

Abstract
Background
Ultra-rare genetic variants, including non-recurrent copy number variations (CNVs) affecting important dosage-sensitive genes, are important contributors to the etiology of neurodevelopmental disorders (NDDs). Pairing family-based whole-genome sequencing (WGS) with detailed phenotype data can enable novel gene associations in NDDs.
Methods
We performed WGS of six members from a three-generation family, where three individuals each had a spectrum of features suggestive of a NDD. CNVs and sequence-level variants were identified and further investigated in disease and control databases.
Results
We identified a novel 252-kb deletion at 15q21 that overlaps the synaptic gene DMXL2 and the gene GLDN. The microdeletion segregated in NDD-affected individuals. Additional rare inherited and de novo sequence-level variants were found that may also be involved, including a missense change in GRIK5. Multiple CNVs and loss-of-function sequence variants affecting DMXL2 were discovered in additional unrelated individuals with a range of NDDs.
Conclusions
Disruption of DMXL2 may predispose to NDDs including autism spectrum disorder. The robust interpretation of private variants requires a multifaceted approach that incorporates multigenerational pedigrees and genome-wide and population-scale data.
Keywords: Copy number variation, Genome sequencing, Autism, ADHD, DMXL2, GRIK5
Background
Rare genetic variants can contribute to the etiology of common neurodevelopmental disorders (NDDs) such as autism spectrum disorder (ASD) [1–7], attention-deficit/hyperactivity disorder (ADHD) [8, 9], intellectual disability (ID) [10–12], and schizophrenia [13–17]. Marked genetic heterogeneity contributed to the recommendation that genome-wide chromosomal microarray (CMA) be a first-tier genetic test for individuals with selected NDDs [10, 14]. CMA along with whole-genome sequencing (WGS) technology have allowed increasing detection of genomic alterations such as copy number variations (CNVs) affecting important developmental genes [10, 18–20]. These variants are difficult to adjudicate through parental testing alone, as even established “genomic disorders” can demonstrate highly variable neurodevelopmental and neuropsychiatric expression [21, 22]. Many are inherited from a putatively unaffected parent, who may have sub-clinical traits or a history of symptoms suggestive of an undiagnosed psychiatric condition [23]. WGS represents a comprehensive platform for detection of coding and non-coding sequence-level, CNV, structural, and mitochondrial variation [1, 24–26].
There are two main contemporary strategies for discovering NDD genes based on CNVs [18, 27]. One begins with a unique variant in a proband and involves a traditional family-based study design with deep phenotyping and curation of all genomic variants. The other involves searching for genes or loci that are overrepresented in large datasets of rare genetic variation assembled through clinical testing or research consortia. In this study, we combined these “depth” and “breadth” approaches to characterize a small non-recurrent CNV of uncertain clinical significance. We sequenced the genomes of six individuals from a single multiplex NDD family and propose that a novel 15q21 microdeletion is the likely genetic lesion involved, because of haploinsufficiency of a synaptic scaffolding protein encoded by DMXL2.
Methods
Family recruitment and phenotyping
The adult proband contacted our research group requesting to participate in our ongoing genetic studies of ASD, after clinical CMA testing had revealed a 15q21 deletion of uncertain significance. History provided by the proband was supplemented by review of all available lifetime medical and psychiatric records. The proband and five family members across three generations provided DNA samples for WGS, and written informed consent was obtained for all participants. After the initial submission of this manuscript, one additional family member provided a saliva sample for targeted genetic testing.
Whole-genome sequencing
WGS methods are as described in detail elsewhere [1, 24]. In brief, WGS was performed at The Centre for Applied Genomics (Toronto, Canada) using DNA extracted from whole blood. Sequencing was performed with the Illumina HiSeq X system and following Illumina’s recommended protocols. Base calling and data analysis were performed using Illumina HiSeq Analysis Software (HAS) version 2-2.5.55.1311. Reads were mapped to the hg19 reference sequence using Isaac alignment software (Isaac alignment software: SAAC00776.15.01.27), and single nucleotide variants (SNVs) and small indel variants were detected using the Isaac variant caller [Isaac Variant Caller (Starling): 2.1.4.2]. These variants were annotated using a custom pipeline based on ANNOVAR [28]. Rare variants were defined as those with ≤ 1% frequency in large public control databases [29–31]. CNVs were detected using the read-depth methods ERDS [32] and CNVnator [33] (using a window size of 500 bp). High-quality CNVs were defined as those greater than 1 kb and detected by both ERDS and CNVnator with more than 50% reciprocal overlap [34], and rare CNVs as those with ≤ 1% frequency in the Autism Speaks MSSNG dataset (probands and parents) [1]. We also annotated CNVs with respect to overlap with gold standard variants in the Database of Genomic Variants (DGV) [35, 36]. For further targeted review of candidate CNVs, we used a control dataset comprised of 10,851 unrelated subjects, with a majority being of European ancestry, who were genotyped on multiple microarray platforms including the Affymetrix Genome-wide Human SNP Array 6.0, Illumina HumanOmni2.5, and Affymetrix CytoScan HD [37, 38]. All genome coordinates in this manuscript refer to NCBI Build 37 (UCSC hg19). Variants of interest were confirmed by PCR and Sanger sequencing (primers available on request) or by CMA.
Review of public and private databases
We conducted a comprehensive review of public, and non-public clinical and research laboratory, databases of genomic variation in disease (primarily NDD) to identify additional individuals with DMXL2, GLDN, and GRIK5 variants. We focused on smaller (< 1 Mb and < 15 genes) CNVs predicted to disrupt any of these genes (i.e., deletions impacting the coding region of the gene, and intragenic duplications), as well as predicted null SNVs and small indels. The denominator of individuals with data available for review is estimated to have been ~ 100,000. This included > 30,000 postnatal cases from Canadian laboratory clinical CMA databases, > 20,000 schizophrenia cases with CMA data from the Psychiatric Genomics Consortium [17], > 18,000 cases with CMA data in DECIPHER [39], > 23,000 cases from the Lineagen Inc. CMA database (Salt Lake City, Utah, USA), and > 6000 cases with CMA data from our ongoing ASD studies and/or WGS data from MSSNG [1].
Results
Family members demonstrated variable features of NDDs
The proband (II-2; Fig. 1) is a 34-year-old female of Dutch ancestry with a reported history of difficulties with sensory processing and physical contact as a child. During adolescence, symptoms across a broad range of domains were reported, including conduct symptoms, attention deficit, anxiety including panic attacks, and fluctuating mood. She completed preparatory middle-level vocational education (voorbereidend middelbaar beroepsonderwijs or VMBO), the lowest level educational stream in the Netherlands. At age 28 years, Wechsler Adult Intelligence Scale—Third Edition (WAIS-III) showed a marked and unusual [40] difference between verbal IQ (VIQ; 79) and performance IQ (PIQ; 100).
Fig. 1.
