPL_Ne10000.settings -ploidy 1 -go) modes with a starting Ne of 500 and 10,000 (56). The following additional settings were specified in the input file: s3iters:100000, s4iters:100000, s1minsnps:100, s1indfrac:0.1, -s1args:-in -iM --emfilesonly -n Ne. Final map states for the tree files were generated with fs fs -X -Y -e X2 chunkfile.out treefile.out mappopchunkfile.csv. The mean coincidence files were generated with fs fs -X -Y -e meancoincidence chunkfile.out mcmcfile.out meancoincidencefile.csv. The provided Rscripts FinestructureLibrary.R, FinestructureDendrogram.R, and FinestructureExample.R were used to draw the dendrogram and heat maps of FineStructure results (https://people.maths.bris.ac.uk/~madjl/finestructure/finestructureR.html).
Accession number(s).
All whole-genome resequencing data for the newly sequenced Australian isolates can be found at NCBI BioProject accession number PRJNA480739. The individual GenBank accession numbers for the cytB gene are MH699681 to MH699754.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded as a coinvestment by the Grains and Research Development Corporation and the NSW Department of Primary Industries (DAN00177). M.C.M. is supported by the Australian National University, Grains and Research Development Corporation, and NSW Department of Primary Industries coinvestment DAN00203 as part of the Grains, Agronomy and Pathology Partnership. D.C. is supported by the Swiss National Science Foundation (grant 31003A_173265).
M.R., M.S., and B.O. performed experiments. M.C.M. and D.C. performed bioinformatic and population genetic analyses. M.C.M., P.S.S., and A.M. conceived of the study, designed the experiments, and wrote the manuscript. All authors read and helped revise the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01908-18.
REFERENCES
- 1.Bebber DP, Gurr SJ. 2015. Crop-destroying fungal and oomycete pathogens challenge food security. Fungal Genet Biol 74:62–64. doi: 10.1016/j.fgb.2014.10.012. [DOI] [PubMed] [Google Scholar]
- 2.Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL, Gurr SJ. 2012. Emerging fungal threats to animal, plant and ecosystem health. Nature 484:186–194. doi: 10.1038/nature10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Figueroa M, Hammond-Kosack KE, Solomon PS. 2018. A review of wheat diseases—a field perspective. Mol Plant Pathol 19:1523–1536. doi: 10.1111/mpp.12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDonald BA, Linde C. 2002. The population genetics of plant pathogens and breeding strategies for durable resistance. Euphytica 124:163–180. [Google Scholar]
- 5.Lucas JA, Hawkins NJ, Fraaije BA. 2015. The evolution of fungicide resistance. Adv Appl Microbiol 90:29–92. doi: 10.1016/bs.aambs.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 6.van den Bosch F, Paveley N, Shaw M, Hobbelen P, Oliver R. 2011. The dose rate debate: does the risk of fungicide resistance increase or decrease with dose? Plant Pathol 60:597–606. doi: 10.1111/j.1365-3059.2011.02439.x. [DOI] [Google Scholar]
- 7.Price CL, Parker JE, Warrilow AG, Kelly DE, Kelly SL. 2015. Azole fungicides—understanding resistance mechanisms in agricultural fungal pathogens. Pest Manag Sci 71:1054–1058. doi: 10.1002/ps.4029. [DOI] [PubMed] [Google Scholar]
- 8.Torriani SFF, Melichar JPE, Mills C, Pain N, Sierotzki H, Courbot M. 2015. Zymoseptoria tritici: a major threat to wheat production, integrated approaches to control. Fung Genet Biol 79:8–12. doi: 10.1016/j.fgb.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 9.Cools HJ, Fraaije BA. 2013. Update on mechanisms of azole resistance in Mycosphaerella graminicola and implications for future control. Pest Manag Sci 69:150–155. doi: 10.1002/ps.3348. [DOI] [PubMed] [Google Scholar]
