

Abstract
Cholesterolysis of Hedgehog family proteins couples endoproteolysis to protein C-terminal sterylation. The transformation is self-catalyzed by HhC, a partially characterized enzymatic domain found in precursor forms of Hedgehog. Here we explore spatial ambiguity in sterol recognition by HhC, using a trio of derivatives where the sterol A-ring is contracted, fused, or distorted. Sterylation assays indicate that these geometric variants react as substrates with relative activity: cholesterol, 1.000> A-ring contracted, 0.100> A-ring fused, 0.020> A-ring distorted, 0.005. Experimental results and computational sterol docking into the first HhC homology model suggest a partially unstructured binding site with substrate recognition governed in large part by hydrophobic interactions.
Graphical Abstract
Proteins in the Hedgehog (Hh) family undergo a self-catalyzed endoproteolytic event called cholesterolysis in which the nucleophilic agent is cholesterol. Cholesterolysis separates precursor Hh into two polypeptides: a sterylated fragment with cell-signaling activity (HhN-sterol), and a partially characterized enzymatic fragment, abbreviated HhC (Fig. 1A). The entire transformation appears to be brought about by HhC through an acyl relay mechanism analogous to self-splicing inteins1. However, the specific interactions involved in cholesterol recruitment and activation by HhC remain largely unresolved. Thus we have a limited biochemical understanding of how congenital mutations in HhC pose risk factors for severe birth defects2; while dysregulated Hh expression promotes the progression of sporadic tumors3.
Figure 1.

Assessing A-ring specificity in HhC catalysis. (A) Hedgehog cholesterolysis. Precursor hedgehog undergoes peptide bond rearrangement at a conserved Gly-Cys motif; the resulting thioester is cleaved by substrate sterol (bracket), releasing HhN-sterol and HhC. Catalytic cysteine and aspartate acid residues of HhC depicted as ball-and-strick.(B) Sterols with variant A-ring structures assayed as alternative HhC substrates. Red circles, oxygen atom.
Cholesterol is not the only substrate accepted by HhC. In Drosophila melanogaster, which lacks the full complement of genes for cholesterol biosynthesis, the native substrate for HhC may be a dietary sterol such as yeast ergosterol4, 5. In humans with the congenital disorder Smith-Lemli-Opitz syndrome (SLOS), cholesterol is likely replaced to some extent by the metabolic precursor, 7-dehydro-cholesterol6. Abiraterone is a synthetic anti-androgen used to treat advanced prostate cancer through inhibition of cytochrome P450-17A17, 8. We reported that abiraterone exhibits robust substrate activity with HhC9. Other synthetic variants bearing modifications to cholesterol’s isooctyl tail also show substrate activity and have been useful for metabolic labeling 10. Together, these studies indicate that the substrate tolerance of HhC is relatively broad, at least toward sterols with alterations distant from the site of bond scission.
Here, geometric tolerance is probed at the reaction center using cholesterol A-ring analogs. The A-ring contains sterol’s nucleophilic substituent and is therefore expected to be under stringent constraints. We investigate the substrate activity of an A-ring contracted sterol (II), a pentacyclic cholesterol derivative (III), and the nonplanar cholesterol analogue, coprostanol (V) (Fig. 1B). We employ a continuous assay to monitor HhC activity in the presence of potential substrates by changes in FRET11, 12. The assay uses a 3-part Hh fusion construct, C-H-Y, where HhC is flanked by cyan fluorescent protein and yellow fluorescent protein (Fig. 2A). In the presence of substrate, FRET signal between CFP and YFP rapidly diminishes with release of sterylated CFP. SDS-PAGE, RP-HPLC and mass spectrometry provide independent means of confirming protein sterylation9, 11, 13.
Figure 2.

