

Abstract
Implantation of an embryo in the endometrium is a critical step for continuation of pregnancy, and implantation failure is a major cause of infertility. In rats, the implantation process involves invasion of the endometrial epithelial lining by the trophoblastic cells in order to reach the underlying stromal cells. Transforming growth factor beta (TGFB) is a multifunctional cytokine that regulates proliferation, differentiation, and invasiveness of multiple cell lineages. We used rat HRP-1 and RCHO-1 placental cell lines to perform this study. HRP-1 cells were derived from midgestation chorioallantoic placental explants of the outbred Holtzman rat, whereas RCHO-1 cells were established from a rat choriocarcinoma. MTT proliferation assays revealed that each TGFB isoform decreased HRP-1 cell growth in a dose-dependent manner, whereas RCHO-1 cells were resistant to the growth-suppressive effect of TGFB1 and TGFB3. Only TGFB2 reduced RCHO-1 cell proliferation. Activation of ERK, MAPK14 (p38 MAPK), or SMAD pathways is known to play a role in cell proliferation, and we found that TGFB activates these pathways in both HRP-1 and RCHO-1 cells in an isoform-specific manner. MTT proliferation assays revealed that ERK pathway is partially implicated in TGFB3-reduced HRP-1 cell proliferation. Hoechst nuclear staining and caspase-3 cleavage demonstrated that TGFB isoforms failed to induce apoptosis in both cell lines. Matrigel invasion assays showed that both HRP-1 and RCHO-1 cells exhibit intrinsic invasive ability under untreated conditions. The capacity of HRP-1 cells to invade the Matrigel was selectively increased by TGFB2 and TGFB3, whereas all TGFB isoforms could increase the invasiveness of RCHO-1 cells. These important functional studies progressively reveal a key role for TGFB in regulating proliferation and invasiveness of placental cells.
Keywords: apoptosis, endometrium, invasion, placenta, pregnancy, proliferation, TGFB, trophoblast
Introduction
Implantation of the endometrium is the process during which the embryo attaches and invades the mother endometrium. Trophoblast cells, which form the placenta, are responsible for endometrial invasion. Trophoblast cell invasion of the uterus is essential for the maintenance of the pregnancy, and this is a very temporally and spatially regulated process. Some factors, such as cytokines, growth factors, and hormones produced by the uterus during pregnancy or by trophoblast themselves [1, 2], are suspected of being responsible for this regulation, and other factors are still undetermined.
Transforming growth factor βs (TGFBs) are members of a large superfamily of cytokines that includes activins, inhibins, bone-morphogenic proteins, and others [3]. In mammals, TGFB exists in three isoforms, called TGFB1, TGFB2, and TGFB3, encoded by three different genes [4]. TGFB is known to regulate a variety of cellular functions, including cell proliferation, differentiation, apoptosis, migration/invasion, matrix synthesis, and the immune response [5–7]. TGFB initiates these processes by binding to specific cell surface receptors with intrinsic serine/threonine kinase activity, designated TGFB receptor (TGFBR) types 1, 2, and 3 [8]. Upon binding with the ligand (TGFB), TGFBR3 phosphorylates and activates TGFBR1, which, in turn, will induce SMAD2 or SMAD3 phosphorylation. Phosphorylated SMAD2/SMAD3 associates with SMAD4 to form a heterodimeric complex. This complex then translocates to the nucleus, where it acts as a transcription factor for various genes [9]. In addition to the SMAD pathway, various signaling pathways, such as PIK3 and MAPK (MAPK14, MAPK3/1, and MAPK8) are activated by TGFB in different cell lines [10]. Activation of these pathways has been shown to play an important role in growth factor-dependent regulation of trophoblast growth differentiation, invasion, and migration [11].
Although it has been shown that TGFB isoforms are present in the uterus during pregnancy [12, 13], little information is available about the specific effect of each isoform in trophoblast cells. It is known that successful invasion of the maternal vascular system by trophoblast cells is a prerequisite for the establishment of a normal placenta and continuation of pregnancy. Previous studies also suggest that TGFB could play a crucial role in the invasion of trophoblastic cells [14–16]. However, the mechanisms of action through which it exerts its effect are still poorly understood. Rat and mouse models have provided valuable insights into the molecular mechanisms that occur during trophoblast differentiation and invasion; however, these models do not necessarily translate to the human, because of different reproductive physiology [17]. The present study offers the rationale for investigating the effect of each TGFB isoform to activate signaling pathways and to modulate the proliferation and invasion of two rat placental cell lines: HRP-1 cells, which were derived from a normal midgestation chorioallantoic placenta [18], and RCHO-1 cells, which were established from a choriocarcinoma [19, 20]. The results from the present study suggest the involvement and the importance of TGFB in the invasion of rat placental cell lines.
