

Abstract
The phosphoinositide 3-kinase (PI3K) pathway plays an integral role in many cellular processes and is frequently altered in cancer, contributing to tumor growth and survival. Small molecule inhibitors have been developed that target the three major nodes of this pathway: PI3K, AKT, and mammalian target of rapamycin. However, because oncogenic PI3K pathway activation is achieved in diverse, potentially redundant ways, the clinical efficacy of these inhibitors as monotherapies has, so far, been limited, despite demonstrating promising preclinical activity. Moreover, pathway activation is associated with resistance to other therapies; thus, in combination, PI3K pathway inhibitors could restore therapeutic sensitivity to these agents. To maximize therapeutic benefit, drug combinations and schedules must be explored to identify those with the highest efficacy and lowest toxicity overlap. In addition, defining appropriate patient subpopulations, for both monotherapy and drug combinations, will be important. However, identifying predictive biomarkers remains a challenge.
INTRODUCTION
The phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway is frequently altered in cancer,1 promoting growth, proliferation, and survival.1,2 Targeting its three major nodes (PI3K, AKT, and mTOR), therefore, represents a key therapeutic opportunity.1
Class IA PI3Ks are heterodimers composing a regulatory (p85) and catalytic (p110) subunit, and exist in four isoforms (α, β, γ, and δ) with differential tissue expression.1 Growth factor stimulation of receptor tyrosine kinases triggers PI3K activation, downstream activation of phosphoinositide-dependent kinase 1 (PDK1) and AKT, and, subsequently, mTOR complex 1 (mTORC1), which promotes cell growth and protein synthesis.2 The mTORC1 substrate ribosomal S6 protein kinase (p70S6K) phosphorylates ribosomal protein S6, stimulating protein synthesis, and feeds back to insulin receptor substrate 1 to downregulate insulin-mediated PI3K pathway activation. The pathway can be activated by G protein-coupled receptors or by oncogenic proteins such as RAS.1
The tumor suppressor phosphatase and tensin homolog (PTEN) is a key negative regulator of the PI3K pathway.2 Others include inositol polyphosphate 4-phosphatase type II (INPP4B)3 and the protein tyrosine phosphatase nonreceptor 12 (PTPN12/PTP-PEST).4 This review summarizes PI3K pathway alterations found in solid tumors and discusses pathway inhibitors, their class-specific toxicities, and the possible challenges underpinning patient selection and drug resistance.
THE PI3K PATHWAY IS FREQUENTLY ALTERED IN SOLID TUMORS
PI3K pathway alterations include somatic amplification, mutation, loss of heterozygosity, or changes in DNA methylation, often in multiple genes (Fig 1).5
Fig 1.

Common PI3K pathway aberrations found in a variety of solid tumor types. Activation of the PI3K pathway contributes to tumor growth, survival, and resistance to anticancer therapies. FGFR2, fibroblast growth factor receptor 2; GBM, glioblastoma multiforme; HER2, human epidermal growth factor receptor 2; HNSCC, head and neck squamous cell carcinoma; INPP4B, inositol polyphosphate 4-phosphatase type II; MET, hepatocyte growth factor receptor; mTORC, mammalian target of rapamycin complex; NSCLC, non–small-cell lung cancer; PI3K, phosphoinositide 3-kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PTEN, phosphatase and tensin homolog; PTPN12, protein tyrosine phosphatase nonreceptor 12; RTK, receptor tyrosine kinase; SCLC, small-cell lung cancer; TNBC, triple-negative breast cancer. Adapted from a figure provided by Ana Maria Gonzalez-Angulo.
Breast Cancer
In breast cancer, most mutations occur in PIK3CA. Three frequent “hotspot” mutations within the helical (E545K and E542K in exon 9) and kinase domains (H1047R in exon 20) result in constitutive p110α activity.6 Approximately 20% to 50% of breast cancers exhibit PIK3CA mutations, including approximately 35% of hormone receptor (HR)–positive and approximately 23% of human epidermal growth factor receptor 2 (HER2)–positive breast cancers.7 PIK3CA mutations occur less frequently (<10%) in triple-negative breast cancer (TNBC), although pathway activation may be driven instead by PIK3CA amplification or genomic loss of PTEN or INPP4B.7,8 In particular, INPP4B is lost in 30% to 56% of TNBC.8,9
PTEN mutations occur in < 3% of breast cancers; however, loss of PTEN protein occurs in approximately 30% of cases.5,7 PTEN protein loss and PIK3CA mutations appear to have different functional effects: PTEN protein loss is associated with elevated AKT phosphorylation, whereas PIK3CA mutations have not been associated with significant differences in the levels of PTEN protein or of phosphorylated downstream substrates compared with wild-type PIK3CA breast tumors.7
Activating mutations in the catalytic domain of AKT have not been observed. However, approximately 3% of HR-positive breast cancers exhibit an E17K substitution in the pleckstrin homology domain, resulting in constitutive activation.7,10
In breast tumors, PTPN12 downregulates growth factor receptor signaling to suppress the transformation of human mammary epithelial cells.4 PTPN12 protein expression is lost in approximately 23% of breast tumors, especially TNBC,4 and is associated with poor patient outcome.11
Lung Cancer
PI3K pathway activation, as demonstrated by AKT phosphorylation, occurs in 50% to 70% of non-small cell lung cancers (NSCLCs).12 This pathway is altered in 47% of squamous cell carcinomas.13 PI3K pathway activation can occur through activating mutations in EGFR, KRAS, PI3K, or AKT, as well as PIK3CA amplification or loss of PTEN expression.12 Somatic mutations in PIK3CA are relatively infrequent,14 whereas genomic amplification is more common, occurring in 43% of lung cancers.15 Mutations in AKT itself are rare; the AKT E17K mutation has been reported in approximately 2% of NSCLCs, restricted to the squamous histotype16; however, the importance of oncogenic AKT activity is underlined by the high incidence of loss of PTEN and INPP4B protein expression (75% and 47% of NSCLCs, respectively).17,18
Head and Neck Cancer
PI3K pathway alterations occur in 30% to 66% of head and neck squamous cell carcinomas (HNSCCs); this rate increases to 90% if changes in mRNA levels are also considered.19,20 Common alterations include reduced PTEN expression (30% of patients) and AKT amplification (5%).20 PIK3CA is the most frequently altered gene (36%); mutation and amplification are mutually exclusive and equally prevalent.20 Human papillomavirus-positive tumors are associated with PIK3CA hotspot mutations.19,20 HNSCC tumors harboring multiple aberrations in the PI3K pathway are linked to advanced disease, suggesting genomic instability contributions to disease progression.19
Gynecologic Cancers
PI3K pathway activation occurs in up to 30% of ovarian cancers, mainly due to PIK3CA or AKT mutation or amplification.21,22 Although it is less common in ovarian cancers, PTEN loss of heterozygosity occurs in up to 45% of the endometrioid subgroup.22 INPP4B has been identified as a tumor suppressor in ovarian cancers, and loss of INPP4B protein correlates with reduced patient survival.3
In endometrial cancers, PIK3CA mutations and PTEN loss (through mutations or reduction of protein expression) occur in up to 30% and 55% of tumors, respectively.23 Endometrial cancers are highly complex, often exhibiting coexistent alterations in PTEN, PIK3CA, PIK3R1, and KRAS.24 Although this may contribute to the difficulty of treatment, PI3K pathway reliance may sensitize tumors to PI3K pathway inhibition.20
Colorectal Cancers
PI3K pathway alterations in colorectal cancers (CRCs) are dominated by PIK3CA and PTEN. Approximately 14% exhibit PIK3CA catalytic domain hotspot mutations and are associated with invasive, progressive disease.25 Amplification and overexpression of PIK3CB (p110β), and mutations in PIK3R1 (p85α) have been observed.25 Loss of PTEN protein occurs in 20% to 40% of cases.25 The AKT E17K mutation has only been observed in 6% of CRCs, and the functional consequences of AKT kinase domain mutations are poorly understood.25 Mutations in INPP4B have been identified.26
Glioblastoma Multiforme
PI3K pathway alterations occur in 50% of glioblastoma multiforme (GBM) tumors and are associated with poor survival.27 Mutations in PTEN occur in 5% to 40% of GBM tumors, whereas loss of heterozygosity occurs in 60% to 80% of all cases.28 PIK3CA and PIK3R1 are mutated in approximately 10% and approximately 8% of tumors, respectively,27 contributing to tumor invasion and migration.29 AKT mutations have not been observed, and there is limited evidence that other PI3K pathway nodes play significant roles.30,31
Prostate Cancers
PI3K pathway activation is associated with metastasis, resistance to therapy, and poor outcome in patients with prostate cancer.32 In one study, 49% of patients with metastatic, castration-resistant prostate cancer harbored somatic alterations in the PI3K pathway, including biallelic loss of PTEN, hotspot mutations, amplifications and activating fusions in PIK3CA, and E17K-activating mutations in AKT1.33 PTEN alterations include deletions and inactivating mutations in approximately 15% of primary tumors and in 50% in hormone-refractory disease.34 Loss of PTEN protein occurs in approximately 20% of localized tumors and is correlated with advanced stage and high Gleason score.34 In contrast, PIK3CA alterations occur in only 6% of primary tumors and in 16% of metastases, although the regulatory subunit PIK3R1 is mutated in 22% of primary and 58% of secondary tumors, providing an alternative route to pathway activation.35 PIK3CB mutations have been observed in PTEN-deficient metastatic cases.33 Tumor suppressor INPP4B has been shown to be altered or downregulated in primary and metastatic disease.35,36 The net result of these diverse alterations is activation of the PI3K/AKT/mTOR pathway, leading to increased tumor growth, survival, and resistance to targeted therapies.