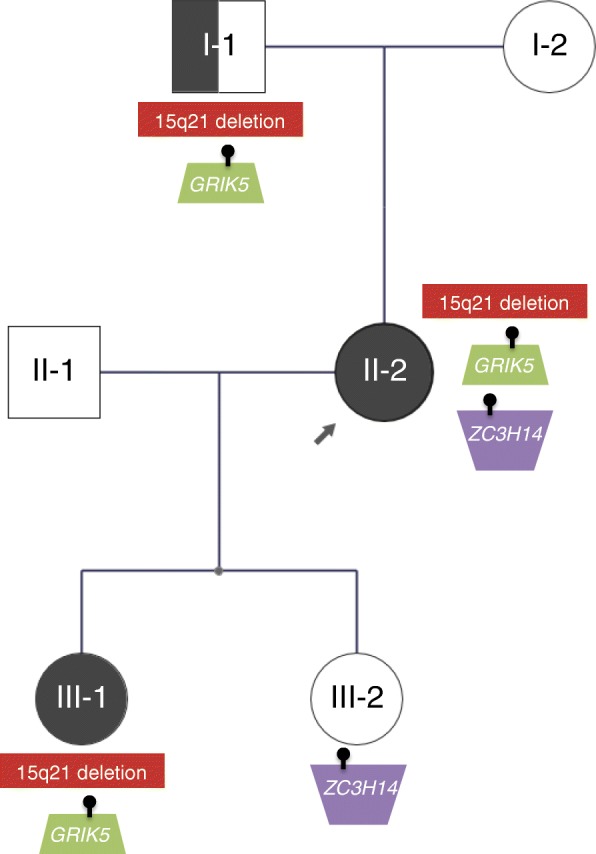
Familial segregation of selected monoallelic rare variants identified by WGS. Individuals II-2 and III-1 have multiple diagnoses including ASD. Individual I-1 was coded as likely affected with a major psychiatric disorder and/or NDD. See text for details. A female paternal first cousin of the proband (not pictured), with possible persistent depressive disorder and no suspected NDD, did not carry the 15q21 deletion or the GRIK5 missense variant
At about age 20, she received pharmacological as well as non-pharmacological treatments for symptoms of anxiety and panic. After the birth of her first child, at age 25, she was treated briefly with an antidepressant for postpartum depression. She was diagnosed with ADHD at age 27 and treated with methylphenidate. At age 33, two independent psychiatric assessments confirmed the previous diagnosis of ADHD. In addition, several aspects of ASD were mentioned and endorsed by questionnaire reports, including reduced empathic abilities, difficulties in social interaction and communication, and difficulties coping with change in her routines. Results of the Social Responsiveness Scale for Adults (reported by self and husband) and the Pervasive Developmental Disorders Screening Questionnaire were in the clinical range. However, these symptoms were evaluated as insufficient to justify a formal diagnosis of ASD. Fluctuations in mood and poor coping skills were interpreted at times as aspects of a borderline personality disorder. A subsequent psychiatric assessment at age 34 years concluded that, in retrospect, ASD was a justified diagnosis based on all available information.
The early developmental trajectory of one of the proband’s daughters (III-1; Fig. 1) was marked by mild cognitive delay and a mixed language disorder (as assessed with the Clinical Evaluation of Language Fundamentals—Fourth Edition). Motor milestones were unremarkable. Her medical history was notable for a febrile seizure at age 3 years and recurrent otitis media, which was associated with temporary hearing loss. She was reported to be a highly anxious infant with a high threshold for pain and hypersensitivity to sensory input. She continued to display moderate behavioral issues as a toddler, both at home and in school, with social and communicative difficulties as well as oppositional, attentional, and anxious features. Early clinical diagnoses included developmental language disorder and disorder of childhood not otherwise specified. At 7 years 11 months, full scale IQ was assessed to be 85, with VIQ 87 and PIQ 86, using the Wechsler Intelligence Scale for Children—Third Edition (WISC-III). The most recent child psychiatric assessment performed at age 9.5 years recorded symptoms including low reciprocity, inadequate social behaviors, and behavioral rituals. Although she scored just below the clinical threshold of the Autism Diagnostic Observation Schedule (ADOS), the overall picture was deemed to justify a clinical diagnosis of ASD, in addition to the language disorder.
The proband’s father (I-1; Fig. 1) was described as having longstanding difficulties with impulsivity/anger, obsessiveness, and other distressing characterological traits that interfered with his functioning. He had similar educational achievement to his daughter and has maintained stable employment without advancement or promotion. He has had at least three “nervous breakdowns” in his life that required him to take a leave of absence from his work. On the basis of this limited history, he was coded prior to WGS as likely affected with a psychiatric disorder and/or NDD. The proband’s other daughter (III-2; Fig. 1) has had normal development, and there are no concerns for ASD. She was diagnosed with Pediatric Acute-onset Neuropsychiatric Syndrome (PANS) after developing motor tics post viral infection. Although the tics initially resolved after a short course of treatment with methylprednisolone, treatment of a recurrence is ongoing. She was coded prior to WGS as unaffected with a NDD. The other two members of the family displayed in Fig. 1 (I-2 and II-1; Fig. 1) are reported to have no major learning difficulties, diagnosed psychiatric conditions, or features of a NDD. There is no known history of unexplained hearing loss in the family [41] nor of infertility or recurrent (> 2) miscarriage, delayed puberty, hypothyroidism, peripheral neuropathy, or short stature [42]. Facial photographs of the proband and her two daughters are available upon request; there are no notable dysmorphic features.
The familial microdeletion implicates the candidate gene DMXL2
WGS identified the novel 262-kb loss at chromosome 15q21.2 (Fig. 2) in the proband that had been detected on CMA. The deletion was inherited from her father and transmitted to her affected daughter (Fig. 1). This deletion overlaps the entire DMXL2 gene (GenBank: NM_001174116.1) and the first three exons of GLDN (GenBank: NM_181789.3). There were no segmental duplication repeats within 200 kb of the breakpoints. No additional rare CNVs of interest were shared amongst these three family members, or between just the proband and her affected daughter.
Fig. 2.

Approximate genomic position of the familial 15q21 deletion and the DECIPHER deletion predicted to disrupt DMXL2, visualized using the DGV genome browser [35]. See text and Table 1 for details
DMXL2 encodes DmX-like protein 2, previously known as Rabconnectin-3. This is a component of the human neocortex post-synaptic density (PSD) proteome [43] that may serve as a scaffold protein on synaptic vesicles, and it is a target of the fragile X mental retardation protein (FMRP) [44, 45]. Multiple lines of evidence, including conditional heterozygous deletion of Dmxl2 in mouse neurons, suggest that the DMXL2 protein functions in the brain in neuronal and endocrinological homeostasis [42]. The Exome Aggregation Consortium (ExAC) probability of loss-of-function intolerance (pLI) of DMXL2 is 1.00, with tenfold fewer observed variants than anticipated [31]. DMXL2 is in a copy number-stable region of the genome; no deletions of DMXL2 were found in the DGV gold standard variants (Fig. 2) or in our CNV control datasets (see the “Methods” section) [36]. The DMXL2 gene contains “brain-critical exons” [7]. Two de novo missense variants in DMXL2 were previously observed in 2517 simplex ASD probands from the Simons Simplex Collection (SSC), with no de novo events in this gene observed in their unaffected siblings [5, 6]. There is also evidence of enrichment for rare sequence variants in DMXL2 in major depressive disorder [46]. With respect to the other gene overlapped by the 15q21 loss CNV, GLDN encodes the protein gliomedin, a secreted cell adhesion molecule involved in the formation of the nodes of Ranvier [47]. In contrast to DMXL2, the ExAC pLI is 0.00 and a deletion was observed in one of our control individuals, suggesting there may not be overt clinical consequences for a heterozygous null allele. Biallelic loss of function of GLDN causes a lethal congenital contracture syndrome (MIM #617194), and as expected, carrier parents were described as unaffected [48].