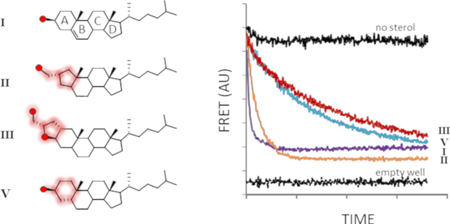
A-ring derivatives retain substrate activity (A) FRET on/off system for continuous monitoring of HhC activity. A tripartite fusion protein C-H-Y comprises cyan fluorescent protein. Drosophila melanogaster HhC and yellow fluorescent protein; sterolysis separates C-H-Y into C-sterol and H-Y, with FRET signal loss. (B) Sterol A-ring variants exhibit varying substrate activity. Representative kinetic traces for reactions of HhC with the indicated sterols at 50 pM. Control traces with C-H-Y with no sterol (top trace) and wells containing buffer only (bottom trace) in black.
We first tested A-nor cholestanol (II), a contracted analogue with a cyclopentyl A-ring and hydroxymethyl substituent at the 2-position. The freely rotating primary alcohol of II replaces cholesterol’s ring-constrained secondary alcohol as the lone nucleophilic group. A-nor cholestanol was prepared in two steps from cholestanone14. Sterol II is reminiscent of naturally occurring A-nor sterols identified in the extracts of certain marine sponges 15.
Despite structural deviation from cholesterol, A-nor cholestanol displayed robust substrate activity in multiple assays with HhC. Shown in Fig. 2B is the kinetic trace for reaction of 50 µM II with the FRET reporter, C-H-Y, where FRET decay reports HhC activity. Controls experiments include reactions with no sterol, with added cholesterol (I), and without the C-H-Y construct. Addition of II resulted in rapid FRET loss, with t1/2 of ~8 min, on par with cholesterol’s t1/2 value of 3 min. Plots of initial velocity versus substrate concentration indicated Km and kmax values for II that deviated from cholesterol by only ~3-fold (Table 1; Sup. Fig. 1A). To establish sterylation with II, we replaced C-H-Y with a closer physiological mimic of Hh precursor, where the human sonic hedgehog signaling domain (SHhN) is translationally fused to Drosophila HhC 12. Addition of substrate sterol activates precursor processing into two products, SHhN-sterol (20 kDa) and DHhC (26 kDa), which readily separate by SDS-PAGE12. As displayed in Sup. Fig. 1B, the reaction of SHhN-DHhC with II generated the expected products. Further, MALDI-TOF analysis identified the sterylated adduct with II (Sup. Table 1). We also assessed the extent of protein sterylation by subjecting the product mixture to reverse-phase HPLC16. In Sup. Fig. 2B, control separations are shown of SHhN-DHhC following incubation in the absence and presence of added cholesterol, where peaks are visible for HhN (minor peak, 8.9 min’), and cholesterylated HhN (13.9 min’), respectively. For samples with A-nor used as substrate, the putative HhN-II product exhibits a retention time (15.1 min’) comparable to the cholesterol adduct, consistent with covalent sterylation. Thus, kinetic and product analysis support the notion that at the ground and transition states for protein sterylation, HhC does not strongly discriminate between cyclohexyl and cyclopentyl A-rings nor is there overwhelming preference for secondary over primary alcohol nucleophiles.
Table 1.
Kinetic parameters for sterol substrates with hedgehog HhC using the FRET reporter assay. Standard errors were less than or equal to ±10% for kmax values and ±16% for KM values, n ≥ 3. Relative proficiency compares second order rate constants (kmax/Km).
