

Abstract
Expansion of a CGG-repeat tract in the 5’UTR of FMR1 is responsible for the Fragile X-related disorders (FXDs), FXTAS, FXPOI and FXS. Previous work in a mouse model of these disorders has implicated proteins in the base excision and the mismatch repair pathways in the expansion mechanism. However, the precise role of these factors in this process is not well understood. The essential role of MutLγ, a complex that plays a minor role in mismatch repair (MMR) but that is essential for resolving Holliday junctions during meiosis, raises the possibility that expansions proceed via a Holliday junction-like intermediate that is processed to generate a double-strand break (DSB). We show here in an FXD mouse model that LIG4, a ligase essential for non-homologous end-joining (NHEJ), a form of DSB repair (DSBR), protects against expansions. However, a mutation in MRE11, a nuclease that is important for several other DSBR pathways including homologous recombination (HR), has no effect on the extent of expansion. Our results suggest that the expansion pathway competes with NHEJ for the processing of a DSB intermediate. Thus, expansion likely proceeds via an NHEJ-independent DSBR pathway that may also be HR-independent.
Keywords: Fragile X related disorders, Fragile X syndrome (FXS), Repeat Expansion Diseases, Double-strand break repair (DSBR), Non-homologous end-joining (NHEJ), DNA ligase IV (Lig4), MRE11
Graphical Abstract
1. Introduction
The CGG-repeat tract just downstream of the transcription start site of the FMR1 gene (MIM* 309550) is expansion-prone with the expansion risk increasing with increasing repeat number. Individuals who inherit alleles with >54 repeats are at risk of 3 different clinical conditions referred to collectively as the Fragile X-related disorders (FXDs) or the Fragile X spectrum disorders [1]. Premutation (PM) alleles, alleles with 55-~200 repeats, confer risk of Fragile X-associated primary ovarian insufficiency (FXPOI; MIM# 300624) and Fragile X-associated tremor/ataxia syndrome (FXTAS; MIM# 300623), while full mutation (FM) alleles, those with >200 repeats, confer risk of a form of intellectual disability and autism, Fragile X syndrome (FXS; MIM# 300624) [1]. While expansions have severe implications for human health, the mechanism of expansion is still largely unclear. Evidence from a knock-in mouse model of a PM allele demonstrate that the mismatch repair (MMR) protein complexes MutSα, MutSβ and MutLγ, all play important roles in generating expansions, with MutSβ and MutLγ being essential [2–4]. The requirement for MutLγ is intriguing since it is the least abundant of the MutL protein complexes in mammals and while it only plays a minor role in MMR, it plays an essential role in the processing of Holliday junctions (HJs) generated during meiosis [5]. Since the individual strands of the FX repeat are both able to form hairpins [6–9], this could result in double-hairpins that are reminiscent of the HJ substrate for MutLγ. It is thus particularly interesting that EXO1 protects against expansions in both a nuclease-dependent and a nuclease-independent manner [4]. The nuclease-dependent role may reflect the normal role of EXO1 in processing MMR intermediates, while the nuclease-independent role may reflect EXOI’s role in determining the origin of cleavage of HJs. Cleavage of an HJ-like intermediate in a manner analogous to the resolution of HJs in meiosis, would give rise to a double-strand break (DSB). If such a break is an intermediate in the expansion process, it would need to be processed by classical non-homologous end-joining (cNHEJ), homologous recombination (HR) or some other form of double strand break repair (DSBR) pathway. We show here that loss of DNA ligase IV (LIG4), a DNA ligase essential for cNHEJ, results in increase in expansions. In contrast, a hypomorphic mutation in the meiotic recombination 11 homolog 1 (MRE11) protein [10], a nuclease important for end resectioning in a number of DSBR pathways including HR, has no effect. The effect of the loss of LIG4 suggests that NHEJ competes with the expansion process for the processing of a DSB intermediate and thus that expansion in the FXD mouse likely involves components of a DSBR pathway other than NHEJ and perhaps HR.