Additional rare sequence variants may shape expression
Considering SNVs and small indels, there were no likely pathogenic or pathogenic variants [49] in established NDD risk genes in any of the six family members. A total of 198 high-quality, rare, predicted damaging variants were shared by the three family members coded as affected (I-1, II-2, III-1; Fig. 1), including 89 that were absent in the proband’s unaffected daughter (III-2). One of these 89 is a missense variant of uncertain significance in GRIK5 [NM_001301030: c.1840G>A: p.(Ala614Thr)]. It is absent in control databases, affects a highly conserved amino acid, and is predicted to be damaging by in silico programs including SIFT (score 0), PolyPhen-2 (score 0.977), MutationTaster (probability 0.999), and CADD (scaled C-score 32). GRIK5 encodes an understudied auxiliary subunit of the kainate receptor (glutamate receptor, ionotropic, kainate 5). Dysregulation of other components of this receptor has been associated with psychiatric and neurodevelopmental diseases [50–54]. In ExAC, GRIK5 is constrained with respect to loss-of-function (pLI = 0.92) and missense (z = 3.85) variants. There are no coding CNVs in our control databases impacting GRIK5. Similar to DMXL2, prior evidence for an association with ASD is derived from the SSC and Autism Sequencing Consortium, where there were three de novo missense variants in the GRIK5 gene in simplex ASD probands and none in the unaffected SSC siblings [4–6].
We also identified three high confidence de novo exonic variants. In the proband (II-2), there was a 2-bp frameshift deletion in exon 10 of ZC3H14 [NM_024824.4: c.1339_1340del: p.(Leu447Alafs*8)] and a synonymous substitution in exon 3 of TRPC4 that was not predicted to affect splicing. ZC3H14 plays a key role in neurodevelopmental processes [55, 56]. The gene is associated with an autosomal recessive form of ID (MIM #617125) [57], but is also constrained against loss-of-function variants (ExAC pLI = 1), suggesting that it may be sensitive to haploinsufficiency. However, the de novo frameshift variant was transmitted only to the proband’s unaffected daughter (III-2; Fig. 1). The affected daughter (III-1) has a novel de novo missense variant in exon 3 of DLGAP3 [NM_001080418.2: c.33 T>A: p.(His11Gln)]. This gene is a member of the DLGAP family that has been studied in relation to various neuropsychiatric disorders [58]. However, in silico predictions suggest the missense change here will have a benign impact. No de novo exonic variants were identified in the unaffected daughter (III-2).
Genetic variants in additional individuals pinpoint DMXL2 as a risk gene for NDD
Much larger deletions encompassing the 15q21.2 locus are associated with ID and other features [59, 60]. A comprehensive review of public databases identified a single comparable 274-kb 15q21 loss (Fig. 2), in DECIPHER [39]. The individual is a 21-year-old Belgian male with diagnoses of mild ID, ASD, and ADHD (case 4 in Table 1). Growth parameters were within normal limits (weight 71.5 kg, height 168 cm, head circumference 56.4 cm), and there were no major congenital anomalies or notable dysmorphic features. Inheritance of the CNV could not be assessed as the proband was adopted. Seven other individuals with CNVs predicted to disrupt DMXL2 were identified in private clinical laboratory databases (cases 5–11 in Table 1), with available phenotype data supporting a primary NDD phenotype in most. Additional CNVs disrupting DMXL2 were identified in schizophrenia probands (cases 12–13 in Table 1) in the PGC [17], and novel loss-of-function SNVs were identified in ASD probands (cases 14–17 in Table 1) in the Autism Speaks MSSNG study [1]. Similar efforts to identify variants in GRIK5 were not successful, with only a single exonic deletion found in the MSSNG cohort (data not shown).
Table 1.
NDD and psychiatric phenotypes in individuals heterozygous for variants disrupting DMXL2
| Case # | Sourcea | Variant type | Variant details | Individual | Sex | Age group | Inheritance | Reported NDD phenotype(s) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ASD | DD/ID | ADHD | SCZ/psychosis | Other | ||||||||
| 1 | This report | Multigene loss | chr15:51,670,601–51,933,000 × 1 262,400 bp (DMXL2, GLDN) |
Proband (II-1) | F | Adult | Pat | + | − | + | − | +b |
| 2 | Daughter (III-1) | F | Child | Mat | + | +/− | − | − | +b | |||
| 3 | Father (I-1) | M | Adult | N.D. | − | − | − | − | +b | |||
| 4 | DECIPHER | Multigene loss | chr15:51,568,830–51,843,305 × 1 274,476 bp (DMXL2, GLDN, CYP19A1) |
Proband | M | Adult | N.D. | + | + | + | − | − |
| 5 | Canadian laboratory | Intragenic loss | chr15:51,806,694–51,843,305 × 1 36,612 bp (DMXL2) |
Proband | M | Child | N.D. | − | + | − | − | − |
| 6 | Lineagen laboratory | Intragenic gain | chr15:51,717,028–51,792,612 × 3 75,585 bp (DMXL2) |
Proband | M | Child | N.D. | + | − | + | − | +c |
| 7 | Intragenic gain | chr15:51,708,028–51,874,928 × 3 166,901 bp (DMXL2) |
Proband | M | Child | Pat | − | + | − | − | +d | |
| 8 | Multigene loss | chr15:51,735,136–52,620,104 × 1 884,969 bp (DMXL2 and 12 other genes) |
Proband | F | Adult | N.D. | − | + | + | − | +e | |
| 9 | Twin sister | F | Adult | N.D. | − | − | − | + | − | |||
| 10 | Multigene gainf | chr15:50,848,381–51,741,314 × 3 892,934 bp (DMXL2, GLDN, and 7 other genes) |
Proband | M | Child | N.D. | − | − | − | − | − | |
| 11 | Siblingg | F | Child | N.D. | − | − | − | − | − | |||
| 12 | PGC CNV data | Multigene gainf | chr15:50,888,568–51,748,611 × 3 860,044 bp (DMXL2, GLDN, and 7 other genes) |
Proband | M | Adult | N.D. | − | − | − | + | − |
| 13 | Multigene gainf | chr15:50,892,945–51,748,611 × 3 855,667 bp (DMXL2, GLDN, and 7 other genes) |
Proband | F | Adult | N.D. | − | − | − | + | − | |
| 14 | Autism Speaks MSSNG WGS data | LoF SNV | c.9081dupT [p.N3028_I3029delinsX] | Proband | M | Child | N.D. | + | − | − | − | − |
| 15 | LoF SNV | c.4387dupC [p.Q1463fs] | Probandh | F | Child | Pat | + | − | − | − | − | |
| 16 | LoF SNV | c.2239C>T [p.R747X] | Proband | M | Child | Mat | + | − | − | − | − | |
| 17 | LoF SNV | c.1618-2A>G | Proband | M | Child | Pat | + | − | − | − | − | |
Reported physical phenotypes not described elsewhere include: case #5 with mildly coarse features, dental caries, pyloric stenosis, bleeding disorder, and undergrowth; case #8 with coarctation of the aorta; case #10 with short stature and growth hormone deficiency; and case #11 with short stature and short fifth metacarpal. See the “Methods” section for details. There is no mention of psychiatric phenotyping of individuals heterozygous for an in-frame deletion in DMXL2 in the family published by Tata and colleagues [42]
ADHD, attention-deficit/hyperactivity disorder; ASD, autism spectrum disorder; CMA, chromosomal microarray; CNV, copy number variation; DD, developmental delay; F, female; ID, intellectual disability; LoF, loss of function; M, male; Mat, maternal; N.D., not determined; Pat, paternal; PGC, Psychiatric Genomics Consortium; SCZ, schizophrenia; SNV, single nucleotide variant; WGS, whole-genome sequencing