| substrate | kmax (s−1) (x10−3) | KM (µM) | Relative proficiency |
|---|---|---|---|
| Cholesterol (I) | 4.7 | 1.9 | 1 |
| II | 1.4 | 4.7 | 0.1 |
| III | 0.49 | 12 | .02 |
| V | 0.27 | 23 | .005 |
| IV | <.005 | nd |
We next probed geometric constraints in the opposite sense using an oversized sterol analog in which the A-ring was fused to a hydroxymethyl tetrahydrofuran 17. Like II, sterylation involves attack by an exocyclic primary alcohol. FRET assays of substrate activity indicated that HhC accepted the pentacyclic α epimer (III) albeit with modest efficiency. The order of reactivity is apparent from comparison of kinetic traces in Fig. 2B and in steady state kinetic parameters in Table 1, where ground state binding of III is 6-fold weaker than cholesterol and the maximum reaction rate is slower by almost 10-fold (see also, Sup. Fig. 1A). Substrate activity of III was corroborated by SDS-PAGE analysis using the chimeric Hh precursor, SHhN-DHhC (Sup. Fig. 1B), and by MALDI-TOF (Sup. Table 1). In addition, the HhN product sterylated with pentacyclic sterol III exhibited the characteristic extended retention time of 13.4 min’ on RP-HPLC (Sup. Fig. 2). Substrate activity with III accords in part with the inference above that a ring constrained alcohol is not imperative, while also revealing a reduced degree of promiscuity in accommodating an extra ring. Likewise, the apparent absence of reactivity with the β epimer (IV) indicates that the substrate range of HhC is not unlimited. Comparison of 3-D models of III, IV and cholesterol shows that the –OH group of the unreactive IV is displaced relative to cholesterol’s –OH group by a distance greater than the –OH group active III epimer. If activation of the sterol –OH group involves a distance dependent mechanism like general base catalysis 18, 19,20, this could explain the observed stereospecificity.
The last analog we evaluated was coprostanol, 5β-cholestan-3β-OL (V), a microbial metabolite of cholesterol 21, which has an A-ring displaced almost 90o (A/B cis) compared with the pseudo planar rings of cholesterol (A/B trans). Of the A-ring analogs accepted by HhC, coprostanol exhibited the weakest substrate activity. We estimate based on kinetic analysis that the Km value for V is 23 µM and the kmax value is 0.27×10−3 sec−1 (Table 1, and Sup. Fig. 1). Thus, coprostanol binds half as tightly and reacts half as slowly with HhC as pentacyclic sterol III. Comparison of coprostanol with cholesterol shows less than 1% activity. Substrate behavior of coprostanol is supported by SDS-PAGE (Sup. Fig. 1B), MALDI-TOF (Sup. Table 1) and analysis of the product mixture by RP-HPLC, where the reaction generates a late eluting, HhN-coprostanol conjugate (Sup. Fig. 2B). The marginal reactivity of HhC toward coprostanol recalls the specificity observed with sterol sterol sulfotransferases SULT2B1b 22 and glucosylating enzymes 23, which exhibit weak or no activity toward V, respectively.
To better understand the substrate ambiguity of HhC, we attempted to integrate the experimental findings with a computational model of HhC-sterol interactions. Early mutagenesis experiments on the Drosophila melanogaster HhC suggest a composite structure, where the first 150 amino acids of HhC is a self-splicing module, called HINT, related structurally to inteins 24, 25; and the final 70 amino acids harbor the sterol binding or recognition region, SRR. While the structural fold of the self-splicing HINT module is well characterized, 3-D structure of the SRR is not yet available. We therefore relied on the published crystal structure of HhC’s HINT 25 and sought a homologous scaffold to construct the adjacent sterol binding SRR. By manually performing pair-wise sequence comparisons a SRR scaffold candidate was identified in the small fungal protein cryptogein (PDB ID: 1LRI) 26. Cryptogein is similar in length to the SRR (90 AA vs. 70 AA) and functions as a sterol binding protein.
Using the cryptogein sequence alignment, we employed MODELLER 27 to generate a provisional homology model of full-length HhC (HINT-SRR) bound to cholesterol. Subsequent optimization through Langevin molecular dynamics (MD) simulations performed using CHARMM program28 with available CHARMM protein 29 and cholesterol 30 force field parameters positioned cholesterol with its attacking -OH group near to HhC catalytic residues. Additional refinements were undertaken based on HhC sequence conservation (See Supporting information).
