

Abstract
Background
Maladaptive remodeling in pressure overload (PO)-induced left ventricular hypertrophy (LVH) may lead to heart failure. Major sex differences have been reported in this process. The steroid hormone 17β-estradiol, along with its receptors ERα and ERβ, is thought to be crucial for sex differences and is expected to be protective, but this may not hold true for males. Increasing evidence demonstrates a major role for microRNAs (miRNAs) in PO-induced LVH. However, little is known about the effects of biological sex and ERβ on cardiac miRNA regulation and downstream mitochondrial targets. We aimed at the analysis of proteins involved in mitochondrial metabolism testing the hypothesis that they are the target of sex-specific miRNA regulation.
Methods
We employed the transverse aortic constriction model in mice and assessed the levels of five mitochondrial proteins, i.e., Auh, Crat, Decr1, Hadha, and Ndufs4.
Results
We found a significant decrease of the mitochondrial proteins primarily in the male overloaded heart compared with the corresponding control group. Following computational analysis to identify miRNAs putatively targeting these proteins, our in vitro experiments employing miRNA mimics demonstrated the presence of functional target sites for miRNAs in the 3′-untranslated region of the messenger RNAs coding for these proteins. Next, we assessed the levels of the functionally validated miRNAs under PO and found that their expression was induced only in the male overloaded heart. In contrast, there was no significant effect on miRNA expression in male mice with deficient ERβ.
Conclusion
We put forward that the male-specific induction of miRNAs and corresponding downregulation of downstream protein targets involved in mitochondrial metabolism may contribute to sex-specific remodeling in PO-induced LVH.
Keywords: Cardiac, ERβ, microRNA, Mitochondrial, Pressure overload, Sex differences
Background
Under pressure overload (PO) conditions as in aortic stenosis or hypertension, left ventricular (LV) hypertrophy (LVH) develops. Following the initial adaptive LV remodeling, when PO persists, maladaptive LV remodeling leads to the development of heart failure. In the process of LVH development and the transition to heart failure, major sex differences have been reported (reviewed in [1]). Briefly, women develop a more concentric form of LVH with less ventricular dilation and wall thinning than men, thereby leading to sex-specific cardiac dysfunction [2, 3]. We and others have reported a stronger induction of fibrosis and fibrotic gene expression in the male versus female overloaded heart [4–6]. Similarly, our previous studies with experimental animals revealed that the female sex is associated with less pronounced ventricular dilation and fibrosis in response to PO [7]. On the basis of the identified sex-specific fibrotic gene regulation, we further reported the sex-specific regulation of six fibrosis-related microRNAs (miRNA) in the mouse overloaded heart [8].
miRNAs are small non-coding RNAs of approximately 22 nucleotides [9] that negatively regulate gene expression pairing to the 3′ untranslated region (3′-UTR) of target messenger RNAs (mRNAs) [10]. The expression of several miRNAs has been described in the mouse heart, many of which are regulated during the development of LVH [11–15]. However, the impact of biological sex on miRNA regulation and the relevance for the documented sex differences in cardiovascular disease are poorly understood.
The steroid hormone 17β-estradiol (E2), along with its receptors ERα and ERβ, is thought to play a major role in the development of sex differences in cardiovascular disease. In fact, the E2/ER axis is expected to be protective, but this may not hold true for males. In females, though, ERβ has been shown to confer cardio-protection under PO [16–18]. The mechanisms underlying these effects have been the subject of intense investigations (reviewed in [1]), and we have shown that deletion of ERβ leads to the modulation of several genes in the overloaded heart [7, 19]. However, it is not clear to what extent mitochondrial proteins might be regulated in a sex-specific manner under PO and what the role of miRNAs might be in this regulation. An aberrant regulation of mitochondrial function and bioenergetics in the heart, a high energy-demanding organ, could be detrimental leading to LV dilation and dysfunction.
On the basis of the aforementioned transcriptomic studies in PO [7, 19], we aimed here at characterizing under PO the modulation of five proteins, namely, AU RNA binding methylglutaconyl-CoA hydratase (Auh), carnitine O-acetyltransferase (Crat), 2,4-dienoyl-CoA reductase 1 (Decr1), hydroxyacyl-CoA dehydrogenase alpha subunit (Hadha), and NADH:ubiquinone oxidoreductase subunit S4 (Ndufs4), due to their importance and involvement in mitochondrial metabolism. We tested the hypothesis that these proteins are the target of sex-specific miRNA regulation.
Methods
Experimental animals
Male and female C57Bl/6J mice at the age of 2 months were employed. To study the effects of ERβ on miRNA regulation in vivo, ERβ knockout (ERβ−/−) mice generated from heterozygous mouse colonies [20] were employed. The genotype of the mice was screened using PCR amplification as described previously [20]. The mice were kept on a 12–12-h light/dark cycle in temperature-controlled rooms with water ad libitum. All experiments were carried out in accordance with the EU Directive 2010/63/EU for animal experiments and approved by the Landesamt für Gesundheit und Soziales, Berlin, Germany.