2. Materials and Methods
2.1. Reagents and services
All reagents were from Sigma-Aldrich (St. Louis, MO) unless otherwise specified. Primers were from Life Technologies (Grand Island, NY). Capillary electrophoresis of fluorescently labeled PCR genotyping products was carried out by the Roy J Carver Biotechnology Center, University of Illinois (Urbana, IL).
2.2. Mouse breeding and maintenance
The generation of the FXD mice was described previously [11]. These mice are on a C57BL/6 background. Mice were maintained in accordance with the guidelines of the NIDDK Animal Care and Use Committee and with the Guide for the Care and Use of Laboratory Animals (NIH publication no. 85-23, revised 1996). Mre11ATLD1/ATLD1 mice were a gift of John Petrini (Sloan Kettering Institute, NY, NY) [12]. Mice with a floxed Lig4 allele [13] were obtained from André Nussenzweig (NIH, Bethesda, MD).
2.3. Breeding strategy
To selectively delete Lig4 in hepatocytes, we crossed the floxed Lig4 mice with FXD mice to create females carrying both the Fmr1 KI and Lig4 floxed alleles. We then crossed these females with Alb-cre transgenic males (Strain 003574 (B6.Cg-Tg(Alb-cre)21Mgn/J)) [14] obtained from Jackson Laboratory (Bar Harbor, ME). The resulting offspring were used for additional breeding to create control mice, carrying the Fmr1 Kl and Alb-cre alleles, and Lig4 mutant mice carrying the Fmr1 Kl and Alb-cre alleles and homozygote to the Lig4 floxed allele.
2.4. DNA isolation
Genomic DNA from tails of 3-week-old mice was extracted using KAPA Mouse Genotyping Kit (KAPA Biosystems, Wilmington, MA). Genomic DNA of different organs was isolated using the Maxwell®16 Mouse Tail DNA Purification kit (Promega, Madison, WI). Genomic DNA from Hepatocytes and NPCs was isolated using the salting-out method. Briefly, cells were resuspended in ATL lysis buffer (Qiagen, Valencia, CA) with 0.55 mg/ml proteinase K solution (Invitrogen, Carlsbad, CA) and incubated at 55°C overnight before the addition of 1.5 M NaCl. The resultant precipitate was pelleted by centrifugation and ethanol was added to supernatant. DNA was pelleted and dissolved in TE by incubation at 60°C. Sorted Hepatocytes genomic DNA was isolated using a standard phenol:chloroform extraction protocol. Sperm was isolated from epididymis using the swim-out technique and pelleted by centrifugation at 300× g for 5 min. Genomic DNA was then isolated using the salting-out method described above after an overnight incubation in ATL lysis buffer containing 0.55 mg/ml proteinase K solution and 30 mM DTT.
