

Abstract
Aromatase (CYP19A1) catalyzes the conversion of C19 steroids to estrogens. Aromatase and its product estradiol (E2) are crucial for the sexually dimorphic development of the fetal brain and the regulation of gonadotropin secretion and sexual interest in adults. The regulation of aromatase expression in the brain is not well understood. The aromatase (Cyp19a1) gene is selectively expressed in distinct neurons of the hypothalamus through a distal brain-specific promoter I.f located ∼36 kb upstream of the coding region. Here, we investigated a short feedback effect of E2 on aromatase mRNA expression and enzyme activity using estrogen receptor alpha (ESR1; also known as ERalpha)-positive or ESR1-negative mouse embryonic hypothalamic neuronal cell lines that express aromatase via promoter I.f. Estradiol regulated aromatase mRNA expression and enzyme activity in a time- and dose-dependent manner, whereas an E2 antagonist reversed these effects. The nucleotide −200/−1 region of promoter I.f conferred E2 responsiveness. Two activator protein 1 (AP-1) elements in this region were essential for induction of promoter activity by E2. ESR1 and JUN (c-Jun) bound to these AP-1 motifs in intact cells and under cell-free conditions. The addition of an ESR1 mutant that interacts with JUN but not directly with DNA enhanced E2-dependent promoter I.f activity. Independently, we demonstrated an interaction between ESR1 and JUN in hypothalamic cells. Knockdown of ESR1 abolished E2-induced aromatase mRNA and enzyme activity. Taken together, E2 regulates Cyp19a1 expression via promoter I.f by enhanced binding of an ESR1/JUN complex to distinct AP-1 motifs in hypothalamic cells. We speculate that this mechanism may, in part, regulate gonadotropin secretion and sexual activity.
Keywords: aromatase, brain, c-Jun, estradiol, estradiol receptor, estrogen receptor 1, hypothalamus, mechanisms of hormone action, neuron, promoter I.f
Introduction
Estrogen is produced in the ovary, testis, and brain. Besides its well-established biologic effects on reproductive neuroendocrine function and recovery after brain injury, estrogen also exerts a wide variety of actions on regions of the developing and adult brain that influence higher cognitive functions, pain mechanisms, fine motor skills, susceptibility to seizures, mood, temperature regulation, and sleep [1, 2]. Estrogen biosynthesis is dependent on the facilitation of the entry of cholesterol into mitochondria followed by six enzymatic steps. Aromatase is the key enzyme and catalyzes the final step; that is, the conversion of C19 steroids to estrone or estradiol (E2) [3–5].
A single gene, Cyp19a1, encodes aromatase, the inhibition of which effectively eliminates estrogen production in the entire body [6, 7]. The mouse aromatase gene is located on the long arm of chromosome 9. This gene spans a region that consists of a 30-kb coding region and a minimum of 75 kb of regulatory region (at least 105-kb total length) [8]. The downstream 30-kb coding region comprises nine coding exons (II–X) [9]. The upstream 40-kb portion of the gene contains multiple promoters that direct transcription of alternative first exons, giving rise to aromatase mRNA species with unique 5′-untranslated regions. This region contains four promoters regulated in a partly tissue-specific manner: the proximal gonad-specific promoter and the far distal adipose promoter, which lies some 75 kb upstream of the ATG translational start site of the coding exon II, mark the ends of this regulatory region. Brain-specific promoter I.f is located at approximately 36 kb upstream of the coding region [8, 10, 11].
In the vertebrate brain, aromatase is expressed primarily in the hypothalamus, hippocampus, and amygdala via a highly conserved promoter I.f [12, 13]. Aromatase expression in the hypothalamus is localized primarily in the medial preoptic area and the ventromedial nucleus of the hypothalamus (VMH), which govern various reproductive functions of both sexes in different species [14–17].
The physiological roles of aromatase in the brain were suggested by studies on mice with a disrupted aromatase gene (Cyp19a1 KO) and men with inactivating mutations of the aromatase gene [6, 18]. In both cases, aromatase deficiency was associated with increased testosterone and gonadotropin levels, indicating that aromatase and E2 were essential for regulating gonadotropin secretion. Additionally, sexual activity and desire were significantly decreased in aromatase-deficient men and male mice, and they could be restored by E2 treatment, thus revealing essential roles for aromatase and E2 in regulating sexual behavior [7, 19, 20].
The signaling pathways or molecular mechanisms that regulate the brain-specific aromatase promoter I.f are not well understood. Several groups of investigators found that protein kinases A and C and cAMP regulate aromatase expression and activity in the brain [21, 22]. Other groups showed that testosterone also upregulates aromatase mRNA and enzyme activity in the brain [17, 23–26]. Moreover, E2 was found to upregulate or downregulate hypothalamic aromatase mRNA and enzyme activity under both in vivo and in vitro circumstances [17, 25, 27, 28]. None of these studies, however, provided a connection between aromatase expression and the regulation of promoter I.f in brain tissue. In the only relevant publication, it was reported that the cis-regulatory element stress response element regulated the aromatase promoter I.f; however, neither the transcription factors nor any exogenous hormones responsible for this regulation have been reported [12]. We conducted this study to demonstrate the regulation of hypothalamic aromatase activity by E2 via the brain-specific aromatase promoter I.f and to identify the cis-regulatory elements and transcriptional activators involved.
Materials and Methods
Cell Culture
Mouse embryonic hypothalamic neuronal cell lines N38 and N42 were purchased from Cellutions Biosystems Inc. (Toronto, ON, Canada) [29] and were cultured in Dulbecco modified Eagle medium (DMEM; Gibco, New York, NY) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin-streptomycin (Gibco). The cells were studied at passages 15 to 16. After 24 h of serum starvation, cells were treated with various doses of 17β-estradiol (Sigma-Aldrich, St. Louis, MO) for different times.
RNA Isolation and Real-Time RT-PCR
Hypothalamic cells were treated with either vehicle (ethanol) or 10−7, 10−8, or 10−9 M E2, and total RNA was extracted after 6, 12, and 24 h of treatment using TRI reagent (Sigma) per the manufacturer's instructions. On column, DNAse digestion was carried out using a DNAse I kit (Qiagen, Valencia, CA). The integrity of the isolated RNA was verified by running 5 μg of total RNA on a 1% formaldehyde gel. Five micrograms of total RNA from vehicle or E2-treated hypothalamic cells was used for reverse transcription in a final volume of 20 μl using Superscript III First Strand RT synthesis kit (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Five microliters of cDNA was used for real-time RT-PCR (Taqman real-time primers and probe for aromatase were designed using ABI primer express software 3.0) on an Applied Biosystems 7000 Sequence Detection System (Applied Biosystems, Foster City, CA). The reactions were carried out using the ABI Taqman assay system for the aromatase promoter I.f-specific sequence (untranslated first exon I.f) and power SYBR green for the aromatase coding region according to the manufacturer's protocol. The primers and probes for aromatase (Cyp19a1) and Esr1 are as follows. Taqman Assay primers for Cyp19a1 exon I.f: forward 5′-aactcaccatcttcaagagtcca-3′, reverse 5′-gagtggcatggcactgacagt-3′, probe 5′-aggtccggttta-3′. Each contained a 6-carboxy-fluorescein phosphoramidite (FAM dye; Applied Biosystems, Foster City, CA) label at the 5′ end and a minor groove binder and nonfluorescent quencher at the 3′ end and were designed to hybridize to the exon I.f-exon II junction. SYBR green assay primers for Cyp19a1: forward 5′-tgtgttgaccctcatgagaca-3′, reverse 5′-cttgacggatcgttcatacttc-3′; for Esr1: forward 5′-atgaaaggcggcatacggaaag-3′, reverse 5′-cacccatttcatttcggccttc-3′; and for Gapdh: forward 5′-accacagtccatgccatcac-3′, reverse 5′-tccaccaccctgttgctgta-3′. Conventional PCR primers for Esr1: forward 5′-tccttctagacccttcagtgaagcc-3′, reverse 5′-acatgtcaaagatctccaccatgcc-3′.