aSee the “Methods” section for details
bSee the “Results” section for details
cEncephalopathy, speech delay, aggression/behavior issues, and vocal tics
dUnilateral ptosis, hypotonia, toe walking, and some sensory and behavioral issues
eBipolar affective disorder, anxiety, and one episode of catatonia
fBreakpoint lies within genomic extent of DMXL2
gAlso with inv(5)(q13.3q33.1)
hAlso with 15q11.2-q13.3 gain
Discussion
We employed WGS and a multifaceted strategy to characterize a CNV of uncertain clinical significance. Diverse and converging lines of evidence suggest that haploinsufficiency of DMXL2 is a risk factor for NDDs. The available data indicate variable expressivity and possibly incomplete penetrance (or at least age-related penetrance). Families exhibiting an apparently heritable but broad phenotype of NDD symptoms, including members with and without a clinical diagnosis of ASD, can expand our knowledge of the genetically related spectrum of disease. One of the most pressing challenges in the field is to understand the typically high degree of variable neuropsychiatric expression associated with risk variants [61, 62]. The range of symptomatology observed in this family is reminiscent of what is often observed with genomic disorders and non-recurrent large CNVs, and it should further highlight the impact of rare inherited genetic variants [18, 22, 63, 64]. In some cases, there is emerging evidence for additional deleterious variants elsewhere in the genome that may act as modifiers [65, 66]. We also identified de novo and inherited sequence variants of potential relevance in this family, including a rare missense change in GRIK5. The latter was not as compelling of a candidate variant as the microdeletion overlapping DMXL2 because of (i) the predicted less-severe nature of the genetic lesion, (ii) the current absence of overt functional or model organism data at the gene level, and (iii) our inability to adequately replicate the finding in the population-scale NDD cohorts. Nonetheless, it could still be a contributor to the risk for NDD in this family. A limitation of our study was that the relatively small nuclear families precluded the identification of an individual with one, but not both, of the variants.
Our findings provide a clinical impetus to now develop and test functional hypotheses regarding disease mechanism(s). The Dmxl2 gene is best studied in the gonadotropin-releasing hormone neurons [42, 67], where low expression in mice impedes normal dendritic development [67]. However, the phenotypes observed in knockout mice are not entirely attributable to deficient Dmxl2 in that neuronal cell population [67]. Dmxl2+/− mice also demonstrate neuroanatomical differences in the corpus callosum [68]. At least four of ten experimentally proven protein interactors of DMXL2 [69] are encoded by established or candidate genes for NDDs: CYFIP2 [70, 71], DYNC1H1 [72, 73], MATR3 [74], and NCKAP1 [5, 75, 76]. Both CYFIP2 and NCKAP1 shape the formation of dendritic spines via the WAVE actin-remodeling complex [77, 78]. DMXL2 protein dosage insufficiency may therefore result in structural (e.g., abnormal dendritic spines morphology) and/or functional (e.g., impaired transmission) synaptic consequences in humans, as have been observed in other genetic forms of ID, ASD, and schizophrenia [79, 80].
It remains difficult in clinical practice to interpret novel inherited CNVs, which are often labeled variants of uncertain clinical significance [49]. Incomplete penetrance and epistasis are likely underappreciated. Most approaches to CNV adjudication focus on the overlapped genes and disregard the remainder of the genome [81], in part because CMA is incapable of identifying smaller and balanced genetic variants. Whereas WGS is more comprehensive in detection [1, 24–26], sometimes also revealing complicating data for clinical interpretation, as was the case with the GRIK5 variant. Updated guidelines are required for interpreting CNVs that take into account sequence variants [81]. Specific consideration will need to be given with respect to how to determine if two or more variants are major contributors to disease within a specific individual. The risk profile from common variants with a polygenic effect may also become clinically relevant in time, even in those individuals with a monogenic diagnosis or genomic disorder [82, 83].
Variant inheritance patterns in multigenerational pedigrees can be highly informative, with the caveat that customary assumptions (e.g., de novo suggests pathogenic, and inherited from putatively unaffected parent suggests benign) are imperfect [84–86]. The availability of samples and detailed phenotype data from three generations of a family, including offspring and parents of an individual with ASD or other NDD phenotypes, is uncommon [23]. There is substantial evidence that different rare disease-causing variants can segregate within the same family [1, 3]. Our approach here of examining multiple rare penetrant variants across generations might reveal new genes for NDD. This may especially be the case for genes involved in higher functioning forms of ASD, perhaps including DMXL2. Our study design also allowed for the observation that the proband’s de novo variant in ZC3H14 was not inherited by her daughter with NDD. In contrast, both the 15q21 deletion and the GRIK5 variant did segregate with the apparent NDD phenotypes, albeit in a simple pedigree where the a priori probability was p = 0.125 for any single variant [87].
Conclusions
These results support DMXL2 as a candidate gene for a spectrum of NDDs that includes ASD. As for virtually all (even highly penetrant) risk variants, additional modifiers of expression remain to be discovered. The robust interpretation of ultra-rare variants identified by CMA or other testing requires incorporation of WGS from affected families and population-scale data. Although currently expensive for clinical diagnostics [88], in time, we expect the same utility will be found as in this research study and WGS will become a first-tier clinical genetic test for NDDs.
Acknowledgements
The authors thank the family for their participation, and staff and trainees at The Centre for Applied Genomics. The authors wish to acknowledge the resources of MSSNG (www.mss.ng), Autism Speaks and The Centre for Applied Genomics at The Hospital for Sick Children, Toronto, Canada. They thank the participating families for their time and contributions to this database, as well as the generosity of the donors who supported this program. This study also makes use of data generated by the DECIPHER community. A full list of centres who contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. Funding for the project was provided by the Wellcome Trust.