Materials and Methods
Cell Lines and Reagents
Established placental HRP-1 [18] and RCHO-1 [19, 20] cells were maintained in RPMI 1640 medium, supplemented with 20% fetal bovine serum (heat inactivated), 100 μg/ml penicillin-streptomycin, 1 mM sodium pyruvate, and 50 μM 2-mercaptoethanol. TGFB1 and TGFB2 were purchased from Biosource (Invitrogen), and TGFB3 was from Calbiochem (San Diego, CA). All of the antibodies were from Cell Signaling Technology (Beverly, MA), except for the anti-rabbit secondary antibody (Bio-Rad) and anti-β-actin antibody (Sigma, St. Louis, MO). PD98059 (MAP2K1 [MEK1] inhibitor) was purchased from Cell Signaling Technology and SB431542 (specific TGFBR inhibitor) was from Sigma. These inhibitors were reconstituted in dimethyl sulfoxide.
RNA Isolation and Analysis
Total RNA was isolated from cells using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. First-strand cDNA was synthesized from 0.4 μg of RNA using Moloney murine leukemia virus reverse transcriptase (Invitrogen). Primers for PCR amplification are listed in Table 1. PCRs were conducted in an MJ Research Thermal cycler (model PTC-100) using the following conditions: 30 sec at 94°C, 30 sec at melting point temperature (Table 1), and 1 min at 72°C for 36 cycles, except for GAPDH (25 cycles). The reaction mixture was size separated on an agarose gel and visualized using SYBR-Safe (Invitrogen) staining upon ultraviolet transillumination.
Table 1.
Primers for PCR amplification.
| Gene | Forward primer | Reverse primer | Tm (°C)* | Cycle | Size (bp) |
|---|---|---|---|---|---|
| Tgfb1 | gatagccatttcctcaccga | ggtggcgttcatgtaggagt | 66 | 40 | 756 |
| Tgfb2 | gcgacgaagagtactacgcc | ctccattgctgagacgtcaa | 58 | 40 | 218 |
| Tgfb3 | ggttttccgcttcaatgtgt | gcagttctcctccaagttgc | 58 | 40 | 556 |
| Tgfbr1 | tcaggaagtggctctggtct | cgtttccctgaaccatgact | 59 | 36 | 180 |
| Tgfbr2 | atgacctggctaacagtggg | gccatggagtagacatcggt | 58 | 36 | 121 |
| Gapdh | ccattcttccacctttgatg | gtctgggatggaattgtgag | 58 | 25 | 265 |
Melting point temperature.
Western Blot Analysis
Cells were washed with PBS and submitted to lysis in cold radioimmune precipitation assay buffer (1× PBS [pH 7.4]; 1% Nonidet P-40 0.5% sodium deoxycholate; 0.1% SDS) containing protease inhibitors (Complete from Roche Applied Science), followed by three freeze-thaw cycles. Homogenates were centrifuged (13 000 rpm for 20 min at 4°C) to remove insoluble material. The supernatant was recovered and stored at −20°C until further analysis. Equal amounts of cell lysates (as determined using Bio-Rad DC protein assay) were separated onto 8% polyacrylamide gels and then transferred onto nitrocellulose membranes (Bio-Rad). The membranes were blocked with 5% bovine serum albumin in PBS with 0.05% Tween 20 for 1 h at room temperature, probed with primary antibody, washed in PBS with 0.05% Tween 20, and incubated with horseradish peroxidase-conjugated secondary antibody (Bio-Rad). Detection was performed using SuperSignal West Femto substrate (Pierce), as described by the manufacturer. Signal was visualized using the Biochemi Imaging System (UVP). Densitometric analyses were performed (protein of interest and β-actin) using Quantity One software (Bio-Rad). Results are expressed as a ratio (protein of interest:β-actin) to correct for loading for each sample.
MTT Proliferation Assay
Cells were plated in 96-well plates at a density of 7 × 103 cells/well for HRP-1 cells and 8.5 × 103 cells/well for RCHO-1 cells in 180 μl of culture medium and incubated overnight at 37°C in a 5% CO2 atmosphere, after which they reached 50% confluence. Recombinant TGFBs were added to selected wells at the indicated concentrations in 100 μl culture medium, and plates were incubated for the indicated times at 37°C. MTT reagent (tetrazolium salt; Sigma) was added to the wells (10 μl of a 0.5% solution in PBS) 3.5 h before the end of the incubation period, after which conversion of yellow tetrazolium salt to blue thiazol crystals by metabolically active cells was stopped by adding 100 μl of 10% SDS, 0.1% HCl in PBS solution to each well. The plates were incubated overnight at 37°C to allow complete solubilization of thiazol crystals, and the intensity of blue emission in each well was measured using a FluoStar multiwell plate reader (BMG Laboratories, Durham, NC). The percentage of proliferating cells was calculated as the ratio of optical densities of treated to nontreated (control) cells.
Hoechst Nuclear Staining
The treated cells were collected, washed twice in 1× PBS, resuspended at an approximate density of 2 × 105 cells/ml in PBS containing Hoechst 33258 (Sigma), and incubated for 24 h at 4°C before fluorescence microscopy analysis of apoptotic cells. At least 200 cells were counted for each sample, and a percentage of apoptotic cells was calculated as the ratio of apoptotic cells (with characteristic apoptotic morphology, such as nuclear shrinkage and condensation) to total cell count.