Polymerase chain reaction was carried out in triplicate in a 50-μl reaction volume using SYBR green PCR mix. The reactions were incubated at 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. The threshold cycle (CT) is defined as the fractional cycle number at which the fluorescence passes the fixed threshold. Relative mRNA units were calculated from CT values as described previously [30]. All RNA samples were normalized to the expression levels of a glyceraldehyde-3-phosphate dehydrogenase (Gapdh) gene endogenous control. No-template and no-RT controls were used to ensure the absence of genomic DNA and reaction specificity. Three independent experiments were performed to demonstrate reproducibility. Real-time RT-PCR product specificity was confirmed by melt-curve analysis, gel electrophoresis, and product sequencing.
Aromatase Enzyme Activity Assay
The aromatase enzyme activity within hypothalamic cells was measured by [3H]water release assay, which is routinely used in our laboratory [31]. In each well, 60 pmol of [3H-1β]androstenedione (PerkinElmer Life Sciences, Waltham, MA) and 240 pmol of cold androstenedione (Sigma) were added to 2 ml of serum-free DMEM covering hypothalamic cells in 35-mm culture dishes. To study the effect of E2, hypothalamic cells at 80%–90% confluency were first serum starved for 24 h, and then treated with vehicle or E2 for 6, 12, and 24 h at 37°C in 95% air and 5% CO2. Each treatment was performed in triplicate. Four hours prior to the end of each time point, a mixture of labeled and cold androstenedione was added to each well and incubated until the completion of each treatment period. [3H-1β]androstenedione conversion to [3H]estrone was stopped by adding 10% (wt/vol) trichloroacetic acid. Steroidal compounds containing unconverted [3H-1β]androstenedione were removed from the mixture by first mixing with 4 ml of chloroform followed by centrifugation at 3000 rpm. The upper aqueous layer was removed and mixed with dextran-coated charcoal (1% wt/vol). Charcoal was precipitated by centrifugation. From each tube, 2 ml of clear solution was placed into a scintillation vial containing 10 ml of scintillation fluid and was counted in a scintillation counter (LS 6500; Beckman Coulter Inc., Fullerton, CA).
Immunoblotting
Hypothalamic neuronal cell lines were cultured in 150-mm dishes in DMEM containing 10% FBS until reaching 80% confluency. Total protein was extracted from cells using the M-PER mammalian protein extraction reagent (Pierce, Rockford, IL). Protein concentration was determined using the BCA protein assay kit (Pierce). Fifty micrograms of cell lysate was mixed with 2× Laemmli Sample Buffer (Bio-Rad Laboratories, Hercules, CA) and fractionated in 4%–15% SDS-PAGE. Proteins from the gel were transferred to a nitrocellulose membrane. The membranes were then incubated with mouse estrogen receptor alpha (ESR1) antibody (1:1000 dilution; Upstate, Charlottesville, VA) in 5% milk solution overnight at 4°C. The membrane was washed for 30 min then incubated with anti-rabbit immunoglobulin G (IgG)-peroxidase conjugate (Upstate) as a secondary antibody (1:3000 dilution). Incubation with the secondary antibody was performed at room temperature for 1 h. The signal was detected using SuperSignal West Femto maximum-sensitivity substrate chemiluminescence kit (Pierce) according to the manufacturer's instructions and exposed to BioMax ML X-ray film (Eastman Kodak, Rochester, NY) for 1–5 min. Then, the membranes were stripped of the first antibody and probed with anti-actin, followed by the same signal detection procedure described above.
Transient Transfections and Luciferase Assays
Transient transfection of N42 hypothalamic neuronal cell lines was carried out in 12-well plates with Fugene HD transfection reagent (Roche Diagnostics, Pleasanton, CA) with the following plasmids: 1) 1 μg of pGL3-basic luciferase reporter plasmid containing the promoter I.f nt −1000/−1 fragment, progressively truncated promoter I.f fragments (−700, −500, −200, −50), or nt −200/−1 constructs with mutations in the activator protein 1 (AP-1) cis-acting elements; 2) 50 ng of pRL-TK plasmid as an internal control (Promega, Madison, WI); and 3) 1 μg of wild-type mouse ESR1 or DNA-binding domain mutant ESR1-AA expression vectors. Cells were serum deprived 1 day before transfection. Wild-type ESR1 or ESR1-AA expression vectors were cotransfected with the nt −200/−1 promoter-luciferase reporter construct in equal amounts. Twenty-four hours after transfection, N42 cells were treated either with vehicle (ethanol) or 10−7 M E2 for 24 h. At the end of the treatment period, transfected cells were washed twice in PBS and lysed in 250 μl of lysis buffer (0.1 M potassium phosphate [pH 7.8], 1% Triton X-100, 1 mM dithiothreitol, and 2 mM ethylenediaminetetraacetic acid [EDTA]). Luciferase assays were performed with 20 μl of cell lysate employing a Dual-Luciferase Reporter Assay System kit (Promega). Luminescence was measured with a LUMAT LB9507 luminometer (EG&G Berthold, Bad Wildbad, Germany). Results are presented as the average of luciferase activity from triplicate experiments and expressed as the ratio to the internal standard Renilla luciferase. Plasmids used in transfection experiments were purified using an EndoFree Plasmid Isolation Kit (Qiagen), and their purity was verified by spectrophotometry and agarose gel electrophoresis. All transfection assays were performed using equimolar amounts of plasmids.
Site-Directed Mutagenesis
The putative AP-1 cis-acting elements were mutated using QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions. A chimeric luciferase construct containing the nt −200/−1 region of promoter I.f was used as a template for site-directed mutagenesis. Briefly, a PCR reaction using the DNA template (50 ng), mutagenesis primers (125 ng), deoxyribonucleotide triphosphate, and PfuTurbo DNA polymerase (2.5 U/μl) was carried out for 15 cycles at 95°C for 30 sec, 55°C for 60 sec, and 68°C for 10 min. After the reaction, DpnI restriction enzyme (10 U/μl) was added to the PCR products and incubated at 37°C for 60 min to eliminate the native DNA template. The mutagenesis reaction was then used to transform DH-5α-competent cells. Desired mutations were confirmed by DNA sequencing (Northwestern University sequencing facility). The oligonucleotides used for mutagenesis, with the mutated nucleotides underlined, are as follows (the complementary strand sequence is not shown): mutant AP-1 (nt −166/−160), 5′-agaggccgagatgaaataaaaaaatttggttataatttatg-3′; mutant AP-1 (nt −130/−124), 5′-tacgctgagatctaggaaaaagccaaacattcagaggccg-3′.