- 10.Stergiopoulos I, van Nistelrooy JGM, Kema GHJ, de Waard MA. 2003. Multiple mechanisms account for variation in base-line sensitivity to azole fungicides in field isolates of Mycosphaerella graminicola. Pest Manag Sci 59:1333–1343. doi: 10.1002/ps.766. [DOI] [PubMed] [Google Scholar]
- 11.Dean R, Van Kan JAL, Pretorius ZA, Hammond-Kosack KE, Di Pietro A, Spanu PD, Rudd JJ, Dickman M, Kahmann R, Ellis J, Foster GD. 2012. The top 10 fungal pathogens in molecular plant pathology. Mol Plant Pathol 13:414–430. doi: 10.1111/j.1364-3703.2011.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suffert F, Sache I. 2011. Relative importance of different types of inoculum to the establishment of Mycosphaerella graminicola in wheat crops in north-west Europe. Plant Pathol 60:878–889. doi: 10.1111/j.1365-3059.2011.02455.x. [DOI] [Google Scholar]
- 13.Eriksen L, Munk L. 2003. The occurrence of Mycosphaerella graminicola and its anamorph Septoria tritici in winter wheat during the growing season. Eur J Plant Pathol 109:253–259. [Google Scholar]
- 14.Linde CC, Zhan J, McDonald BA. 2002. Population structure of Mycosphaerella graminicola: from lesions to continents. Phytopathology 92:946–955. doi: 10.1094/PHYTO.2002.92.9.946. [DOI] [PubMed] [Google Scholar]
- 15.Orton ES, Deller S, Brown JKM. 2011. Mycosphaerella graminicola: from genomics to disease control. Mol Plant Pathol 12:413–424. doi: 10.1111/j.1364-3703.2010.00688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Razavi M, Hughes GR. 2004. Microsatellite markers provide evidence for sexual reproduction of Mycosphaerella graminicola in Saskatchewan. Genome 47:789–794. doi: 10.1139/g04-036. [DOI] [PubMed] [Google Scholar]
- 17.Suffert F, Sache I, Lannou C. 2011. Early stages of Septoria tritici blotch epidemics of winter wheat: build-up, overseasoning, and release of primary inoculum. Plant Pathol 60:166–177. doi: 10.1111/j.1365-3059.2010.02369.x. [DOI] [Google Scholar]
- 18.Cools HJ, Hawkins NJ, Fraaije BA. 2013. Constraints on the evolution of azole resistance in plant pathogenic fungi. Plant Pathol 62:36–42. doi: 10.1111/ppa.12128. [DOI] [Google Scholar]
- 19.Blake JJ, Gosling P, Fraaije BA, Burnett FJ, Knight SM, Kildea S, Paveley ND. 2018. Changes in field dose-response curves for demethylation inhibitor (DMI) and quinone outside inhibitor (QoI) fungicides against Zymoseptoria tritici, related to laboratory sensitivity phenotyping and genotyping assays. Pest Manag Sci 74:302–313. doi: 10.1002/ps.4725. [DOI] [PubMed] [Google Scholar]
- 20.Cools HJ, Fraaije BA. 2008. Are azole fungicides losing ground against Septoria wheat disease? Resistance mechanisms in Mycosphaerella graminicola. Pest Manag Sci 64:681–684. doi: 10.1002/ps.1568. [DOI] [PubMed] [Google Scholar]
- 21.Fraaije BA, Cools HJ, Kim S-H, Motteram J, Clark WS, Lucas JA. 2007. A novel substitution I381V in the sterol 14alpha-demethylase (CYP51) of Mycosphaerella graminicola is differentially selected by azole fungicides. Mol Plant Pathol 8:245–254. doi: 10.1111/j.1364-3703.2007.00388.x. [DOI] [PubMed] [Google Scholar]
- 22.Leroux P, Albertini C, Gautier A, Gredt M, Walker A-S. 2007. Mutations in the CYP51 gene correlated with changes in sensitivity to sterol 14α-demethylation inhibitors in field isolates of Mycosphaerella graminicola. Pest Manag Sci 63:688–698. doi: 10.1002/ps.1390. [DOI] [PubMed] [Google Scholar]
- 23.Jørgensen LN, Matzen N, Hansen JG, Semaskiene R, Korbas M, Danielewicz J, Glazek M, Maumene C, Rodemann B, Weigand S, Hess M, Blake J, Clark B, Kildea S, Batailles C, Ban R, Havis N, Treikale O. 2018. Four azoles’ profile in the control of Septoria, yellow rust and brown rust in wheat across Europe. Crop Protec 105:16–27. doi: 10.1016/j.cropro.2017.10.018. [DOI] [Google Scholar]