THE ROLE OF PI3K PATHWAY ACTIVATION IN TREATMENT RESISTANCE
PI3K pathway activation is implicated in de novo and acquired treatment resistance in various tumor types treated with targeted therapies (Fig 2).37 Genetic resistance mechanisms can arise through genomic instability.38 Resistance of tumor-initiating cells to apoptosis and epigenetic mechanisms may also contribute to relapse after therapy.39,40 Genetic and epigenetic mechanisms are associated with PI3K pathway activation.37 A large-scale RNA interference screen in HER2-positive breast cancer found that loss of the PTEN transcript conferred trastuzumab resistance.41 Oncogenic mutations in PIK3CA also conferred trastuzumab resistance in vitro, and activation of the PI3K pathway predicted poor trastuzumab response.41 In addition, patients with HER2-positive breast cancer whose tumors harbor PIK3CA-activating mutations derive less benefit from neoadjuvant HER2-targeted therapies than patients without a PIK3CA-activating mutation.42 In KRAS-mutant CRC cell lines, PIK3CA and PTEN mutations were associated with resistance to MEK inhibition.43 Moreover, PI3K pathway activation promotes resistance to BRAF inhibitors in BRAF-mutant melanoma, with 22% of progressive tumors harboring mutations that upregulate PI3K pathway activity.44 Loss of PTEN expression has been associated with reduced number and function of tumor-infiltrating T cells and resistance to anti-PD-1 immunotherapy in patients with melanoma.45 These findings have raised the possibility that targeted PI3K pathway inhibitors could potentially restore sensitivity to existing treatments.
Fig 2.

Common nodes of resistance to targeted therapies within the PI3K pathway. PI3K pathway alterations that confer resistance to targeted therapies across various tumor types are shown. CRC, colorectal cancer; EGFR, epidermal growth factor receptor; HER2, human epidermal growth factor receptor 2; HR, hormone receptor; INPP4B, inositol polyphosphate 4-phosphatase type II; MET, hepatocyte growth factor receptor; mTORC, mammalian target of rapamycin complex; PDK1, phosphoinositide-dependent kinase 1; PI3K, phosphoinositide 3-kinase; PIK3CA, phosphatidylinositol 3-kinase catalytic subunit alpha; PIK3CG, phosphatidylinositol 3-kinase catalytic subunit gamma; PIK3R2, phosphatidylinositol 3-kinase regulatory subunit beta; PIP2, phosphatidylinositol 4,5- bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PTEN, phosphatase and tensin homolog; RTK, receptor tyrosine kinase.
INHIBITORS OF THE PI3K PATHWAY
The PI3K pathway is dysregulated in many solid tumors, supporting the use of PI3K pathway inhibitors in the clinic. Oncogenic PI3K pathway activation is achieved in different (and potentially redundant) ways, requiring rational, tailored strategies to inhibit appropriate pathway nodes in each tumor type. Recently developed small-molecule inhibitors are presented in Tables 1 and 2, and Figure 3.
Table 1.
PI3K Pathway Inhibitor Potencies

Table 2.
AKT Inhibitor Potencies
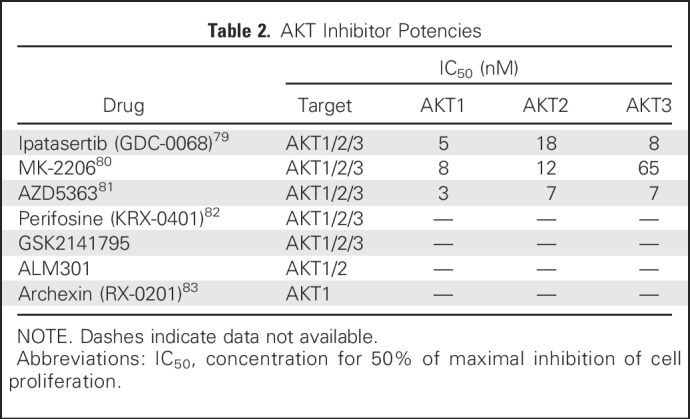
Fig 3.

Targeting the PI3K pathway in cancer with small-molecule inhibitors. Inhibitors discussed in the text are included in the figure. IGF-1R, insulin-like growth factor 1 receptor; InsR, insulin receptor; IRS1, insulin receptor substrate 1; mTORC, mammalian target of rapamycin complex; PDK1, phosphoinositide-dependent kinase 1; PI3K, phosphoinositide 3-kinase; RTK, receptor tyrosine kinase.
Pan-Class I PI3K Inhibitors
Pan-class I PI3K inhibitors target all four isoforms of p110. Buparlisib (BKM120), the most advanced agent in this class, inhibits all class I PI3K isoforms, with little activity toward other classes of PI3K or mTOR, and has demonstrated strong antiproliferative activity in more than 400 cancer cell lines (data on file; Novartis, Basel, Switzerland).46 Given its ability to penetrate the blood-brain barrier, it may represent a therapeutic option for GBM tumors or brain metastases, as shown in early-phase trials in refractory GBM84,85 and breast cancer-derived brain metastases.86 Buparlisib and letrozole have demonstrated antitumor activity in HR-positive breast cancer.87
Pictilisib (GDC-0941) is being investigated in HER2-positive metastatic breast cancer and advanced NSCLC. It has equipotent activity in vitro against p110α and -δ isoforms, and also inhibits p110β and -γ.47 Phase I studies demonstrated signs of clinical activity in advanced solid tumors.88,89
Copanlisib (BAY 80-6946) is an intravenously administered pan-class I PI3K inhibitor.48 It inhibits p110α and -δ, and, therefore, may suit T-cell malignancies; although, interestingly, concomitant p110δ inhibition in solid tumors may contribute to an immune environment that facilitates cytotoxic T-cell responses in addition to the cell-intrinsic antiproliferative effects of p110α inhibition.90
Isoform-Specific PI3K Inhibitors
The rationale for PI3K isoform-specific inhibition was validated in p110δ-driven hematologic malignancies by combined idelalisib and rituximab treatment.91 In solid tumors, isoform-specific PI3K inhibitors might have fewer toxicities compared with pan-PI3K inhibitors, allowing higher doses and resulting in more complete inhibition.1 Alpelisib (BYL719) was the first potent, p110α-selective inhibitor. A first-in-human phase I study in patients with advanced solid tumors demonstrated a manageable safety profile and antitumor activity, notably in patients with PIK3CA-mutant HR-positive breast cancer.92 Alpelisib has shown preliminary efficacy in combination with cetuximab in patients with recurrent or metastatic HNSCC.93
Taselisib (GDC-0032) inhibits p110α, -γ, and -δ equally, and p110β with 30-fold lower potency.51 Greater isoform selectivity is predicted to translate into improved efficacy in PIK3CA-mutant-driven tumors compared with pan-PI3K inhibitors. Preliminary results have shown activity in a PIK3CA-mutant xenograft model,94 as well as in PIK3CA-mutant HER2-positive and HR-positive breast tumors.95-97 Taselisib combined with letrozole has shown activity in PIK3CA-mutant breast cancers.98
The rationale for p110β inhibition is less straightforward than p110α. Although p110β might be a valid target in some tumors exhibiting PTEN loss, preclinical studies suggest that p110α and p110β have overlapping roles. The success of p110β inhibition may depend on the absence of concomitant p110α-activating mutations.90,99 Selective inhibitors of p110β that have undergone phase I investigation include GSK2636771 and SAR260301.59,100
DUAL-SPECIFICITY PI3K/mTOR INHIBITORS
Dual-specificity inhibitors were designed to inhibit the structurally similar PI3K and mTOR kinase domains simultaneously and have the benefit of interrupting the feedback loop that activates AKT when mTORC1 is inhibited in isolation (Fig 3).101 Compounds in this class include BEZ235, GDC-0980, and SAR245409 (XL765).
Although phase I studies initially reported clinical activity,102,103 subsequent studies revealed that controlling bioavailability and limiting toxicities are challenging. For example, issues with dosing and bioavailability of BEZ235 led to the development of different oral formulations,102 and the phase II FERGI trial (GDC-0980 with fulvestrant in breast cancer) was stopped because of toxicity concerns.
mTOR Inhibitors: Rapalogs
mTOR inhibitors were the first agents developed to target the PI3K pathway. The earliest compound in this class, rapamycin (sirolimus), inhibits the activity of mTORC1 but not mTORC2.104 Limitations in the pharmacokinetic (PK) properties of sirolimus led to the development of analogs (called “rapalogs”) with improved characteristics, including temsirolimus, everolimus, and ridaforolimus.104 Both temsirolimus and everolimus were approved for use in renal cell carcinoma, highlighting PI3K pathway activation importance in this setting.104 Improvements in sirolimus PK continue to be investigated, with a nanoparticle albumin-bound formulation in development.70
mTOR Inhibitors: Catalytic
Catalytic mTOR inhibitors improve on rapalogs by inhibiting both mTORC1 and mTORC2, thus suppressing the feedback-mediated activation of AKT by mTORC2.105 In a phase I trial, AZD2014 shows signs of clinical activity in advanced solid tumors,106 and CC-223 shows indications of activity in patients with neuroendocrine tumors, NSCLC, GBM, and hepatocellular carcinoma.107,108 Trials of MLN0128 in advanced prostate cancer and GBM are ongoing.109,110
AKT Inhibitors
As one of the key effector nodes in the PI3K pathway, AKT could be a promising target in PI3K pathway-activated tumors. Pan-AKT inhibitors under development are either allosteric (MK-2206) or adenosine triphosphate (ATP)-competitive (AZD5363, ipatasertib [GDC-0068]). MK-2206, AZD5363, and ipatasertib have shown preliminary activity in phase I trials, and are being tested in a range of solid tumors.111-114 Two more ATP-competitive AKT inhibitors, afuresertib (GSK2110183) and GSK2141795, have undergone clinical investigation.115,116
p70S6 Kinase Inhibitors
p70S6K is activated downstream of AKT and regulates translation by phosphorylating ribosomal protein S6. p70S6K amplification confers a proliferative advantage on tumor cells, is correlated with poor prognosis and reduced survival,117 and is, therefore, under investigation as a drug target in several phase I clinical trials.118,119
CHALLENGES FOR PI3K INHIBITOR DEVELOPMENT
Recognition of the PI3K pathway’s contributions to tumorigenesis has stimulated the development of numerous targeted agents (Table 1); however, the efficacy of monotherapy inhibition has been disappointing.90 Insufficient target inhibition, due to toxicity or suboptimal dosing schedules, represents one potential explanation.1 Evaluating alternative dosing schedules may help overcome toxicities and enable maximal target inhibition.90 Incorporation of pharmacodynamic assessments of validated, robust biomarkers into clinical trials is needed to identify patients who may derive therapeutic benefit.90,120,121
The limited clinical efficacy with these agents may also result from a feedback response whereby PI3K pathway inhibition stimulates compensatory activation of a complementary pathway such as growth factor receptor signaling, bypassing the effects of targeted blockade.90 Data suggest that PTEN loss represents a convergent evolutionary mechanism of treatment resistance.122 Schwartz et al showed that p110α activity was suppressed in PTEN-mutant tumors, and that PI3K signaling was instead driven by p110β.123 However, p110β inhibition only transiently inhibited AKT/mTOR signaling due to feedback inhibition of insulin-like growth factor 1 receptor and other receptors, resulting in p110α activation and rebound downstream signaling.123 Combined p110α and p110β inhibition suppressed this rebound effect.123 In PTEN knockdown models, PTEN loss contributed to alpelisib resistance, an effect that was reversed by concurrent p110α and p110β inhibition.122 Similarly, prolonged treatment with pictilisib resulted in PTEN loss, leading to the development of pictilisib resistance in CRC cell lines, which was overcome by concurrent PI3K and MAPK inhibition.124
Another explanation for the lack of pan-PI3K inhibitor single-agent activity stems from the relative lack of p110β inhibition in vitro.1 Residual p110β activity during pan-PI3K inhibitor treatment may provide sufficient downstream PI3K signaling for continued growth, particularly in PTEN-deficient tumors where p110β is the major isoform mediating tumorigenesis.1
Similarly, isoform-selective inhibitors may not achieve sustained benefit due to rebound activation of the uninhibited isoforms, which has been observed in luminal breast cancer cell lines harboring PIK3CA or HER2 amplifications treated with alpelisib,125 and in PTEN-mutated breast and prostate tumors treated with AZD8186. In these models, concomitant p110α and p110β inhibition had a synergistic antitumor effect, similar to that reported by Schwartz et al.123,125 Interestingly, p110β-mediated PI3K pathway reactivation in PIK3CA-mutated breast cancer cells occurred independently of AKT activation, indicating that other signals downstream of PI3K might contribute to lack of efficacy in these cells.125 Indeed, many PIK3CA-mutant cancer cell lines and human breast tumors appear to rely on activation of PDK1 and its substrate, serum/glucocorticoid regulated kinase 3 (SGK3), rather than AKT, for growth and tumorigenicity.126 These findings demonstrate that a variety of feedback mechanisms can reactivate the PI3K pathway in response to PI3K inhibitor treatment.