Transverse aortic constriction
To induce PO in mice, the transverse aortic constriction (TAC) method was used as described previously [7]. Two-month-old mice were anesthetized with ketamine hydrochloride (80 mg/ml)/xylazine hydrochloride (12 mg/ml) administered by intraperitoneal injection at a dose of 1 mg/kg. Sham animals underwent an identical surgical procedure without placement of a suture. Animals recovered from anesthesia under warming conditions and normal ventilation. After 9 weeks, the mice were sacrificed by isoflurane overdose followed by laryngotomy. Following weight measurements, the hearts were snap frozen in liquid nitrogen and stored at − 80 °C until further analysis.
Immunoblotting
Protein lysates were isolated from LV tissue samples as described previously [21, 22]. Independent biological replicates for each group were run separately on SDS-PAGE gels and transferred to nitrocellulose membranes using standard procedures. Primary antibodies against Auh (ab155980, Abcam), Crat (ab153699, Abcam), Decr1 (sc-366484, Santa Cruz), Hadha (sc-82185, Santa Cruz), Ndufs4 (ab139178, Abcam), Tim23 (611222, BD; control for mitochondrial structural integrity), Tom40 (sc-365466, Santa Cruz; control for mitochondrial structural integrity), and tubulin (T9026, Sigma-Aldrich; loading control) were used. For immunodetection, secondary antibody donkey anti-rabbit, anti-goat, or anti-mouse (Dianova) and ECL™ Prime Western Blotting Reagent (Amersham) were used. Data were quantified with the ImageJ 1.41 version software (http://rsbweb.nih.gov/ij/).
Computational analysis
TargetScan (http://www.targetscan.org) was used for the identification of putative miRNA binding sites in the 3′-UTRs of the mRNAs coding for the mitochondrial targets as previously described [8, 23].
Cell culture experiments
The cardiac muscle cell line HL-1 was used for target validation studies and cultured as described previously [24, 25]. For luciferase/renilla reporter assays, 3′-UTRs [Auh NM_016709.2, Crat NM_007760.3, Decr1 NM_026172.3, Hadha NM_178878.2, Ndufs4 NM_010887.2] or putative target sites were site directed cloned (XhoI/NotI) in psiCheck™-2 (Promega) behind the renilla coding region and a Dual-Glo® Luciferase Assay (Promega) was performed. Transient transfection was performed with FuGene HD (Promega) or Attractene (Qiagen) following the provider’s instructions [26]. The assays including the addition of specific miRNA mimic (1 μl, 20 μM/well) were done by electroporation using the Amaxa™ Cell Line Nucleofector™ Kit V (Lonza, protocol T020) followed by a second transfection with the reporter clones 24 h later. The following miRNA mimics were used: Syn-mmu-miR-106a-5p, MSY0000385; Syn-mmu-miR-130a-3p, MSY0000141; Syn-mmu-miR-133a-3p, MSY0000145; Syn-mmu-miR-143-3p, MSY0000247; Syn-mmu-miR-19b-3p, MSY0000513; Syn-mmu-miR-199b-5p, MSY0000672; Syn-mmu-miR-23a-3p, MSY0000532; Syn-mmu-miR-24, MSY0000219; Syn-mmu-miR-27b-3p, MSY0000126; Syn-mmu-miR-29a-3p, MSY0000535; Syn-mmu-miR-497a-5p, MSY0003453; Syn-mmu-let-7e-5p, MSY0000524 (Qiagen).
Real-time quantitative RT-PCR
RNA was isolated using the miRNeasy kit (Qiagen). Purified miRNAs were reverse transcribed with the miScript Reverse Transcription kit (Qiagen), followed by real-time PCR using the QuantiTect SYBR Green PCR kit (Qiagen) using the primers let-7e: TGAGGTAGGAGGTTGTATAGTT; miR-23a: TCACATTGCCAGGGATTT; miR-27b: TCACAGTGGCTAAGTTCTGC; miR-130a: AGTGCAATGTTAAAAGGGC; miR-133a: CCCCTTCAACCAGCTG; miR-TGAGATGAAGCACTGTAGCTC. The amount of miRNA was normalized using the average of the expression of RNU6B and RNU1A (Qiagen internal references).
Statistical analysis
All data are presented as mean ± SEM. Statistical significance was assessed using the R 2.14.2 software. Comparisons among multiple groups were made with two-way ANOVA and Tukey’s post hoc test adjusting for multiple comparisons. P ≤ 0.05 was considered significant.