Matrigel Invasion Assay
The invasive properties of TGFB-treated HRP-1 and RCHO-1 cells were measured using Matrigel-coated Transwell inserts (Costar, Corning, NY). Briefly, Transwell inserts with an 8-μm pore size were coated with 2 mg/ml Matrigel, and the cells were collected after 24 h of treatment, washed, and resuspended in basal medium without serum. The lower chambers were filled with 700 μl of culture medium, and 1 × 105 treated cells were added to the upper chamber inserts. The plates were incubated for 48 h at 37°C. After the incubation period, noninvading cells were removed from the top of the Matrigel using a cotton swab. Cells that had reached the filter by degrading the Matrigel were fixed in methanol, washed, and stained with Hoechst. The filter was then removed and mounted on a glass slide with invasive cells facing up, and submitted for densitometric analysis using Quantity One software. Results are expressed as a ratio (invasive, treated cells:invasive, nontreated cells) to show the fold increase in invasion for each treatment.
Statistical Analysis
All the experiments were repeated at least three times. Results subjected to statistical analysis were expressed as means ± SEM. Data were subjected to one-way analysis of variance (PRISM software version 3.03; GraphPad, San Diego, CA). Differences between experimental groups were determined by post hoc Student-Newman-Keuls test. Statistical significance was accepted at P < 0.05.
Results
TGFB Isoforms and Their Receptors Are Expressed by Both HRP-1 and RCHO-1 Trophoblast Cells
We examined whether HRP-1 and RCHO-1 rat placental cell lines express TGFB isoforms and their receptors. The results show that these cell lines express TGFBR1 and TGFBR2 at both transcript and protein levels (Fig. 1A). At the transcript level, TGFB1 was found only in RCHO-1 cells, whereas TGFB3 was found in both cell lines. However, TGFB2 was not detected in either of the placental cell lines used in the present study (Fig. 1B).
Fig. 1.

TGFBR and TGFB isoforms in HRP-1 and RCHO-1 cells. TGFBR transcript levels (A) and protein levels (B) were measured by RT-PCR and Western blot analysis, respectively. Endogenous TGFB (C) transcript levels in HRP-1 and RCHO-1 cells. Total RNA from MCF-7 cells was used as positive control (C+). GAPDH and β-actin was included as a loading control. The results shown are representative of three independent experiments.
TGFB Isoforms Activate SMAD, ERK, and MAPK14 Signaling Pathways in HRP-1 and RCHO-1 Trophoblast Cells
As mentioned previously, the TGFB signaling pathway involves phosphorylation and thereby activation of SMAD proteins. In the present study, total levels of SMAD2, SMAD3, and their phosphorylated forms (pSMAD2 and pSMAD3) were determined in HRP-1 (Fig. 2, A and B) and RCHO-1 cells (Fig. 2, C and D) by Western blot analysis. The results show that both cell lines have high levels of total SMAD2 as compared with total SMAD3, and the levels of total SMAD2 remain unchanged with TGFB treatment in HRP-1 and RCHO-1 cells. Furthermore, there was no effect on the levels of total SMAD3 in HRP-1 cells. However, the levels of SMAD3 were undetectable in RCHO-1 cells with the antibody used. We observed a similar increase in the phosphorylation of SMAD2 in both cell lines in response to all the TGFB isoforms. SMAD3 was also found to be phosphorylated in both cell lines in the presence of TGFB.
Fig. 2.

SMAD signaling is activated in TGFB-treated HRP-1 and RCHO-1 trophoblastic cells. The levels of total pSMAD2, total pSMAD3, and pSMAD2/3 protein were analyzed by Western blotting in HRP-1 (A) and RCHO-1 (B) cells treated for 1 h with the three TGFB isoforms at a concentration of 1 or 10 ng/ml. Beta-actin was used as a loading control. The results shown are representative of three independent experiments.
In addition, we examined whether ERK and MAPK14 signaling pathways could be triggered by TGFB isoforms in both model cell lines. Results show an increase in the phosphorylation of pERK and pMAPK14 by TGFB1 in HRP-1 and RCHO-1 cells (Fig. 3). TGFB2 and TGFB3 also activated MAPK14 and ERK signaling pathways in a similar manner (data not shown). Therefore, all three TGFB isoforms have the ability to phosphorylate, and thus activate, the SMAD, ERK, and MAPK14 signaling pathways in both HRP-1 and RCHO-1 cells.
Fig. 3.

Time course activation of MAPK14 and ERK pathways in HRP-1 and RCHO-1 cells. Phosphorylated MAPK14 and MAPK3/1 (ERK) protein levels were measured in trophoblastic cells after treatment with 10 ng/ml TGFB1 for the indicated times (15 min to 24 h) using Western blotting. Beta-actin was used as a loading control. The results shown are representative of three independent experiments.
Effect of TGFB on HRP-1 and RCHO-1 Cell Growth
We compared the growth-inhibitory effects of TGFB isoforms on each cell line using the MTT proliferation assays. We found that TGFB has a growth-suppressive effect on HRP-1 cells (Fig. 4A, 24 h; Fig. 4B, 48 h). It was observed that cell growth was inhibited by all TGFB isoforms, and TGFB2 showed the highest inhibitory activity. On the other hand, no such growth-inhibitory effect was observed in RCHO-1 cells with TGFB1 and TGFB3 treatments, while TGFB2 had a slight but significant inhibitory effect (Fig. 4C, 24 h; Fig. 4D, 48 h).
