

Abstract
Glucose in our body is maintained within a narrow range by the humoral control and a ‘lipostat’ system regulated by leptin from adipose tissues, which keep our accumulated fat stores in check. Any disturbance in this delicately poised homeostasis could be disastrous as it can lead to obesity and its associated metabolic manifestations. Laboratory animals, especially rodents, have contributed to our knowledge in understanding this physiological mechanism through an array of genetic and non-genetic animals developed over the years. Two rat mutant obese models-Wistar inbred at National Institute of Nutrition (WNIN)/Ob-obese rats with normal glucose levels and WNIN/GR-Ob-obese with impaired glucose tolerance were developed in the National Centre for Laboratory Animal Sciences (Now ICMR-National Animal Resource Facility for Biomedical Research) at Hyderabad, India. These animals are unique, as, unlike the earlier models, they show all types of degenerative disorders associated with obesity, within a single system. Thus they show impairment in all the major organs of the body - liver, pancreas, kidney, bones, muscles, gonads, brain, eyes, and are sensitive to diet manipulations, have compromised immunity, often develop tumours and have reduced life span. One may argue that there are limitations to one's interpretations from animal studies to human application, but then one cannot shut one's eyes to the new lessons they have taught us in modifying our life styles.
Keywords: Animal models, energy, glucose, homeostasis, leptin, immunity obesity, obesity genes, retinal disorders
Introduction
Glucose is the main source of fuel for the cells in human body and its normal homeostasis is the pre-requisite for maintaining health. Blood sugar levels are tightly regulated and maintained within a narrow range by interplay of hormones - insulin and glucagon. In normal course, excess glucose gets stored in the liver as glycogen and if the levels are more it gets converted into fat and gets stored in liver as well as in adipose tissues. However, as more and more glucose is pumped into the system through high intake of caloric food, insulin keeps pace with it for some time, but at some point, the cells become resistant to insulin resulting in pre-diabetes and ultimately diabetes and excess weight. There are vegetative controls in the central nervous system, which keep the food intake and energy expenditure in check, so that energy reserves remain constant and thus the body weight as well. When the fat cells increase their fat storage, the adipose tissue releases leptin, the circulating satiety hormone, which signals the hypothalamus to regulate the food intake and inhibition of fatty acid synthesis. Abnormality in glucose and energy homeostasis can exist independent of each other, or these can co-exist as well.
Obesity has become a huge epidemic problem in the West affecting on an average 25-30 per cent men and 30-40 per cent woman1. The scenario is similar for developing countries, like South America, South East Asia and even India where States like Punjab and Kerala are leading, with the trend very much visible in all metro cities. This modern malady is now considered as a forerunner of many metabolic disorders and thus aptly called as the metabolic syndrome1. It is debated as to what degree obesity is genetically linked versus acquired due to environmental factors. Looking at the frequency with which obesity occurs, it seems likely that aetiology is heterogeneous across human population. Thus, both environment and genetic factors are equally important, but the relative significance of each or the interplay between the two may vary. Investigations using laboratory animals are valuable in such a scenario. They are either manipulated physically (altering environment and injecting drugs or chemicals) or genetically (mutants, knockouts and transgenic) to produce the desired disease conditions seen in human beings2.
Animals in the forefront of obesity and diabetes
The first observation was made in 1905 by Cuenot, who observed that laboratory mice with yellow coat colour were obese3. It took another nine decades to identify the defective gene and then clone it. Another important breakthrough happened in 1942, when Hetherington and Ranson4 published an article on experimentally producing obesity by electrolytically lesioning the ventromedial hypothalamus of the rat. This showed the central role of hypothalamus in regulating body weight. Since food is the prime agent of abnormalities of glucose and energy handling, diet-induced form of experimental obesity among vulnerable strains of mice and rats - C3H and A, OM, LEW and SD - were done using high carbohydrate and fat diets alone or together5,6,7. Selafani and Gorman8 fed the Sprague-Dawley (SD) rats with what they called cafeteria diets (the so-called junk food), which resulted in hyperphagia, excess weight gain and adiposity. All these studies on diet-induced obesity showed the importance of environmental factors for developing the syndrome. Overfeeding of the rat pups was done by reducing the number of pups in the litter (resulting in higher weaning weights, which persisted in later life) or by meal feeding (tube feeding by force and training the rats to finish all the diet in two hours) after weaning8. The results from these studies gave credence to fat cell theory, which claimed that overfeeding of human infants led to induction of adipocytes, which predisposed them to obesity in later life9. While work was going on in this direction, some investigators pursued the relative importance of genetic disposition towards the development of the metabolic syndrome. This resulted in the identification, isolation and establishment of genetic models of obesity, mainly from rodents and specifically from mice and rats10. They can be either monogenic (specific single-gene mutation) or an assortment of polygenic (more gene loci) models as well. Unlike the non-genetic models, where overweight was produced experimentally, these are heritable forms of obesity stressing the importance of genetic factors for the development of the syndrome (Table).
Table.
Genetic models of obesity with and without diabetes
| Type | Species/strains |
|---|---|
| Polygenic | Mouse - C3H, A, NZO, KK, PBB, BRSUNT/N, L/IRE |
| Rat - BHE/Cdb | |
| Others - Sand rat, Spiny mouse, Tuco-tuco, Djungarian hamster | |
| Monogenic | Mouse - Yellow, Ob/ob, db/db, fat/fat/tub/tub, Ad/Ad |
| Rat - ZDF, WDF/Ta-fa, WDF/Ta-Drt-fa, BB/ZDR/Wor (Zucker and its variants) SHROB, SHR/N-cp, Jcr: LA, SHHF/Mec.fa, LA/N-cp (Koletsky and its variants) OLTEFF WNIN/Ob, WNIN/GR-Ob |
Source: Refs 11-30
In polygenic models, no single genetic lesion is involved, but they have an array of genes that give them higher growth and metabolic rates. These are the most realistic models of glucose and energy imbalance and can be manipulated easily by diet and environment and are reversible also. These include inbred stains of mice and rats and hamsters. Amongst these, NZO and KK mice were more popular, with the former showing the classical abdominal obesity seen in humans, but the complications seen in humans such as increased gluconeogenesis and adrenal hyperactivity with hyperinsulinaemia and insulin resistance were milder11. They were sensitive to dietary manipulations. The KK mouse developed at Japan showed moderate obesity, which got exaggerated by diet, thioglucose administration and hybridizing it with other mouse strains such as C57, BL/6J and C, BL/Ks J23. Amongst the rat strains, the BHE strain was found to be carbohydrate sensitive and developing obesity with hyperinsulinaemia and mild glucose intolerance12. The Israeli sand rat (Psammomys obesus) is a classic example of how drastic environmental changes (from wild to protected laboratory environment) bring about abnormalities in glucose as well as energy homeostasis. This model was actually developed to explain how the people in Israel developed obesity and diabetes after rapid urbanization through generations13.