Results
Sex-specific regulation of the mitochondrial targets under pressure overload
Our previous human and animal studies on PO have indicated the sex-specific regulation of mitochondrial metabolism-related genes [4, 7]. Based on this, among the identified regulated candidates, we selected five proteins, due to their importance and involvement in mitochondrial metabolism, and assessed their levels in response to PO. For this purpose, we analyzed LV samples from male and female mice subjected to PO (TAC vs. sham), which showed significant sex differences in the development of LVH (Table 1). In line and as we had hypothesized, we found that the selected mitochondrial proteins were regulated in a sex-specific manner (Fig. 1). In particular, the abundance of all five proteins was significantly decreased in male mice subjected to TAC compared with the corresponding sham group (Fig. 1). In contrast, there was no significant effect between TAC and sham in female mice for four of these proteins (Fig. 1). Only the abundance of Auh was decreased in female mice subjected to TAC compared with the corresponding sham group. However, it is worth noting that the downregulation of Auh in female mice was not as pronounced as in male mice.
Table 1.
Data on body and heart weight
| Male | Female | |||
|---|---|---|---|---|
| Sham | TAC | Sham | TAC | |
| Body weight (g) | 27.0 ± 0.6e | 28.2 ± 0.7f | 23.4 ± 0.4 | 23.1 ± 0.2 |
| Heart weight (g) | 0.117 ± 0.004 | 0.206 ± 0.014c,e | 0.097 ± 0.003 | 0.154 ± 0.013b |
| Tibia length (mm) | 17.3 ± 0.1 | 17.5 ± 0.2 | 17.3 ± 0.1 | 17.1 ± 0.2 |
| Heart weight/tibia length | 6.74 ± 0.2 | 11.83 ± 0.9c,d | 5.62 ± 0.1 | 9.04 ± 0.8a |
Data present mean ± SEM; n = 6–10/group
aP < 0.05
bP < 0.01
cP < 0.001 vs. corresponding sham group
dP < 0.05
eP < 0.01
fP < 0.001 vs. female
Fig. 1.
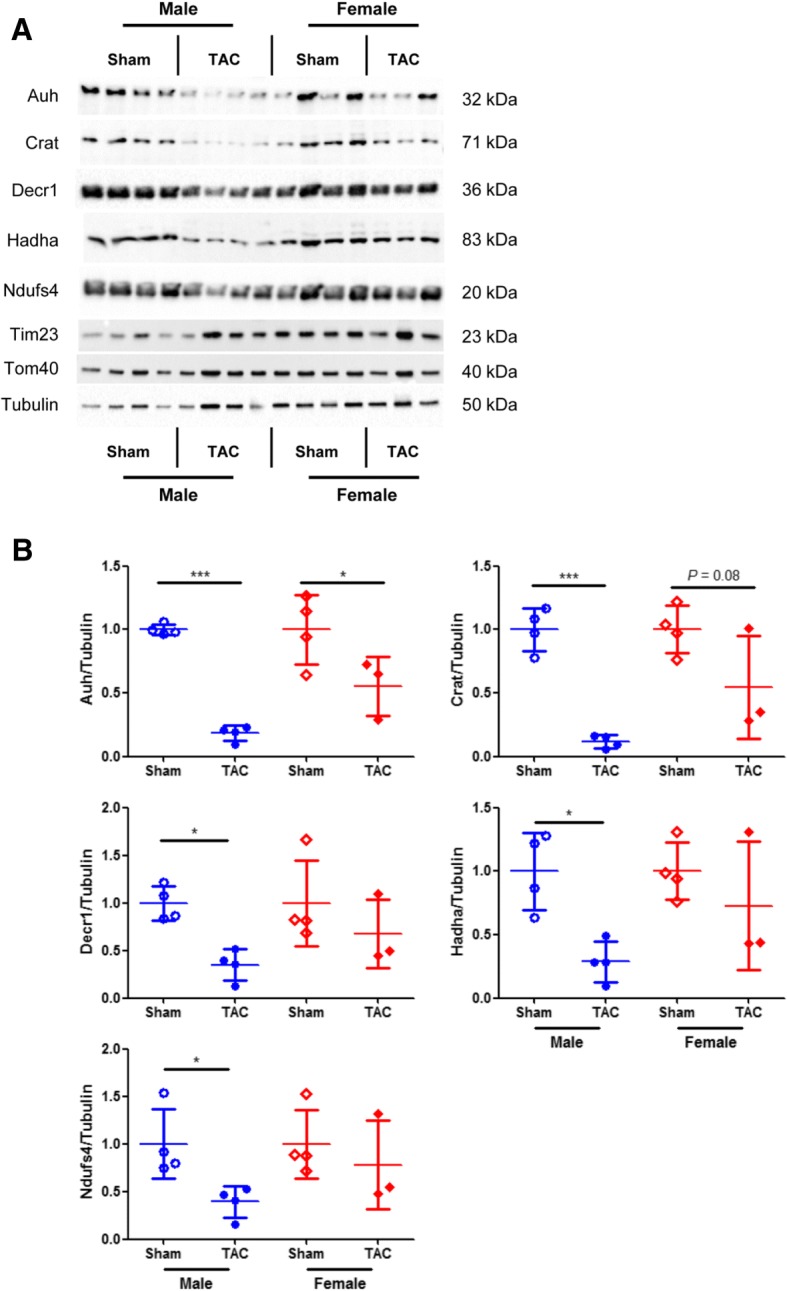
Sex-specific mitochondrial protein regulation under pressure overload. Representative immunoblots (a) and quantification of protein abundance (b) in LV samples from male sham (n = 4), male TAC (n = 4), female sham (n = 4), and female TAC (n = 3) mice. Tim23 and Tom40 were used as controls for mitochondrial structural integrity and tubulin as loading control. Data are presented in scatter dot plots including mean with SD. ***P < 0.001, *P < 0.05
Identification of miRNAs putatively regulating the mitochondrial targets
Because miRNAs are important regulators of transcription, we sought to identify those miRNAs that could putatively target the mRNA of the analyzed mitochondrial proteins. In order to do this, we exploited miRNA target prediction algorithms performing a TargetScan analysis [23]. As expected, we identified several putative binding sites for various miRNAs in the mRNA of each of these mitochondrial proteins (Table 2).