Fig. 4.

Effect of TGFB isoforms on HRP-1 and RCHO-1 cell proliferation. Cell proliferation rates were measured using the MTT assay. Cells were treated with 0, 0.1, 1, and 10 ng/ml TGFB isoforms in HRP-1 cells for 24 h (A) or 48 h (B), and in RCHO-1 cells for 24 h (C) or 48 h (D). The results are the mean ± SEM of eight independent experiments, each performed in duplicate. *P < 0.05 compared to untreated cells.
SMAD Pathway Is Not Implicated in HRP-1 Cell Proliferation, While ERK Pathway Is Partially Implicated in TGFB3 Action
In order to investigate which particular pathway is involved in the growth-inhibitory effects of TGFB isoforms, we pretreated HRP-1 cells for 1 h with SB431542, a specific TGFBR inhibitor, or PD98059, a MAP2K1 inhibitor, and cell proliferation was monitored using MTT proliferation assays. After a pretreatment with SB431542, TGFB isoforms still showed a growth-suppressive effect on HRP-1 cells, indicating that the SMAD pathway is probably not responsible for the inhibition of cell proliferation (Fig. 5A), in spite of the fact that SMAD2 and SMAD3 were found to be activated with TGFB treatment. On the other hand, we found that PD98059 partially reversed TGFB3-induced growth suppression in HRP-1 cells after a 48-h exposure (Fig. 5B). To confirm the efficiency of pathway inhibition, pSMAD2 and pERK were both measured. It was observed that the levels of pSMAD2 and pERK decreased in the presence of SB431542 and PD98059, respectively (data not shown).
Fig. 5.
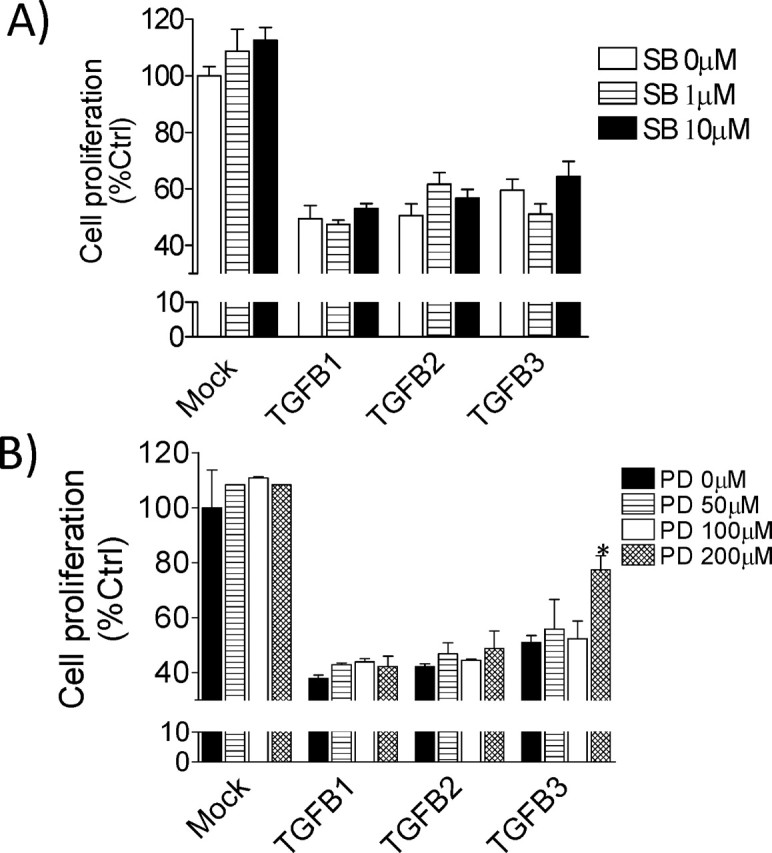
Inhibition of SMAD receptor or MAPK signaling pathway in TGFB-treated HRP-1 cell proliferation. HRP-1 cellular proliferation was assessed using MTT proliferation assay. Cells were pretreated with SB431542 (A) or PD98059 (B) for 1 h, followed by treatment with 10 ng/ml TGFB isoforms for 48 h. The mock group received equal amounts of vehicle (dimethyl sulfoxide [DMSO]). The results are the mean ± SEM of three independent experiments, each performed in duplicate. *P < 0.05 compared to untreated cells.
TGFB Isoforms Are Unable to Trigger Apoptosis
In order to determine if the growth inhibitory effect of TGFB on HRP-1 and RCHO-1 cell growth was due to increased apoptosis, we stained cells using Hoechst nuclear staining following TGFB treatment. There was no significant increase in the apoptosis of HRP-1 (Fig. 6A) and RCHO-1 (Fig. 6B) cells in the presence of TGFB isoforms. In contrast, TGFB3 significantly decreased the percentage of apoptotic cells as compared with control. Furthermore, we examined cleaved caspase 3 by Western blot analysis. The results demonstrate that TGFB isoforms failed to activate caspase 3 (data not shown).