Chromatin-Immunoprecipitation-PCR Assay
The chromatin-immunoprecipitation PCR (ChIP) assay was performed with the Acetyl-Histone H4 Immunoprecipitation (ChIP) Assay Kit (Upstate Biotechnology, Lake Placid, NY) following the manufacturer's instructions with minor modifications. Briefly, after reaching 80%–90% confluency in 150-mm culture dishes (∼2 × 107 cells), hypothalamic cells were serum starved for 24 h, then treated with either vehicle (ethanol) or 10−7 M E2 for 6 h. Cells were then cross-linked with 1% formaldehyde for 10 min at room temperature on an orbital shaker, followed by the addition of 1 M glycine for 5 min. Cells were washed with cold PBS containing protease inhibitors and were lysed using a lysis buffer (50 mM Tris-HCl, pH 8.1, containing 1% SDS and 10 mM EDTA). The cells were sheared using a Sonic Dismembrator Model 100 (Fisher Scientific, Hampton, NH) at 70% maximum amplitude (15 sec on, 60 sec off) for five pulses to obtain DNA fragments in the range of 200 to 1000 bp. The sonicated cell supernatant was diluted 10-fold in ChIP dilution buffer, and 40 μl was used as input DNA (positive control). Precleared chromatin was used for immunoprecipitation (IP) reactions with 5 μg of the following specific antibodies: ESR1 (Upstate), c-Jun (JUN; Calbiochem, San Diego, CA), c-Fos (FOS; Calbiochem), or nonspecific rabbit IgG (Upstate). The reactions were incubated on a rotator overnight at 4°C. The immune complexes were collected using protein A-agarose/DNA beads and washed five times using low salt, high salt, and lithium chloride, and washed twice with Tris-EDTA buffers. The absorbed immune complexes were recovered by incubation with elution buffer (1% SDS and 0.1 M NaHCO3). Part of the eluate was saved and checked for the targeted protein IP by immunoblotting (IB). After reverse cross-linking at 65°C for 4 h, the genomic DNA was purified using a DNA purification kit (Qiagen). The promoter I.f region was scanned for possible ESR1 binding by PCR amplification using seven different pairs of primer sets representing the nt −1000/−1 region with ∼150-bp intervals. The PCR reactions were optimized according to each primer set, but the best amplification conditions for all primer sets were the following: 35 cycles (30 sec of denaturation at 94°C, 1 min of annealing at 57.5°C, and 1 min of elongation at 72°C). Chip primers were designed using the Primer3 Software (http://frodo.wi.mit.edu/primer3/input.htm) and are as follows: forward 5′-ggcttctcttggtacgctga-3′ and reverse 5′-ttgttgctaagagatcagttgctt-3′, which amplified the nt −190/−40 region of promoter I.f to yield a 150-bp amplicon; forward 5′-accacagagagtgaaagtttgag-3′ and reverse 5′-gcgtaccaagagaagccaat-3′, which amplified the nt −334/−173 region of promoter I.f to yield a 162-bp amplicon; forward 5′-gcattcaagtttgctcagagg-3′ and reverse 5′-ttcttttgatggggttgcac-3′, which amplified the nt −431/−281 region of promoter I.f to yield a 151-bp amplicon; forward 5′-cctgcctaaaggctaagatcc-3′ and reverse 5′-acacacatacacctctgagcaaa-3′, which amplified the nt −549/−399 region of promoter I.f to yield a 151-bp amplicon; forward 5′-cccaaaccttatcaacttagcc-3′ and reverse 5′-ggatcttagcctttaggcaggt-3′, which amplified the nt −692/−528 region of promoter I.f to yield a 165-bp amplicon; forward 5′-tgagatgtaaacattatgtgtgtgct-3′ and reverse 5′-ggtttgggtttaggggaat-3′, which amplified the nt −793/−685 region of promoter I.f to yield a 109-bp amplicon; and forward 5′-agcagagatggcttgtggtt-3′ and reverse 5′-gcacacacataatgtttacatctca-3′, which amplified the nt −925/−768 region of promoter I.f to yield a 158-bp amplicon. All ChIP buffers used in this protocol contained a 1× protease inhibitor cocktail (Sigma).
Electrophoretic Mobility Shift Assay
Nuclear protein extracts used for electrophoretic mobility shift assay (EMSA) were prepared as described previously [32, 33]. The sequences of the double-stranded oligonucleotides used for EMSA were: 5′-GCTGAGATCTATTACAAAGCCAAAC-3′, which is identical to a 25-bp-long sequence (nt −175/−151) within promoter I.f and contains one AP-1 cis-acting element at nt −166/−160; and 5′-AGATGAAATGACACAATTTGGTTAT-3′, which is identical to a 25-bp-long sequence (nt −138/−114) within promoter I.f and contains one AP-1 cis-acting element at nt −130/−124. The mutant forms of the oligonucleotides, with the mutated bases underlined, were: 5′-GCTGAGATCTGGGACAAAGCCAAAC-3′ and 5′-AGATGAAATTTTACAATTTGGTTAT-3′. All oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA). The sequence for the AP-1 cis-acting element consensus sequence oligonucleotide was 5′-CGCTTGATGACTCAGCCGGGA-3′, and its mutant form, with the mutated bases underlined, was 5′-CGCTTGATGACTTGGCCGGAA-3′. Both oligonucleotides were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). After renaturing two single-stranded oligonucleotides to a duplex oligonucleotide, we end labeled the AP-1 consensus cis-acting element and promoter I.f putative AP-1 cis-acting element containing oligonucleotides with [γ-32P]ATP using T4 kinase. Free [γ-32P]ATP was removed by Bio-Spin 6 chromatography columns (Bio-Rad). Briefly, 4-μg samples of nuclear extracts were incubated with the radiolabeled double-stranded oligonucleotide probe (50 0000 cpm per reaction) for 20 min at room temperature in a reaction buffer containing 20 mM HEPES (pH 7.6), 75 mM KCl, 0.2 mM EDTA, 20% glycerol, and 2 μg of poly(dI-dC)-poly(dI-dC) as a nonspecific competitor. For competition EMSA, a nonradiolabeled double-stranded oligonucleotide was added simultaneously with the labeled probe at 100× concentration. Immunodepletion was performed after the addition of 2 μg of an antibody against JUN (Cell Signaling Technology, Danvers, MA), ESR1, or IgG to the binding reaction, followed by a 30-min incubation at room temperature before electrophoresis. Then, protein-DNA complexes were resolved on 6% nondenaturing polyacrylamide gels with 0.5× Tris-borate-EDTA running buffer. Gels were covered with cellophane membranes (Bio-Rad) and dried on a Bio-Rad gel air dryer for 2 h. Gels were then exposed to BioMax ML x-ray film overnight for visualization.
IP/IB Assay
Immunoprecipitation was performed with nuclear protein preparations of hypothalamic cells prepared by N-PER nuclear extraction kit (Pierce). Protein concentration was determined using BCA protein assay kit according to the manufacturer's instructions. Nuclear protein preparations (200 μg) were immunoprecipitated with either of anti-ESR1, anti-JUN, anti-FOS, or IgG antibodies by incubating overnight on a rotator at 4°C. Protein-antibody complexes were recovered by protein A-Sepharose. The isolated immune complexes were washed five times with PBS and resolved onto SDS-PAGE. Proteins were transferred to nitrocellulose membrane and were probed with antibodies against c-Jun or ESR1. We performed the IP assay for ESR1 and IB assay for JUN, and vice versa, to confirm the ESR1-JUN interaction.
Small-Interfering RNA
Nonspecific (NS) small-interfering RNA (siRNA) and ESR1 siRNA were purchased from Dharmacon (Chicago, IL). ESR1 knockdown was verified by real-time RT-PCR and IB. One day before transfection, hypothalamic cells were plated in six-well plates to achieve 50% confluency at the time of transfection, and they were then transfected with 100 nM NS siRNA or ESR1 siRNA in triplicate using lipofectamide RNAiMAX reagent (Invitrogen Life Technologies Inc., New York, NY) according to the manufacturer's protocol. Forty-eight hours after transfection, cells were serum starved for 24 h, followed by treatment with 10−7 M E2 for 6 h. Total mRNA was extracted for real-time RT-PCR assay, and total protein was isolated by M-PER reagent for immunoblot analysis.
Statistical Analysis
Statistical analyses were performed by Welch paired t-test and one-way ANOVA followed by Tukey multiple-comparisons test using the StatView 5.0 statistical software package (SAS Institute, Cary, NC). Significance was determined at α = 0.05 and β = 0.20. The values for mRNA and aromatase activity were provided as the mean ± SEM.