Table 2.
Putative miRNAs regulating the mRNA of mitochondrial proteins
| Mitochondrial protein | Putative miRNA regulator |
|---|---|
| Auh | miR-19b, miR-23a, miR-133a |
| Crat | miR-23a, miR-24, miR-27b, miR-29a, miR-143, let-7e |
| Decr1 | miR-106a, miR-130a, miR-199b, miR-497a, let-7e |
| Hadha | miR-19b, miR-27b, miR-130a, miR-143, let-7e |
| Ndufs4 | miR-23a, miR-27b, miR-106a, miR-130a |
Validation of miRNA binding sites in the mRNA of the mitochondrial targets
To confirm the functionality of the binding sites for the identified miRNAs and to examine the possible involvement of these miRNAs in the regulation of the mitochondrial targets, we determined the effect of miRNA mimics in clones carrying the 3′-UTR from each protein-coding mouse mRNA of the mitochondrial targets as part of gain of function experiments in cardiac muscle cells (HL-1). The functional binding of miRNA mimics to the cloned 3′-UTR would lead to reduced expression compared with the no mimic group. To discard false positive findings due to unspecific effects, we employed a miRNA mimic that had no predicted target sites in each corresponding 3′-UTR as negative control (black bars in Fig. 2). As expected, not every putative miRNA binding site was biologically functional (Fig. 2). Only miR-133a reduced Auh expression; miR-23a, miR-27b, and let-7e reduced Crat; miR-130a reduced Decr1; miR-143 reduced Hadha; and miR-27b reduced Ndufs4 (Fig. 2). Taken together, all mRNAs coding for the analyzed mitochondrial proteins carry functional binding sites for at least one of the identified miRNAs.
Fig. 2.

Active miRNA binding sites in the 3′-UTRs of mitochondrial targets. Effect of specific miRNA mimics on complete 3′-UTR clones of Auh, Crat, Decr1, Hadha, and Ndufs4 in cardiac muscle cells (HL-1). Black bars indicate the respective non-specific mimic controls. Data present mean ± SEM. n = 5/group; ***P < 0.001, **P < 0.01, *P < 0.05
Sex-specific miRNA regulation under pressure overload
Since the levels of the analyzed mitochondrial proteins were decreased in male mice under PO, we tested the hypothesis that this should be associated with a male-specific induction of the functionally confirmed miRNAs targeting the mRNA of these mitochondrial proteins. On the basis of this, we assessed the levels of these miRNAs and found that their expression was significantly induced in male mice subjected to TAC compared with the corresponding sham group (Fig. 3). In contrast, there was no major effect between TAC and sham in female mice (Fig. 3).
Fig. 3.

Sex-specific miRNA regulation under pressure overload. The levels of miRNAs were assessed in LV samples from male sham (n = 10), male TAC (n = 10), female sham (n = 7), and female TAC (n = 10) mice with intact ERβ. Data are presented in scatter dot plots including mean with SD. ***P < 0.001, **P < 0.01, *P < 0.05
Involvement of ERβ in miRNA regulation
The sex-specific miRNA expression together with our previous findings on the effects of ERβ in PO [4, 7, 27] led us to the hypothesis that ERβ may play a role in the regulation of the identified miRNAs. For this purpose, we determined the effect of ERβ genetic deletion (ERβ−/−) on cardiac miRNA expression assessing the levels of the miRNAs in LV samples of male and female ERβ−/− mice in response to PO. There was no significant effect between TAC and sham in male or female ERβ−/− mice for five of these miRNAs (Fig. 4). Only the expression of miR-143 was decreased in male mice subjected to TAC compared with the corresponding sham group (Fig. 4).
Fig. 4.