Additional acknowledgements regarding the control dataset are as follows:
(1) Funding support for the Study of Addiction: Genetics and Environment (SAGE) was provided through the NIH Genes, Environment and Health Initiative [GEI] (U01 HG004422). SAGE is one of the genome-wide association studies funded as part of the Gene Environment Association Studies (GENEVA) under GEI. Assistance with phenotype harmonization and genotype cleaning, as well as with general study coordination, was provided by the GENEVA Coordinating Center (U01 HG004446). Assistance with data cleaning was provided by the National Center for Biotechnology Information. Support for collection of datasets and samples was provided by the Collaborative Study on the Genetics of Alcoholism (COGA; U10 AA008401), the Collaborative Genetic Study of Nicotine Dependence (COGEND; P01 CA089392), and the Family Study of Cocaine Dependence (FSCD; R01 DA013423). Funding support for genotyping, which was performed at the Johns Hopkins University Center for Inherited Disease Research, was provided by the NIH GEI (U01HG004438), the National Institute on Alcohol Abuse and Alcoholism, the National Institute on Drug Abuse, and the NIH contract “High throughput genotyping for studying the genetic contributions to human disease” (HHSN268200782096C). The datasets used for the analyses described in this manuscript were obtained from dbGaP at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000092.v1.p1 through dbGaP accession number phs000092.v1.p1.
(2) The authors acknowledge the contribution of data from Genetic Architecture of Smoking and Smoking Cessation accessed through dbGaP. Funding support for genotyping, which was performed at the Center for Inherited Disease Research (CIDR), was provided by 1 X01 HG005274-01. CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN268200782096C. Assistance with genotype cleaning, as well as with general study coordination, was provided by the Gene Environment Association Studies (GENEVA) Coordinating Center (U01 HG004446). Funding support for collection of datasets and samples was provided by the Collaborative Genetic Study of Nicotine Dependence (COGEND; P01 CA089392) and the University of Wisconsin Transdisciplinary Tobacco Use Research Center (P50 DA019706, P50 CA084724). The datasets used for the analyses described in this manuscript were obtained from dbGaP at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000404.v1.p1 through dbGaP accession number phs000404.v1.p1.
(3) A dataset used for the analyses described in this manuscript were obtained from the NEI Refractive Error Collaboration (NEIREC). Funding support for NEIREC was provided by the National Eye Institute. We would like to thank NEIREC participants and the NEIREC Research Group for their valuable contribution to this research. The datasets used for the analyses described in this manuscript were obtained from dbGaP at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000303.v1.p1 through dbGaP accession number phs000303.v1.p1.
(4) Funding support for the “CIDR Visceral Adiposity Study” was provided through the Division of Aging Biology and the Division of Geriatrics and Clinical Gerontology, NIA. The CIDR Visceral Adiposity Study includes a genome-wide association study funded as part of the Division of Aging Biology and the Division of Geriatrics and Clinical Gerontology, NIA. Assistance with phenotype harmonization and genotype cleaning, as well as with general study coordination, was provided by Heath ABC Study Investigators. The datasets used for the analyses described in this manuscript were obtained from dbGaP at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000169.v1.p1 through dbGaP accession number phs000169.v1.p1.
Funding
This study was funded by The Centre for Applied Genomics, The Hospital for Sick Children, Genome Canada, Autism Speaks, the Canadian Institutes of Health Research (CIHR), the Canadian Institutes for Advanced Research (CIFAR), Ontario Brain Institute, the Government of Ontario, Canada Foundation for Innovation (CFI), and the University of Toronto McLaughlin Centre. S.W.S. holds the GlaxoSmithKline-CIHR Endowed Chair in Genome Sciences at The Hospital for Sick Children and the University of Toronto. The funding bodies played no role in the design of the study, the collection, analysis, and interpretation of data, or the writing of the manuscript.
Availability of data and materials
The dataset generated during the current study will be deposited in an open-access repository.
Abbreviations
- ADHD
Attention-deficit/hyperactivity disorder
- ADOS
Autism Diagnostic Observation Schedule
- ASD
Autism spectrum disorder
- CMA
Chromosomal microarray
- CNV
Copy number variation
- DGV
Database of Genomic Variants
- ID
Intellectual disability
- NDD
Neurodevelopmental disorder
- PGC
Psychiatric Genomics Consortium
- PIQ
Performance intelligence quotient
- SNV
Single nucleotide variant
- VIQ
Verbal intelligence quotient
- WAIS-III
Wechsler Adult Intelligence Scale—Third Edition
- WGS
Whole-genome sequencing
- WISC-III
Wechsler Intelligence Scale for Children—Third Edition
Authors’ contributions
GC, SW, and SWS designed the study and analyzed and interpreted the data. The other co-authors made substantial contributions to conception and design, acquisition of data, and/or analysis and interpretation of data. GC, SW, and SWS drafted the manuscript, and the remaining authors were involved in revising it critically for important intellectual content. All authors have given final approval of the version to be published.
Ethics approval and consent to participate
This study was approved by our local institutional Research Ethics Board, and consent to participate was provided by all participants.
Consent for publication
The participants provided consent for publication of this manuscript.
Competing interests
S.W.S. is on the Scientific Advisory Committees of Deep Genomics and Population Bio, and Lineagen licenses intellectual property based on his discoveries from The Hospital for Sick Children. A.P., M.A.S., and H.T. are employees of Lineagen. The remaining authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Gregory Costain, Email: gregory.costain@sickkids.ca.
Susan Walker, Email: susan.walker@sickkids.ca.
Bob Argiropoulos, Email: bob.argiropoulos@ahs.ca.
Danielle A. Baribeau, Email: danielle.baribeau@mail.utoronto.ca
Anne S. Bassett, Email: anne.bassett@utoronto.ca
Erik Boot, Email: erik.boot@uhn.ca.
Koen Devriendt, Email: koenraad.devriendt@uzleuven.be.
Barbara Kellam, Email: bkellam@sickkids.ca.
Christian R. Marshall, Email: crm@sickkids.ca
Aparna Prasad, Email: aprasad@lineagen.com.
Moises A. Serrano, Email: mserrano@lineagen.com
D. James Stavropoulos, Email: james.stavropoulos@sickkids.ca.
Hope Twede, Email: htwede@lineagen.com.