Results
Determination of Aromatase and Esr1 Expression Levels in N42 and N38 Hypothalamic Neuronal Cell Lines
Conventional RT-PCR, real-time RT-PCR, and IB analyses were performed to measure aromatase and Esr1 expression in N42 and N38 hypothalamic neuronal cell lines. Both promoter I.f-driven aromatase expression (Fig. 1A) and Esr1 expression (Fig. 1, B and C) were remarkably higher in N42 cells compared with N38 cells. These results were reproducibly observed in three independent experiments.
Fig. 1.

N42 and N38 hypothalamic cells use promoter I.f to regulate aromatase mRNA expression. Esr1 mRNA and ESR1 protein expression levels in N42 hypothalamic cells are higher than N38 hypothalamic cells. A) Real-time RT-PCR was performed employing a probe complementary to the exon I.f-exon II junction to measure promoter I.f-driven aromatase mRNA expression. Relative units are shown as the ratio of aromatase mRNA:Gapdh mRNA; (presented as l.f-specific aromatase mRNA/Gapdh mRNA). Results are expressed as the mean ± SEM (n = 3); **P < 0.01, paired t-test. Conventional RT-PCR (B) and IB (C) were performed to measure ESR1 expression in brain tissue (positive control) and N42 or N38 hypothalamic cells. Gapdh and actin were used as loading controls. The figures represent one of three independently performed experiments.
Effect of E2 on Aromatase mRNA Expression and Enzyme Activity
Real-time RT-PCR and aromatase enzyme activity assays were performed to determine whether E2 had any effects on aromatase mRNA expression and enzyme activity. We treated N42 and N38 hypothalamic cells with 10−7 M, 10−8 M, and 10−9 M E2 for 6, 12, and 24 h. At 10−8 M and 10−9 M, E2 had no effect on aromatase mRNA expression and enzyme activity in either cell line (data not shown). Estradiol at 10−7 M, however, had a biphasic effect on aromatase mRNA expression and enzyme activity in N42 hypothalamic cells (Fig. 2, A and B). In N38 hypothalamic cells, 10−7 M E2 had no effect on aromatase mRNA expression or enzyme activity at any time point (Fig. 2C). Our results suggested that high baseline levels of aromatase expression with a remarkable response to E2 in N42 cells compared with lower aromatase expression without any E2 response in N38 cells may, in part, be due to much higher ESR1 levels in N42 cells.
Fig. 2.

Estradiol regulates aromatase mRNA expression and enzyme activity in N42, but not N38, hypothalamic cells. A) Real-time RT-PCR was performed to measure aromatase mRNA expression after 6, 12, and 24 h of E2 (10−7 M) treatment. Aromatase mRNA levels were normalized to Gapdh mRNA levels. Results are expressed as the mean ± SEM (n = 3); **P < 0.01, paired t-test. Aromatase activity assays were performed in N42 (B) and N38 (C) hypothalamic cells after 6, 12, and 24 h of E2 (10−7 M) treatment. The results are expressed as the mean ± SEM (n = 3); **P < 0.01, paired t-test. Aromatase enzyme activity in each neuronal line treated with the aromatase enzyme inhibitor letrozole (LET) was undetectable (data not shown).
The ESR Antagonist ICI 182,780 Inhibits E2-Induced Aromatase mRNA Expression and Enzyme Activity
To clarify whether the mechanism of E2 action on aromatase expression and enzyme activity is ESR dependent, we treated N42 hypothalamic cells with the E2 antagonist ICI 182,780 (ICI). Treatment with ICI only reduced baseline aromatase mRNA levels significantly (Fig. 3A). ICI interfered with E2-induced aromatase mRNA expression and enzyme activity, as assessed by real-time RT-PCR and aromatase enzyme activity assays (Fig. 3, A and B).
Fig. 3.

The E2 antagonist ICI inhibits baseline and E2-induced aromatase mRNA expression and enzyme activity in N42 hypothalamic cells. A) Real-time RT-PCR was performed after cells were incubated in the presence or absence of 10−7 M E2 plus or minus 10−5 M ICI for 6 h. Aromatase mRNA levels were normalized to Gapdh mRNA levels. The results are expressed as the mean ± SEM (n = 3); **P < 0.01, ANOVA. B) Aromatase activity assays were performed to measure enzyme activity in cells incubated in the presence or absence of 10−7 M E2 plus or minus 10−5 M ICI for 6 h. The results are expressed as the mean ± SEM (n = 3); **P < 0.01, ANOVA. Aromatase activity in the N42 neuronal line treated with the aromatase enzyme inhibitor LET was undetectable (data not shown).
The nt −200/−1 Region of Promoter I.f Confers Responsiveness to E2
To identify specific cis-regulatory elements within the −1000/−1 region of aromatase promoter I.f, we generated a series of truncated reporter constructs and transiently transfected them into N42 hypothalamic cells. The −500/−1 construct conferred the highest baseline promoter activity, suggesting that −500/−200-bp region contains critical cis-regulatory elements for baseline gene expression (Fig. 4A). On the other hand, only the nt −200/−1 promoter-luciferase reporter construct was responsive to E2 (∼2.3-fold induction; Fig. 4A).
Fig. 4.

AP-1 cis-regulatory elements within the nt −200/−1 region of promoter I.f are essential for E2-dependent induction of aromatase expression in N42 hypothalamic cells. A) Serial deletion analysis was performed to localize the E2-responsive regions of aromatase promoter I.f. The promoter I.f-luciferase (LUC) reporter constructs were named according to the nucleotide positions of their 5′ termini. All constructs were transiently transfected into N42 hypothalamic cells. Luciferase assays were performed a minimum of three times in the presence or absence of 10−7 M E2. Mean luciferase activity of each construct is given relative to the nt −1000/−1 promoter-luciferase reporter construct. Summary data for three independent experiments are shown. Results are expressed as the mean ± SEM (**P < 0.01, paired t-test). B) Mutational analysis of promoter I.f was performed to verify functional cis-regulatory elements. Substitution mutations were made within two putative AP-1 cis-regulatory elements in the −200/−1 promoter-luciferase reporters. Constructs were transiently cotransfected with ESR1 expression vector into N42 hypothalamic cells, and luciferase activities were measured after treatment with vehicle or 10−7 M E2. Results shown are the mean ± SEM of three independent experiments (**P < 0.01, ANOVA).
It was proposed that ESR1 exerts some of its genomic actions via indirect binding to AP-1 or SP1 motifs in target promoters [34, 35]. ESR1 binding to AP-1 cis-regulatory elements may occur as a complex with JUN and/or FOS transcription factors [35]. We could not identify any palindromic estrogen response elements (EREs) or ERE half-sites in the nt −1000/−1 region, but there were two putative AP-1 motifs at nt −166/−160 and nt −130/−124. Introduction of substitution mutations into these putative AP-1 motifs abolished promoter activity of the nt −200/−1 promoter-reporter luciferase construct, even in the presence of overexpressed ESR1 (Fig. 4B).
ESR1 and JUN, but Not FOS, Interact with Promoter I.f
We performed ChIP assays to confirm that ESR1 or JUN binds to aromatase promoter I.f. We used seven different sets of overlapping primer pairs representing the nt −1000/−1 region of promoter I.f, with ∼150-bp intervals. As shown in Figure 5A, the most consistent and intense transcription factor binding occurred at the nt −200/−1 region. Under serum-starved conditions, we observed ESR1 binding in N42 but not in N38 hypothalamic cells, and binding of ESR1 to the nt −193/−47 region was enhanced by E2 treatment (10−7 M; 6 h) in both N42 and N38 hypothalamic cells. Estradiol-induced recruitment of ESR1 was observed at the E2-responsive nt −193−47 but not at a more distal (nt −925/−768) region (Fig. 4). We also found that JUN, but not FOS, bound to the nt −193/−47 region in N42 hypothalamic cells, and JUN bound only in the presence of E2 (Fig. 5B).