Sex-specific miRNA regulation under pressure overload in ERβ−/− mice. The levels of miRNAs were assessed in LV samples from male sham, male TAC, female sham, and female TAC mice with deficient ERβ (ERβ−/−). Data are presented in scatter dot plots including mean with SD. n = 8/group; *P < 0.05
Discussion
In the present study, we found that biological sex plays a major role in the regulation of miRNAs targeting proteins involved in mitochondrial metabolism in the heart, thereby leading to sex-specific cardiac protein regulation (Fig. 5).
Fig. 5.
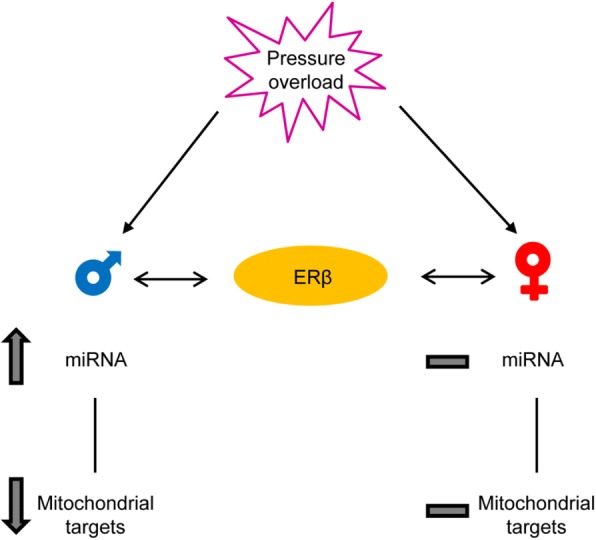
Schematic representation showing the sex-specific effects on miRNA levels and mitochondrial targets in response to pressure overload
There are pronounced differences between males and females in the development of LVH and the transition to heart failure. Although several factors may influence disease progression and outcome [28], the female sex is generally thought to be protective to a certain degree. The effects are prominent at the structural and functional level. Mitochondrial metabolism and bioenergetics in the heart, which is a high energy-demanding organ, are of utmost importance. Based on our previous human and animal studies [4, 7], where we found sex-specific regulation of mitochondrial metabolism-related genes, we report here the significant downregulation of five proteins involved in mitochondrial metabolism in response to PO primarily in male mice. These include Auh, whose aberrant regulation leads to changes in mitochondrial morphology and decreased mitochondrial biogenesis and respiratory function [29]; Crat, Decr1, and Hadha, which play a crucial role in the metabolism and transport of fatty acids for β-oxidation [30–32]; and Ndufs4, an important accessory subunit of the mitochondrial membrane respiratory chain NADH dehydrogenase (complex I) [33]. The overall downregulation of these mitochondrial proteins could have important consequences for sex differences in LVH and male-specific dysfunction documented previously [34, 35], and it requires further research.
Since miRNAs have been identified as novel important regulators of transcription with an ability to simultaneously regulate multiple elements of relevant pathways and several of them have been shown to be regulated in cardiovascular disease [36], including PO [11–15] and cardiac arrhythmias [37], we sought to identify those miRNAs targeting the modulated mitochondrial proteins. Our computational analysis and in vitro experiments identified and validated six miRNAs targeting the mRNAs of the five mitochondrial proteins. Since little is known about the role of sex in the regulation of cardiac miRNAs, we hypothesized and confirmed that these miRNAs are regulated in a sex-specific manner in response to PO. In particular, we found their expression to be induced only in male overloaded hearts. We further ensured that the male-specific downregulation of the target proteins was due to the induction of the regulating miRNAs and not simply due to a general inhibition of mitochondrial proteins in males, since there was no major effect in the levels of Tim23 and Tom40, two important mitochondrial structural proteins involved in the translocation of proteins into the mitochondria. Previous reports corroborate our findings on miR-23a and miR-27b showing induced expression in response to PO [13, 14]. However, others have reported the opposite direction, i.e., downregulation, of miR-133a in response to PO [12, 15], while a third study corroborates our findings on this miRNA [14]. Such discrepancies might be due to differences in the sex, which is not always declared, the strain [38], and age of the mice employed, as well as the time of induction after TAC and its duration before tissue collection [11–15].
The E2/ER axis is expected to be one of the major factors mediating sex-specific effects in the heart. In fact, we have previously reported direct effects of E2 in the heart [19, 38–41], which may differ significantly between the sexes [8, 27, 42, 43]. Notably, we found that E2 exerts deleterious effects in males increasing pro-fibrotic gene expression or impairing contractile function [5, 43]. Along this line, increased circulating E2 levels are a significant predictor of poor prognosis and higher mortality in men with chronic heart failure and reduced ejection fraction [44]. However, the underlying mechanisms and any role of ERβ are incompletely understood. The present data show that in male mice with intact ERβ, there was an increase in miRNA expression in response to PO. In contrast, in male mice with deficient ERβ, there was no significant effect in all miRNAs but miR-143. Overall, these data suggest that ERβ may contribute to a significantly altered expression of cardiac miRNAs.