2.5. Genotyping
All genotyping PCRs were carried out using KAPA2G Fast HotStart Genotyping Mix (KAPA Biosystems) according to the manufacturer’s instructions. Alb-cre genotyping was carried out with primers Alb-cre-20239-F (specific for the WT allele; 5’-TGCAAACATCACATGCACAC-3’), Alb-cre-olMR5374-F (specific for the transgenic allele; 5’-GAAGCAGAAGCTTAGGAAGATGG-3’) and Alb-cre-20240-R (common to both alleles; 5’-TTGGCCCCTTACCATAACTG-3’). The PCR parameters were: 94°C for 2 min, 10× (94°C for 20 sec, 65°C (−0.5°C /cycle) for 15 sec, 68° for 10 sec), 28× (94°C for 15 sec, 60°C for 15 sec, 72°for 10 sec), 72°C for 2 min (https://www2.jax.org/protocolsdb/f?p=116:5:0::NO:5:P5_MASTER_PROTOCOL_ID,P5_JRS_CODE:26917,003574). Genotyping of the floxed Lig4 allele was carried out using primers: mLig4-3_F (5’-ATCGCTCTTGTCCCAGTACACCTGC-3’) and mLig2-2_R (5’-GTGCATTAAATGGAGTGCTGTGC-3’) and PCR parameters: 95°C for 3 min, 35× (95°C for 15 sec, 60°C for 15 sec, 72°C for 30 sec), 72°C for 1 min. Lig4 KO genotyping was carried out using primers: Lig4loxp1 (5’-CACCAGTTCCATCCTGTAGC-3’) and mLig2-2_R (5’-GTGCATTAAATGGAGTGCTGTGC-3’) and PCR parameters: 95°C for 3 min, 30× (95°C for 15 sec, 63°C for 15 sec, 72°C for 30 sec), 72°C for 5 min. Mre11 ATLD1 genotyping was carried out with primers MRE11-18576-F (specific for the WT allele; 5’-TCACGTGGAGGTGTTCCTTC-3’), MRE11-18578-F (specific for the mutant allele; 5’-ATCAGCAGCCTCTGTTCC-3’) and MRE11-18626-R (common to both alleles; 5’-GAATCCCAAGTGGCAGAAAG-3’). The PCR parameters were: 94°C for 2 min, 10× (94°C for 20 sec, 65°C (−0.5°C /cycle) for 15 sec, 68° for 10 sec), 28× (94°C for 15 sec, 60°C for 15 sec, 72°for 10 sec), 72°C for 2 min (https://www.jax.org/jax-mice-and-services/customer-support/technical-support/genotyping-resources).
Fmr1 PM allele genotyping and repeat number evaluation was performed using a fluorescent PCR assay using Not_FRAX_M8-F (5’-AGTTCAGCGGCCGCGCTGCCGGGGGGCGTGCGGTAACG-3’) and FAM-labeled Not_FRAX_M9-R (5’-CAAGTCGCGGCCGCGGGCTGCAGGCGCTTGAGGCCCAG-3’) primer pair. PCR was carried out using the KAPA2G Fast FlotStart Genotyping Mix (KAPA Biosystems) supplemented with 2.5M betaine (Sigma-Aldrich), 2% DMSO (americanBIO, Natick, MA), 0.12mM dGTP and dCTP (Thermo scientific, Waltham, MA) and PCR parameters: 95°C for 5 min, 35× (95°C for 30 sec, 67°C for 30 sec, 72°C, for 2 min), 72°C for 10 min. Small pool PCR was used to analyze sperm DNA as previously described [15]. Briefly, diluted DNA was subjected to nested PCR. The first round of PCR was carried out using the primers FraxC (5’-GCTCAGCTCCGTTTCGGTTTCACTTCCGGT-3’) and FraxF (5’-AGCCCCGCACTTCCACCACCAGCTCCTCCA-3’). One microliter of this PCR mix was used in second round of PCR with FAM-labeled FraxM4 (5’-CTTGAGGCCCAGCCGCCGTCGGCC-3’) and FraxM5 (5’-CGGGGGGCGTGCGGTAACGGCCCAA-3’) primer pair. Repeat PCR reactions were resolved by capillary electrophoresis on an ABI Genetic Analyzer. The resultant fsa file was then displayed using a custom R script that is available upon request [16].
2.6. Analysis of Repeat Number
To quantitate somatic expansions, we calculated the repeat number added to the expanded allele as previously described [17]. Briefly, the average size of the expanded alleles was subtracted from the repeat number of the inherited allele, determined from heart, an organ that we have previously shown does not show expansions [18]. Since stable and expanded alleles have an approximately normal distribution, we calculated the standard deviation (SD) of expanded alleles using the right side of the relevant PCR profile, while the standard deviation for stable alleles was calculated using the left side of the appropriate PCR profile. This strategy avoids problems resulting from the region of overlap between the PCR profiles from the 2 allele size classes. In some cases, the SD was used to generate a normal distribution profile for the stable and expanded allele populations that was superimposed on the electrophoretogram. Statistical analysis was carried out using Student’s t-test, Fisher’s exact test (http://www.graphpad.com/quickcalcs) and Mann-Whitney U test (vassarstats.net).