Fig. 6.
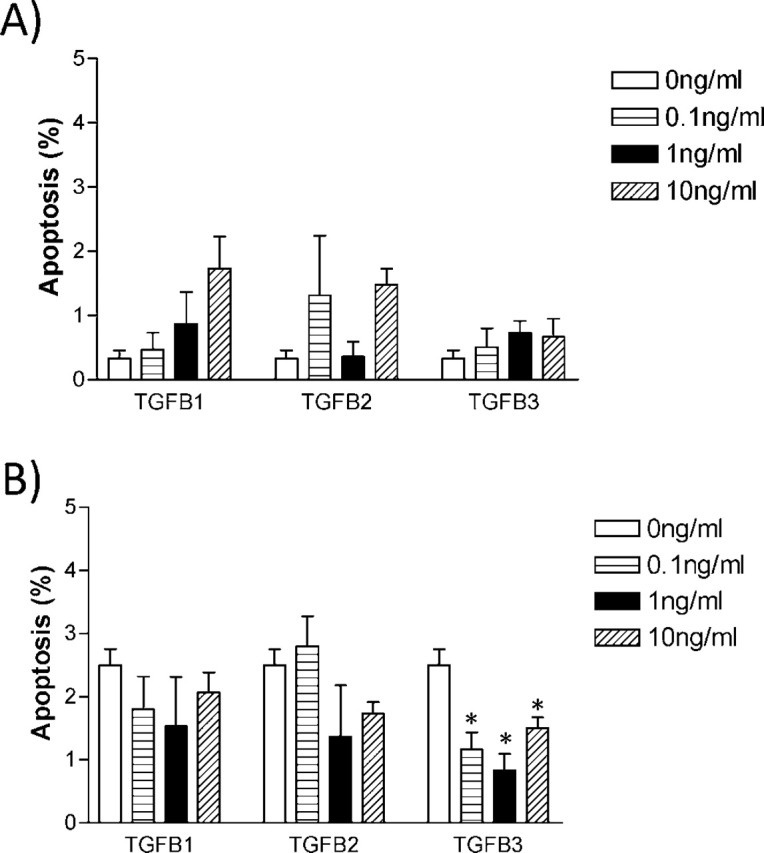
TGFB has no effect on apoptosis in either HRP-1 or RCHO-1 cells. The apoptotic effect of TGFB isoforms on HRP-1 (A) and RCHO-1 (B) was determined by treatment with 0, 0.1, 1, and 10 ng/ml TGFB isoforms for 24 h, followed by nuclear staining with Hoechst dye. Apoptotic and nonapoptotic cells were counted under a fluorescence microscope. The results are presented as the mean ± SEM of three independent experiments. *P < 0.05.
TGFB Increases HRP-1 and RCHO-1 Cell Invasion
We next tested the effect of TGFB isoforms on the invasiveness of HRP-1 and RCHO-1 cells using the Matrigel invasion assay. HRP-1 cells exhibited low intrinsic invasive properties, but invasiveness was selectively increased with TGFB2 and TGFB3 treatment (Fig. 7A). RCHO-1 cells also exhibited low intrinsic invasive properties, but all three TGFB isoforms could induce cell invasion (Fig. 7B).
Fig. 7.

Invasiveness of placental cells is selectively increased by TGFB isoforms. The effect of TGFB isoforms (1 ng/ml) on the invasive capacity of HRP-1 (A) and RCHO-1 (B) cells was determined using the Matrigel invasion assay. The results are presented as the mean ± SEM of three independent experiments, each performed in duplicate. *P < 0.05 compared to untreated cells.
Discussion
Trophoblast cells undergo various differential stages, including proliferation, differentiation, and migration/invasion, in order to guarantee successful placental development. During these phases, trophoblast cells are constantly exposed to the uterine fluid, which is rich in growth factors, cytokines, and hormones [21]. These molecules play a role in regulating trophoblast differentiation along the invasive pathway in both an autocrine and a paracrine fashion. Signaling cascades initiated by the aforementioned stimuli need to be precisely coordinated and integrated at all times. Because different isoforms of TGFB are expressed by the uterus during pregnancy [13], and several studies suggest that TGFB has a role to play in the invasion of the endometrium by the trophoblast [14, 15, 22], we sought to explore the role of each TGFB isoform on their ability to regulate placental cell invasion. This is the first report to demonstrate that the three TGFB isoforms can increase invasiveness of placental cells: TGFB2 and TGFB3 increase the invasiveness of HRP-1 cells, while RCHO-1 invasiveness is increased by all three TGFB isoforms.