Fig. 5.
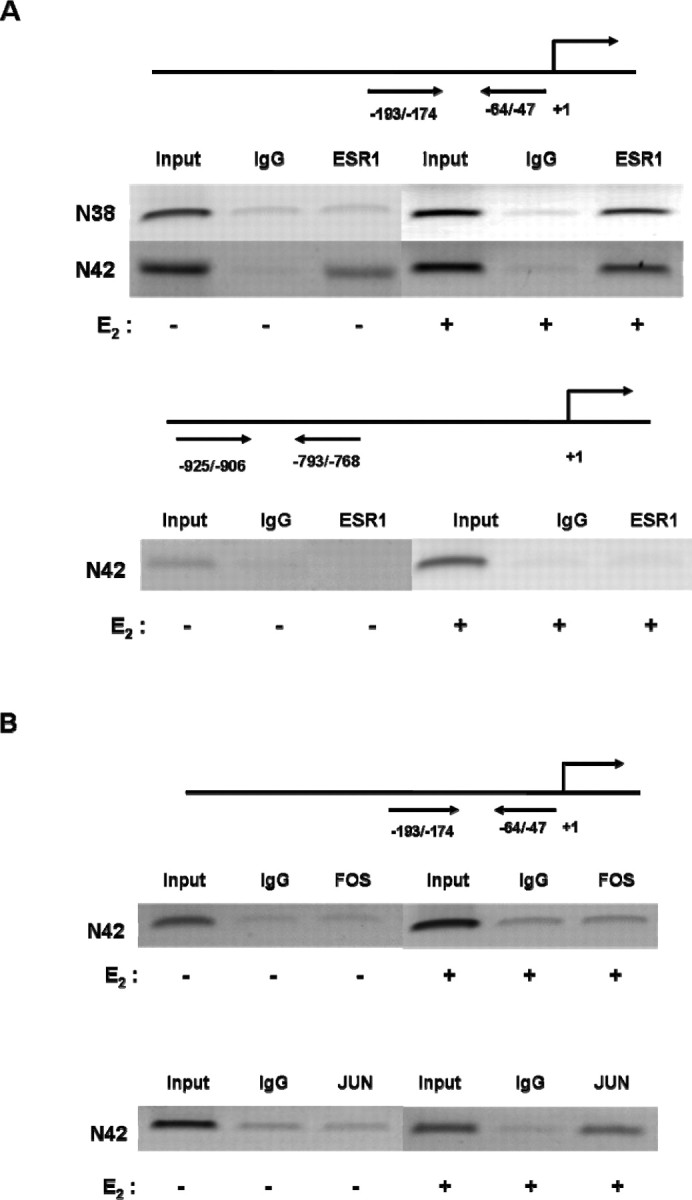
ChIP analysis reveals ESR1 and JUN, but not FOS, bind to promoter I.f in N42 hypothalamic cells. We performed ChIP assays using N42 hypothalamic cell extracts treated with vehicle or 10−7 M E2 for 6 h. Sonicated cell supernatant was used as input DNA (positive control). Precleared chromatin was used for IP reactions with a rabbit polyclonal antibody directed against human (A) ESR1 and (B) JUN or FOS, as well as normal rabbit IgG. ESR1 was recruited to the E2-responsive nt −193/−47 but not to a more distal (nt −925/−768) region (see Fig. 4). Estradiol induced the recruitment of JUN but not FOS to the nt −193/−47 region. The figures represent one of three independently performed experiments.
The nt −130/−124 and nt −166/−160 Regions of Promoter I.f Are the Major ESR1-JUN Binding Sites
To determine whether the putative AP-1 cis-acting elements at nt −130/−124 or nt −166/−160 interact with JUN and ESR1, we performed EMSAs using probes representing either AP-1 site in the I.f promoter. A probe representing the consensus AP-1 sequence was used as a control. Nuclear protein extracts prepared from E2-treated (10−7 M; 6 h) N42 hypothalamic cells formed complexes on DNA fragments containing the nt −130/−124 (Fig. 6A) and the nt −166/−160 (Fig. 6B) regions of the aromatase I.f promoter. Antibodies against ESR1 or JUN immunodepleted and supershifted the specific complexes formed on the probes containing the nt −130/−124 or nt −166/−160 sequences (Fig. 6).
Fig. 6.

Putative AP-1 sites at nt −130/−124 and nt −166/−160 regions of promoter I.f are the major c-Jun-ESR1 binding sites. An EMSA was used to visualize JUN and ESR1 binding to AP-1 sites within promoter I.f. The EMSAs were performed using 5 μg of nuclear extract from N42 hypothalamic cells treated with E2 (10−7 M; 6 h) and radiolabeled oligonucleotide probes containing critical AP-1 sites and flanking sequences. A) Binding to an oligonucleotide identical to the nt −138/−114 sequence in promoter I.f and containing the nt −130/−124 AP-1 element (native; lane 2). Competition studies were performed using 100-fold excess unlabeled wild-type (consensus; lanes 3 and 4) or native oligonucleotides (lane 5) or mutant native oligonucleotides (lane 6). Immunodepletion of complexes was performed using 2 μg of anti-c-Jun (lanes 7 and 9) or 0.5 μg of anti-ESR1 (lanes 8 and 10) antibody (ab). Normal rabbit IgG was added as a negative control (lanes 11 and 12). B) Binding to an oligonucleotide identical to the nt −175/−151 sequence in promoter I.f and containing the nt −166/−160 AP-1 element (native) (lane 2). Competition studies were performed using 100-fold excess unlabeled consensus or native probes (lanes 5 and 7) or mutant consensus or mutant native probes (lanes 6 and 8). Immunodepletion of complexes was performed using 2 μg of anti-c-Jun (lanes 10 and 12) or 0.5 μg of anti-ESR1 (lanes 9 and 11) antibody (ab). Normal rabbit IgG was added as a negative control (lanes 13 and 14). Black arrows indicate JUN-ESR1 complexes, and white arrows indicate supershifted complexes by either c-Jun or ESR1 antibodies. The figures represent one of three independently performed experiments.
ESR1 DNA-Binding Domain Is Not Necessary for ESR1-Mediated Regulation of Aromatase mRNA Expression
To determine whether the ESR1 DNA-binding domain is required for ESR1 action on aromatase mRNA expression, we cotransfected N42 hypothalamic cells with an nt −200/−1 promoter I.f-luciferase reporter and expression plasmids containing wild-type ESR1 or ESR1 carrying a mutation within the DNA-binding domain. Cells were treated with 10−7 M E2. Estradiol-stimulated I.f promoter activity increased in the presence of overexpressed ESR1 or ESR1 with a defective DNA-binding domain (ESR1-AA) but not in the presence of the empty expression vector (Fig. 7).
Fig. 7.

The ESR1 DNA-binding domain is not required for mediating E2-dependent aromatase expression in N42 hypothalamic cells. The nt −200/−1 I.f promoter-luciferase reporter construct was transiently cotransfected with either wild-type ESR1 or ESR1 carrying a mutation at DNA-binding domain (ESR1-AA) into N42 hypothalamic cells. Luciferase assays were performed a minimum of three times in the presence of E2 (10−7 M). Summary data for three independent experiments are shown. Results are expressed as the mean ± SEM (**P < 0.01, ANOVA).