2.7. Isolation of mice liver parenchymal and non-parenchymal Cells
Mouse hepatocytes and non-parenchymal cells (NPCs) were isolated from livers of mice using a two-step collagenase perfusion protocol [19]. Briefly, mice were terminally anesthetized, and the liver was perfused with Krebs Ringer buffer with glucose, followed by continuous perfusion with the same buffer containing 1.4mM CaCl2 and 100u/ml collagenase type 1 (Worthington Biochemical Corporation, Lakewood, NJ). Following dissection of the liver, cells were filtered through a 70 μm nylon cell strainer (Corning, Corning, NY). Low speed centrifugation at 50 ×g for 4 min at 4°C was used to harvest hepatocytes. The hepatocytes were further purified by centrifugation in a 50% Percoll solution at 50 ×g [19]. NPCs were then harvested and purified from the supernatant essentially as previously described [20, 21]. Briefly, the supernatant was first centrifuged at 72 ×g for 5 min at 4°C to eliminate the remaining erythrocytes and hepatocytes. The resultant supernatant was centrifuged at 400 ×g for 5 min at 4°C. NPCs were then purified by a 2-step Percoll gradient [25%/50% (vol/vol)] and centrifugation at 1,500 ×g for 10 min at 4°C. NPCs were collected from the interphase between the 2 Percoll layers.
2.8. Hepatocytes FACS sorting based on DNA content
Isolated hepatocytes were fixed in 70% ethanol overnight at −20°C. Following a wash with PBS, cells were stained in 10mM Tris (pH 7.4-7.5) and 10mM NaCl containing 50 μg/ml propidium iodide (PI) and 0.2 mg/ml RNase overnight at 4°C. Cell sorting was carried out using BD FACSAria™ Fusion cell sorter with the 2n, 4n and 8n hepatocytes populations being identified based on their PI signal.
3. Results
3.1. LIG4 protects against repeat expansions.
To investigate whether DSBR plays a role in CGG repeat instability we studied the effect of the loss of LIG4, a DNA ligase critical for NHEJ, on CGG-repeat expansions in the FXD mouse model. Lig4 null mice are not viable [22], thus we could not examine the effect of the ubiquitous loss of LIG4 on expansion. Since mice are particularly sensitive to the loss of LIG4 in the brain [22], we decided to conditionally delete Lig4 in liver, which like brain, is an expansion-prone organ [18]. To do this we took advantage of the Cre-LoxP system and the availability of mice expressing Cre recombinase under the control of the albumin promoter. This promoter is expressed specifically in hepatocytes, cells that constitute the majority of the liver mass, and Cre recombinase expressed under the control of this promoter has been shown to have close to 100% excision efficiency for a number of different floxed alleles (https://www.jax.org/strain/003574). Hepatocyte-specific Lig4 knock-out was achieved by generating mice homozygous for the Lig4 floxed allele that also carry the Alb-Cre transgene (hereafter referred to as Lig4fl/fl) (Fig. S1). We then compared the repeat PCR profiles of the livers of these animals to those of control mice that were homozygous for the wild type Lig4 gene and carry the Alb-Cre transgene (Lig4+/+). Repeat profiles in heart were used to determine the original inherited allele of each animal as heart shows no expansions even in very old animals [18]. While the repeat PCR profiles in heart are unimodal, consistent with the fact that the repeat is stable in this organ, liver repeat PCR profiles are typically bimodal (Fig. 1 and Fig. S2). The smaller peak represents a cell population with a stable allele displaying the same number of repeats as the heart (Fig. 1A). The larger peak represents a cell population with expanding alleles. This bimodality becomes more apparent with age as the expanding allele increases in size and diverges further from the stable allele (Fig. S2).