In monogenic models, obesity with or without diabetes arises as a result of point mutation causing alteration or loss of a single peptide (primary biochemical lesion). Although different models exhibit variable metabolic defects, the major defects appear to occur in the following three areas - hypersecretion of insulin, hyperphagia and decreased thermogenesis - resulting in increased storage of lipids in liver and adipose tissues. With loss of control of lipolysis, the serum lipids are also very high. Pituitary and adrenals play a significant role in genetic obesity and hence altered adrenal and gonadal functions (defective testis and ovary) are common in these mutants, leading to infertility. So often, adrenelectomy, or injection of high quantities of steroid hormones, or even food restriction brings about reduction in weight gain and in most cases infertility could be reversed. Mutations at eight genetic loci contributing to obesity are now identified in rodents; six of them in mice and three in rats1.
The ‘obese’ and ‘diabetic’ mice developed from C57 strain maintained at Jackson laboratory, USA, are the most extensively studied single-gene mutants14. The obese (ob/ob) mice are deficient in leptin gene, which is the regulator of energy balance. These leptin-deficient mice become obese at a very early age (at one month) accompanied by hyperinsulinaemia, hyperglycaemia, impaired glucose tolerance (IGT) and fatty liver. However, they do not develop hypertension or dyslipidaemia15. The diabetic (db/db) mice have a mutation of the leptin receptor gene16, which impairs leptin signalling. Like ob/ob mice, they also fail to develop hypertension, yet they develop cardiovascular complications and dyslipidaemia. Food restriction and a diet low in carbohydrate can prolong their life span and improve their reproduction rate. Self-selection studies17 showed that they prefer a high portion of a high fat (HF) diet as the main source of energy and a lower portion of high carbohydrate diet compared to control. This appears to be an adaptation to overcome the deleterious effect of insulin resistance. Like in humans, db/db mice also succumb to diabetic neuropathy and nephropathy18. While a high carbohydrate diet over a sustained period seems to be deleterious to them, a high fibre diet significantly helps in achieving higher glycaemic control19.
Amongst rats, Zucker and Koletsky strains are universally used in obesity and diabetic research worldwide and they in combination with other rat strains have contributed over 11 strains20. Zucker rats, deficient in leptin receptor gene21, show visible obesity by five weeks and by the 14th wk, their body will have more than 40 per cent fat22. Like ob mice, they are hyperlipaemic hypercholesterolemic, hyperinsulinemic, and develop adipocyte atrophy23. Impaired renal function with proteinuria is quite common and the homozygous ones are infertile which can be reversed by food restriction and injection of testosterone and adrenelectomy24. Unlike ob/ob and db/db mice, they are euglycaemic, but by selective breeding, a diabetic strain is developed from it called Zucker diabetic fatty rat25, having the highest level of glucose (500 mg% by 10 wk) amongst similar rat models. In combination with WKY strain, a Wistar diabetic fatty rat was developed with increased fatness, glucose intolerance, decreased insulin sensitivity, hepatic insulin resistance, neuropathic complications and sometimes with autoimmune insulinitis26. Unlike Zucker, Koletsky rats are obese with hypertension, having developed from spontaneous hypertensive and SD parents. They are leptin receptor deficient and sub-strains within them often show atherosclerotic and myocardial lesions as well27. The Ostuka-Long Evans Tokushima fatty (OLETF) rats are deficient in cholecystokinin (CSK)-1 receptor (CSK secreted from intestine regulates digestion and food intake) and has dyslipidaemia, hyperglycaemia, IGT and insulin resistance, along with hypertension and cardiovascular complications28. Goto-Kakizaki (GK) rats are non-obese and develop diabetes at later stage of life. Hyperglycaemia is seen at an early age, with cardiac hypertrophy and systolic dysfunction leading to kidney damage. It shows IGT, dyslipidaemia and insulin resistance, but no hypertension29.
Two rat mutant obese models were added to this illustrious list: the first one was Wistar inbred at National Institute of Nutrition (WNIN)/Ob-obese with normal glucose levels (euglycaemia) and the second one was WNIN/GR-Ob - obese with defect in glucose handling (impaired glucose tolerant or IGT)30. These were collectively referred as Indian ‘Sumo’ rats as these were the fattest rats hitherto reported in literature, the fattest one recorded in the colony was in the range of 1.4 kg (Figure).
Figure.
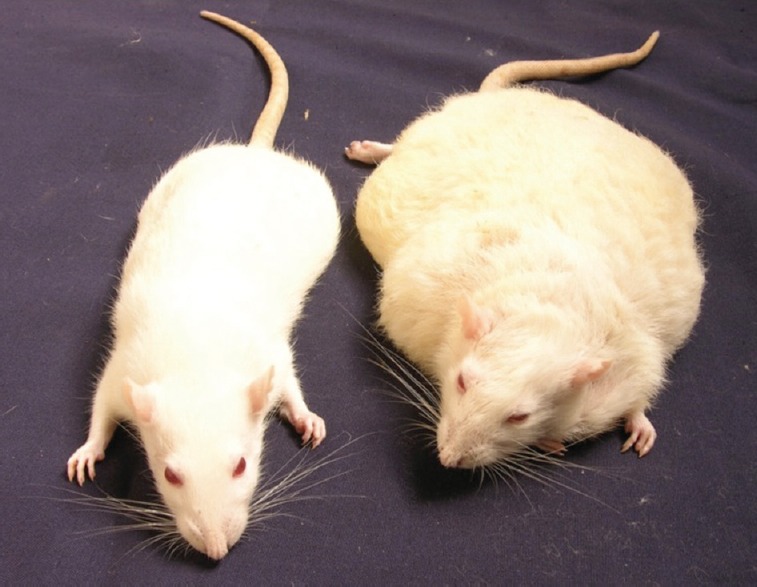
WNIN/Ob Rat. Adult (left) lean (right) obese rats of same age.