Additional mechanisms of resistance have been investigated. Cell lines sensitive to alpelisib treatment were associated with complete inhibition of TORC1 pathway signaling, while resistant cell lines had persistently active mTORC1; combining alpelisib with everolimus overcame resistance in vitro and in vivo.127 In the clinic, pS6 (a biomarker of mTORC1 signaling) was expressed in tumors that initially responded and subsequently progressed.127 A combinatorial screen demonstrated the efficacy of the cyclin-dependent kinase (CDK) 4/6 inhibitor ribociclib (LEE011) combined with alpelisib, particularly in tumors with phosphorylated retinoblastoma protein (pRB).128 Notably, CDK4/6 inhibition emerged as the strongest sensitizer for PI3K inhibition, exerting its activity by binding and activating cyclin D1, whose expression is often regulated by TORC1.128-130 Other resistance mechanisms include MYC overexpression or amplification, matrix-associated resistance, and activity of ribosomal S6 kinase 3/4.48,128,131-133 CDK4/6 and mTOR inhibitors should be tested in the clinic to understand feedback alteration of various pathways and provide novel strategies in response to treatment resistance. Improved clinical responses might be achieved by combining PI3K inhibitors with other agents targeting known resistance pathways that interact with PI3K, such as the estrogen receptor pathway.134
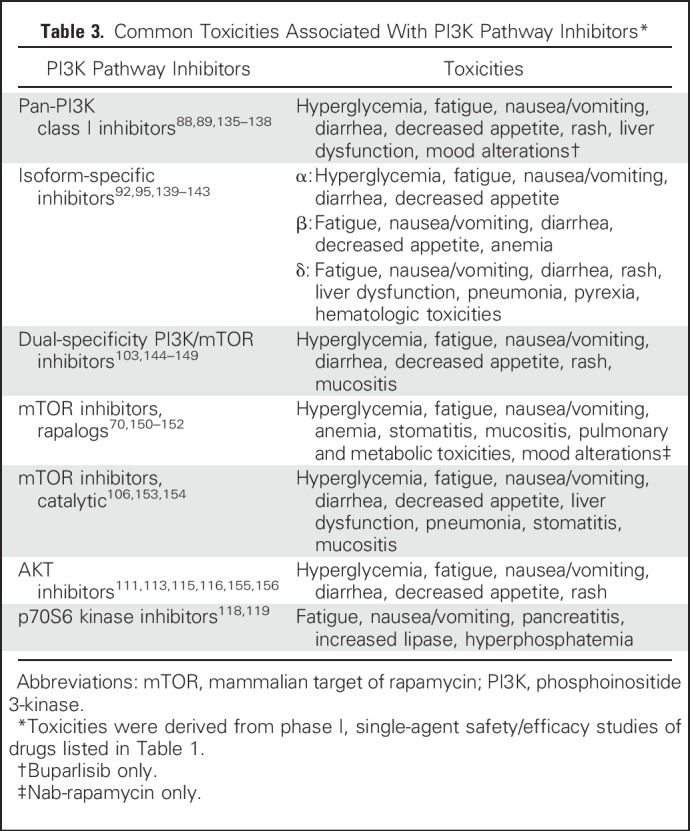
MANAGEMENT OF CLASS-SPECIFIC TOXICITIES
Toxicity profiles of PI3K pathway inhibitors are related to their mechanism of action and have become more favorable with the advent of second-generation agents (Table 3).157 Managing and minimizing toxicity risks, particularly in susceptible patients (eg, hyperglycemia in diabetic patients), are important considerations when designing treatment strategies. Several management guidelines for commonly encountered toxicities have been published and may be helpful to clinicians using this class of agent, namely those for hyperlipidemia, hyperglycemia, rash, stomatitis, and noninfectious pneumonitis.158-161
Table 3.
Common Toxicities Associated With PI3K Pathway Inhibitors*
BIOMARKERS FOR THERAPY SELECTION
The promise of personalized therapy depends on the availability of predictive biomarkers for treatment response. Despite preclinical evidence that PIK3CA-mutant cell lines and tumors are sensitive to PI3K inhibition,121 correlations between molecular status and clinical efficacy are less clear. Retrospective analyses of patients suggest that PIK3CA H1047R correlates with response,162,163 although in the phase I trial of alpelisib with letrozole in HR-positive breast cancer, clinical responses were also seen in PIK3CA wild-type patients.121,164 Although this pathway is important, other factors are probably involved in determining sensitivity to PI3K pathway inhibition.
Work must continue to validate biomarkers to inform clinical treatment decisions, including factors predictive of treatment response or resistance. As an example, a phase II trial of alpelisib or buparlisib with letrozole is ongoing in patients with confirmed PIK3CA mutation status.165 Strategies to monitor the status of other biomarkers, such as pRB, will be important for combination regimens.128
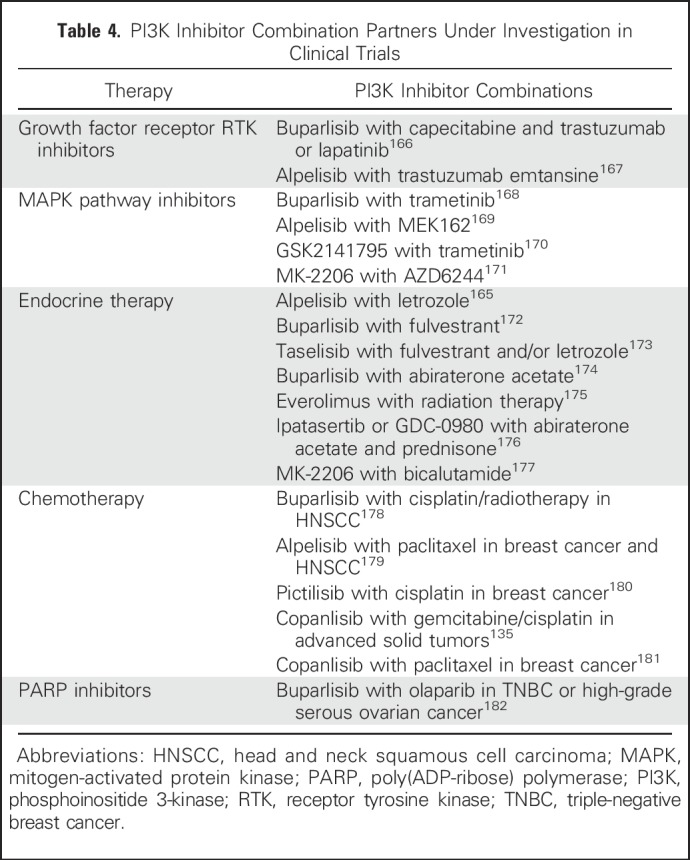
COMBINATION THERAPY STRATEGIES
Single-agent activity of pan-PI3K inhibitors has been limited. Isoform-specific p110δ (idelalisib) and p110α inhibitors (alpelisib, taselisib) have had greater success, and will be important as combination partners to maximize therapeutic benefit. A key challenge is developing effective drug combinations with complementary modes of action, targeting the most relevant pathways in each tumor type (Table 4).
Table 4.
PI3K Inhibitor Combination Partners Under Investigation in Clinical Trials
Combinations With Growth Factor Receptor Inhibitors
Growth factor receptors are common nodes of oncogenic alteration; for example, HER2 amplification is frequently observed in breast cancer.183 Inhibition of the growth factor receptor has several advantages: By blocking initiation of signaling and cross-talk with complementary pathways, tumor cells are sensitized to treatment with chemotherapy and radiotherapy.183 As examples, combinations of buparlisib with paclitaxel and trastuzumab in HER2-positive breast cancer184 and alpelisib with cetuximab in HNSCC93 have shown early indications of antitumor activity, supporting the rationale for this combination approach.
Combinations With Mitogen-Activated Protein Kinase Inhibitors
The PI3K and mitogen-activated protein kinase (MAPK) pathways interact at multiple levels, providing a potential escape mechanism for tumors treated with single-agent PI3K or MAPK pathway inhibitors, suggesting that simultaneous inhibition may be required to achieve adequate tumor control.185 In support of this, a retrospective analysis of patients receiving PI3K and MAPK pathway inhibitors in phase I trials showed combination treatment can improve efficacy relative to single-pathway inhibition, albeit with increased toxicity that can limit treatment exposure.186 Examples include combinations of copanlisib and refametinib, buparlisib and vemurafenib, or pictilisib and GDC-0973.187-189
In phase I trials, promising efficacy signals have been observed in KRAS- or BRAF-mutant tumors treated with combinations of pictilisib and GDC-0973,187 SAR245409 and pimasertib,190 and buparlisib and trametinib.191 Likely indications for PI3K and MAPK inhibitor combinations will include solid tumors reliant on MAPK pathway activation, such as BRAF- and KRAS-mutant melanoma, CRC, and ovarian cancer. Continued investigation of combinations of agents from both of these pathways will be necessary, because toxicity has delayed the development of these regimens.