ESR1 and JUN Form a Complex
We performed IP/IB assays to determine whether ESR1 forms complexes with JUN or FOS in E2-treated hypothalamic N42 cells. We performed IP using antibodies against JUN or FOS, followed by IB using an antibody against ESR1. After 6 h of E2 (10−7 M) treatment, ESR1 formed a complex with JUN but not with FOS (Fig. 8A). We verified these findings by reversing the order in which the antibodies were used. Immunoprecipitation with a human antibody raised in mouse (mESR1) or rabbit (rESR1) followed by IP using an antibody against JUN confirmed formation of a complex between ESR1 and JUN in N42 hypothalamic cells (Fig. 8B).
Fig. 8.

ESR1 and JUN, but not FOS, form a complex in N42 hypothalamic cells. A) Immunoprecipitation with antibodies against human JUN or FOS and IB with an antibody against ESR1 (mESR1; raised in mouse, monoclonal) were performed in N42 hypothalamic cells treated with E2 (10−7 M; 6 h). B) Immunoprecipitation with two different ESR1 antibodies, mESR1 or rESR1 (raised in rabbit, polyclonal), followed by IB with an antibody against JUN was performed in N42 hypothalamic cells treated with E2 (10−7 M; 6 h). Immunoprecipitation with a nonspecific rabbit IgG was the negative control. The figures represent one of three independently performed experiments.
E2-Stimulated Aromatase Expression Is Mediated by ESR1
The siRNA technology was used to determine whether endogenous ESR1 in N42 hypothalamic cells mediates E2-stimulated aromatase expression. As shown in Figure 9, A and B, siRNA knockdown of Esr1 in N42 hypothalamic cells abolished induction of aromatase mRNA levels and enzyme activity by E2 (10−7 M) treatment. The knockdown of Esr1 was confirmed by real-time RT-PCR and IB (Fig. 9, C and D).
Fig. 9.

ESR1 mediates E2-stimulated aromatase mRNA expression in N42 hypothalamic cells. N42 hypothalamic cells were transfected with Esr1 siRNA or with nonspecific siRNA as a negative control (NS) and were cultured for an additional 48 h. A) Aromatase mRNA levels were analyzed by real-time RT-PCR in the presence or absence of E2 (10−7 M; 6 h). Aromatase mRNA levels were normalized to Gapdh mRNA levels. The results are expressed as the mean ± SEM (n = 3; **P < 0.01, ANOVA. B) Aromatase activity assays were performed in the presence or absence of E2 (10−7 M; 6 h). The results are expressed as the mean ± SEM (n = 3); **P < 0.01, ANOVA. Aromatase enzyme activity in N42 neuronal line treated with the aromatase enzyme inhibitor LET was undetectable (data not shown). Real-time RT-PCR (C) and IB (D) were performed to measure Esr1 mRNA and protein levels, respectively. Actin was used as a loading control. The IB figure represents one of three independently performed experiments. Real-time RT-PCR results are expressed as the mean ± SEM (n = 3), and Gapdh was used as a loading control. **P < 0.01, ANOVA. Representative results from three independent experiments are shown.
Discussion
Aromatase deficiency in humans disrupts normal gonadotropin regulation and libido [6]. Likewise, Cyp19a1KO mice exhibit disrupted reproductive behavior, including defective lordosis (females), low copulatory behavior, reduced sexual interest in the opposite sex, and deficiency in pup care [36–39]. The gonadotropin secretion profile is also disrupted in Cyp19a1 KO mice, which have increased luteinizing hormone, follicle-stimulating hormone, testosterone, and prolactin, decreased E2, and unchanged dihydrotestosterone (DHT) [6, 18]. The reproductive phenotypes manifested in Cyp19a1 KO mice are similar to those observed in Esr1 knockout mice [1, 2, 40, 41], suggesting that aromatase and ESR1 are important in mediating reproductive behavior controlled by the brain, and in particular by the hypothalamus.
Aromatase is expressed primarily in gonads and the brain [3, 6, 14]. Loss of local E2 synthesis in the brain likely leads to the hormonal and behavioral phenotypes observed in aromatase-deficient humans and mice. In fact, alterations in brain aromatase expression have been directly linked to changes in sexual behavior in birds, rodents, sheep, and humans [6, 38, 39, 42, 43].
Gonadectomy reduces aromatase expression and enzyme activity in the brain; however, treatment of gonadectomized animals with testosterone or E2 markedly restores aromatase activity in several brain regions, including the preoptic/hypothalamic area in birds and mammals [17, 21, 23–26]. Moreover, the positive effects of testosterone on copulatory behavior can be blocked by inhibiting ESR1 action or aromatase. These observations suggest that not only ESR1, but also endogenously synthesized E2 may play a role in the regulation of aromatase in the brain. On the contrary, DHT by itself has almost no effect on aromatase expression, suggesting that testosterone needs to be converted to E2 to exert its effects on the brain [14, 23, 24, 40]. Consistent with these published data, we did not observe any time- or dose-dependent effect of DHT on aromatase mRNA expression or enzyme activity in hypothalamic neuronal cell lines (data not shown).
Estradiol has been reported to alternately increase or decrease brain aromatase expression and enzyme activity [17, 25, 27, 28, 44, 45]. These reports are consistent with our current findings that E2 showed a biphasic stimulatory effect on aromatase mRNA levels and enzyme activity in hypothalamic cells. Moreover, our observation that E2-induced aromatase expression is dependent on the presence of ESR1 is also consistent with the findings of other groups employing similar E2 concentrations under various in vivo and in vitro settings [28, 46–49].
The varying intracellular concentration of E2 and possible alterations in the composition of the multimeric transcriptional complex at promoter I.f over time may explain the mechanism of biphasic response observed in this study. These considerations will be investigated as future directions.
Estradiol is synthesized in specific brain regions, including the hypothalamus and hippocampus, where tissue E2 concentrations are found to be higher than circulating E2 levels [1, 2, 46–50]. Because E2 is synthesized within the cytoplasm of neurons in these regions, it is possible that supraphysiologic levels of E2 mediate intracrine and paracrine effects on neighboring steroidogenic and neuronal cells, comparable to effects of supraphysiologic quantities of endocrine E2 supplied to the brain via the bloodstream [6, 46, 48]. This notion has been raised by numerous studies, which have reported that the biologic effects of E2 could only be triggered by concentrations in the nanomolar to micromolar range [5, 6, 27, 28, 46–49]. In this study, we used micromolar to nanomolar (10−7 to 10−9) E2 concentrations. Although these E2 concentrations are much higher than those observed in the plasma, they might appropriately mimic the local conditions in target areas of the brain.
Interestingly, it was reported that E2 induces aromatase expression in the VMH but not in the medial preoptic area, and there is less frequent ESR1 and aromatase colocalization compared with the VMH [14, 41, 42, 51]. These findings suggest a critical in vivo role for ESR1 in regulating aromatase expression [14, 41, 42, 51]. In parallel with these findings, we observed that E2 induced aromatase mRNA expression and enzyme activity in N42 but not in N38 hypothalamic cells, and the N38 cells expressed markedly lower levels of ESR1 compared with N42 cells (Figs. 1 and 2). It is likely that basal aromatase expression in N42 cells may be regulated by locally produced E2, because basal aromatase expression was suppressed by an estradiol antagonist (Figs. 1A and 3A).
The developmental and tissue-, region-, and neuron-specific expression of aromatase in the brain has been reported to be transcriptionally regulated through promoter I.f, with the region 202 bp upstream of the transcriptional initiation site being essential and sufficient for basal transcriptional activity in cultured neurons [12, 52]. Our current findings are largely in agreement with these previous findings (Fig. 4A).