Indian ‘SUMO’ rat
History
Laboratory rat as a model biomedical research was introduced in 1906, and the first strain to get that distinction was Wistar, a randomly maintained strain from the Wistar institute, Philadelphia, USA31. From the original Wistar institute, several lines of this strain spread across the world, and at present, there are several biomedical institutions in the world, where this is maintained mostly random bred, or in some strictly inbred. Wistar contributed some prime models such as Wistar BB, Worcester BB, Wistar fatty and GK for obese and diabetes studies. A breeding nucleus of the original Wistar stock was brought to the NIN, Hyderabad in 1920 and, ever since been maintained by strict sister X brother (SXB) mating. A pedigree analysis of this Wistar inbred stock in 1978 showed the presence of four inbred parallel lines and these could be traced to four pairs of parents dating back to 196530. The strain was re-designated as WNIN and the four lines as A, B, C and D. In 1993, from lines C and D, we identified a few animals with IGT, which were then isolated and maintained as a separate strain called WNIN/GR32. Around the same time, a male rat was observed in the one of the litters of the ‘A’ sub line, which showed abnormal increase in weight with age, reaching a weight of 800 g by 3-4 months. It turned to be infertile, as all attempts to mate them did not yield any result. The parents of these rats were traced and by diligent selection of ‘carrier’ heterozygous animals, a colony of obese rats was established in 1996 and designated as WNIN/Ob33. In 1998, by an accidental mixing of WNIN/GR (lean with IGT) and WNIN/Ob (obese with euglycaemia), a third strain emerged, precipitating the traits of the parents, IGT and obesity. This was designated as WNIN/GR-Ob34. Both the strains have completed over 90 generations and the mutations were quite stable. They are fully characterized for their obesity and IGT traits, physically, physiologically and biochemically.
Though these new models share most of the characteristics shown by similar models identified so far, there are some unique features, which keep them apart from the rest. The salient features are given below: (i) These are the first spontaneous mutant rats that evolved from a Wistar inbred colony across the world and thus the first obese rat with an inbred status; (ii) They can be easily propagated as the ‘carrier’ animals and can be easily identified much earlier by physical and biochemical traits. The mutation seen here is autosomal incomplete dominance, unlike other well-known models, majority of them being autosomal recessive; (iii) Tail ‘Kinkiness’ is a signature trait of the model and seen only in obese and carriers and this is often used as a physical marker to assist breeding. No other model shows such a trait; (iv) Infertility seen in male obese rats is fully reversible by food restriction35 without losing the biochemical traits of obesity; (v) The average life span is reduced to 1.5 yr from the normal range of three years and these show all signs of accelerated ageing by as early as one year: (vi) The animals also develop high blood pressure (BP) as early as eight months36; (vii) An average 15-20 per cent of the obese rats show retinal degeneration and cataract; (viii) The immunity is totally compromised and they develop tumours and catch opportunistic infections by one year; and (ix) Animals show hyperleptinaemia, but the defect(s) are not due to either leptin or leptin receptor genes, but at a locus close to the latter on chromosome no. 537.
The WNIN obese rats are unique as they fulfil all the requirements to study the metabolic syndrome seen in humans, as all defects are seen in one model per se. The animals apart from being obese, are insulin and leptin resistant, osteoporotic, infertile with polycystic ovaries and have cataract and retinal degeneration, high BP and accelerated ageing with defective immune surveillance.
Highlights of recent studies in WNIN/ob rats
Influence of diet
The Wistar stock of rats are being maintained in an inbred status (WNIN) over 98 yr approximately in NIN, Hyderabad30. Their diet is based on recommended nutrient composition for laboratory rats using locally available diet ingredients. In 1976, experiments using these rats turned disastrous, as most of the animals in both control and experimental groups showed high levels of glucose on oral glucose test. A systematic cross-sectional study was launched covering all the animals in the colony and it was observed that 30 per cent of the colony suffers from glucose intolerance32. This accidental discovery finally led to the establishment of two obese mutant rats (WNIN/Ob and GR-Ob)1.
The cost of maintaining the obese rats is higher than that of normal rats. Hence, a preliminary study was conducted using WNIN/GR-Ob rats to see whether they could be maintained on a cheaper laboratory chow (containing 14% protein) than the conventional rat chow (containing 20% protein), which is normally given to pregnant and locating mothers. However, it was found that the metabolic status of these animals suffered in terms of growth and feed efficiency resulting in increased triglyceride and plasma glucose levels. WNIN/GR-Ob animals were diet sensitive, especially with reference to glucose handling38. This observation was probed further by feeding the animals differing in source of carbohydrates, to see how far the situation could get worsened. The diet contained 65 per cent of carbohydrate in the form of glucose/sucrose/starch, 20 per cent of protein, 7 per cent of fat, 5 per cent of fibre, 3.5 per cent of mineral mixture and one per cent of vitamin mixture. Lean animals were used as the control, and the experimental period was eight weeks. Glucose and insulin levels increased significantly with the duration of experiment, maximum been achieved in starch-based diets, followed by sucrose and glucose in that order38. But, unlike other obese fatty rats such as WDF, glycosuria was only a trace, requiring a longer feeding schedule beyond eight weeks38.
The spin off from this observation was 2-fold. One, a natural way of producing frank diabetes in an animal model without resorting to injecting alloxan or streptozotocin (STZ), which often produces experimental diabetes in animals, by total destruction of beta cells. The diabetes produced by such chemical means is severe and sudden and cannot strictly be compared to type II diabetes seen in humans, wherein the transition is gradual and not severe. Second, the GR-Ob model mimics the human situation, wherein IGT or pre-diabetes status, if continued without any diet control, often results in full-blown diabetes by five years. The GR-Ob strain maintains its IGT status on normal laboratory rat chow and never becomes fully diabetic due to the high fibre (7%) content in our diet. It is only when switched to purified diets with high carbohydrate content (especially starch and sucrose) that animal develops frank diabetes, underlying the importance of fibre in the diet38.
Impact on eye disorders
Epidemiological studies have shown that obese people are twice likely to develop age-associated macular degeneration (AMD)39. Association between body mass index (BMI) and earlier macular degeneration has also been reported40. Furthermore, abdominal obesity in patients with early and intermediate stages of AMD increases the risk to advanced level. At NIN, Hyderabad, 15-20 per cent of both WNIN/Ob and GR-Ob rats developed cataract as they became adults, indicating that eye problems existed in this mutant rat population, unlike the parental stock41. Hence, a systematic study was initiated to find out whether these mutant obese rats have any retinal regeneration, as they become one year old. Retina from 2 to 12 month old WNIN-Ob rats were compared with the age-matched lean controls for their morphology using histology and immunohistochemistry. RNA from retinas of 12 month old rats was subjected to microarray and real-time polymerase chain reaction (PCR) gene analysis. The study showed retinal degeneration setting in 4-6 month old rats with a systematic worsening as the animals neared one year. Retinal morphology by histology (transmission electronic microscopy) showed thickness of the outer layer reduced by four months of age, which worsened further by 12 months of age. While photoreceptor cells in central area were affected qualitatively (in terms of histology), those of the peripheral area suffered quantitatively (number reduction). These image analyses were well supported by gene expression studies, wherein genes involved in stress and tissue remodelling were overexpressed, while those of phototransduction and photoreception were underexpressed42. The study was repeated using GR-Ob animals to know the additional effect of IGT if any. The retinal degeneration observed was similar, but greater compared to WNIN-Ob (degeneration occurs in three months and there is 90% reduction in rhodopsin)42.