In addition, the present study raises points that require further research. For example, it is not understood how ERβ might regulate miRNAs in a sex-specific manner. Sex-specific co-factor binding or ER activation may be potential mechanisms involved in this process. Furthermore, miR-143 was downregulated upon genetic deletion of ERβ. This indicates that ERα might also regulate cardiac miRNA expression, which, however, was not within the focus of this study. Similarly, the levels of Auh were decreased in response to PO in female mice, but there was no major effect on miR-133a. This may be due to another miRNA affecting the levels of Auh in females, which, however, was not identified by the present pipeline. To this extent, limitations in the computational analysis for the identification of putative miRNA binding sites are well known in the field. Moreover, due to the previously unrecognized role in the development of PO-induced LVH, the male-specific ERβ-mediated regulation of miRNAs and downstream mitochondrial proteins requires further investigation to identify potential functional consequences on mitochondrial metabolism and contribution to sex-specific cardiac remodeling and function.
Perspectives and significance
In response to PO, a set of proteins involved in mitochondrial metabolism were regulated in a sex-specific manner, targeted by sex-specific miRNA regulation. A better understanding of the sex differences in the regulation of miRNAs and downstream targets, as well as the elucidation of the underlying mechanisms, will contribute towards the development of more appropriate therapeutic approaches for both sexes.
Acknowledgements
We thank the Sonnenfeld Stiftung for providing a spectrophotometer, as well as J. Jansen, N. Haritonow and V. Riese for technical assistance.
Funding
This work was supported by the DSHF (Deutsche Stiftung für Herzforschung), the DZHK (German Centre for Cardiovascular Research), the BMBF (German Ministry for Education and Research), the DFG (German Research Foundation) and the Open Access Publication Fund of Charité – Universitätsmedizin Berlin. The funding bodies had no role in the design of the study, collection, analysis and interpretation of the data, and in writing the manuscript.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors’ contributions
HSR, VRZ, and GK designed the research. HSR, AMQ, DF, and CE performed the experiments. HSR, AMQ, and GK analyzed the data. GK drafted the manuscript. HSR, AMQ, DF, CE, and VRZ revised critically the manuscript. All authors read and approved of the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hugo Sanchez-Ruderisch, Email: hugo.sanchez@charite.de.
Ana Maria Queirós, Email: ana.m.queiros@gmail.com.
Daniela Fliegner, Email: daniela.fliegner@pfizer.com.
Claudia Eschen, Email: claudiaeschen@gmx.de.
Georgios Kararigas, Phone: +49-30-450525355, Email: georgekararigas@gmail.com.
Vera Regitz-Zagrosek, Email: vera.regitz-zagrosek@charite.de.
References
- 1.Regitz-Zagrosek V, Kararigas G. Mechanistic pathways of sex differences in cardiovascular disease. Physiol Rev. 2017;97(1):1–37. doi: 10.1152/physrev.00021.2015. [DOI] [PubMed] [Google Scholar]
- 2.Carroll JD, Carroll EP, Feldman T, Ward DM, Lang RM, McGaughey D, et al. Sex-associated differences in left ventricular function in aortic stenosis of the elderly. Circulation. 1992;86(4):1099–1107. doi: 10.1161/01.CIR.86.4.1099. [DOI] [PubMed] [Google Scholar]
- 3.Villari B, Campbell SE, Schneider J, Vassalli G, Chiariello M, Hess OM. Sex-dependent differences in left ventricular function and structure in chronic pressure overload. Eur Heart J. 1995;16(10):1410–1419. doi: 10.1093/oxfordjournals.eurheartj.a060749. [DOI] [PubMed] [Google Scholar]
- 4.Kararigas G, Dworatzek E, Petrov G, Summer H, Schulze TM, Baczko I, et al. Sex-dependent regulation of fibrosis and inflammation in human left ventricular remodelling under pressure overload. Eur J Heart Fail. 2014;16(11):1160–1167. doi: 10.1002/ejhf.171. [DOI] [PubMed] [Google Scholar]
- 5.Petrov G, Regitz-Zagrosek V, Lehmkuhl E, Krabatsch T, Dunkel A, Dandel M, et al. Regression of myocardial hypertrophy after aortic valve replacement: faster in women? Circulation. 2010;122(11 Suppl):S23–S28. doi: 10.1161/CIRCULATIONAHA.109.927764. [DOI] [PubMed] [Google Scholar]
- 6.Villar AV, Llano M, Cobo M, Exposito V, Merino R, Martin-Duran R, et al. Gender differences of echocardiographic and gene expression patterns in human pressure overload left ventricular hypertrophy. J Mol Cell Cardiol. 2009;46(4):526–535. doi: 10.1016/j.yjmcc.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 7.Fliegner D, Schubert C, Penkalla A, Witt H, Kararigas G, Dworatzek E, et al. Female sex and estrogen receptor-beta attenuate cardiac remodeling and apoptosis in pressure overload. Am J Physiol Regul Integr Comp Physiol. 2010;298(6):R1597–R1606. doi: 10.1152/ajpregu.00825.2009. [DOI] [PubMed] [Google Scholar]