In the rat and mouse, trophoblast cells located at the maternal interface achieve a unique phenotype, and are referred to as trophoblast giant cells. These cells arise from precursors by a process referred to as endoreduplication, and possess endocrine activities and the potential for invasion [23–25]. Trophoblast giant cells express and secrete MMP9, and have been shown to be involved in the induction of the invasive phenotype of RCHO-1 cells [26]. Thus, RCHO-1 cells are a well-described model to study trophoblast cell differentiation [27, 28], and the current results support the hypothesis that TGFB isoforms might be important factors involved in this process. TGFB signaling occurs through the interaction of TGFB with two different types of serine/threonine kinase receptor: type I and type II. First, we assessed the transcript levels of TGFBR and endogenous TGFB in HRP-1 and RCHO-1 cells. The present data indicate that both HRP-1 and RCHO-1 cells express TGFBRs and TGFB isoforms. Furthermore, the results suggest a possible autocrine and/or paracrine action for TGFB in rat placental cell lines. Therefore, these cells offer an excellent model to study the impact of TGFB isoforms on the regulation of placental cell invasion during the implantation process and/or later in pregnancy.
SMAD proteins are downstream effectors of the TGFB superfamily [29]. To test whether TGFB modulates SMAD activation through phosphorylation in placental cells, we treated HRP-1 and RCHO-1 cells with different TGFB isoforms. Our results show that, upon TGFB treatment, SMAD2 and SMAD3 were phosphorylated, suggesting that the TGFB pathway is active in these cells. We highlighted earlier the importance of the SMAD pathway in TGFB-induced cellular invasiveness in endometrial cancer cells [30]. It is logical to suggest that a similar pathway could be involved in the increased invasiveness of trophoblast cells.
Given the fact that MAPKs regulate almost all cellular activities, it is therefore not surprising that these enzymes also control cell invasion/migration. Moreover, TGFB activates non-SMAD pathways, including the three distinct MAPK pathways: the ERK, MAPK8 (c-Jun N-terminal kinase), and MAPK14 kinase pathways [31–33]. The results of the present study also show that, upon TGFB treatment, ERK and MAPK14 pathways were triggered in the form of increased phosphorylation. These pathways were previously shown to regulate migration in different cell types using multiple mechanisms, including phosphorylation of the focal adhesion adaptor paxillin [34], activation of myosin light chain kinase [35], upregulation of MMP9 [36], and processing of MMP2 [37]. As both cell lines (HRP-1 and RCHO-1) express endogenous TGFB, this could explain why SMAD2, MAPK14, and MAPK3/1 (p44/42) are constitutively phosphorylated in these cells.
We further examined the effect of TGFB isoforms on HRP-1 and RCHO-1 cell growth. MTT proliferation assays revealed a growth-suppressive effect on HRP-1 placental cell lines. In a previous report, TGFB1 was shown to be able to reduce RCHO-1 cell proliferation after 8 days of culture [38]. Previously, members of the Src family kinases were found to be involved in human trophoblast differentiation [39]. In the current study, although we could see a trend toward growth inhibition, we did not see a significant inhibitory effect of either TGFB isoform at the same dose used in the previous study, and this may be explained by the short exposure time (48 h) to TGFB isoforms. It is well known that trophoblast cells differentiate into extravillous trophoblasts in order to invade the endometrium. Moreover, inhibition in cell proliferation and apoptosis is a prerequisite for any cell to differentiate. Given the timing of trophoblast invasion in rats, which is between Day 6 and Day 7, the growth-inhibitory effect of TGFB on trophoblastic cells in our study suggests that this differentiation process could be more pronounced with TGFB treatment. A longer period of treatment with TGFBs is probably necessary to induce a significant growth inhibition. In order to verify whether apoptosis is involved in this phenomenon, we performed Hoechst nuclear staining and caspase 3 cleavage measurements, and our results show that TGFB isoforms were unable to trigger apoptosis. In a previous study, we reported the growth-suppressive effect of TGFB in endometrial cells [30]. The latter study highlighted the involvement of XIAP in the growth inhibitory effect of TGFB; nonetheless, this study supports our current findings that TGFB could be involved in inhibiting trophoblast cell growth.
Over the years, TGFB isoforms have been widely studied in association with cancer progression and metastases [40, 41], but there is limited information on their role in trophoblast invasion. Our results show that TGFB2 and TGFB3 increase the invasiveness of HRP-1 cells, while RCHO-1 invasiveness was not influenced by TGFB isoforms. Indeed, we have previously reported that TGFB isoforms differ in their ability to regulate invasive behavior, as only TGFB3 could increase the invasiveness of endometrial cancer cells [30]. This is, however, the first evidence of an increased invasive phenotype of placental cells that is induced by TGFB isoforms. In contrast, others have reported an inhibition of trophoblast cell invasion by TGFB isoforms [14, 42]. This discrepancy could be well explained by the fact that these authors used a lower dose of TGFB to treat the trophoblast cells. Furthermore, in those studies, the trophoblast cells were derived from placental explants obtained from humans, and the difference could arise from species specificity, as reviewed in Ref. 17. During implantation in rats, the luminal epithelium forms an invagination to surround the trophoblast, whereas, in humans, interstitial implantation occurs where the trophoblast passes through the luminal epithelium to invade the endometrial stroma and to become imbedded in the wall of the uterus.