- 24.Stammler G, Semar M. 2011. Sensitivity of Mycosphaerella graminicola (anamorph: Septoria tritici) to DMI fungicides across Europe and impact on field performance. EPPO Bull 41:149–155. doi: 10.1111/j.1365-2338.2011.02454.x. [DOI] [Google Scholar]
- 25.Cools HJ, Mullins JGL, Fraaije BA, Parker JE, Kelly DE, Lucas JA, Kelly SL. 2011. Impact of recently emerged sterol 14 alpha-demethylase (CYP51) variants of Mycosphaerella graminicola on azole fungicide sensitivity. Appl Environ Microbiol 77:3830–3837. doi: 10.1128/AEM.00027-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cools HJ, Parker JE, Kelly DE, Lucas JA, Fraaije BA, Kelly SL. 2010. Heterologous expression of mutated eburicol 14α-demethylase (CYP51) proteins of Mycosphaerella graminicola to assess effects on azole fungicide sensitivity and intrinsic protein function. Appl Environ Microbiol 76:2866–2872. doi: 10.1128/AEM.02158-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mullins JGL, Parker JE, Cools HJ, Togawa RC, Lucas JA, Fraaije BA, Kelly DE, Kelly SL. 2011. Molecular modelling of the emergence of azole resistance in Mycosphaerella graminicola. PLoS One 6:e20973. doi: 10.1371/journal.pone.0020973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huf A, Rehfus A, Lorenz KH, Bryson R, Voegele RT, Stammler G. 2018. Proposal for a new nomenclature for CYP51 haplotypes in Zymoseptoria tritici and analysis of their distribution in Europe. Plant Pathol 67:1706–1712. doi: 10.1111/ppa.12891. [DOI] [Google Scholar]
- 29.Brunner PC, Stefanato FL, McDonald BA. 2008. Evolution of the CYP51 gene in Mycosphaerella graminicola: evidence for intragenic recombination and selective replacement. Mol Plant Pathol 9:305–316. doi: 10.1111/j.1364-3703.2007.00464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Estep LK, Torriani SFF, Zala M, Anderson NP, Flowers MD, McDonald BA, Mundt CC, Brunner PC. 2015. Emergence and early evolution of fungicide resistance in North American populations of Zymoseptoria tritici. Plant Pathol 64:961–971. doi: 10.1111/ppa.12314. [DOI] [Google Scholar]
- 31.Vagndorf N, Heick TM, Justesen AF, Andersen JR, Jahoor A, Jørgensen LN, Orabi J. 2018. Population structure and frequency differences of CYP51 mutations in Zymoseptoria tritici populations in the Nordic and Baltic regions. Eur J Plant Pathol 41:226. [Google Scholar]
- 32.Boukef S, McDonald BA, Yahyaoui A, Rezgui S, Brunner PC. 2012. Frequency of mutations associated with fungicide resistance and population structure of Mycosphaerella graminicola in Tunisia. Eur J Plant Pathol 132:111–122. doi: 10.1007/s10658-011-9853-8. [DOI] [Google Scholar]
- 33.Heick TM, Justesen AF, Jørgensen LN. 2017. Resistance of wheat pathogen Zymoseptoria tritici to DMI and QoI fungicides in the Nordic-Baltic—a status. Eur J Plant Pathol 149:669–682. doi: 10.1007/s10658-017-1216-7. [DOI] [Google Scholar]
- 34.Wellings CR. 2007. Puccinia striiformis in Australia: a review of the incursion, evolution, and adaptation of stripe rust in the period 1979–2006. Aust J Agric Res 58:567–575. doi: 10.1071/AR07130. [DOI] [Google Scholar]
- 35.Murray GM, Brennan JP. 2009. The current and potential costs from diseases of wheat in Australia. GRDC, Canberra, Australia. [Google Scholar]
- 36.Milgate A, Adorada D, Orchard B, Pattemore J. 2016. First report of resistance to DMI fungicides in Australian populations of the wheat pathogen Zymoseptoria tritici. Plant Dis 100:522. doi: 10.1094/PDIS-06-15-0704-PDN. [DOI] [Google Scholar]