There are a few other rodent models such as Tubby mouse, Bbs4 (Bardet - Bidel) knockout mouse, Tubby-like protein-1 knockout mice (TULP-1) and ZDF rat, all of them showing retinal degeneration with or without obesity. The tubby mouse develops neurosensory abnormalities (retinal degeneration and neurosensory hearing loss) much earlier than the onset of obesity43. Bbs4 mice are similar to this, but have additional phenotype traits44. TULP-1 mouse has retinal degeneration but is not obese45. WNIN obese rats are unique because retinal degeneration kicks in as soon as the onset of obesity and both obesity and retinal degeneration are severe. Another interesting point is the preservation of cones even after the severe loss of rods. Normally, in humans and other animal models, one observes rod cell death preceding cone loss42. The expression of genes associated with WNIN/Ob retina is similar to what is reported for Bbs4 knockout mice, thus showing a direct association with obesity42. However, whether it is due to obesity per se or through the metabolic abnormalities it produces needs to be answered.
As mentioned earlier, 15-20 per cent WNIN obese mutant rats develop cataract by 12 months of age41, which has been not reported in other known animal models. Unlike other eye disorders, cataract seems to be more strongly linked to obesity as evidenced by large-scale epidemiological studies39. Hence, detailed studies were undertaken to see first the biochemical mechanism(s) involved in the cataract formation in these rats and later study the susceptibility of these animals for cataract formation in vitro and in vivo. Lens protein (soluble) levels and aggregation, crystalline distribution, non-enzymatic glycation of lens, oxidative stress and antioxidation systems and polyol pathways (systems involved in catractogenesis) were studied in 3 to 12 month old obese animals, using lean animals of the same age as controls41. No difference was seen in any of the parameters, except for alteration in polyol pathway in terms of activity of aldose reductase (ALR2), sorbitol dehydrogenase and sorbitol levels in lens. Sorbitol levels were found to increase from three months onwards with concomitant increase in ALR2. ALR2 expression (by immunodetection and PCR) was also found to be high in obese rat lenses compared to lean controls. GR-Ob animals had higher sorbitol levels than WNIN-Ob, suggesting synergistic effect of IGT with obesity. The involvement of sorbitol pathway as a risk factor in cataract development under obesity conditions so far was not shown in any other model and this adds to its uniqueness41.
It was still intriguing as to why only 20 per cent of the rats developed cataract and for this, susceptibility of lenses of obese and lean rats to develop cataract was studied using environmental [heat and ultraviolet (UV) irradiation] and physiological insults (glucose mediated, hyperglycaemia in vivo). Heat and UV-induced aggregation studies46 showed that the light-scattering ability of lens-soluble proteins was high in six month and 12 month old obese rats than in lean animals. While lens opacification by glucose in lens organ culture ex vivo required seven days in lean animals, this was accomplished in four days in obese lenses. Between Ob and GR-Ob animals, the latter accumulated twice the quantity of sorbitol in the tissue. It also showed a critical level of 650 nmoles/g tissue of sorbitol will result in cataract formation, and this was found to be true when sorbitol levels were measured in animals that showed cataract in vivo46. Galactose-induced and STZ-induced diabetes was studied in three-month-old Ob and GR-Ob rats, and it was seen that the stage of cataract and onset were faster in obese animals compared to that of lean controls. As expected, GR-Ob developed the syndrome earlier and moreover the STZ dose required to produce hyperglycaemia was much less (27 mg/kg body weight) compared to Ob (30 mg/kg body weight) and lean (35 mg/kg body weight) animals46. These studies unequivocally showed that accumulation of sorbitol in retina could structurally change eye lenses, making them vulnerable to aggregation by physical and environmental inducers, ultimately precipitating cataract. In diabetic retinopathy cases, sorbitol seems to accumulate in retina and this is well supported by STZ rat model (having IGT and insulin resistance)45. Ob/Ob mouse on STZ injection (produces hyperglycaemia, but no cataract) did not show any change in sorbitol levels, while STZ-induced db/db mouse showed increased sorbitol, but no cataract47. The present model is unique to show increased sorbitol levels in the lens, which causes cataract in some percentage of obese animals when its level reaches a critical threshold value.
Impact on neurodegeneration
WNIN obese rats in comparison to their lean litter mates have low brain weights and there is as much as 50 per cent reduction in organ to body weight ratio48. Their average life span is 18-20 months compared to 30-36 months of normal rats. As they age faster, they develop tumours, impaired vision, impaired immunity and neurodegeneration. The ubiquitin-proteasome (UPS) pathway is the principal mechanism in protein catabolism in mammalian cytosol and nucleus and any defects in the system can lead to several diseases including neurodegeneration49. Ultrastructural analysis of WNIN/Ob rat's brain showed neuronal cell abnormalities in the peripheral cortex region. In the same animals, the status of UPS was investigated, as UPS is implicated in various neurodegenerative diseases. Results showed altered UPS (upregulation of UCH 1 and 3 and downregulation of UCH 5, reduced proteasome activity), existence of endoplasmic reticulum (ER) stress and upregulation of apoptosis in the central cortex of obese rats. It is possible that neuronal cell death observed in obese rats could be due to apoptosis-mediated UCH-1, by stabilizing P53. Since diet restriction is an economical and effective intervention strategy for obesity, these rats were diet restricted (DR) from 45 days to a period of 6.5 months. DR alleviated all changes observed earlier viz. alteration of UPS, increased ER stress and increased expression of apoptosis, which resulted in the neonatal cell death seen in obese rats50.