Docking calculations successfully positioned the A-ring sterol variants at the putative cholesterol binding site in the SRR of the HhC homology model (see overlay Fig. 3). Hydroxyl groups of the sterols also pointed toward the catalytic center, consistent with the proposed mechanism. On the other hand, there was variability among docked positions of sterols in the putative binding site. An obvious molecular explanation for the observed 100-fold range of reactivity among the A-ring variants was not revealed by these initial models. Comparison between the Autodock binding scores and experimental ranking of the sterol analog affinity based on Km values also did not show full agreement. It is possible that initial sterol-HhC encounter promotes a change in HhC conformation, and that our present modeling does not capture this induced fit. The HhC structure should therefore be considered as a testable hypothesis for the molecular basis of sterol recognition, the first that we are aware of, which is to be improved through further experimental validation and refinement.
Figure 3.

Computational model of HhC docks sterols near catalytic cysteine residue. Result combines the solved crystal structure of the HINT module (green) with homology model of sterol recognition region (yellow). Sterols positioned using AutoDock. Right, expanded view of sterol binding site . Hydrophobic residues of the SRR displayed as spheres. Key catalytic cysteine, C258, residue displayed as sticks with sulfur as sphere. Sterol coloring: cholesterol, orange; II, purple; III, red; V, cyan. Nucleophilic hydroxyl group of each sterol displayed as colored sphere.
Carboxyl terminal sterylation is a defining feature of Hh signaling proteins, with the lipid providing a membrane anchor for cell signaling that is spatially restricted31. In the present report, we combined experimental and computational studies of HhC to probe the catalyst’s substrate selectivity, expanding on earlier work that established tolerance of HhC toward sterols bearing modifications of the lipid’s isooctyl side chain. The activity observed here of a trio of A-ring cholesterol analogues suggest that the active site of HhC is spatially accommodating even at the reaction center. The nucleophilic substituent can be switched from secondary to primary alcohol; its position altered by contracting the A-ring or installing an extra ring, or by swapping the A-ring configuration, all while retaining activity. We find these observations consistent with the notion that sterol binding by HhC is governed in large part through a primitive selectivity mechanism. Hydrophobic binding, where nonpolar HhC residues simply collapse around the aliphatic substrate through dispersion forces, offers one possibility. This hypothesis would accord with modeling that suggests hydropathic segments of the sterol binding site lack defined secondary structure.
Supplementary Material
Acknowledgements
We acknowledge generous support from the National Cancer Institute (Grant R01 CA206592) and Department of Defense (Grant W81XWH-14-1-0155). We also acknowledge the upgrade of the 600 MHz NMR spectrometer at SUNY-ESF under NSF grant CHE-1048516 and NIH grant S10 OD012254. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), supported by National Science Foundation grant number ACI-1548562.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Porter JA, Young KE and Beachy PA, Science, 1996, 274, 255–259. [DOI] [PubMed] [Google Scholar]
- 2.Roessler E, Belloni E, Gaudenz K, Vargas F, Scherer SW, Tsui LC and Muenke M, Hum. Mol. Genet, 1997, 6, 1847–1853. [DOI] [PubMed] [Google Scholar]