Joris R. Vermeesch, Email: joris.vermeesch@uz.kuleuven.ac.be
Jacob A. S. Vorstman, Email: jacob.vorstman@sickkids.ca
Stephen W. Scherer, Email: stephen.scherer@sickkids.ca
References
- 1.Yuen RKC, Merico D, Bookman M, Howe LJ, Thiruvahindrapuram B, Patel RV, Whitney J, Deflaux N, Bingham J, Wang Z, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. 2017;20(4):602–611. doi: 10.1038/nn.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tammimies K, Marshall CR, Walker S, Kaur G, Thiruvahindrapuram B, Lionel AC, Yuen RK, Uddin M, Roberts W, Weksberg R, et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA. 2015;314(9):895–903. doi: 10.1001/jama.2015.10078. [DOI] [PubMed] [Google Scholar]
- 3.Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, Chrysler C, Nalpathamkalam T, Pellecchia G, Liu Y, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med. 2015;21(2):185–191. doi: 10.1038/nm.3792. [DOI] [PubMed] [Google Scholar]
- 4.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, Raja A, Coe BP, Stessman HA, He ZX, et al. Excess of rare, inherited truncating mutations in autism. Nat Genet. 2015;47(6):582–588. doi: 10.1038/ng.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uddin M, Tammimies K, Pellecchia G, Alipanahi B, Hu P, Wang Z, Pinto D, Lau L, Nalpathamkalam T, Marshall CR, et al. Brain-expressed exons under purifying selection are enriched for de novo mutations in autism spectrum disorder. Nat Genet. 2014;46(7):742–747. doi: 10.1038/ng.2980. [DOI] [PubMed] [Google Scholar]
- 8.Williams NM, Zaharieva I, Martin A, Langley K, Mantripragada K, Fossdal R, Stefansson H, Stefansson K, Magnusson P, Gudmundsson OO, et al. Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet. 2010;376(9750):1401–1408. doi: 10.1016/S0140-6736(10)61109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lionel AC, Crosbie J, Barbosa N, Goodale T, Thiruvahindrapuram B, Rickaby J, Gazzellone M, Carson AR, Howe JL, Wang Z, et al. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci Transl Med. 2011;3(95):95ra75. doi: 10.1126/scitranslmed.3002464. [DOI] [PubMed] [Google Scholar]
- 10.Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43(9):838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, Kwint M, Janssen IM, Hoischen A, Schenck A, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511(7509):344–347. doi: 10.1038/nature13394. [DOI] [PubMed] [Google Scholar]
- 13.Xu B, Ionita-Laza I, Roos JL, Boone B, Woodrick S, Sun Y, Levy S, Gogos JA, Karayiorgou M. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat Genet. 2012;44(12):1365–1369. doi: 10.1038/ng.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costain G, Lionel AC, Merico D, Forsythe P, Russell K, Lowther C, Yuen T, Husted J, Stavropoulos DJ, Speevak M, et al. Pathogenic rare copy number variants in community-based schizophrenia suggest a potential role for clinical microarrays. Hum Mol Genet. 2013;22(22):4485–4501. doi: 10.1093/hmg/ddt297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, Rippey C, Shahin H, Consortium on the Genetics of S, Group PS et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013;154(3):518–529. doi: 10.1016/j.cell.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506(7487):179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, Antaki D, Shetty A, Holmans PA, Pinto D, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet. 2017;49(1):27–35. doi: 10.1038/ng.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cook EH, Jr, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455(7215):919–923. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- 19.Riggs ER, Ledbetter DH, Martin CL. Genomic variation: lessons learned from whole-genome CNV analysis. Curr Genet Med Rep. 2014;2:146–150. doi: 10.1007/s40142-014-0048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hehir-Kwa JY, Pfundt R, Veltman JA. Exome sequencing and whole genome sequencing for the detection of copy number variation. Expert Rev Mol Diagn. 2015;15(8):1023–1032. doi: 10.1586/14737159.2015.1053467. [DOI] [PubMed] [Google Scholar]
- 21.Vorstman JA, Ophoff RA. Genetic causes of developmental disorders. Curr Opin Neurol. 2013;26(2):128–136. doi: 10.1097/WCO.0b013e32835f1a30. [DOI] [PubMed] [Google Scholar]
- 22.Lowther C, Costain G, Baribeau DA, Bassett AS. Genomic disorders in psychiatry-what does the clinician need to know? Curr Psychiatry Rep. 2017;19(11):82. doi: 10.1007/s11920-017-0831-5. [DOI] [PubMed] [Google Scholar]
- 23.Woodbury-Smith M, Paterson AD, Thiruvahindrapduram B, Lionel AC, Marshall CR, Merico D, Fernandez BA, Duku E, Sutcliffe JS, O'Conner I, et al. Using extended pedigrees to identify novel autism spectrum disorder (ASD) candidate genes. Hum Genet. 2015;134(2):191–201. doi: 10.1007/s00439-014-1513-6. [DOI] [PubMed] [Google Scholar]
- 24.Lionel AC, Costain G, Monfared N, Walker S, Reuter MS, Hosseini SM, Thiruvahindrapuram B, Merico D, Jobling R, Nalpathamkalam T, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med. 2018;20(4):435–443. doi: 10.1038/gim.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stavropoulos DJ, Merico D, Jobling R, Bowdin S, Monfared N, Thiruvahindrapuram B, Nalpathamkalam T, Pellecchia G, Yuen RKC, Szego MJ, et al. Whole genome sequencing expands diagnostic utility and improves clinical management in pediatric medicine. NPJ Genom Med. 2016;1:15012. [DOI] [PMC free article] [PubMed]
- 26.Noll AC, Miller NA, Smith LD, Yoo B, Fiedler S, Cooley LD, Willig LK, Petrikin JE, Cakici J, Lesko J, et al. Clinical detection of deletion structural variants in whole-genome sequences. NPJ Genom Med. 2016;1:16026. doi: 10.1038/npjgenmed.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buchanan JA, Scherer SW. Contemplating effects of genomic structural variation. Genet Med. 2008;10(9):639–647. doi: 10.1097/GIM.0b013e318183f848. [DOI] [PubMed] [Google Scholar]
- 28.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Genomes Project C. Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337(6090):64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu M, Need AC, Han Y, Ge D, Maia JM, Zhu Q, Heinzen EL, Cirulli ET, Pelak K, He M, et al. Using ERDS to infer copy-number variants in high-coverage genomes. Am J Hum Genet. 2012;91(3):408–421. doi: 10.1016/j.ajhg.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21(6):974–984. doi: 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trost B, Walker S, Wang Z, Thiruvahindrapuram B, MacDonald JR, Sung WWL, Pereira SL, Whitney J, Chan AJS, Pellecchia G, et al. A comprehensive workflow for read depth-based identification of copy-number variation from whole-genome sequence data. Am J Hum Genet. 2018;102(1):142–155. doi: 10.1016/j.ajhg.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42(Database issue):D986–D992. doi: 10.1093/nar/gkt958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet. 2015;16(3):172–183. doi: 10.1038/nrg3871. [DOI] [PubMed] [Google Scholar]