Impact on immunity
Both epidemiological data51 and studies in rodent models52 demonstrate that the incidence and severity of infectious diseases will increase under obese conditions. Susceptibility to infections in obesity indicates a total derangement in immune dysfunction. Both genetically and diet-induced obese animals exhibited low response to T-cells, reduced capacity to respond to mitogen, altered dendrite cell and altered natural killer (NK) cell function53,54. White adipose tissue (WAT) was initially thought to be an inert organ primarily for energy storage. But, in the last decade, several studies indicated that these tissues have a major role in maintaining many physiological processes in the body including immunity55. Incidence and severity of infectious diseases seemed to increase with increase in weight in both humans and rodents. Both genetically and DIO animals exhibit low response to T-cells, reduced capacity to mitogens, altered dendrite cells and altered NK cell functions- all indicative of a total derangement in immune function. At the same time, a reduction in total body weight correlated well with impaired immunity, thus suggesting a major role for adipose tissue in maintaining immunity. The puzzle as to how this could be achieved was resolved with the discovery of leptin, which in circulation fits the role of as an endocrine mediator between adipose tissue and immune cells. Leptin receptors are expressed in neutrophils, monocytes and lymphocytes and exert its effect on processes such as development, maturation and activation of immune cells55. Leptin mediates through pro-inflammatory cells, promoting T-helper cells to produce cytokines such as interleukin-2 (IL-2) and IL-6. Low levels are observed in starvation and malnutrition associated with thymus and splenic weights and decrease in lymphocyte number and function, and leptin administration reversed starvation- or malnutrition-induced immunosuppression. At the other end of the spectrum, high serum leptin levels are observed in autoimmune disorders including type 1 diabetes as well as in obesity55. Leptin is thus the key molecule involved in the pro-inflammatory status observed in obesity. It looks now very clear that obesity can make one susceptible to infectious diseases in the short run and also to autoimmune disorder if obesity persists without correction leading to total derangement in immune function even to the extent of affecting long-term memory like vaccination. WNIN obese rats were seen to get opportunistic infections as they reach one year of age and they also develop tumours. Hence, innate and adaptive immune responses were studied in these rats by using hepatitis B vaccine as an immunogen and the underlying mechanisms involved in the altered immunity was also explored56,57.
The innate response of both WNIN/Ob and GR-Ob (three months old) was found to be impaired. Obese animals from both genders from WNIN/Ob and GR-Ob showed low spleen weight/g body weight, and differences in splenic CD4+ helper T-cells, total CD3+T cells and CD8+ T cytotoxic cells, along with differences in the splenic lymphocyte, proliferate response to con A compared to lean litter mates. There were differences in each of the parameters, between genders and between strains. This was true of humoral immunity as shown by serum IgG and IgM levels. Serum leptin levels were high in both the strains and in both sexes. As the basal immune response was thus impaired in these obese animals, humoral, cell-mediated and innate responses were analysed by challenging the animals with hepatitis B vaccine56,57. Upon vaccination, the HBsAg-specific IgG response and the splenic lymphocyte proliferative response to HBsAg were low in both WNIN/Ob and WNIN/GR-Ob obese animals compared to their lean animals, suggesting loss of long-term protective and immunological memory in response to vaccination56,57. Further, the splenic lymphocyte proliferative response to con A and nitric oxide (NO) and tumour necrosis factor-α production by peritoneal macrophages upon HBV was altered in WNIN/GR-Ob obese animals, only suggesting that IGT worsens the immune response to HBV under obese conditions57.
It is intriguing to note that despite the presence of high leptin levels, the immune functions are impaired in these obese animals. The resistance or non-responsiveness to leptin was probed further in both the mutants by intraperitoneal injection of recombinant leptin under starvation for 48 h, and looking for immunity parameters along with the expression of phosphorylated form of leptin receptor (OBR) and the signalling molecules (JAk2 proteins) by Western blots. In WNIN/Ob lean animals, leptin administration significantly increased cell-mediated immunity in terms of CD4/CD8 ratio and innate immunity in terms of lipopolysaccharide (LPS)-stimulated peritoneal macrophage NO production. Further, in lean animals, there was a significant trend towards increased JAK2 production upon leptin treatment. Thus, we can attribute the non-responsiveness to leptin in obese animals to reduced JAK2 protein expression58.
Similar to WNIN/Ob, in WNIN/GR-Ob lean animals also, leptin administration significantly increased cell-mediated immunity in terms of CD4/CD8 ratio and lymphocyte proliferative response to con A and innate immunity in terms of LPS-stimulated peritoneal macrophage NO production. Leptin despite being a pro-inflammatory molecule, led to the down regulation of lymphocyte proliferative response to con A in obese animals. Consistent with this, leptin treatment resulted in the downregulation of OBR expression also and thus induces leptin resistance in these obese animals58. Since the defects seem to be due to leptin resistance, either by low OBR expression (as in WNIN/GR-Ob) or by defective signalling (as in WNIN/OB), strategies that could overcome this can be developed to overcome this. Nature showed the way as caloric restriction for a month was shown to induce leptin responsiveness59 in both genetic and diet induced obese animals by increasing OBR expression. Another way could be by injecting amylin (peptide hormone co-secreted with insulin in pancreatic beta cells), which in conjugation with leptin was seen to increase the basic OBR expression as well as the signalling systems60.
Nature versus nurture
Normally in humans, the fatness is basically due to a quantitative trait, reflecting the interactions of environment (nurture) with that of genotype (nature). Studies in twins, adoptees and family show that 80 per cent of the variance in BMI basically stems from one's genotypes. Obesity and non-insulin-dependent diabetes mellitus were shown to be linked to a region in chromosome no. 1, containing leptin receptor61. But, though some exons of leptin receptor genes showed sequence variations, none of these variations could be seen linked to the observed fat mass62. However, individual human obese cases were reported with defective leptin, leptin receptor and even the fat mouse type of mutation63,64.
May be one has to go as far as down the foetal stage to explain all the metabolic abnormalities that comes later in adult life, as hypothesized by Baker65. According to him, a susceptible gene could be altered epigenetically at the foetal stage in an uterine environment, leading to a major alteration later in adult life. Such a possibility was well established in animal studies, especially in the ‘pup in a cup’ model, wherein rat pups reared on artificial milk containing high fat (normal rat milk is low in fat) developed IGT as they are weaned and went on to become obese and diabetic. But, the most unexpected thing was that when such pups became adults, their pups also became obese without rearing on the artificial milk66. That was a case of epigenetics where the susceptible gene was switched on to develop a trait that was induced in the parents earlier.