- 3.Owens AE, de Paola I, Hansen WA, Liu YW, Khare SD and Fasan R, J. Am. Chem. Soc, 2017, 139, 12559–12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karpen HE, Bukowski JT, Hughes T, Gratton JP, Sessa WC and Gailani MR, J Biol Chem, 2001, 276, 19503–19511. [DOI] [PubMed] [Google Scholar]
- 5.Mann RK and Beachy PA, Annu. Rev. Biochem, 2004, 73, 891–923. [DOI] [PubMed] [Google Scholar]
- 6.Cooper MK, Wassif CA, Krakowiak PA, Taipale J, Gong R, Kelley RI, Porter FD and Beachy PA, Nat. Genet, 2003, 33, 508–513. [DOI] [PubMed] [Google Scholar]
- 7.Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, Settatree S, Barrett M, Parker C, Martins V, Folkerd E, Clark J, Cooper CS, Kaye SB, Dearnaley D, Lee G and de Bono JS, J Clin. Oncol, 2008, 26, 4563–4571. [DOI] [PubMed] [Google Scholar]
- 8.DeVore NM and Scott EE, Nature, 2012, 482, 116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bordeau BM, Ciulla DA and Callahan BP, ChemMedChem, 2016, 11, 1983–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ciepla P, Magee AI and Tate EW, Biochem. Soc. Trans, 2015, 43, 262–267. [DOI] [PubMed] [Google Scholar]
- 11.Owen TS, Ngoje G, Lageman TJ, Bordeau BM, Belfort M and Callahan BP, Anal. Biochem, 2015, 488, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Owen TS, Xie XJ, Laraway B, Ngoje G, Wang C and Callahan BP, Chembiochem, 2015, 16, 55–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie J, Owen T, Xia K, Singh AV, Tou E, Li L, Arduini B, Li H, Wan LQ, Callahan B and Wang C, J. Biol. Chem, 2015, 290, 11591–11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Balakrishnan P and Bhattacharyya SC, Indian J. of Chem., Section B: Organic Chemistry Including Medicinal Chemistry, 1986, 25B, 1050–1051. [Google Scholar]
- 15.Minale L and Sodano G, J. Chem. Soc., Perkin I 1974. 2380–2384.
- 16.Baker DP, Taylor FR and Pepinsky RB, Methods Mol Biol, 2007, 397, 1–22. [DOI] [PubMed] [Google Scholar]
- 17.Giner JL, J. Org. Chem, 2005, 70, 721–724. [DOI] [PubMed] [Google Scholar]
- 18.Xie J, Owen T, Xia K, Callahan B and Wang C, J. Am. Chem. Soc, 2016, 138, 10806–10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ciulla DA, Jorgensen MT, Giner JL and Callahan BP, J. Am. Chem. Soc, 2018, 140, 916–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schowen KB, Limbach HH, Denisov GS and Schowen RL, Biochim. Biophys. Acta, 2000, 1458, 43–62. [DOI] [PubMed] [Google Scholar]
- 21.Garcia JL, Uhia I and Galan B, Microb. Biotechnol, 2012, 5, 679–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wojciechowski ZA, Zimowski J, Zimowski JG and Lyznik A, Biochim. Biophys. Acta, 1979, 570, 363–370. [DOI] [PubMed] [Google Scholar]
- 23.Strott CA, Endocr. Rev, 2002, 23, 703–732. [DOI] [PubMed] [Google Scholar]
- 24.Xie J, Du Z, Callahan B, Belfort M and Wang C, Biomol. NMR Assign, 2014, 8, 279–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hall TM, Porter JA, Young KE, Koonin EV, Beachy PA and Leahy DJ, Cell, 1997, 91, 85–97. [DOI] [PubMed] [Google Scholar]
- 26.Lascombe MB, Ponchet M, Venard P, Milat ML, Blein JP and Prange T, Acta. Crystallogr. D Biol. Crystallogr, 2002, 58, 1442–1447. [DOI] [PubMed] [Google Scholar]
- 27.Webb B and Sali A, Curr. Protoc. Bioinformatics, 2014, 47, 561–5632. [DOI] [PubMed] [Google Scholar]
- 28.Brooks BR, Brooks CL 3rd, Mackerell AD Jr., Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM and Karplus M, J. Comp. Chem, 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M and Mackerell AD Jr., J Chem Theory Comput, 2012, 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lim JB, Rogaski B and Klauda JB, J. Phys. Chem. B, 2012, 116, 203–210. [DOI] [PubMed] [Google Scholar]
- 31.Petrov K, Wierbowski BM and Salic A, Annu. Rev. Cell. Dev. Biol, 2017, 33, 145–168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.