- 37.Uddin M, Thiruvahindrapuram B, Walker S, Wang Z, Hu P, Lamoureux S, Wei J, MacDonald JR, Pellecchia G, Lu C, et al. A high-resolution copy-number variation resource for clinical and population genetics. Genet Med. 2015;17(9):747–752. doi: 10.1038/gim.2014.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uddin M, Pellecchia G, Thiruvahindrapuram B, D’Abate L, Merico D, Chan A, Zarrei M, Tammimies K, Walker S, Gazzellone MJ, et al. Indexing effects of copy number variation on genes involved in developmental delay. Sci Rep. 2016;6:28663. doi: 10.1038/srep28663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, Carter NP. DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am J Hum Genet. 2009;84(4):524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iverson GL, Woodward TS, Green P. Base rates of WAIS-R VIQ-PIQ differences in 1593 psychiatric inpatients. J Clin Psychol. 2001;57(12):1579–1587. doi: 10.1002/jclp.1120. [DOI] [PubMed] [Google Scholar]
- 41.Chen DY, Liu XF, Lin XJ, Zhang D, Chai YC, Yu DH, Sun CL, Wang XL, Zhu WD, Chen Y, et al. A dominant variant in DMXL2 is linked to nonsyndromic hearing loss. Genet Med. 2017;19(5):553–558. doi: 10.1038/gim.2016.142. [DOI] [PubMed] [Google Scholar]
- 42.Tata B, Huijbregts L, Jacquier S, Csaba Z, Genin E, Meyer V, Leka S, Dupont J, Charles P, Chevenne D, et al. Haploinsufficiency of Dmxl2, encoding a synaptic protein, causes infertility associated with a loss of GnRH neurons in mouse. PLoS Biol. 2014;12(9):e1001952. doi: 10.1371/journal.pbio.1001952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bayes A, van de Lagemaat LN, Collins MO, Croning MD, Whittle IR, Choudhary JS, Grant SG. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci. 2011;14(1):19–21. doi: 10.1038/nn.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ascano M, Jr, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492(7429):382–386. doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146(2):247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pirooznia M, Wang T, Avramopoulos D, Potash JB, Zandi PP, Goes FS. High-throughput sequencing of the synaptome in major depressive disorder. Mol Psychiatry. 2016;21(5):650–655. doi: 10.1038/mp.2015.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eshed Y, Feinberg K, Poliak S, Sabanay H, Sarig-Nadir O, Spiegel I, Bermingham JR, Jr, Peles E. Gliomedin mediates Schwann cell-axon interaction and the molecular assembly of the nodes of Ranvier. Neuron. 2005;47(2):215–229. doi: 10.1016/j.neuron.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 48.Maluenda J, Manso C, Quevarec L, Vivanti A, Marguet F, Gonzales M, Guimiot F, Petit F, Toutain A, Whalen S, et al. Mutations in GLDN, encoding Gliomedin, a critical component of the nodes of Ranvier, are responsible for lethal arthrogryposis. Am J Hum Genet. 2016;99(4):928–933. doi: 10.1016/j.ajhg.2016.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Griswold AJ, Ma D, Cukier HN, Nations LD, Schmidt MA, Chung RH, Jaworski JM, Salyakina D, Konidari I, Whitehead PL, et al. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum Mol Genet. 2012;21(15):3513–3523. doi: 10.1093/hmg/dds164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Casey JP, Magalhaes T, Conroy JM, Regan R, Shah N, Anney R, Shields DC, Abrahams BS, Almeida J, Bacchelli E, et al. A novel approach of homozygous haplotype sharing identifies candidate genes in autism spectrum disorder. Hum Genet. 2012;131(4):565–579. doi: 10.1007/s00439-011-1094-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takenouchi T, Hashida N, Torii C, Kosaki R, Takahashi T, Kosaki K. 1p34.3 deletion involving GRIK3: further clinical implication of GRIK family glutamate receptors in the pathogenesis of developmental delay. Am J Med Genet A. 2014;164A(2):456–460. doi: 10.1002/ajmg.a.36240. [DOI] [PubMed] [Google Scholar]
- 53.Aller MI, Pecoraro V, Paternain AV, Canals S, Lerma J. Increased dosage of high-affinity kainate receptor gene grik4 alters synaptic transmission and reproduces autism spectrum disorders features. J Neurosci. 2015;35(40):13619–13628. doi: 10.1523/JNEUROSCI.2217-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Micheau J, Vimeney A, Normand E, Mulle C, Riedel G. Impaired hippocampus-dependent spatial flexibility and sociability represent autism-like phenotypes in GluK2 mice. Hippocampus. 2014;24(9):1059–1069. doi: 10.1002/hipo.22290. [DOI] [PubMed] [Google Scholar]
- 55.Bienkowski RS, Banerjee A, Rounds JC, Rha J, Omotade OF, Gross C, Morris KJ, Leung SW, Pak C, Jones SK, et al. The conserved, disease-associated RNA binding protein dNab2 interacts with the fragile X protein ortholog in Drosophila neurons. Cell Rep. 2017;20(6):1372–1384. doi: 10.1016/j.celrep.2017.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rha J, Jones SK, Fidler J, Banerjee A, Leung SW, Morris KJ, Wong JC, Inglis GAS, Shapiro L, Deng Q, et al. The RNA-binding protein, ZC3H14, is required for proper poly(A) tail length control, expression of synaptic proteins, and brain function in mice. Hum Mol Genet. 2017;26(19):3663–3681. doi: 10.1093/hmg/ddx248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pak C, Garshasbi M, Kahrizi K, Gross C, Apponi LH, Noto JJ, Kelly SM, Leung SW, Tzschach A, Behjati F, et al. Mutation of the conserved polyadenosine RNA binding protein, ZC3H14/dNab2, impairs neural function in Drosophila and humans. Proc Natl Acad Sci U S A. 2011;108(30):12390–12395. doi: 10.1073/pnas.1107103108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rasmussen AH, Rasmussen HB, Silahtaroglu A. The DLGAP family: neuronal expression, function and role in brain disorders. Mol Brain. 2017;10(1):43. doi: 10.1186/s13041-017-0324-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tempesta S, Sollima D, Ghezzo S, Politi V, Sinigaglia B, Balducci F, Celso B, Restuccia A, Stefani M, Cernetti R, et al. Mild mental retardation in a child with a de novo interstitial deletion of 15q21.2q22.1: a comparison with previously described cases. Eur J Med Genet. 2008;51(6):639–645. doi: 10.1016/j.ejmg.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 60.Velazquez-Wong AC, Ruiz Esparza-Garrido R, Velazquez-Flores MA, Huicochea-Montiel JC, Cardenas-Conejo A, Miguez-Munoz CP, Araujo-Solis MA, Salamanca-Gomez F, Arenas-Aranda DJ. Clinical and molecular characterization of a patient with 15q21.2q22.2 deletion syndrome. Cytogenet Genome Res. 2014;144(3):183–189. doi: 10.1159/000370081. [DOI] [PubMed] [Google Scholar]
- 61.Vorstman JAS, Parr JR, Moreno-De-Luca D, Anney RJL, Nurnberger JI, Jr, Hallmayer JF. Autism genetics: opportunities and challenges for clinical translation. Nat Rev Genet. 2017;18(6):362–376. doi: 10.1038/nrg.2017.4. [DOI] [PubMed] [Google Scholar]