- 37.McDonald MC, McGinness L, Hane JK, Williams AH, Milgate A, Solomon PS. 2016. Utilizing gene tree variation to identify candidate effector genes in Zymoseptoria tritici. G3 (Bethesda) 6:779–791. doi: 10.1534/g3.115.025197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartmann FE, Croll D. 2017. Distinct trajectories of massive recent gene gains and losses in populations of a microbial eukaryotic pathogen. Mol Biol Evol 34:2808–2822. doi: 10.1093/molbev/msx208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartmann FE, Sánchez-Vallet A, McDonald BA, Croll D. 2017. A fungal wheat pathogen evolved host specialization by extensive chromosomal rearrangements. ISME J 11:1189–1204. doi: 10.1038/ismej.2016.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leroux P, Walker A-S. 2011. Multiple mechanisms account for resistance to sterol 14α‐demethylation inhibitors in field isolates of Mycosphaerella graminicola. Pest Manag Sci 67:44–59. doi: 10.1002/ps.2028. [DOI] [PubMed] [Google Scholar]
- 41.Torriani SF, Brunner PC, McDonald BA, Sierotzki H. 2009. QoI resistance emerged independently at least 4 times in European populations of Mycosphaerella graminicola. Pest Manag Sci 65:155–162. doi: 10.1002/ps.1662. [DOI] [PubMed] [Google Scholar]
- 42.Hagerty CH, Anderson NP, Mundt CC. 2017. Temporal dynamics and spatial variation of azoxystrobin and propiconazole resistance in Zymoseptoria tritici: a hierarchical survey of commercial winter wheat fields in the Willamette Valley, Oregon. Phytopathology 107:345–352. doi: 10.1094/PHYTO-06-16-0237-R. [DOI] [PubMed] [Google Scholar]
- 43.Stammler G, Carstensen M, Koch A, Semar M, Strobel D, Schlehuber S. 2008. Frequency of different CYP51-haplotypes of Mycosphaerella graminicola and their impact on epoxiconazole-sensitivity and -field efficacy. Crop Protec 27:1448–1456. doi: 10.1016/j.cropro.2008.07.007. [DOI] [Google Scholar]
- 44.Gilmore AR, Gogel BJ, Cullis BR, Thompson R. 2009. ASReml user guide 3.0. VSN International Ltd, Hemel Hempstead, United Kingdom. [Google Scholar]
- 45.Verbyla AP, Cullis BR, Kenward MG, Welham SJ. 1999. The analysis of designed experiments and longitudinal data by using smoothing splines. J R Stat Soc Series C Appl Stat 48:269–311. doi: 10.1111/1467-9876.00154. [DOI] [Google Scholar]
- 46.Hall T. 2001. BioEdit version 5.0.6. http://jwbrown.mbio.ncsu.edu/Bioedit/BioDoc.pdf.
- 47.Torriani SFF, Stukenbrock EH, Brunner PC, McDonald BA, Croll D. 2011. Evidence for extensive recent intron transposition in closely related fungi. Curr Biol 21:2017–2022. doi: 10.1016/j.cub.2011.10.041. [DOI] [PubMed] [Google Scholar]
- 48.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, 1000 Genomes Project Analysis Group. 2011. The variant call format and VCFtools. Bioinformatics 27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McDonald MC, Williams AH, Milgate A, Pattemore JA, Solomon PS, Hane JK. 2015. Next-generation re-sequencing as a tool for rapid bioinformatic screening of presence and absence of genes and accessory chromosomes across isolates of Zymoseptoria tritici. Fungal Genet Biol 79:71–75. doi: 10.1016/j.fgb.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 52.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 53.Clement M, Posada D, Crandall KA. 2000. TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659. [DOI] [PubMed] [Google Scholar]
- 54.Leigh JW, Bryant D. 2015. popart: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116. doi: 10.1111/2041-210X.12410. [DOI] [Google Scholar]
- 55.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lawson DJ, Hellenthal G, Myers S, Falush D. 2012. Inference of population structure using dense haplotype data. PLoS Genet 8:e1002453. doi: 10.1371/journal.pgen.1002453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ritz C, Baty F, Streibig JC, Gerhard D. 2015. Dose-response analysis using R. PLoS One 10:e0146021. doi: 10.1371/journal.pone.0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.