We have seen earlier, that while some inbred mice and rats could be made overweight or obese by feeding high-energy dense food, others seem to resist such a tendency. This is certainly due to interaction of nature with nurture. In our animal facilities, four rat strains have been maintained and it is only from WNIN, the obese mutants (WNIN-Ob and GR-Ob) are developed, giving a doubt whether this strain is obese or overweight prone. Wistar is certainly different from the other two strains, SD and F-344, in terms of percentage body fat (WNIN>SD>F-344) as well as in their physical activity (WNIN<SD< F-344). The response of these three strains to high calorie dense diets was explored to see whether any epigenetic mechanism was in operation with respect to their ability to become overweight or obese67. This will also answer the common question, why some individuals are more prone than others, when all are exposed to same environmental excess? The animals received HF, high sucrose (HS) and both HSF for a period of 10 wk from weaning onwards. WNIN showed significantly high energy consumption, and decreased energy expenditure on HF and HSF diets, resulting in increased body weight gain, BMI, fat per cent, WAT weights and adiposity index - all hallmarks of central obesity. SD and F-344 rat strains under the same high calorie environment resisted the tendency to put on weight despite high-energy consumption67. This is a direct reflection of their metabolic profiles, as well as their physical fitness. WNIN in HF and HSF diets showed hepatic lipogenesis, decreased fatty acid oxidation, while the other two increased their fatty acid oxidation, thus more thermogenesis on same diets, which prevented them from putting more weight. Further, increased adipocyte hypertrophy, visceral adiposity and hepatic oxidation stress, which resulted in low-grade systemic inflammation, were seen in WNIN strain. Insulin levels were equally elevated subsequently leading to insulin resistance, and then to IGT. The animals showed increased triglycerides, decreased high-density lipoprotein cholesterol and even deposition of hepatic lipid droplets (fatty liver), clearly indicating the vulnerability of WNIN rats to develop obesity67. Transcriptome analysis of liver and adipose tissues are in progress, and preliminary data revealed some interesting insights into the obesogenic nature of WNIN rats (unpublished data). It is no wonder why both the mutant obese strains (WNIN/Ob and WNIN/GR-Ob) were evolved from this strain only. It is often said that ‘what you eat is what you are’, but one should also be genetically predisposed to develop the final phenotype expression.
Lessons to be learned
In the wild, one hardly comes across animals exhibiting abnormalities in glucose/energy homeostasis leading to obesity and other metabolic abnormalities that follow. By natural instinct, the animals are diligent to what they eat as per the availability of the season and they keep themselves busy, actively foraging for food of their choice. But laboratory animals are a class apart, since they are deliberately bred under controlled and defined conditions, in terms of space, food and inherent traits. Here, they are fully confined to their respective cages which allows them only limited movement, and fed on highly processed animal chow. WNIN rat first showed IGT, later developed obesity with and without IGT, incorporating various aspects of metabolic syndrome and then affecting almost every organ or tissue in the body. It is also to be seen that once obesity has set in it could lead to infertility, though it is possible to restore it by food restriction from weaning onwards. The only way to correct a genetic obesity is by intervening in utero by dietary means, an exciting prospect, yet to be tested.
The ‘Indian Sumo rat’ thus teaches us an important lesson – the need for a regulated life style with moderate physical activity, consumption of wholesome food against processed food to keep one's glucose/energy homeostasis intact, lest one develops overweight/obesity and associated degenerative diseases, later into adulthood. With ageing setting in earlier, premature death is a certainty. It is pathetic to see that the food these animals are so fond of has only resulted in their premature death and there is lesson in this for all of us.
Acknowledgment
Author acknowledges his collaborators (Drs Bhanuprakash Reddy, P. N. Yadagiri Reddy, A. Vajreswari, R. Rajender Rao, Hemalatha Rajkumar, Jeffry M. Friedman) and his students (Drs C. N. Lakshmi, N. Harishankar, B. Kirankumar, B. Prathibha, Shri M. N. Muralidhar and Dr S. Sreenivasa Reddy), and thank his colleague Dr N. Sathya Vani, who was the breeding supervisor of the colony, and animal technician late M. Ganesh for technical excellence in maintaining the WNIN obese rat colony and the timely supply of the required animals for experiments.
Footnotes
Financial support & sponsorship: Author acknowledges the financial help obtained from the Indian Council of Medical Research and Department of Biotechnology, Government of India, New Delhi, for carrying out the work done in WNIN obese rats.
Conflicts of Interest: None.
References
- 1.Giridharan NV. Animal models of obesity & their usefulness in molecular approach to obesity. Indian J Med Res. 1998;108:225–42. [PubMed] [Google Scholar]
- 2.Mitruka BM, Rawnsley HM, Vadehra DN, editors. London: John Wiley & Sons; 1976. Animals for medical research. [Google Scholar]
- 3.Cuenot L. Pure strains and their contributions in the mouse. Arch Zool Exptl Gene Serc. 1905;122:123–6. [Google Scholar]
- 4.Hetherington AW, Ranson AW. The relation of various biochemical lesions to adiposity in the rat. J Comp Neurol. 1942;76:475–99. [Google Scholar]
- 5.Fenton PF, Dowling MT. Studies on obesity. I. Nutritional obesity in mice. J Nutr. 1953;49:319–31. doi: 10.1093/jn/49.2.319. [DOI] [PubMed] [Google Scholar]
- 6.Miller DS. Non genetic models of obesity. In: Festing MFW, editor. Animal models of obesity. London: Macmillan; 1979. pp. 131–40. [Google Scholar]
- 7.Kanmark KB, Ortilian-Gambil N. Dietary induced obesity in experimental animals. In: Beyman AC, West CE, editors. Use of animal models for research in human nutrition. Vol. 18. Basel: Karger; 1988. pp. 1021–6. [Google Scholar]