Honda et al. [12] demonstrated interactions between nuclear proteins and several sequences within the nt −200/−1 region of promoter I.f. Of these sequences, nt −171/−147, nt −136/−109, and nt −84/−60 were reported to form complexes exhibiting a similar pattern of mobility on gel shift assays, suggesting that all three sites bind to the same transcription factor(s). Indeed, these three sequences were shown previously to interact with an identical nuclear factor whose molecular mass was indicated to be around 42 kDa [12], but the identity of these nuclear factors has never been revealed. Interestingly, we showed that both JUN and ESR1 bound to AP-1 motifs within the region that encompasses these first two sequences. We have not characterized binding of JUN or ESR1 to the nt −84/−60 sequence, but we have identified two putative AP-1 motifs within this region. Since the molecular mass of JUN is approximately 42 kDa, we postulate that the unknown transcription factor in the previous study was JUN. The expression profile of JUN in the brain of mice and rats is similar to that of aromatase, with a transient peak followed by a gradual decrease during fetal development [14, 35, 53]. Thus, these data collectively suggest that JUN may be one of the critical transcription factors responsible for regulating prenatal aromatase expression in the fetal brain.
In summary, we demonstrated a nonclassical action of ESR1 in mediating estrogen-dependent regulation of aromatase expression in hypothalamic neuronal cell lines. Our future directions include verification of these findings using in vivo models. We also plan to further investigate the in vitro and in vivo effects of other hormones (e.g., progesterone) on aromatase expression and activity to elucidate the molecular mechanisms mediating local E2 synthesis in the hypothalamus.
References
- 1. Beyer C. Estrogen and the developing mammalian brain. Anat Embryol (Berl)1999; 199:379–390.. [DOI] [PubMed] [Google Scholar]
- 2. Li R, Shen Y. Estrogen and brain: synthesis, function and diseases. Front Biosci 2005; 10:257–267.. [DOI] [PubMed] [Google Scholar]
- 3. Bulun SE, Lin Z, Imir G, Amin S, Demura M, Yilmaz B, Martin R, Utsunomiya H, Thung S, Gurates B, Tamura M, Langoi D et al. Regulation of aromatase expression in estrogen-responsive breast and uterine disease: from bench to treatment. Pharmacol Rev 2005; 57:359–383.. [DOI] [PubMed] [Google Scholar]
- 4. Kamat A, Hinshelwood MM, Murry BA, Mendelson CR. Mechanisms in tissue-specific regulation of estrogen biosynthesis in humans. Trends Endocrinol Metab 2002; 13:122–128.. [DOI] [PubMed] [Google Scholar]
- 5. Simpson ER, Davis SR. Minireview: aromatase and the regulation of estrogen biosynthesis–some new perspectives. Endocrinology 2001; 142:4589–4594.. [DOI] [PubMed] [Google Scholar]
- 6. Jones ME, Boon WC, Proietto J, Simpson ER. Of mice and men: the evolving phenotype of aromatase deficiency. Trends Endocrinol Metab 2006; 17:55–64.. [DOI] [PubMed] [Google Scholar]
- 7. Simpson ER. Models of aromatase insufficiency. Semin Reprod Med 2004; 22:25–30.. [DOI] [PubMed] [Google Scholar]
- 8. Zhao H, Innes J, Brooks DC, Reierstad S, Yilmaz MB, Lin Z, Bulun SE. A novel promoter controls Cyp19a1 gene expression in mouse adipose tissue. Reprod Biol Endocrinol 2009; 7:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Golovine K, Schwerin M, Vanselow J. Three different promoters control expression of the aromatase cytochrome p450 gene (cyp19) in mouse gonads and brain. Biol Reprod 2003; 68:978–984.. [DOI] [PubMed] [Google Scholar]
- 10. Honda S, Harada N, Takagi Y. The alternative exons 1 of the mouse aromatase cytochrome P-450 gene. Biochim Biophys Acta 1996; 1305:145–150.. [DOI] [PubMed] [Google Scholar]
- 11. Basic Local Alignment Search Tool (BLAST),National Center for Biotechnology Information, U.S. National Library of Medicine, Bethesda, Maryland. World Wide Web (http://www.ncbi.nlm.nih.gov/BLAST/). (February 2008).
- 12. Honda S, Harada N, Abe-Dohmae S, Takagi Y. Identification of cis-acting elements in the proximal promoter region for brain-specific exon 1 of the mouse aromatase gene. Brain Res Mol Brain Res 1999; 66:122–132.. [DOI] [PubMed] [Google Scholar]
- 13. Lephart ED. Molecular aspects of brain aromatase cytochrome P450. J Steroid Biochem Mol Biol 1997; 61:375–380.. [PubMed] [Google Scholar]
- 14. Lephart ED. A review of brain aromatase cytochrome P450. Brain Res Brain Res Rev 1996; 22:1–26.. [PubMed] [Google Scholar]
- 15. Peterson RS, Yarram L, Schlinger BA, Saldanha CJ. Aromatase is pre-synaptic and sexually dimorphic in the adult zebra finch brain. Proc Biol Sci 2005; 272:2089–2096.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Voigt C, Ball GF, Balthazart J. Neuroanatomical specificity of sex differences in expression of aromatase mRNA in the quail brain. J Chem Neuroanat 2007; 33:75–86.. [DOI] [PubMed] [Google Scholar]
- 17. Zhao C, Fujinaga R, Tanaka M, Yanai A, Nakahama K, Shinoda K. Region-specific expression and sex-steroidal regulation on aromatase and its mRNA in the male rat brain: immunohistochemical and in situ hybridization analyses. J Comp Neurol 2007; 500:557–573.. [DOI] [PubMed] [Google Scholar]
- 18. Fisher CR, Graves KH, Parlow AF, Simpson ER. Characterization of mice deficient in aromatase (ArKO) because of targeted disruption of the cyp19 gene. Proc Natl Acad Sci U S A 1998; 95:6965–6970.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bakker J, Baillien M, Honda S, Harada N, Balthazart J. Relationships between aromatase activity in the brain and gonads and behavioural deficits in homozygous and heterozygous aromatase knockout mice. J Neuroendocrinol 2004; 16:483–490.. [DOI] [PubMed] [Google Scholar]
- 20. Bakker J, Honda S, Harada N, Balthazart J. Restoration of male sexual behavior by adult exogenous estrogens in male aromatase knockout mice. Horm Behav 2004; 46:1–10.. [DOI] [PubMed] [Google Scholar]
- 21. Balthazart J, Baillien M, Ball GF. Phosphorylation processes mediate rapid changes of brain aromatase activity. J Steroid Biochem Mol Biol 2001; 79:261–277.. [DOI] [PubMed] [Google Scholar]
- 22. Lavaque E, Mayen A, Azcoitia I, Tena-Sempere M, Garcia-Segura LM. Sex differences, developmental changes, response to injury and cAMP regulation of the mRNA levels of steroidogenic acute regulatory protein, cytochrome p450scc, and aromatase in the olivocerebellar system. J Neurobiol 2006; 66:308–318.. [DOI] [PubMed] [Google Scholar]
- 23. Abdelgadir SE, Resko JA, Ojeda SR, Lephart ED, McPhaul MJ, Roselli CE. Androgens regulate aromatase cytochrome P450 messenger ribonucleic acid in rat brain. Endocrinology 1994; 135:395–401.. [DOI] [PubMed] [Google Scholar]
- 24. Lephart ED, Simpson ER, McPhaul MJ, Kilgore MW, Wilson JD, Ojeda SR. Brain aromatase cytochrome P-450 messenger RNA levels and enzyme activity during prenatal and perinatal development in the rat. Brain Res Mol Brain Res 1992; 16:187–192.. [DOI] [PubMed] [Google Scholar]