Combinations With Endocrine Therapy
Aberrant hormone receptor signaling plays an important role in HR-positive breast and prostate cancers. Patients who initially respond to endocrine therapy often develop resistance and progress. The PI3K pathway is activated in approximately 35% of HR-positive breast cancers,7 and PI3K pathway blockade can restore sensitivity to endocrine therapy in breast cancer cell lines.192 Findings that estrogen deprivation triggers apoptosis in PI3K pathway-inhibited cells provide further rationale for combination therapy, perhaps even as a first-line option.193 Final results of the BOLERO-2 (Breast Cancer Trials of Oral Everolimus-2) phase III trial of everolimus in HR-positive breast cancer showed the everolimus combination was more effective than exemestane alone.194
Likewise, PI3K pathway alterations are prevalent in prostate cancer, contributing to the development of castration-resistant prostate cancer.32 Cross-talk between PI3K and androgen receptor pathways suggests combined inhibition may provide improved benefit over hormone therapy alone,195 and preclinical studies show p110β inhibition and hormone therapy have synergistic antitumor activity.196
COMBINATIONS WITH CHEMOTHERAPY
Preclinical studies suggest that PI3K pathway inhibitors might sensitize tumors to chemotherapy by altering surrounding vasculature and tumor perfusion,197 thereby increasing exposure to systemic therapies and synergistically inducing apoptosis.198-200 Early clinical studies showed PI3K pathway inhibitors were well tolerated with chemotherapy: Antitumor activity was observed in patients with NSCLC treated with pictilisib, carboplatin, and paclitaxel, with or without bevacizumab.201 Preliminary positive results of ipatasertib with chemotherapy have supported the initiation of a phase II trial in gastric cancers.202
Combination With Poly(ADP-Ribose) Polymerase Inhibitors
Targeting the connection between the PI3K pathway and DNA repair is emerging as a therapeutic strategy in BRCA1-deficient tumors, based on findings that PI3K pathway inhibitors increase DNA damage and sensitize cell lines to poly(ADP-ribose) polymerase inhibitors.203,204 A phase I study of buparlisib with olaparib is ongoing in patients with TNBC or high-grade serous ovarian cancer.182,205 Results from these trials will lead to a better understanding of this combination, potentially lending insight into strategies to overcome resistance.
CHALLENGES FOR COMBINATION THERAPY
Overlapping toxicity profiles with many PI3K inhibitor-containing combinations is challenging, often preventing sufficient dose administration to achieve the necessary exposure of each drug.187 A notable exception is the reduction in hyperproliferative cutaneous events for combined MEK and BRAF inhibition, which is a mechanistic effect of suppressing paradoxical MAPK pathway activation.206 However, PI3K inhibitor combinations generally result in cumulative, nonspecific toxicities, and the most tolerable dose ratios and drug sequences will have to be found empirically.
Toxicities need to be closely monitored, adjusting drug doses appropriately to prolong treatment of as long as possible. Compared with monotherapy, toxicity management for combination regimens could be increasingly complex.
In conclusion, the importance of the PI3K pathway in solid tumors is well established; however, treatments with single-agent PI3K inhibitors have been disappointing. Several questions must be investigated to guide the design of effective treatment regimens. First, which PI3K pathway components contribute the most to particular tumor types? Second, how do different pathways interact to support tumor growth, and how should they be targeted (individually or in tandem, and at what potencies)?
Finally, can treatment strategies be designed to respond to the emergence of drug resistance? These questions will be addressed by continued investigation through intelligently designed, biomarker-driven clinical trials, using treatment combinations tailored toward defined molecular alterations.
ACKNOWLEDGMENT
Ana Maria Gonzalez-Angulo contributed to content discussions of this manuscript and provided an initial draft of Figure 1. Novartis Pharmaceuticals provided financial support for medical editorial assistance. I thank Nirmal Jethwa, PhD, for medical editorial assistance.
Footnotes
Supported by National Cancer Institute Grant No. UM1 CA186689.
Author’s disclosures of potential conflicts of interest are found in the article online at www.jco.org.
AUTHOR’S DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Inhibition of the PI3K/AKT/mTOR Pathway in Solid Tumors
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Patricia Mucci LoRusso
Consulting or Advisory Role: Alexion Pharmaceuticals, Ariad, Celgene, Genentech, Novartis, Pfizer
Research Funding: Abbvie (Inst), Alexion Pharmaceuticals (Inst), Genentech (Inst), Incyte (Inst), Novartis (Inst), Tensha (Inst), Stemline Therapeutics (Inst)
REFERENCES
- 1.Dienstmann R, Rodon J, Serra V, et al. Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 2014;13:1021–1031. doi: 10.1158/1535-7163.MCT-13-0639. [DOI] [PubMed] [Google Scholar]
- 2.Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28:1075–1083. doi: 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agoulnik IU, Hodgson MC, Bowden WA, et al. INPP4B: The new kid on the PI3K block. Oncotarget. 2011;2:321–328. doi: 10.18632/oncotarget.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun T, Aceto N, Meerbrey KL, et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell. 2011;144:703–718. doi: 10.1016/j.cell.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 6.Baselga J. Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. Oncologist. 2011;16(suppl 1):12–19. doi: 10.1634/theoncologist.2011-S1-12. [DOI] [PubMed] [Google Scholar]
- 7.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084–6091. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gewinner C, Wang ZC, Richardson A, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16:115–125. doi: 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carpten JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 11.Wu M-Q, Hu P, Gao J, et al. Low expression of tyrosine-protein phosphatase nonreceptor type 12 is associated with lymph node metastasis and poor prognosis in operable triple-negative breast cancer. Asian Pac J Cancer Prev. 2013;14:287–292. doi: 10.7314/apjcp.2013.14.1.287. [DOI] [PubMed] [Google Scholar]
- 12.Cooper WA, Lam DCL, O’Toole SA, et al. Molecular biology of lung cancer. J Thorac Dis. 2013;5(suppl 5):S479–S490. doi: 10.3978/j.issn.2072-1439.2013.08.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. doi: 10.1038/nature11404. Cancer Genome Atlas Research Network: Comprehensive genomic characterization of squamous cell lung cancers [Erratum: Nature 491:288, 2012]. Nature 489:519-525, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarris EG, Saif MW, Syrigos KN. The biological role of PI3K pathway in lung cancer. Pharmaceuticals (Basel) 2012;5:1236–1264. doi: 10.3390/ph5111236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Massion PP, Taflan PM, Shyr Y, et al. Early involvement of the phosphatidylinositol 3-kinase/Akt pathway in lung cancer progression. Am J Respir Crit Care Med. 2004;170:1088–1094. doi: 10.1164/rccm.200404-487OC. [DOI] [PubMed] [Google Scholar]
- 16.Malanga D, Scrima M, De Marco C, et al. Activating E17K mutation in the gene encoding the protein kinase AKT1 in a subset of squamous cell carcinoma of the lung. Cell Cycle. 2008;7:665–669. doi: 10.4161/cc.7.5.5485. [DOI] [PubMed] [Google Scholar]
- 17.Marsit CJ, Zheng S, Aldape K, et al. PTEN expression in non-small-cell lung cancer: Evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum Pathol. 2005;36:768–776. doi: 10.1016/j.humpath.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 18.Stjernström A, Karlsson C, Fernandez OJ, et al. Alterations of INPP4B, PIK3CA and pAkt of the PI3K pathway are associated with squamous cell carcinoma of the lung. Cancer Med. 2014;3:337–348. doi: 10.1002/cam4.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lui VWY, Hedberg ML, Li H, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013;3:761–769. doi: 10.1158/2159-8290.CD-13-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vander Broek R, Mohan S, Eytan D, et al. The PI3K/Akt/mTOR axis in head and neck cancer: functions, aberrations, cross-talk, and therapies. Oral Dis. 2013;21:815–825. doi: 10.1111/odi.12206. [DOI] [PubMed] [Google Scholar]
- 21.Banerjee S, Kaye SB. New strategies in the treatment of ovarian cancer: Current clinical perspectives and future potential. Clin Cancer Res. 2013;19:961–968. doi: 10.1158/1078-0432.CCR-12-2243. [DOI] [PubMed] [Google Scholar]
- 22.Carden CP, Stewart A, Thavasu P, et al. The association of PI3 kinase signaling and chemoresistance in advanced ovarian cancer. Mol Cancer Ther. 2012;11:1609–1617. doi: 10.1158/1535-7163.MCT-11-0996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: Variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weigelt B, Warne PH, Lambros MB, et al. PI3K pathway dependencies in endometrioid endometrial cancer cell lines. Clin Cancer Res. 2013;19:3533–3544. doi: 10.1158/1078-0432.CCR-12-3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, Roberts TM, Shivdasani RA. Targeting PI3K signaling as a therapeutic approach for colorectal cancer. Gastroenterology. 2011;141:50–61. doi: 10.1053/j.gastro.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 26.Shanmugam V, Ramanathan RK, Lavender NA, et al. Whole genome sequencing reveals potential targets for therapy in patients with refractory KRAS mutated metastatic colorectal cancer. BMC Med Genomics. 2014;7:36. doi: 10.1186/1755-8794-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carico C, Nuño M, Mukherjee D, et al. Loss of PTEN is not associated with poor survival in newly diagnosed glioblastoma patients of the temozolomide era. PLoS One. 2012;7:e33684. doi: 10.1371/journal.pone.0033684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weber GL, Parat M-O, Binder ZA, et al. Abrogation of PIK3CA or PIK3R1 reduces proliferation, migration, and invasion in glioblastoma multiforme cells. Oncotarget. 2011;2:833–849. doi: 10.18632/oncotarget.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson JP, Vanbrocklin MW, McKinney AJ, et al. Akt signaling is required for glioblastoma maintenance in vivo. Am J Cancer Res. 2011;1:155–167. [PMC free article] [PubMed] [Google Scholar]
- 31.Bleeker FE, Lamba S, Zanon C, et al. Absence of AKT1 mutations in glioblastoma. PLoS One. 2009;4:e5638. doi: 10.1371/journal.pone.0005638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bitting RL, Armstrong AJ. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr Relat Cancer. 2013;20:R83–R99. doi: 10.1530/ERC-12-0394. [DOI] [PubMed] [Google Scholar]
- 33. doi: 10.1016/j.cell.2015.05.001. Robinson D, Van Allen EM, Wu Y-M, et al: Integrative clinical genomics of advanced prostate cancer [Erratum: Cell 162:454, 2015]. Cell 161:1215-1228, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Majumder PK, Sellers WR. Akt-regulated pathways in prostate cancer. Oncogene. 2005;24:7465–7474. doi: 10.1038/sj.onc.1209096. [DOI] [PubMed] [Google Scholar]
- 35.Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hodgson MC, Shao LJ, Frolov A, et al. Decreased expression and androgen regulation of the tumor suppressor gene INPP4B in prostate cancer. Cancer Res. 2011;71:572–582. doi: 10.1158/0008-5472.CAN-10-2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burris HA., III Overcoming acquired resistance to anticancer therapy: Focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol. 2013;71:829–842. doi: 10.1007/s00280-012-2043-3. [DOI] [PubMed] [Google Scholar]
- 38.Burrell RA, Swanton C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol Oncol. 2014;8:1095–1111. doi: 10.1016/j.molonc.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frank NY, Schatton T, Frank MH. The therapeutic promise of the cancer stem cell concept. J Clin Invest. 2010;120:41–50. doi: 10.1172/JCI41004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Obenauf AC, Zou Y, Ji AL, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 2015;520:368–372. doi: 10.1038/nature14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 42. Majewski IJ, Nuciforo P, Mittempergher L, et al: PIK3CA mutations are associated with decreased benefit to neoadjuvant human epidermal growth factor receptor 2–targeted therapies in breast cancer. J Clin Oncol 33:1334-1339, 2015. [DOI] [PMC free article] [PubMed]
- 43.Wee S, Jagani Z, Xiang KX, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69:4286–4293. doi: 10.1158/0008-5472.CAN-08-4765. [DOI] [PubMed] [Google Scholar]
- 44.Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peng W, Chen JQ, Liu C, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6:202–216. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maira S-M, Pecchi S, Huang A, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11:317–328. doi: 10.1158/1535-7163.MCT-11-0474. [DOI] [PubMed] [Google Scholar]
- 47.Folkes AJ, Ahmadi K, Alderton WK, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51:5522–5532. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- 48.Liu N, Rowley BR, Bull CO, et al. BAY 80-6946 is a highly selective intravenous PI3K inhibitor with potent p110α and p110δ activities in tumor cell lines and xenograft models. Mol Cancer Ther. 2013;12:2319–2330. doi: 10.1158/1535-7163.MCT-12-0993-T. [DOI] [PubMed] [Google Scholar]
- 49. Edelman G, Bedell C, Shapiro G, et al: A phase I dose-escalation study of XL147 (SAR245408), a PI3K inhibitor administered orally to patients (pts) with advanced malignancies. J Clin Oncol 28, 2010 (suppl; abstr 3004) http://meetinglibrary.asco.org/content/53257-74.