- 8.Sclafani A, Gorman AN. Effects of age, sex, and prior body weight on the development of dietary obesity in adult rats. Physiol Behav. 1977;18:1021–6. doi: 10.1016/0031-9384(77)90006-3. [DOI] [PubMed] [Google Scholar]
- 9.Bjorntorp P, Sjostrom L. Number and size of adipose tissue fat cells in relation to metabolism in human obesity. Metab. 1971;20:703–13. doi: 10.1016/0026-0495(71)90084-9. [DOI] [PubMed] [Google Scholar]
- 10.York AD. Festing MFW, editor. The characteristics of genetically obese mutants. Animal models of obesity London: Macmillian. 1979:39–64. [Google Scholar]
- 11.Bielschowsky F, Bielschowsky M. The New Zealand strain of obese mice; their response to stilboestrol and to insulin. Aust J Exp Biol Med Sci. 1956;34:181–98. doi: 10.1038/icb.1956.22. [DOI] [PubMed] [Google Scholar]
- 12.Berdainer CD. Hepatic metabolism in glycemic/hyperlypedemic BHE rats: Evidence for a membrane error in mature onset diabetes. In: Shaffrier E, Renold AE, editors. Lessons from animal diabetes. London: John Wiley; 1984. pp. 2010–2. [Google Scholar]
- 13.Zahand GR, Adler JH. Sand rat as a model of diabetic cataract - A major blinding condition. In: Shafrir E, Ronald AE, editors. Lessons from animal diabetes. London: John Libbey; 1984. pp. 500–2. [Google Scholar]
- 14.Coleman DL. Obese and diabetes: Two mutant genes causing diabetes obese syndrome in mice. Diabetiolgia. 1978;14:141–8. doi: 10.1007/BF00429772. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM, et al. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–32. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 16.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–71. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 17.Romsos DR, Ferguson D. Self-selected intake of carbohydrate, fat, and protein by obese (ob/ob) and lean mice. Physiol Behav. 1982;28:301–5. doi: 10.1016/0031-9384(82)90079-8. [DOI] [PubMed] [Google Scholar]
- 18.Sima AA, Robertson DM. Peripheral neuropathy in the diabetic mutant mouse. An ultrastructural study. Lab Invest. 1979;40:627–32. [PubMed] [Google Scholar]
- 19.Lee SM. The effect of a high fibre diet on diabetic nephropathy in the db/db mouse. Diabetologia. 1982;22:349–53. doi: 10.1007/BF00253580. [DOI] [PubMed] [Google Scholar]
- 20.Special issue: New rat models of obesity and type II diabetes. [accessed on September 9, 2018];ILAR J. 1990 32:2–38. Available from: https://academic.oup.com/ilarjournal/issue/32/3 . [Google Scholar]
- 21.Truett GE, Bahary N, Friedman JM, Leibel RL. Rat obesity gene fatty (fa) maps to chromosome 5: Evidence for homology with the mouse gene diabetes (db) Proc Natl Acad Sci U S A. 1991;88:7806–9. doi: 10.1073/pnas.88.17.7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zucker TF, Zucker LM. Fat accretion and growth in the rat. J Nutr. 1963;80:6–19. doi: 10.1093/jn/80.1.6. [DOI] [PubMed] [Google Scholar]
- 23.Kava R, Greenwood MRC, Johnson PR. Zucker (fa/fa) rat. ILAR J. 1990;32:4–8. [Google Scholar]
- 24.Matuso T, Shimkawa K, Omoni Y, Okeda H, Ikeda H. Fertility improvement by adrenelectomy of genetically obese-diabetic rodents. Zucker fa/fa, and Wistar fa/fa rats and Ob/Ob mice. In: Shafrir E, Renold AE, editors. Frontiers of diabetic research, Lessons from Animal diabetes II. London: John Libbey; 1988. pp. 374–7. [Google Scholar]
- 25.Peterson RG, Shaw WN, Neel MA, Little LA, Eichberg J. Zucker diabetic fatty rat as a model for non-insulin dependent diabetes mellitus. ILAR J. 1990;32:16–9. doi: 10.1093/ilar.32.3.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kava R, Peteron RG, West DB, Greenwood MRC. Wistar diabetic fatty rat. ILAR J. 1990;32:9–13. [Google Scholar]
- 27.Koletsky S. Obese spontaneously hypertensive rats - A model for study of atherosclerosis. Exp Mol Pathol. 1973;19:53–60. doi: 10.1016/0014-4800(73)90040-3. [DOI] [PubMed] [Google Scholar]
- 28.Panchal SK, Brown L. Rodent models for metabolic syndrome research. J Biomed Biotechnol. 2011;2011:351982. doi: 10.1155/2011/351982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Portha B, Giroix MH, Tourrel-Cuzin C, Le-Stunff H, Movassat J. The GK rat: A prototype for the study of non-overweight type 2 diabetes. Methods Mol Biol. 2012;933:125–59. doi: 10.1007/978-1-62703-068-7_9. [DOI] [PubMed] [Google Scholar]
- 30.Giridharan N, Lakshmi CN, Satyavani M, Harishankar M. WISTAR-NIN-IGT, WISTAR-NIN-OB, WISTAR-OB-1GT - three mutant rat models for diabetes and obesity from a Wistar inbred colony. Rat News Lett. 1993;29:14–5. [Google Scholar]
- 31.Hans J, Hedrich . Part 1: The history and development of rat as a laboratory model. In: Georg J Krinke., editor. The Laboratory Rat. New York, Boston, London: Academic Press; 2000. pp. 3–8. [Google Scholar]
- 32.Giridharan NV, Lakshmi CN, Raghuramulu N. Identification of impaired-glucose-tolerant animals from a wistar inbred rat colony. Lab Anim Sci. 1997;47:428–31. [PubMed] [Google Scholar]
- 33.Giridharan NV, Harishankar N, Satyavani M. A new rat model for the study of obesity. Scand J Lab Anim Sci. 1996;23:131–7. [Google Scholar]
- 34.Harishankar N, Vajreswari A, Giridharan NV. WNIN/GR-Ob - An insulin-resistant obese rat model from inbred WNIN strain. Indian J Med Res. 2011;134:320–9. [PMC free article] [PubMed] [Google Scholar]
- 35.Dinesh Yadav DM, Muralidhar MN, Prasad SMVK, Rajender Rao K. Pre-pubertal diet restriction reduces reactive oxygen species and restores fertility in male WNIN/Obese rat. Andrologia. 2018;50 doi: 10.1111/and.12849. doi:10.1111/and.12849. [DOI] [PubMed] [Google Scholar]
- 36.Giridharan NV, Sailaja P, Harishankar N. A new obese rat model to study obesity and cardiovascular risks. CMR J. 2010;3:96–7. [Google Scholar]
- 37.Kalashikam RR, Battula KK, Kirlampalli V, FriedmanJM, Nappanveettil G. Obese locus in WNIN/obese rat maps on chromosome 5 upstream of leptin receptor. PLoS One. 2013;8:e77679. doi: 10.1371/journal.pone.0077679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harishankar N, Seshadri E, Kalyanasundaram S, Giridharan N. Impaired glucose tolerance to frank diabetes: Diet manipulations in WNIN /GR-Ob rats. J Diabetes Mellitus. 2012;2:52–8. [Google Scholar]
- 39.Chung N, Wong TY. Obesity and eye disorders. Serv Opthalmol. 52:180–95. [Google Scholar]