- 25. Negri-Cesi P, Colciago A, Motta M, Martini L, Celotti F. Aromatase expression and activity in male and female cultured rat hypothalamic neurons: effect of androgens. Mol Cell Endocrinol 2001; 178:1–10.. [DOI] [PubMed] [Google Scholar]
- 26. Roselli CE, Abdelgadir SE, Resko JA. Regulation of aromatase gene expression in the adult rat brain. Brain Res Bull 1997; 44:351–357.. [DOI] [PubMed] [Google Scholar]
- 27. Kretz O, Fester L, Wehrenberg U, Zhou L, Brauckmann S, Zhao S, Prange-Kiel J, Naumann T, Jarry H, Frotscher M, Rune GM. Hippocampal synapses depend on hippocampal estrogen synthesis. J Neurosci 2004; 24:5913–5921.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iivonen S, Heikkinen T, Puolivali J, Helisalmi S, Hiltunen M, Soininen H, Tanila H. Effects of estradiol on spatial learning, hippocampal cytochrome P450 19, and estrogen alpha and beta mRNA levels in ovariectomized female mice. Neuroscience 2006; 137:1143–1152.. [DOI] [PubMed] [Google Scholar]
- 29. Belsham DD, Cai F, Cui H, Smukler SR, Salapatek AM, Shkreta L. Generation of a phenotypic array of hypothalamic neuronal cell models to study complex neuroendocrine disorders. Endocrinology 2004; 145:393–400.. [DOI] [PubMed] [Google Scholar]
- 30. Imir AG, Lin Z, Yin P, Deb S, Yilmaz B, Cetin M, Cetin A, Bulun SE. Aromatase expression in uterine leiomyomata is regulated primarily by proximal promoters I.3/II. J Clin Endocrinol Metab 2007; 92:1979–1982.. [DOI] [PubMed] [Google Scholar]
- 31. Shozu M, Sebastian S, Takayama K, Hsu WT, Schultz RA, Neely K, Bryant M, Bulun SE. Estrogen excess associated with novel gain-of-function mutations affecting the aromatase gene. N Engl J Med 2003; 348:1855–1865.. [DOI] [PubMed] [Google Scholar]
- 32. Cheng YH, Imir A, Suzuki T, Fenkci V, Yilmaz B, Sasano H, Bulun SE. SP1 and SP3 mediate progesterone-dependent induction of the 17beta hydroxysteroid dehydrogenase type 2 gene in human endometrium. Biol Reprod 2006; 75:605–614.. [DOI] [PubMed] [Google Scholar]
- 33. Yang S, Fang Z, Gurates B, Tamura M, Miller J, Ferrer K, Bulun SE. Stromal PRs mediate induction of 17beta-hydroxysteroid dehydrogenase type 2 expression in human endometrial epithelium: a paracrine mechanism for inactivation of E2. Mol Endocrinol 2001; 15:2093–2105.. [DOI] [PubMed] [Google Scholar]
- 34. Glidewell-Kenney C, Hurley LA, Pfaff L, Weiss J, Levine JE, Jameson JL. Nonclassical estrogen receptor alpha signaling mediates negative feedback in the female mouse reproductive axis. Proc Natl Acad Sci U S A 2007; 104:8173–8177.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Raivich G, Behrens A. Role of the AP-1 transcription factor c-Jun in developing, adult and injured brain. Prog Neurobiol 2006; 78:347–363.. [DOI] [PubMed] [Google Scholar]
- 36. Bakker J, Honda S, Harada N, Balthazart J. The aromatase knock-out mouse provides new evidence that estradiol is required during development in the female for the expression of sociosexual behaviors in adulthood. J Neurosci 2002; 22:9104–9112.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bakker J, Honda S, Harada N, Balthazart J. The aromatase knockout (ArKO) mouse provides new evidence that estrogens are required for the development of the female brain. Ann N Y Acad Sci 2003; 1007:251–262.. [DOI] [PubMed] [Google Scholar]
- 38. Matsumoto T, Honda S, Harada N. Neurological effects of aromatase deficiency in the mouse. J Steroid Biochem Mol Biol 2003; 86:357–365.. [DOI] [PubMed] [Google Scholar]
- 39. Matsumoto T, Honda S, Harada N. Alteration in sex-specific behaviors in male mice lacking the aromatase gene. Neuroendocrinology 2003; 77:416–424.. [DOI] [PubMed] [Google Scholar]
- 40. Kudwa AE, Michopoulos V, Gatewood JD, Rissman EF. Roles of estrogen receptors alpha and beta in differentiation of mouse sexual behavior. Neuroscience 2006; 138:921–928.. [DOI] [PubMed] [Google Scholar]
- 41. Ogawa S, Eng V, Taylor J, Lubahn DB, Korach KS, Pfaff DW. Roles of estrogen receptor-alpha gene expression in reproduction-related behaviors in female mice. Endocrinology 1998; 139:5070–5081.. [DOI] [PubMed] [Google Scholar]
- 42. Balthazart J, Baillien M, Charlier TD, Cornil CA, Ball GF. Multiple mechanisms control brain aromatase activity at the genomic and non-genomic level. J Steroid Biochem Mol Biol 2003; 86:367–379.. [DOI] [PubMed] [Google Scholar]
- 43. Perkins A, Roselli CE. The ram as a model for behavioral neuroendocrinology. Horm Behav 2007; 52:70–77.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pasmanik M, Callard GV. Changes in brain aromatase and 5 alpha-reductase activities correlate significantly with seasonal reproductive cycles in goldfish (Carassius auratus). Endocrinology 1988; 122:1349–1356.. [DOI] [PubMed] [Google Scholar]
- 45. Reddy VV, Naftolin F, Ryan KJ. Aromatization in the central nervous system of rabbits: effects of castration and hormone treatment. Endocrinology 1973; 92:589–594.. [DOI] [PubMed] [Google Scholar]
- 46. Hojo Y, Hattori TA, Enami T, Furukawa A, Suzuki K, Ishii HT, Mukai H, Morrison JH, Janssen WG, Kominami S, Harada N, Kimoto T et al. Adult male rat hippocampus synthesizes estradiol from pregnenolone by cytochromes P45017alpha and P450 aromatase localized in neurons. Proc Natl Acad Sci U S A 2004; 101:865–870.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Prange-Kiel J, Wehrenberg U, Jarry H, Rune GM. Para/autocrine regulation of estrogen receptors in hippocampal neurons. Hippocampus 2003; 13:226–234.. [DOI] [PubMed] [Google Scholar]
- 48. Rune GM, Lohse C, Prange-Kiel J, Fester L, Frotscher M. Synaptic plasticity in the hippocampus: effects of estrogen from the gonads or hippocampus? Neurochem Res 2006; 31:145–155.. [DOI] [PubMed] [Google Scholar]
- 49. Rune GM, Wehrenberg U, Prange-Kiel J, Zhou L, Adelmann G, Frotscher M. Estrogen up-regulates estrogen receptor alpha and synaptophysin in slice cultures of rat hippocampus. Neuroscience 2002; 113:167–175.. [DOI] [PubMed] [Google Scholar]
- 50. Rune GM, Frotscher M. Neurosteroid synthesis in the hippocampus: role in synaptic plasticity. Neuroscience 2005; 136:833–842.. [DOI] [PubMed] [Google Scholar]
- 51. Tsuruo Y, Ishimura K, Osawa Y. Presence of estrogen receptors in aromatase-immunoreactive neurons in the mouse brain. Neurosci Lett 1995; 195:49–52.. [DOI] [PubMed] [Google Scholar]
- 52. Nausch N, Manteuffel G, Vanselow J. 0.2kb promoter sequence of the murine Cyp19 gene target beta-galactosidase expression to specific brain areas of transgenic mice. J Steroid Biochem Mol Biol 2007; 103:119–128.. [DOI] [PubMed] [Google Scholar]
- 53. Abe-Dohmae S, Tanaka R, Takagi Y, Harada N. In vitro increase of aromatase mRNA in diencephalic neurons. Neuroendocrinology 1996; 63:46–52.. [DOI] [PubMed] [Google Scholar]