- 50.Ihle NT, Paine-Murrieta G, Berggren MI, et al. The phosphatidylinositol-3-kinase inhibitor PX-866 overcomes resistance to the epidermal growth factor receptor inhibitor gefitinib in A-549 human non-small cell lung cancer xenografts. Mol Cancer Ther. 2005;4:1349–1357. doi: 10.1158/1535-7163.MCT-05-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ndubaku CO, Heffron TP, Staben ST, et al. Discovery of 2-3-[2-(1-isopropyl-3-methyl-1H-1,2-4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl-2-methylpropanamide (GDC-0032): A β-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity. J Med Chem. 2013;56:4597–4610. doi: 10.1021/jm4003632. [DOI] [PubMed] [Google Scholar]
- 52.Fritsch C, Huang A, Chatenay-Rivauday C, et al. Characterization of the novel and specific PI3Kα inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014;13:1117–1129. doi: 10.1158/1535-7163.MCT-13-0865. [DOI] [PubMed] [Google Scholar]
- 53. Jessen K, Kessler L, Kucharski J, et al: A potent and selective PI3K inhibitor, INK1117, targets human cancers harboring oncogenic PIK3CA mutations. Mol Cancer Ther 10, 2011(abstr A171)
- 54.So L, Yea SS, Oak JS, et al. Selective inhibition of phosphoinositide 3-kinase p110α function. J Biol Chem. 2013;288:5718–5731. doi: 10.1074/jbc.M112.379446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu N, Scott WJ, Haegebarth A, et al: BAY 1082439, a highly selective and balanced PI3K α/β inhibitor demonstrated potent activity in tumors with activated PI3K and loss-of- function of PTEN. Cancer Res 72, 2012 (suppl; abstr 2799)
- 56.Tanaka H, Yoshida M, Tanimura H, et al. The selective class I PI3K inhibitor CH5132799 targets human cancers harboring oncogenic PIK3CA mutations. Clin Cancer Res. 2011;17:3272–3281. doi: 10.1158/1078-0432.CCR-10-2882. [DOI] [PubMed] [Google Scholar]
- 57. Rivero RA, Hardwicke MA: Identification of GSK2636771, a potent and selective, orally bioavailable inhibitor of phosphatidylinositol 3-kinase-beta (PI3K) for the treatment of PTEN deficient tumors. Cancer Res 72, 2012 (suppl; abstr 2913)
- 58. Hancox U, Cosulich S, Dry H, et al: AZD8186: a potent selective inhibitor of PI3K beta targeting PTEN-deficient tumours dependent on dysregulated PI3K beta signalling. Cancer Res 73, 2013 (suppl; abstr 3264)
- 59. Virone-Oddos A, Bonnevaux H, Lemaitre O, et al: Discovery and characterization of SAR260301, a novel PI3K beta-selective inhibitor in clinical development for the treatment of PTEN-deficient tumors. Cancer Res 73, 2013 (abstr 3258)
- 60.Lannutti BJ, Meadows SA, Herman SEM, et al. CAL-101, a p110δ selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117:591–594. doi: 10.1182/blood-2010-03-275305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dunbar J, Nevejans J, McKee C, et al: Pharmacokinetics and pharmacodynamics of IPI- 145, a potent inhibitor of phosphoinositide-3-kinase-{delta},{gamma}, following single- and multiple-dose administration in healthy subjects and patients with advanced hematologic malignancies. Blood 120:4853, 2012. [Google Scholar]
- 62. Sinclair A, Metz D, Cushing T, et al: Phosphatidylinositol-3 kinase delta (PI3K{delta}) inhibitor AMG 319 is a potent, selective and orally bioavailable small molecule inhibitor that suppresses PI3K-mediated signaling and viability in neoplastic B cells. Blood 118:4964, 2011. [Google Scholar]
- 63.Maira S-M, Stauffer F, Brueggen J, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 64.Sutherlin DP, Bao L, Berry M, et al. Discovery of a potent, selective, and orally available class I phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) kinase inhibitor (GDC-0980) for the treatment of cancer. J Med Chem. 2011;54:7579–7587. doi: 10.1021/jm2009327. [DOI] [PubMed] [Google Scholar]
- 65.Mallon R, Feldberg LR, Lucas J, et al. Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res. 2011;17:3193–3203. doi: 10.1158/1078-0432.CCR-10-1694. [DOI] [PubMed] [Google Scholar]
- 66.Yuan J, Mehta PP, Yin M-J, et al. PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol Cancer Ther. 2011;10:2189–2199. doi: 10.1158/1535-7163.MCT-11-0185. [DOI] [PubMed] [Google Scholar]
- 67.Knight SD, Adams ND, Burgess JL, et al. Discovery of GSK2126458, a highly potent inhibitor of PI3K and the mammalian target of rapamycin. ACS Med Chem Lett. 2010;1:39–43. doi: 10.1021/ml900028r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Brana I, LoRusso P, Baselga J, et al: A phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics of XL765 (SAR245409), a PI3K/TORC1/TORC2 inhibitor administered orally to patients (pts) with advanced malignancies. J Clin Oncol 28, 2010 (suppl; abstr 3030)
- 69.Edwards SR, Wandless TJ. The rapamycin-binding domain of the protein kinase mammalian target of rapamycin is a destabilizing domain. J Biol Chem. 2007;282:13395–13401. doi: 10.1074/jbc.M700498200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gonzalez-Angulo AM, Meric-Bernstam F, Chawla S, et al. Weekly nab-Rapamycin in patients with advanced nonhematologic malignancies: Final results of a phase I trial. Clin Cancer Res. 2013;19:5474–5484. doi: 10.1158/1078-0432.CCR-12-3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shor B, Zhang W-G, Toral-Barza L, et al: A new pharmacologic action of CCI-779 involves FKBP12-independent inhibition of mTOR kinase activity and profound repression of global protein synthesis. Cancer Res 68, 2008 (abstr 2943) [DOI] [PubMed]
- 72.Sedrani R, Cottens S, Kallen J, et al. Chemical modification of rapamycin: The discovery of SDZ RAD. Transplant Proc. 1998;30:2192–2194. doi: 10.1016/s0041-1345(98)00587-9. [DOI] [PubMed] [Google Scholar]
- 73.Rivera VM, Squillace RM, Miller D, et al. Ridaforolimus (AP23573; MK-8669), a potent mTOR inhibitor, has broad antitumor activity and can be optimally administered using intermittent dosing regimens. Mol Cancer Ther. 2011;10:1059–1071. doi: 10.1158/1535-7163.MCT-10-0792. [DOI] [PubMed] [Google Scholar]
- 74.Falcon BL, Barr S, Gokhale PC, et al. Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res. 2011;71:1573–1583. doi: 10.1158/0008-5472.CAN-10-3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pike KG, Malagu K, Hummersone MG, et al. Optimization of potent and selective dual mTORC1 and mTORC2 inhibitors: The discovery of AZD8055 and AZD2014. Bioorg Med Chem Lett. 2013;23:1212–1216. doi: 10.1016/j.bmcl.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 76.Hsieh AC, Liu Y, Edlind MP, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Apsel B, Blair JA, Gonzalez B, et al. Targeted polypharmacology: Discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol. 2008;4:691–699. doi: 10.1038/nchembio.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mortensen DS, Fultz KE, Hickman M, et al: The discovery and preclinical characterization of CC-223, a novel mTOR kinase inhibitor under clinical investigation. Eur J Cancer 48, 2012 (abstr 337)