- 8.Queiros AM, Eschen C, Fliegner D, Kararigas G, Dworatzek E, Westphal C, et al. Sex- and estrogen-dependent regulation of a miRNA network in the healthy and hypertrophied heart. Int J Cardiol. 2013;169(5):331–338. doi: 10.1016/j.ijcard.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 9.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 10.Lai EC. Micro RNAs are complementary to 3' UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet. 2002;30(4):363–364. doi: 10.1038/ng865. [DOI] [PubMed] [Google Scholar]
- 11.Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13(5):613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 12.Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, et al. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol. 2007;170(6):1831–1840. doi: 10.2353/ajpath.2007.061170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. 2007;100(3):416–424. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- 14.Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, et al. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2007;42(6):1137–1141. doi: 10.1016/j.yjmcc.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. 2006;103(48):18255–18260. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Babiker FA, Lips D, Meyer R, Delvaux E, Zandberg P, Janssen B, et al. Estrogen receptor beta protects the murine heart against left ventricular hypertrophy. Arterioscler Thromb Vasc Biol. 2006;26(7):1524–1530. doi: 10.1161/01.ATV.0000223344.11128.23. [DOI] [PubMed] [Google Scholar]
- 17.Pedram A, Razandi M, Lubahn D, Liu J, Vannan M, Levin ER. Estrogen inhibits cardiac hypertrophy: role of estrogen receptor-beta to inhibit calcineurin. Endocrinology. 2008;149(7):3361–3369. doi: 10.1210/en.2008-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skavdahl M, Steenbergen C, Clark J, Myers P, Demianenko T, Mao L, et al. Estrogen receptor-beta mediates male-female differences in the development of pressure overload hypertrophy. Am J Physiol Heart Circ Physiol. 2005;288(2):H469–H476. doi: 10.1152/ajpheart.00723.2004. [DOI] [PubMed] [Google Scholar]
- 19.Kararigas G, Fliegner D, Gustafsson JA, Regitz-Zagrosek V. Role of the estrogen/estrogen-receptor-beta axis in the genomic response to pressure overload-induced hypertrophy. Physiol Genomics. 2011;43(8):438–446. doi: 10.1152/physiolgenomics.00199.2010. [DOI] [PubMed] [Google Scholar]
- 20.Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, et al. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc Natl Acad Sci U S A. 1998;95(26):15677–15682. doi: 10.1073/pnas.95.26.15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dworatzek E, Baczko I, Kararigas G. Effects of aging on cardiac extracellular matrix in men and women. Proteomics Clin Appl. 2016;10(1):84–91. doi: 10.1002/prca.201500031. [DOI] [PubMed] [Google Scholar]
- 22.Octavia Y, Kararigas G, de Boer M, Chrifi I, Kietadisorn R, Swinnen M, et al. Folic acid reduces doxorubicin-induced cardiomyopathy by modulating endothelial nitric oxide synthase. J Cell Mol Med. 2017;21(12):3277–3287. doi: 10.1111/jcmm.13231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 24.Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A. 1998;95(6):2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mahmoodzadeh S, Fritschka S, Dworatzek E, Pham TH, Becher E, Kuehne A, et al. Nuclear factor-kappaB regulates estrogen receptor-alpha transcription in the human heart. J Biol Chem. 2009;284(37):24705–24714. doi: 10.1074/jbc.M109.000463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai S, Collins BC, Colson BA, Kararigas G, Lowe DA. Estradiol modulates myosin regulatory light chain phosphorylation and contractility in skeletal muscle of female mice. Am J Physiol Endocrinol Metab. 2016;310(9):E724–E733. doi: 10.1152/ajpendo.00439.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kararigas G, Fliegner D, Forler S, Klein O, Schubert C, Gustafsson JA, et al. Comparative proteomic analysis reveals sex and estrogen receptor beta effects in the pressure overloaded heart. J Proteome Res. 2014;13(12):5829–5836. doi: 10.1021/pr500749j. [DOI] [PubMed] [Google Scholar]
- 28.Gaignebet L, Kararigas G. En route to precision medicine through the integration of biological sex into pharmacogenomics. Clin Sci (Lond) 2017;131(4):329–342. doi: 10.1042/CS20160379. [DOI] [PubMed] [Google Scholar]