In conclusion, this study suggests, for the first time, that not only do the isoforms of TGFB have a different role on the ability to induce trophoblast invasion, but they also activate the cellular signaling pathways, such as SMAD, ERK, and MAPK14, in these two placental cell lines. Further studies are needed to determine the impact of SMAD, ERK, and MAPK14 pathway in trophoblast and placental cell invasion. This study also provides evidence that the RCHO-1 cell line is a valuable model for studying trophoblast invasion in vitro, and further analyses are required to determine the molecular mechanisms involved in TGFB-regulated invasion. The HRP-1 placental stem cell line also represents an important model for the continued study of the role of TGFB-regulated placental development.
References
- 1. Hill JA. Maternal-embryonic cross-talk. Ann N Y Acad Sci 2001; 943:17–25. [DOI] [PubMed] [Google Scholar]
- 2. Spencer TE, Bazer FW. Biology of progesterone action during pregnancy recognition and maintenance of pregnancy. Front Biosci 2002; 7:d1879–d1898. [DOI] [PubMed] [Google Scholar]
- 3. Herpin A, Lelong C, Favrel P. Transforming growth factor-beta-related proteins: an ancestral and widespread superfamily of cytokines in metazoans. Dev Comp Immunol 2004; 28:461–485. [DOI] [PubMed] [Google Scholar]
- 4. Khalil N. TGF-beta: from latent to active. Microbes Infect 1999; 1:1255–1263. [DOI] [PubMed] [Google Scholar]
- 5. Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer 2006; 6:506–520. [DOI] [PubMed] [Google Scholar]
- 6. Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 2000; 103:295–309. [DOI] [PubMed] [Google Scholar]
- 7. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003; 113:685–700. [DOI] [PubMed] [Google Scholar]
- 8. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002; 298:1912–1934. [DOI] [PubMed] [Google Scholar]
- 9. Miyazono K. TGF-beta signaling by Smad proteins. Cytokine Growth Factor Rev 2000; 11:15–22. [DOI] [PubMed] [Google Scholar]
- 10. Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res 2009; 19:128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pollheimer J, Knofler M. Signalling pathways regulating the invasive differentiation of human trophoblasts: a review. Placenta 2005; 26(suppl A):S21–S30. [DOI] [PubMed] [Google Scholar]
- 12. Shooner C, Caron PL, Frechette-Frigon G, Leblanc V, Dery MC, Asselin E. TGF-beta expression during rat pregnancy and activity on decidual cell survival. Reprod Biol Endocrinol 2005; 3:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simpson H, Robson SC, Bulmer JN, Barber A, Lyall F. Transforming growth factor beta expression in human placenta and placental bed during early pregnancy. Placenta 2002; 23:44–58. [DOI] [PubMed] [Google Scholar]
- 14. Lash GE, Otun HA, Innes BA, Bulmer JN, Searle RF, Robson SC. Inhibition of trophoblast cell invasion by TGFB1, 2, and 3 is associated with a decrease in active proteases. Biol Reprod 2005; 73:374–381. [DOI] [PubMed] [Google Scholar]
- 15. Caniggia I, Grisaru-Gravnosky S, Kuliszewsky M, Post M, Lye SJ. Inhibition of TGF-beta 3 restores the invasive capability of extravillous trophoblasts in preeclamptic pregnancies. J Clin Invest 1999; 103:1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Belkacemi L, Lash GE, Macdonald-Goodfellow SK, Caldwell JD, Graham CH. Inhibition of human trophoblast invasiveness by high glucose concentrations. J Clin Endocrinol Metab 2005; 90:4846–4851. [DOI] [PubMed] [Google Scholar]
- 17. Lee KY, DeMayo FJ. Animal models of implantation. Reproduction 2004; 128:679–695. [DOI] [PubMed] [Google Scholar]
- 18. Soares MJ, Schaberg KD, Pinal CS, De SK, Bhatia P, Andrews GK. Establishment of a rat placental cell line expressing characteristics of extraembryonic membranes. Dev Biol 1987; 124:134–144. [DOI] [PubMed] [Google Scholar]
- 19. Hunt JS, Deb S, Faria TN, Wheaton D, Soares MJ. Isolation of phenotypically distinct trophoblast cell lines from normal rat chorioallantoic placentas. Placenta 1989; 10:161–177. [DOI] [PubMed] [Google Scholar]
- 20. Faria TN, Soares MJ. Trophoblast cell differentiation: establishment, characterization, and modulation of a rat trophoblast cell line expressing members of the placental prolactin family. Endocrinology 1991; 129:2895–2906. [DOI] [PubMed] [Google Scholar]
- 21. Pijnenborg R. Implantation and immunology: maternal inflammatory and immune cellular responses to implantation and trophoblast invasion. Reprod Biomed Online 2002; 4(suppl 3):14–17. [DOI] [PubMed] [Google Scholar]
- 22. Xu G, Chakraborty C, Lala PK. Reconstitution of Smad3 restores TGF-beta response of tissue inhibitor of metalloprotease-1 upregulation in human choriocarcinoma cells. Biochem Biophys Res Commun 2003; 300:383–390. [DOI] [PubMed] [Google Scholar]