- 79. Lin K: GDC-0068: A novel, selective, ATP-competitive inhibitor of Akt. Cancer Res 71, 2011 (abstr DDT02–01)
- 80. Yan L: MK-2206: A potent oral allosteric AKT inhibitor. Proc Am Assoc Cancer Res 69, 2009 (abstr DDT01–1) [Google Scholar]
- 81.Davies BR, Greenwood H, Dudley P, et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther. 2012;11:873–887. doi: 10.1158/1535-7163.MCT-11-0824-T. [DOI] [PubMed] [Google Scholar]
- 82.Gills JJ, Dennis PA. Perifosine: Update on a novel Akt inhibitor. Curr Oncol Rep. 2009;11:102–110. doi: 10.1007/s11912-009-0016-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yoon H, Kim DJ, Ahn EH, et al. Antitumor activity of a novel antisense oligonucleotide against Akt1. J Cell Biochem. 2009;108:832–838. doi: 10.1002/jcb.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wen PY, Yung WKA, Mellinghoff IK, et al. Phase II trial of the phosphatidyinositol-3 kinase (PI3K) inhibitor buparlisib (BKM120) in recurrent glioblastoma conducted by the Ivy Foundation Early Phase Clinical Trials Consortium. Neuro-oncol. 2014;16:iii47–iii52. [Google Scholar]
- 85. Shih KC, Acs P, Burris HA, et al: Phase I study of the combination of BKM120 and bevacizumab in patients with relapsed/refractory glioblastoma multiforme (GBM) or other refractory solid tumors. J Clin Oncol 31, 2013 (suppl: abstr e13045)
- 86. Maira M, Schnell C, Lollini P, et al: Preclinical and preliminary clinical activity of NVP- BKM120, an oral pan-class I PI3K inhibitor, in the brain. ESMO Conference, Vienna, Austria, September 20, 2012 (abstr) [Google Scholar]
- 87. Mayer IA, Abramson VG, Balko JM, et al: SU2C phase Ib study of pan-PI3K inhibitor BKM120 with letrozole in ER+/HER2-metastatic breast cancer (MBC). J Clin Oncol 30, 2012 (suppl; abstr 510)
- 88. Garcia VM, Baird RD, Shah KJ, et al: A phase I study evaluating GDC-0941, an oral phosphoinositide-3 kinase (PI3K) inhibitor, in patients with advanced solid tumors or multiple myeloma. J Clin Oncol 29, 2011 (suppl; abstr 3021)
- 89. Von Hoff DD, LoRusso P, Demetri GD, et al: A phase I dose-escalation study to evaluate GDC-0941, a pan-PI3K inhibitor, administered QD or BID in patients with advanced or metastatic solid tumors. J Clin Oncol 29, 2011 (suppl; abstr 3052)
- 90.Fruman DA, Rommel C. PI3K and cancer: Lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370:997–1007. doi: 10.1056/NEJMoa1315226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Gonzalez-Angulo A, Juric D, Argilés G, et al: Safety, pharmacokinetics, and preliminary activity of the αspecific PI3K inhibitor BYL719: Results from the first-in-human study. J Clin Oncol 31, 2013 (suppl; abstr 2531)
- 93. Razak ARA, Ahn M-J, Yen C-J, et al: Phase Ib/II study of the PI3Kα inhibitor BYL719 in combination with cetuximab in recurrent/metastatic squamous cell cancer of the head and neck (SCCHN). J Clin Oncol 32, 2014 (5 suppl; abstr 6044)
- 94. Olivero AG, Heffron TP, Baumgardner M, et al: Discovery of GDC-0032: A beta-sparing PI3K inhibitor active against PIK3CA mutant tumors. Cancer Res 73, 2013 (abstr DDT02–01)
- 95.Juric D, Infante JR, Krop IE, et al. Evaluation of tolerability and anti-tumor activity of GDC-0032, a PI3K inhibitor with enhanced activity against PIK3CA mutant tumors, administered to patients with advanced solid tumors. Eur J Cancer. 2013;49(suppl 2):S168. [Google Scholar]
- 96. Juric D, Krop I, Ramanathan RK, et al: GDC-0032, a beta isoform-sparing PI3K inhibitor: Results of a first-in-human phase Ia dose escalation study. Cancer Res 73, 2013 (abstr LB–64)
- 97. Juric D, Argiles G, Burris H, et al: Phase I study of BYL719, an alpha-specific PI3K inhibitor, in patients with PIK3CA mutant advanced solid tumors: Preliminary efficacy and safety in patients with PIK3CA mutant ER-positive (ER+) metastatic breast cancer (MBC). Cancer Res 72, 2012 (abstr P6–10)
- 98. Saura C, Sachdev J, Patel MR, et al: Ph1b study of the PI3K inhibitor taselisib (GDC-0032) in combination with letrozole in patients with hormone receptor-positive advanced breast cancer. Cancer Res 75, 2015 (abstr PD5–2)
- 99.Berenjeno IM, Guillermet-Guibert J, Pearce W, et al. Both p110α and p110β isoforms of PI3K can modulate the impact of loss-of-function of the PTEN tumour suppressor. Biochem J. 2012;442:151–159. doi: 10.1042/BJ20111741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. de Bono J, Arkenau H-T, Mateo J, et al: Exploratory genetic analysis of tumors from a phase I/II dose escalation study of GSK2636771 in patients (pts) with PTEN deficient advanced tumors. Cancer Res 75, 2015 (abstr CT328)
- 101.O’Reilly KE, Rojo F, She Q-B, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Peyton JD, Ahnert JR, Burris H, et al: A dose-escalation study with the novel formulation of the oral pan-class I PI3K inhibitor BEZ235, solid dispersion system (SDS) sachet, in patients with advanced solid tumors. J Clin Oncol 29, 2011 (suppl; abstr 3066)
- 103.Dolly S, Wagner A, Bendell J, et al. A first-in-human, phase l study to evaluate the dual PI3K/mTOR inhibitor GDC-0980 administered QD in patients with advanced solid tumors or non-Hodgkin’s lymphoma. J Clin Oncol. 2010;28:3079. [Google Scholar]
- 104. doi: 10.3389/fonc.2014.00064. Porta C, Paglino C, Mosca A: Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol 4:64, 2014. [DOI] [PMC free article] [PubMed]
- 105. Dowling RJO, Topisirovic I, Fonseca BD, et al: Dissecting the role of mTOR: Lessons from mTOR inhibitors. Biochim Biophys Acta 1804:433–439, 2010. [DOI] [PubMed]
- 106. Banerji U, Dean EJ, Gonzalez M, et al: First-in-human phase I trial of the dual mTORC1 and mTORC2 inhibitor AZD2014 in solid tumors. J Clin Oncol 30, 2012 (suppl; abstr 3004)
- 107. Mita MM, Wolin EM, Meyer T, et al: Phase I expansion trial of an oral TORC1/TORC2 inhibitor (CC-223) in nonpancreatic neuroendocrine tumors (NET). J Clin Oncol 31, 2012 (suppl; abstr e15004)
- 108. Varga A, Mita MM, Wu JJ, et al: Phase I expansion trial of an oral TORC1/TORC2 inhibitor (CC-223) in advanced solid tumors. J Clin Oncol 31, 2013 (suppl; abstr 2606)
- 109. ClinicalTrials.gov: Dual mTOR inhibitor MLN0128 in advanced castration-resistant prostate cancer (CRPC) patients. http://clinicaltrials.gov/show/NCT02091531.
- 110. ClinicalTrials.gov: MLN0128 and bevacizumab in treating patients with recurrent glioblastoma or advanced solid tumors. http://clinicaltrials.gov/show/NCT02142803.