- 62.Takumi T, Tamada K. CNV biology in neurodevelopmental disorders. Curr Opin Neurobiol. 2018;48:183–192. doi: 10.1016/j.conb.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 63.Sato D, Lionel AC, Leblond CS, Prasad A, Pinto D, Walker S, O'Connor I, Russell C, Drmic IE, Hamdan FF, et al. SHANK1 deletions in males with autism spectrum disorder. Am J Hum Genet. 2012;90(5):879–887. doi: 10.1016/j.ajhg.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lowther C, Costain G, Melvin R, Stavropoulos DJ, Lionel AC, Marshall CR, Scherer SW, Bassett AS. Adult expression of a 3q13.31 microdeletion. Mol Cytogenet. 2014;7(1):23. doi: 10.1186/1755-8166-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, Filipink RA, McConnell JS, Angle B, Meschino WS, et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med. 2012;367(14):1321–1331. doi: 10.1056/NEJMoa1200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bassett AS, Lowther C, Merico D, Costain G, Chow EWC, van Amelsvoort T, McDonald-McGinn D, Gur RE, Swillen A, Van den Bree M, et al. Rare genome-wide copy number variation and expression of schizophrenia in 22q11.2 deletion syndrome. Am J Psychiatry. 2017;174(11):1054–1063. doi: 10.1176/appi.ajp.2017.16121417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tata BK, Harbulot C, Csaba Z, Peineau S, Jacquier S, de Roux N. Rabconnectin-3alpha is required for the morphological maturation of GnRH neurons and kisspeptin responsiveness. Sci Rep. 2017;7:42463. doi: 10.1038/srep42463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kannan M, Bayam E, Wagner C, Rinaldi B, Kretz PF, Tilly P, Roos M, McGillewie L, Bar S, Minocha S, et al. WD40-repeat 47, a microtubule-associated protein, is essential for brain development and autophagy. Proc Natl Acad Sci U S A. 2017;114(44):E9308–E9317. doi: 10.1073/pnas.1713625114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li KW, Chen N, Klemmer P, Koopmans F, Karupothula R, Smit AB. Identifying true protein complex constituents in interaction proteomics: the example of the DMXL2 protein complex. Proteomics. 2012;12(15–16):2428–2432. doi: 10.1002/pmic.201100675. [DOI] [PubMed] [Google Scholar]
- 70.Abekhoukh S, Bardoni B. CYFIP family proteins between autism and intellectual disability: links with Fragile X syndrome. Front Cell Neurosci. 2014;8:81. doi: 10.3389/fncel.2014.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nakashima M, Kato M, Aoto K, Shiina M, Belal H, Mukaida S, Kumada S, Sato A, Zerem A, Lerman-Sagie T, et al. De novo hotspot variants in CYFIP2 cause early-onset epileptic encephalopathy. Ann Neurol. 2018;83(4):794–806. doi: 10.1002/ana.25208. [DOI] [PubMed] [Google Scholar]
- 72.Poirier K, Lebrun N, Broix L, Tian G, Saillour Y, Boscheron C, Parrini E, Valence S, Pierre BS, Oger M, et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet. 2013;45(6):639–647. doi: 10.1038/ng.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jamuar SS, Lam AT, Kircher M, D'Gama AM, Wang J, Barry BJ, Zhang X, Hill RS, Partlow JN, Rozzo A, et al. Somatic mutations in cerebral cortical malformations. N Engl J Med. 2014;371(8):733–743. doi: 10.1056/NEJMoa1314432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, Pliner HA, Abramzon Y, Marangi G, Winborn BJ, et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci. 2014;17(5):664–666. doi: 10.1038/nn.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Anazi S, Maddirevula S, Salpietro V, Asi YT, Alsahli S, Alhashem A, Shamseldin HE, AlZahrani F, Patel N, Ibrahim N, et al. Expanding the genetic heterogeneity of intellectual disability. Hum Genet. 2017;136(11–12):1419–1429. doi: 10.1007/s00439-017-1843-2. [DOI] [PubMed] [Google Scholar]
- 76.Freed D, Pevsner J. The contribution of mosaic variants to autism spectrum disorder. PLoS Genet. 2016;12(9):e1006245. doi: 10.1371/journal.pgen.1006245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Han K, Chen H, Gennarino VA, Richman R, Lu HC, Zoghbi HY. Fragile X-like behaviors and abnormal cortical dendritic spines in cytoplasmic FMR1-interacting protein 2-mutant mice. Hum Mol Genet. 2015;24(7):1813–1823. doi: 10.1093/hmg/ddu595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Rubeis S, Pasciuto E, Li KW, Fernandez E, Di Marino D, Buzzi A, Ostroff LE, Klann E, Zwartkruis FJ, Komiyama NH, et al. CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron. 2013;79(6):1169–1182. doi: 10.1016/j.neuron.2013.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jiang YH, Ehlers MD. Modeling autism by SHANK gene mutations in mice. Neuron. 2013;78(1):8–27. doi: 10.1016/j.neuron.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sudhof TC. Synaptic neurexin complexes: a molecular code for the logic of neural circuits. Cell. 2017;171(4):745–769. doi: 10.1016/j.cell.2017.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST, Working Group of the American College of Medical Genetics Laboratory Quality Assurance C American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011;13(7):680–685. doi: 10.1097/GIM.0b013e3182217a3a. [DOI] [PubMed] [Google Scholar]
- 82.Merico D, Zarrei M, Costain G, Ogura L, Alipanahi B, Gazzellone MJ, Butcher NJ, Thiruvahindrapuram B, Nalpathamkalam T, Chow EW, et al. Whole-genome sequencing suggests schizophrenia risk mechanisms in humans with 22q11.2 deletion syndrome. G3 (Bethesda) 2015;5(11):2453–2461. doi: 10.1534/g3.115.021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Niemi MEK, Martin HC, Rice DL, Gallone G, Gordon S, Kelemen M, McAloney K, McRae J, Radford EJ, Yu S, et al. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature. 2018;562(7726):268–271. doi: 10.1038/s41586-018-0566-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vermeesch JR, Balikova I, Schrander-Stumpel C, Fryns JP, Devriendt K. The causality of de novo copy number variants is overestimated. Eur J Hum Genet. 2011;19(11):1112–1113. doi: 10.1038/ejhg.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Costain G. Parental expression is overvalued in the interpretation of rare inherited variants. Eur J Hum Genet. 2015;23(1):4–7. doi: 10.1038/ejhg.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Giorgio E, Ciolfi A, Biamino E, Caputo V, Di Gregorio E, Belligni EF, Calcia A, Gaidolfi E, Bruselles A, Mancini C, et al. Whole exome sequencing is necessary to clarify ID/DD cases with de novo copy number variants of uncertain significance: two proof-of-concept examples. Am J Med Genet A. 2016;170(7):1772–1779. doi: 10.1002/ajmg.a.37649. [DOI] [PubMed] [Google Scholar]
- 87.Jarvik GP, Browning BL. Consideration of cosegregation in the pathogenicity classification of genomic variants. Am J Hum Genet. 2016;98(6):1077–1081. doi: 10.1016/j.ajhg.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tsiplova K, Zur RM, Marshall CR, Stavropoulos DJ, Pereira SL, Merico D, Young EJ, Sung WWL, Scherer SW, Ungar WJ. A microcosting and cost-consequence analysis of clinical genomic testing strategies in autism spectrum disorder. Genet Med. 2017;19(11):1268–1275. doi: 10.1038/gim.2017.47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The dataset generated during the current study will be deposited in an open-access repository.