- 40.Schumberg DA, Christen WG, Hankenson SE, Glynn RJ. Body mass index and incidence of visually significant age related maculopathy in man. Arch Opthalmol. 2001;119:1259–65. doi: 10.1001/archopht.119.9.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reddy PY, Giridharan NV, Reddy GB. Activation of sorbitol pathway in metabolic syndrome and increased susceptibility to cataract in wistar-obese rats. Mol Vis. 2012;18:495–503. [PMC free article] [PubMed] [Google Scholar]
- 42.Reddy GB, Vasireddy V, Mandal MN, Tiruvalluru M, Wang XF, Jablonski MM, et al. A novel rat model with obesity-associated retinal degeneration. Invest Ophthalmol Vis Sci. 2009;50:3456–63. doi: 10.1167/iovs.08-2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kleyn PW, Fan W, Kovats SG, Lee JJ, Pulido JC, Wu Y, et al. Identification and characterization of the mouse obesity gene tubby: A member of a novel gene family. Cell. 1996;85:281–90. doi: 10.1016/s0092-8674(00)81104-6. [DOI] [PubMed] [Google Scholar]
- 44.Nishimura DY, Fath M, Mullins RF, Searby C, Andrews M, Davis R, et al. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci U S A. 2004;101:16588–93. doi: 10.1073/pnas.0405496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ikeda S, Shiva N, Ikeda A, Smith RS, Nusinowitz S, Yan G, et al. Retinal degeneration but not obesity is observed in null mutants of the tubby-like protein 1 gene. Hum Mol Genet. 2000;9:155–63. doi: 10.1093/hmg/9.2.155. [DOI] [PubMed] [Google Scholar]
- 46.Reddy PY, Giridharan NV, Balakrishna N, Validandi V, Pullakhandam R, Reddy GB, et al. Increased risk of cataract development in WNIN-obese rats due to accumulation of intralenticular sorbitol. IUBMB Life. 2013;65:472–8. doi: 10.1002/iub.1163. [DOI] [PubMed] [Google Scholar]
- 47.Vicario PP, Slater EE, Saperstein R. The effect of ponalrestat on sorbitol levels in the lens of obese and diabetic mice. Biochem Int. 1989;19:553–61. [PubMed] [Google Scholar]
- 48.Harishankar N, Kumar PU, Sesikeran B, Giridharan N. Obesity associated pathophysiological & histological changes in WNIN obese mutant rats. Indian J Med Res. 2011;134:330–40. [PMC free article] [PubMed] [Google Scholar]
- 49.Ying H, Wang H, Wang G. The ubiquitin proteasome system as a potential target for the treatment of neurodegenerative diseases. Curr Pharm Des. 2013;19:3305–14. doi: 10.2174/1381612811319180013. [DOI] [PubMed] [Google Scholar]
- 50.Reddy SS, Shruthi K, Reddy VS, Raghu G, Suryanarayana P, Giridharan NV, et al. Altered ubiquitin-proteasome system leads to neuronal cell death in a spontaneous obese rat model. Biochim Biophys Acta. 2014;1840:2924–34. doi: 10.1016/j.bbagen.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 51.Falagas ME, Kompoti M. Obesity and infection. Lancet Infect Dis. 2006;6:438–46. doi: 10.1016/S1473-3099(06)70523-0. [DOI] [PubMed] [Google Scholar]
- 52.Ikejema S, Sasaki S, Sahinami H, Mori F, Ogawa Y, Nakmura T, et al. Impairement of host resistance to Listeria monocytogenes infection in liver of db/db and ob/ob mice. Diabetes. 2005;54:182–9. doi: 10.2337/diabetes.54.1.182. [DOI] [PubMed] [Google Scholar]
- 53.Martí A, Marcos A, Martínez JA. Obesity and immune function relationships. Obes Rev. 2001;2:131–40. doi: 10.1046/j.1467-789x.2001.00025.x. [DOI] [PubMed] [Google Scholar]
- 54.Lamasa O, Martineza J, Marti A. T helper lymphopenia and decreased mitogen response in cafeteria diet-induced obese rats. Nutr Res. 2002;22:497–506. [Google Scholar]
- 55.Matarese G, Moschos S, Mantzoros CS. Leptin in immunology. J Immunol. 2005;174:3137–42. doi: 10.4049/jimmunol.174.6.3137. [DOI] [PubMed] [Google Scholar]
- 56.Bandaru P, Rajkumar H, Nappanveettil G. Altered or impaired immune response upon vaccination in WNIN/Ob rats. Vaccine. 2011;29:3038–42. doi: 10.1016/j.vaccine.2011.01.107. [DOI] [PubMed] [Google Scholar]
- 57.Bandaru P, Rajkumar H, Nappanveettil G. Altered or impaired immune response to hepatitis B vaccine in WNIN/GR-ob rat: An obese rat model with impaired glucose tolerance. ISRN Endocrinol. 2011;2011:980105. doi: 10.5402/2011/980105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bandaru P, Rajkumar H, Upsdrasta VP, Nappanveettil G. Role of leptin in immune dysfunction in WNIN obese rats. Endocrinol Metaab Syndr. 2013;2:116. [Google Scholar]
- 59.Witsey J, Scarpace PJ. Calorie restriction reverses defects in leptin receptor protein and leptin signalling capacity with diet induced obesity: role of leptin in the regulation of hypothalamic expression of long form leptin receptor expression. J Endocrinol. 2004;181:297–306. doi: 10.1677/joe.0.1810297. [DOI] [PubMed] [Google Scholar]
- 60.Moon HS, Chamberland JP, Diakopoulos KN, Fiorenza CG, Ziemke F, Schneider B, et al. Leptin and amylin act in an additive manner to activate overlapping signaling pathways in peripheral tissues: in vitro and ex vivo studies in humans. Diabetes Care. 2011;34:132–8. doi: 10.2337/dc10-0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM. Genetic Investigation of ANthropometric Traits Consortium. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thorleifsson G, Walters GB, Gudbjartsson DF, Steinthorsdottir V, Sulem P, Helgadottir A, et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet. 2009;41:18–24. doi: 10.1038/ng.274. [DOI] [PubMed] [Google Scholar]
- 63.Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–8. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 64.Vaisse C, Clement K, Cabro S, Lahlou N, Basdevant A, Bougneres P, et al. An homozygous mutation in the human leptin receptor leads to morbid obesity and pituitary dysfunction (abstract 0055) Diabetes. 1998;47((Suppl 1)):A14. doi: 10.1038/32911. [DOI] [PubMed] [Google Scholar]
- 65.Barker DJ. Fetal origins of coronary heart disease. BMJ. 1995;311:171–4. doi: 10.1136/bmj.311.6998.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Patel MS, Srinivasan M. Metabolic programming: Causes and consequences. J Biol Chem. 2002;277:1629–32. doi: 10.1074/jbc.R100017200. [DOI] [PubMed] [Google Scholar]
- 67.Muralidhar MN, Prasad SMVK, Battula KK, Giridharan NV, Kalashikam RR. Differential response of rat strains to obesogenic diets underlines the importance of genetic makeup of an individual towards obesity. Sci Rep. 2017;7:9162. doi: 10.1038/s41598-017-09149-6. [DOI] [PMC free article] [PubMed] [Google Scholar]