- 111.Yap TA, Yan L, Patnaik A, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol. 2011;29:4688–4695. doi: 10.1200/JCO.2011.35.5263. [DOI] [PubMed] [Google Scholar]
- 112.Molife LR, Yan L, Vitfell-Rasmussen J, et al. Phase 1 trial of the oral AKT inhibitor MK-2206 plus carboplatin/paclitaxel, docetaxel, or erlotinib in patients with advanced solid tumors. J Hematol Oncol. 2014;7:1. doi: 10.1186/1756-8722-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Banerji U, Ranson M, Schellens JH, et al: Results of two phase I multicenter trials of AZD5363, an inhibitor of AKT1, 2 and 3: Biomarker and early clinical evaluation in Western and Japanese patients with advanced solid tumors. AACR 73, 2013 (abstr LB–66) [Google Scholar]
- 114. Yan Y, Wagle M-C, Punnoose E, et al: A first-in-human trial of GDC-0068: A novel, oral, ATP-competitive Akt inhibitor, demonstrates robust suppression of the Akt pathway in surrogate and tumor tissues. Mol Cancer Ther 10, 2011 (abstr B154)
- 115. Spencer A, Yoon S-S, Harrison SJ, et al: Novel AKT inhibitor GSK2110183 shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Preliminary results from a phase I first-time-in-human study. Blood 118:1856, 2011. [DOI] [PMC free article] [PubMed]
- 116. Burris HA, Siu LL, Infante JR, et al: Safety, pharmacokinetics (PK), pharmacodynamics (PD), and clinical activity of the oral AKT inhibitor GSK2141795 (GSK795) in a phase I first-in- human study. J Clin Oncol 29, 2011 (suppl; abstr 3003)
- 117.van der Hage JA, van den Broek LJCM, Legrand C, et al. Overexpression of P70 S6 kinase protein is associated with increased risk of locoregional recurrence in node-negative premenopausal early breast cancer patients. Br J Cancer. 2004;90:1543–1550. doi: 10.1038/sj.bjc.6601741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Calvo E, Benhadji KA, Azaro A, et al: First-in-human phase I study of LY2780301, an oral P70S6K/AKT inhibitor, in patients with refractory solid tumors. J Clin Oncol 30, 2012 (suppl; abstr 3005)
- 119.Tolcher A, Goldman J, Patnaik A, et al. A phase I trial of LY2584702 tosylate, a p70 S6 kinase inhibitor, in patients with advanced solid tumours. Eur J Cancer. 2014;50:867–875. doi: 10.1016/j.ejca.2013.11.039. [DOI] [PubMed] [Google Scholar]
- 120.Doroshow JH, Parchment RE. Oncologic phase 0 trials incorporating clinical pharmacodynamics: From concept to patient. Clin Cancer Res. 2008;14:3658–3663. doi: 10.1158/1078-0432.CCR-07-4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gonzalez-Angulo AM, Blumenschein GR., Jr Defining biomarkers to predict sensitivity to PI3K/Akt/mTOR pathway inhibitors in breast cancer. Cancer Treat Rev. 2013;39:313–320. doi: 10.1016/j.ctrv.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Juric D, Castel P, Griffith M, et al. PI Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature. 2015;518:240–244. doi: 10.1038/nature13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schwartz S, Wongvipat J, Trigwell CB, et al. Feedback suppression of PI3Kα signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kβ. Cancer Cell. 2015;27:109–122. doi: 10.1016/j.ccell.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Edgar KA, Crocker L, Cheng E, et al. Amphiregulin and PTEN evoke a multimodal mechanism of acquired resistance to PI3K inhibition. Genes Cancer. 2014;5:113–126. doi: 10.18632/genesandcancer.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Costa C, Ebi H, Martini M, et al. Measurement of PIP3 levels reveals an unexpected role for p110β in early adaptive responses to p110α-specific inhibitors in luminal breast cancer. Cancer Cell. 2015;27:97–108. doi: 10.1016/j.ccell.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vasudevan KM, Barbie DA, Davies MA, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16:21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Elkabets M, Vora S, Juric D, et al. mTORC1 inhibition is required for sensitivity to PI3K p110α inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med. 2013;5:196ra99. doi: 10.1126/scitranslmed.3005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vora SR, Juric D, Kim N, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014;26:136–149. doi: 10.1016/j.ccr.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Averous J, Fonseca BD, Proud CG. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene. 2008;27:1106–1113. doi: 10.1038/sj.onc.1210715. [DOI] [PubMed] [Google Scholar]
- 130.Takuwa N, Fukui Y, Takuwa Y. Cyclin D1 expression mediated by phosphatidylinositol 3-kinase through mTOR-p70(S6K)-independent signaling in growth factor-stimulated NIH 3T3 fibroblasts. Mol Cell Biol. 1999;19:1346–1358. doi: 10.1128/mcb.19.2.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ilic N, Utermark T, Widlund HR, et al. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci USA. 2011;108:E699–E708. doi: 10.1073/pnas.1108237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Muranen T, Selfors LM, Worster DT, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell. 2012;21:227–239. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Serra V, Eichhorn PJA, García-García C, et al. RSK3/4 mediate resistance to PI3K pathway inhibitors in breast cancer. J Clin Invest. 2013;123:2551–2563. doi: 10.1172/JCI66343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Bosch A, Li Z, Bergamaschi A, et al: PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor–positive breast cancer. Sci Transl Med 7:283ra51, 2015. [DOI] [PMC free article] [PubMed]
- 135. Kim RD, Alberts SR, Renshaw FG, et al: Phase 1 dose escalation study of copanlisib (BAY 80-6946) in combination with gemcitabine or gemcitabine-cisplatin in advanced cancer patients. J Clin Oncol 32, 2014 (suppl; abstr 2610)
- 136.Ando Y, Inada-Inoue M, Mitsuma A, et al. Phase I dose-escalation study of buparlisib (BKM120), an oral pan-class I PI3K inhibitor, in Japanese patients with advanced solid tumors. Cancer Sci. 2014;105:347–353. doi: 10.1111/cas.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rodon J, Braña I, Siu LL, et al. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2014;32:670–681. doi: 10.1007/s10637-014-0082-9. [DOI] [PubMed] [Google Scholar]
- 138.Shapiro GI, Rodon J, Bedell C, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2014;20:233–245. doi: 10.1158/1078-0432.CCR-13-1777. [DOI] [PubMed] [Google Scholar]
- 139. Arkenau H-T, Mateo J, Lemech CR, et al: A phase I/II, first-in-human dose-escalation study of GSK2636771 in patients (pts) with PTEN-deficient advanced tumors. J Clin Oncol 32, 2014 (suppl; abstr 2514)
- 140.Kahl BS, Spurgeon SE, Furman RR, et al. A phase 1 study of the PI3Kδ inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL) Blood. 2014;123:3398–3405. doi: 10.1182/blood-2013-11-537555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Horwitz SM, Flinn I, Patel MR, et al: Preliminary safety and efficacy of IPI-145, a potent inhibitor of phosphoinositide-3-kinase-δ,γ, in patients with relapsed/refractory lymphoma. J Clin Oncol 31, 2013 (suppl; abstr 8518)
- 142.Lanasa MC, Glenn M, Mato AR, et al. First-in-human study of AMG 319, a highly selective, small molecule inhibitor of PI3Kδ, in adult patients with relapsed or refractory lymphoid malignancies. Blood. 2013;122:678. [Google Scholar]
- 143. Suder A: Final results of phase I study of the oral class I PI3k inhibitor CH5132799 in patients with advanced solid tumours. Ann Oncol 24, 2013 (abstr P06.08)
- 144.Wunderle L, Badura S, Lang F, et al. Safety and efficacy of BEZ235, a dual PI3-kinase /mTOR inhibitor, in adult patients with relapsed or refractory acute leukemia: results of a phase I study. Blood. 2013;122:2675. [Google Scholar]
- 145.Markman B, Tabernero J, Krop I, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann Oncol. 2012;23:2399–2408. doi: 10.1093/annonc/mds011. [DOI] [PubMed] [Google Scholar]
- 146. Millham R, Houk B, Borzillo G, et al: First-in-patient study of PF-05212384, a small molecule intravenous dual inhibitor of PI3K and mTOR in patients with advanced cancer: Update on safety, efficacy, and pharmacology. Mol Cancer Ther 10, 2011 (abstr A167)
- 147. doi: 10.1007/s10637-013-0062-5. Britten CD, Adjei AA, Millham R, et al: Phase I study of PF-04691502, a small-molecule, oral, dual inhibitor of PI3K and mTOR, in patients with advanced cancer. Invest New Drugs 32:510-517, 2014 [Erratum: Invest New Drugs, 32:575 2014. [DOI] [PubMed] [Google Scholar]
- 148. Munster PN, Noll R van der, Voest EE, et al: Phase I first-in-human study of the PI3 kinase inhibitor GSK2126458 (GSK458) in patients with advanced solid tumors (study P3K112826). J Clin Oncol 29, 2011 (suppl; abstr 3018)
- 149.Papadopoulos KP, Tabernero J, Markman B, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245409 (XL765), a novel, orally administered PI3K/mTOR inhibitor in patients with advanced solid tumors. Clin Cancer Res. 2014;20:2445–2456. doi: 10.1158/1078-0432.CCR-13-2403. [DOI] [PubMed] [Google Scholar]
- 150.Bellmunt J, Szczylik C, Feingold J, et al. Temsirolimus safety profile and management of toxic effects in patients with advanced renal cell carcinoma and poor prognostic features. Ann Oncol. 2008;19:1387–1392. doi: 10.1093/annonc/mdn066. [DOI] [PubMed] [Google Scholar]
- 151. Sellami DB, Urva SR, Grosch K, et al: Meta-analysis on the relationship between everolimus exposure and safety and efficacy. J Clin Oncol 30, 2012 (suppl; abstr 3099)
- 152.Hartford CM, Desai AA, Janisch L, et al. A phase I trial to determine the safety, tolerability, and maximum tolerated dose of deforolimus in patients with advanced malignancies. Clin Cancer Res. 2009;15:1428–1434. doi: 10.1158/1078-0432.CCR-08-2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Tan DS, Dumez H, Olmos D, et al: First-in-human phase I study exploring three schedules of OSI-027, a novel small molecule TORC1/TORC2 inhibitor, in patients with advanced solid tumors and lymphoma. J Clin Oncol 28, 2010 (suppl; abstr 3006)
- 154. Tabernero J, Cervantes A, Gordon MS, et al: A phase I, open label, dose escalation study of oral mammalian target of rapamycin inhibitor INK128 administered by intermittent dosing regimens in patients with advanced malignancies. Cancer Res 72, 2012 (abstr CT–02)
- 155. Tabernero J, Saura C, Perez DR, et al: First-in-human phase I study evaluating the safety, pharmacokinetics (PK), and intratumor pharmacodynamics (PD) of the novel, oral, ATP-competitive Akt inhibitor GDC-0068. J Clin Oncol 29, 2011 (suppl; abstr 3022)
- 156. Marshall J, Posey J, Hwang J, et al: A phase I trial of RX-0201 (AKT anti-sense) in patients with an advanced cancer. J Clin Oncol 25, 2007 (suppl; abstr 3564) [Google Scholar]
- 157.Kurtz J-E, Ray-Coquard I. PI3 kinase inhibitors in the clinic: An update. Anticancer Res. 2012;32:2463–2470. [PubMed] [Google Scholar]
- 158.Busaidy NL, Farooki A, Dowlati A, et al. Management of metabolic effects associated with anticancer agents targeting the PI3K-Akt-mTOR pathway. J Clin Oncol. 2012;30:2919–2928. doi: 10.1200/JCO.2011.39.7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Schindler K, Abraham R, Shah PD, et al: Clinical and histologic characterization of dermatologic adverse events from the pan-PI3K inhibitor buparlisib (BKM-120). J Clin Oncol 32, 2014 (suppl; abstr e20639)
- 160.Kaplan B, Qazi Y, Wellen JR. Strategies for the management of adverse events associated with mTOR inhibitors. Transplant Rev (Orlando) 2014;28:126–133. doi: 10.1016/j.trre.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 161.Albiges L, Chamming’s F, Duclos B, et al. Incidence and management of mTOR inhibitor-associated pneumonitis in patients with metastatic renal cell carcinoma. Ann Oncol. 2012;23:1943–1953. doi: 10.1093/annonc/mds115. [DOI] [PubMed] [Google Scholar]
- 162.Janku F, Tsimberidou AM, Garrido-Laguna I, et al. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther. 2011;10:558–565. doi: 10.1158/1535-7163.MCT-10-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Janku F, Wheler JJ, Naing A, et al. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res. 2013;73:276–284. doi: 10.1158/0008-5472.CAN-12-1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Mayer IA, Abramson VG, Balko JM, et al: SU2C phase Ib study of the PI3K{alpha} inhibitor BYL719 with letrozole in ER+/HER2- metastatic breast cancer (MBC). J Clin Oncol 32, 2014 (suppl; abstr 516) [Google Scholar]
- 165. Mayer IA, Fasching PA, Gnant M, et al: Phase II, randomized, placebo-controlled study of BYL719 or buparlisib (BKM120) with letrozole for neoadjuvant treatment of postmenopausal women with HR+/HER2−, PIK3CA mutant or wild-type, breast cancer (BC). J Clin Oncol 32, 2014 (suppl; abstr TPS655)
- 166. ClinicalTrials.gov: Study of BKM120 or BYL719 and capecitabine in patients with metastatic breast cancer. http://clinicaltrials.gov/show/NCT01300962.