- 29.Richman TR, Davies SM, Shearwood AM, Ermer JA, Scott LH, Hibbs ME, et al. A bifunctional protein regulates mitochondrial protein synthesis. Nucleic Acids Res. 2014;42(9):5483–5494. doi: 10.1093/nar/gku179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jogl G, Hsiao YS, Tong L. Structure and function of carnitine acyltransferases. Ann N Y Acad Sci. 2004;1033:17–29. doi: 10.1196/annals.1320.002. [DOI] [PubMed] [Google Scholar]
- 31.Alphey MS, Yu W, Byres E, Li D, Hunter WN. Structure and reactivity of human mitochondrial 2,4-dienoyl-CoA reductase: enzyme-ligand interactions in a distinctive short-chain reductase active site. J Biol Chem. 2005;280(4):3068–3077. doi: 10.1074/jbc.M411069200. [DOI] [PubMed] [Google Scholar]
- 32.L IJ, Ruiter JP, Hoovers JM, Jakobs ME, Wanders RJ. Common missense mutation G1528C in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Characterization and expression of the mutant protein, mutation analysis on genomic DNA and chromosomal localization of the mitochondrial trifunctional protein alpha subunit gene. J Clin Invest. 1996;98(4):1028–1033. doi: 10.1172/JCI118863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scacco S, Petruzzella V, Budde S, Vergari R, Tamborra R, Panelli D, et al. Pathological mutations of the human NDUFS4 gene of the 18-kDa (AQDQ) subunit of complex I affect the expression of the protein and the assembly and function of the complex. J Biol Chem. 2003;278(45):44161–44167. doi: 10.1074/jbc.M307615200. [DOI] [PubMed] [Google Scholar]
- 34.Douglas PS, Katz SE, Weinberg EO, Chen MH, Bishop SP, Lorell BH. Hypertrophic remodeling: gender differences in the early response to left ventricular pressure overload. J Am Coll Cardiol. 1998;32(4):1118–1125. doi: 10.1016/S0735-1097(98)00347-7. [DOI] [PubMed] [Google Scholar]
- 35.Weinberg EO, Thienelt CD, Katz SE, Bartunek J, Tajima M, Rohrbach S, et al. Gender differences in molecular remodeling in pressure overload hypertrophy. J Am Coll Cardiol. 1999;34(1):264–273. doi: 10.1016/S0735-1097(99)00165-5. [DOI] [PubMed] [Google Scholar]
- 36.Wronska A, Kurkowska-Jastrzebska I, Santulli G. Application of microRNAs in diagnosis and treatment of cardiovascular disease. Acta Physiol (Oxf) 2015;213(1):60–83. doi: 10.1111/apha.12416. [DOI] [PubMed] [Google Scholar]
- 37.Santulli G, Iaccarino G, De Luca N, Trimarco B, Condorelli G. Atrial fibrillation and microRNAs. Front Physiol. 2014;5:15. doi: 10.3389/fphys.2014.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kararigas G, Nguyen BT, Zelarayan LC, Hassenpflug M, Toischer K, Sanchez-Ruderisch H, et al. Genetic background defines the regulation of postnatal cardiac growth by 17beta-estradiol through a beta-catenin mechanism. Endocrinology. 2014;155(7):2667–2676. doi: 10.1210/en.2013-2180. [DOI] [PubMed] [Google Scholar]
- 39.Kararigas G, Nguyen BT, Jarry H. Estrogen modulates cardiac growth through an estrogen receptor alpha-dependent mechanism in healthy ovariectomized mice. Mol Cell Endocrinol. 2014;382(2):909–914. doi: 10.1016/j.mce.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 40.Schubert C, Raparelli V, Westphal C, Dworatzek E, Petrov G, Kararigas G, et al. Reduction of apoptosis and preservation of mitochondrial integrity under ischemia/reperfusion injury is mediated by estrogen receptor beta. Biol Sex Differ. 2016;7:53. doi: 10.1186/s13293-016-0104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schuster I, Mahmoodzadeh S, Dworatzek E, Jaisser F, Messaoudi S, Morano I, et al. Cardiomyocyte-specific overexpression of oestrogen receptor beta improves survival and cardiac function after myocardial infarction in female and male mice. Clin Sci (Lond). 2016;130(5):365–376. doi: 10.1042/CS20150609. [DOI] [PubMed] [Google Scholar]
- 42.Kararigas G, Becher E, Mahmoodzadeh S, Knosalla C, Hetzer R, Regitz-Zagrosek V. Sex-specific modification of progesterone receptor expression by 17beta-oestradiol in human cardiac tissues. Biol Sex Differ. 2010;1(1):2. doi: 10.1186/2042-6410-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kararigas G, Bito V, Tinel H, Becher E, Baczko I, Knosalla C, et al. Transcriptome characterization of estrogen-treated human myocardium identifies myosin regulatory light chain interacting protein as a sex-specific element influencing contractile function. J Am Coll Cardiol. 2012;59(4):410–417. doi: 10.1016/j.jacc.2011.09.054. [DOI] [PubMed] [Google Scholar]
- 44.Jankowska EA, Rozentryt P, Ponikowska B, Hartmann O, Kustrzycka-Kratochwil D, Reczuch K, et al. Circulating estradiol and mortality in men with systolic chronic heart failure. JAMA. 2009;301(18):1892–1901. doi: 10.1001/jama.2009.639. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.