- 23. De SK, Larsen DB, Soares MJ. Trophoendodermal stem cell-derived extracellular matrices: absence of detectable entactin and presence of multiple laminin species. Placenta 1995; 16:701–718. [DOI] [PubMed] [Google Scholar]
- 24. Enders AC, Welsh AO. Structural interactions of trophoblast and uterus during hemochorial placenta formation. J Exp Zool 1993; 266:578–587. [DOI] [PubMed] [Google Scholar]
- 25. Gardner RL, Beddington RS. Multi-lineage ‘stem' cells in the mammalian embryo. J Cell Sci Suppl 1988; 10:11–27. [DOI] [PubMed] [Google Scholar]
- 26. Peters TJ, Albieri A, Bevilacqua E, Chapman BM, Crane LH, Hamlin GP, Seiki M, Soares MJ. Differentiation-dependent expression of gelatinase B/matrix metalloproteinase-9 in trophoblast cells. Cell Tissue Res 1999; 295:287–296. [DOI] [PubMed] [Google Scholar]
- 27. Sahgal N, Canham LN, Canham B, Soares MJ. Rcho-1 trophoblast stem cells: a model system for studying trophoblast cell differentiation. Methods Mol Med 2006; 121:159–178. [PubMed] [Google Scholar]
- 28. Sahgal N, Canham LN, Konno T, Wolfe MW, Soares MJ. Modulation of trophoblast stem cell and giant cell phenotypes: analyses using the Rcho-1 cell model. Differentiation 2005; 73:452–462. [DOI] [PubMed] [Google Scholar]
- 29. Schiller M, Javelaud D, Mauviel A. TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci 2004; 35:83–92. [DOI] [PubMed] [Google Scholar]
- 30. Van Themsche C, Mathieu I, Parent S, Asselin E. Transforming growth factor-beta3 increases the invasiveness of endometrial carcinoma cells through phosphatidylinositol 3-kinase-dependent up-regulation of X-linked inhibitor of apoptosis and protein kinase C-dependent induction of matrix metalloproteinase-9. J Biol Chem 2007; 282:4794–4802. [DOI] [PubMed] [Google Scholar]
- 31. Hocevar BA, Brown TL, Howe PH. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J 1999; 18:1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adachi-Yamada T, Nakamura M, Irie K, Tomoyasu Y, Sano Y, Mori E, Goto S, Ueno N, Nishida Y, Matsumoto K. p38 mitogen-activated protein kinase can be involved in transforming growth factor beta superfamily signal transduction in Drosophila wing morphogenesis. Mol Cell Biol 1999; 19:2322–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Atfi A, Djelloul S, Chastre E, Davis R, Gespach C. Evidence for a role of Rho-like GTPases and stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) in transforming growth factor beta-mediated signaling. J Biol Chem 1997; 272:1429–1432. [DOI] [PubMed] [Google Scholar]
- 34. Huang C, Jacobson K, Schaller MD. A role for JNK-paxillin signaling in cell migration. Cell Cycle 2004; 3:4–6. [PubMed] [Google Scholar]
- 35. Cho SY, Klemke RL. Extracellular-regulated kinase activation and CAS/Crk coupling regulate cell migration and suppress apoptosis during invasion of the extracellular matrix. J Cell Biol 2000; 149:223–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Turchi L, Chassot AA, Bourget I, Baldescchi C, Ortonne JP, Meneguzzi G, Lemichez E, Ponzio G. Cross-talk between RhoGTPases and stress activated kinases for matrix metalloproteinase-9 induction in response to keratinocytes injury. J Invest Dermatol 2003; 121:1291–1300. [DOI] [PubMed] [Google Scholar]
- 37. Takino T, Miyamori H, Watanabe Y, Yoshioka K, Seiki M, Sato H. Membrane type 1 matrix metalloproteinase regulates collagen-dependent mitogen-activated protein/extracellular signal-related kinase activation and cell migration. Cancer Res 2004; 64:1044–1049. [DOI] [PubMed] [Google Scholar]
- 38. Hamlin GP, Soares MJ. Regulation of deoxyribonucleic acid synthesis in proliferating and differentiating trophoblast cells: involvement of transferrin, transforming growth factor-beta, and tyrosine kinases. Endocrinology 1995; 136:322–331. [DOI] [PubMed] [Google Scholar]
- 39. Daoud G, Rassart E, Masse A, Lafond J. Src family kinases play multiple roles in differentiation of trophoblasts from human term placenta. J Physiol 2006; 571:537–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shigeoka Y, Igishi T, Matsumoto S, Nakanishi H, Kodani M, Yasuda K, Hitsuda Y, Shimizu E. Sulindac sulfide and caffeic acid phenethyl ester suppress the motility of lung adenocarcinoma cells promoted by transforming growth factor-beta through Akt inhibition. J Cancer Res Clin Oncol 2004; 130:146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Farina AR, Coppa A, Tiberio A, Tacconelli A, Turco A, Colletta G, Gulino A, Mackay AR. Transforming growth factor-beta1 enhances the invasiveness of human MDA-MB-231 breast cancer cells by up-regulating urokinase activity. Int J Cancer 1998; 75:721–730. [DOI] [PubMed] [Google Scholar]
- 42. Graham CH. Effect of transforming growth factor-beta on the plasminogen activator system in cultured first trimester human cytotrophoblasts. Placenta 1997; 18:137–143. [DOI] [PubMed] [Google Scholar]