- 167. ClinicalTrials.gov: BYL719 + T-DM1 in HER2(+) Metastatic breast cancer pts who progress on prior trastuzumab & taxane tx. http://clinicaltrials.gov/ct2/show/NCT02038010.
- 168. ClinicalTrials.gov: A study to investigate safety, pharmacokinetics (PK) and pharmacodynamics (PD) of BKM120 Plus GSK1120212 in selected advanced solid tumor patients. http://clinicaltrials.gov/ct2/show/NCT01155453.
- 169. ClinicalTrials.gov: A phase Ib study of MEK162 plus BYL719 in adult patients with selected advanced solid tumors. http://clinicaltrials.gov/ct2/show/NCT01449058.
- 170. ClinicalTrials.gov: Trametinib with GSK2141795 in BRAF wild-type melanoma. http://clinicaltrials.gov/ct2/show/NCT01941927.
- 171. ClinicalTrials.gov: MK-2206 and AZD6244 in patients with advanced colorectal carcinoma. http://clinicaltrials.gov/ct2/show/NCT01333475.
- 172. Iwata H, Baselga J, Campone M, et al: Ph III randomized studies of the oral pan-PI3K inhibitor buparlisib (BKM120) with fulvestrant in postmenopausal women with HR+/HER2– locally advanced or metastatic breast cancer (BC) after aromatase inhibitor (AI; BELLE-2) or AI and mTOR inhibitor (BELLE-3) treatment. J Clin Oncol 31, 2013 (suppl; abstr TPS650)
- 173. ClinicalTrials.gov: A dose escalation study evaluating the safety and tolerability of GDC-0032 in patients with locally advanced or metastatic solid tumors and in combination with endocrine therapy in patients with locally advanced or metastatic hormone receptor-positive breast cancer. http://clinicaltrials.gov/show/NCT01296555.
- 174. ClinicalTrials.gov: Phase Ib of abiraterone acetate plus BEZ235 or BKM120 in castration-resistant prostate cancer (CRPC) patients. http://clinicaltrials.gov/show/NCT01634061.
- 175. ClinicalTrials.gov: Phase 1 trial of the mammalian target of rapamycin (mTOR) inhibitor everolimus plus radiation therapy (rt) for salvage treatment of biochemical recurrence in prostate cancer patients following prostatectomy. http://clinicaltrials.gov/show/NCT01548807.
- 176. ClinicalTrials.gov: Study of GDC-0068 Or GDC-0980 with abiraterone acetate versus abiraterone acetate in patients with castration-resistant prostate cancer previously treated with docetaxel chemotherapy. http://clinicaltrials.gov/show/NCT01485861.
- 177. ClinicalTrials.gov: Bicalutamide with or without akt inhibitor MK2206 in treating patients with previously treated prostate cancer. http://clinicaltrials.gov/show/NCT01251861.
- 178. ClinicalTrials.gov: Phase Ib Study of BKM120 with cisplatin and XRT in high risk locally advanced squamous cell cancer of head and neck. http://clinicaltrials.gov/ct2/show/NCT02113878.
- 179. ClinicalTrials.gov: A study to evaluate the potential benefit of the addition of BYL719 to paclitaxel in the treatment of breast cancer and head-and-neck cancer. http://clinicaltrials.gov/ct2/show/NCT02051751.
- 180. ClinicalTrials.gov: GDC-0941 and cisplatin in treating patients with androgen receptor-negative triple negative metastatic breast cancer. http://clinicaltrials.gov/show/NCT01918306.
- 181. ClinicalTrials.gov: Phase I study of PI3: (Phosphoinositol 3)-kinase inhibitor BAY80-6946 with paclitaxel in patients with advanced cancer. http://clinicaltrials.gov/ct2/show/NCT01411410.
- 182. ClinicalTrials.gov: Phase I of BKM120/Olaparib for triple negative breast cancer or high grade serous ovarian cancer. http://clinicaltrials.gov/ct2/show/NCT01623349.
- 183.Zhang H, Berezov A, Wang Q, et al. ErbB receptors: From oncogenes to targeted cancer therapies. J Clin Invest. 2007;117:2051–2058. doi: 10.1172/JCI32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Cruz C, Schuler MH, Machiels J-PH, et al: Phase lb study of buparlisib (BKM120) plus either paclitaxel (PTX) in advanced solid tumors (aST) or PTX plus trastuzumab (TZ) in HER2+ breast cancer (BC). J Clin Oncol 32, 2014 (5 suppl; abstr 627)
- 185.Britten CD. PI3K and MEK inhibitor combinations: Examining the evidence in selected tumor types. Cancer Chemother Pharmacol. 2013;71:1395–1409. doi: 10.1007/s00280-013-2121-1. [DOI] [PubMed] [Google Scholar]
- 186.Shimizu T, Tolcher AW, Papadopoulos KP, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18:2316–2325. doi: 10.1158/1078-0432.CCR-11-2381. [DOI] [PubMed] [Google Scholar]
- 187. LoRusso P, Shapiro G, Pandya SS, et al: A first-in-human phase Ib study to evaluate the MEK inhibitor GDC-0973, combined with the pan-PI3K inhibitor GDC-0941, in patients with advanced solid tumors. J Clin Oncol 30, 2012 (suppl; abstr 2566)
- 188. Ramanathan RK, Hoff DDV, Eskens F, et al: A phase 1b trial of PI3K inhibitor copanlisib (BAY 80-6946) combined with the allosteric-MEK inhibitor refametinib (BAY 86-9766) in patients with advanced cancer. J Clin Oncol 32, 2014 (suppl; abstr 2588)
- 189. Algazi AP, Posch C, Ortiz-Urda S, et al: A phase I trial of BKM120 combined with vemurafenib in BRAFV600E/k mutant advanced melanoma. J Clin Oncol 32, 2014 (suppl: abstr 9101)
- 190. Heist RS, Gandhi L, Shapiro G, et al: Combination of a MEK inhibitor, pimasertib (MSC1936369B), and a PI3K/mTOR inhibitor, SAR245409, in patients with advanced solid tumors: Results of a phase Ib dose-escalation trial. J Clin Oncol 31, 2013 (suppl; abstr 2530)
- 191. Bedard P, Tabernero J, Kurzrock R, et al: A phase lb, open-label, multicenter, dose- escalation study of the oral pan-PI3K inhibitor BKM120 in combination with the oral MEK1/2 inhibitor GSK1120212 in patients (pts) with selected advanced solid tumors. J Clin Oncol 30, 2012 (suppl; abstr 3003)
- 192.deGraffenried LA, Friedrichs WE, Russell DH, et al. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clin Cancer Res. 2004;10:8059–8067. doi: 10.1158/1078-0432.CCR-04-0035. [DOI] [PubMed] [Google Scholar]
- 193.Sanchez CG, Ma CX, Crowder RJ, et al. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res. 2011;13:R21. doi: 10.1186/bcr2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. doi: 10.1007/s12325-013-0060-1. Yardley DA, Noguchi S, Pritchard KI, et al: Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther 30:870-884, 2013 [Erratum: Adv Ther 31:1008-1009, 2013] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Edlind MP, Hsieh AC. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J Androl. 2014;16:378–386. doi: 10.4103/1008-682X.122876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Marques RB, Aghai A, de Ridder CMA, et al. High efficacy of combination therapy using PI3K/AKT inhibitors with androgen deprivation in prostate cancer preclinical models. Eur Urol. 2015;67:1177–1185. doi: 10.1016/j.eururo.2014.08.053. [DOI] [PubMed] [Google Scholar]
- 197.Fokas E, Im JH, Hill S, et al. Dual inhibition of the PI3K/mTOR pathway increases tumor radiosensitivity by normalizing tumor vasculature. Cancer Res. 2012;72:239–248. doi: 10.1158/0008-5472.CAN-11-2263. [DOI] [PubMed] [Google Scholar]
- 198.Bender A, Opel D, Naumann I, et al. PI3K inhibitors prime neuroblastoma cells for chemotherapy by shifting the balance towards pro-apoptotic Bcl-2 proteins and enhanced mitochondrial apoptosis. Oncogene. 2011;30:494–503. doi: 10.1038/onc.2010.429. [DOI] [PubMed] [Google Scholar]
- 199.Opel D, Westhoff M-A, Bender A, et al. Phosphatidylinositol 3-kinase inhibition broadly sensitizes glioblastoma cells to death receptor- and drug-induced apoptosis. Cancer Res. 2008;68:6271–6280. doi: 10.1158/0008-5472.CAN-07-6769. [DOI] [PubMed] [Google Scholar]
- 200.Kilic-Eren M, Boylu T, Tabor V. Targeting PI3K/Akt represses hypoxia inducible factor-1α activation and sensitizes rhabdomyosarcoma and Ewing’s sarcoma cells for apoptosis. Cancer Cell Int. 2013;13:36. doi: 10.1186/1475-2867-13-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Besse B, Soria J, Gomez-Roca C, et al: A phase Ib study to evaluate the PI3-kinase inhibitor GDC-0941 with paclitaxel (P) and carboplatin (C), with and without bevacizumab (BEV), in patients with advanced non-small cell lung cancer (NSCLC). J Clin Oncol 29, 2011 (suppl; abstr 3044)
- 202. Bang Y-J, Kang Y-K, Alsina M, et al: JAGUAR: A randomized phase II study of the AKT inhibitor ipatasertib (GDC-0068) versus placebo in combination with mFOLFOX6 chemotherapy in patients (pts) with locally advanced or metastatic HER2-negative gastric (G) or gastroesophageal junction (GEJ) adenocarcinoma. J Clin Oncol 32 2014 (suppl; abstr TPS4147)
- 203.Ibrahim YH, García-García C, Serra V, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;2:1036–1047. doi: 10.1158/2159-8290.CD-11-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Juvekar A, Burga LN, Hu H, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012;2:1048–1063. doi: 10.1158/2159-8290.CD-11-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Matulonis U, Wulf GM, Birrer MJ, et al: Phase I study of oral BKM120 and oral olaparib for high-grade serous ovarian cancer (HGSC) or triple-negative breast cancer (TNBC). J Clin Oncol 32, 2014 (suppl; abstr 2510)
- 206.Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877–1888. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]