Fig. 1. LIG4 protects against expansions in liver.
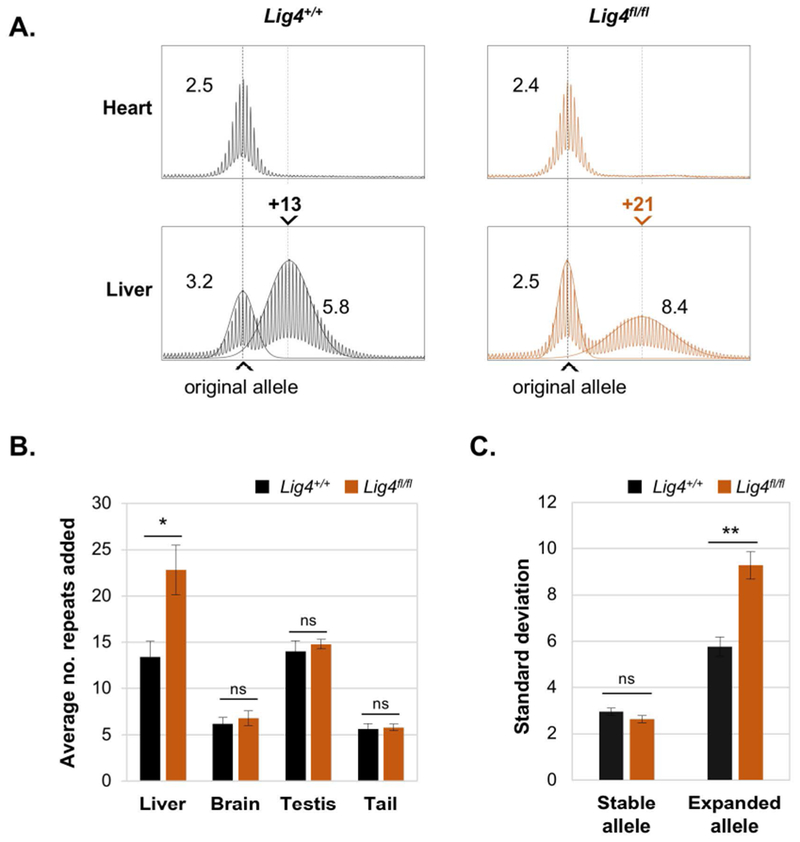
A. Representative repeat PCR profiles from the livers of 10-month old Lig4+/+ and Lig4fl/fl mice with 150 repeats. The repeat PCR profiles from heart, an organ that does not show expansions, are included to indicate the original inherited allele size. Superimposed on the liver profiles is the calculated normal distribution profile of each allele. The numbers above each liver panel indicate the number of repeats added relative to the original allele. The black dotted line in each panel indicates the size of the original inherited allele, while the gray dotted line indicates the average size of the expanded alleles. The numbers adjacent to each peak in the PCR profile are the respective SDs. B. Average repeats added to different tissues of Lig4+/+ and Lig4fl/fl mice. Each panel shows the average of five 10-month old animals with ~150 repeats. C. Average standard deviations of the stable and expanded PM alleles in the livers of the Lig4+/+ and Lig4fl/fl mice shown in Panel B. The error bars represent the standard error. * p= 0.02, ** p= 0.002, ns: not significant.
We used the number of repeats added to the expanded alleles as a metric for the extent of expansion rather than the more commonly used somatic instability index, since the latter metric can be misleading under some circumstances [17]. As can be seen Fig. 1A and B, loss of Lig4 results in an increase in the number of repeats added in liver. For example, a Lig4+/+ mouse with an inherited allele of 150 repeats gained a total of 13 repeats in liver, while a Lig4fl/fl mouse with a similar inherited allele size gained a total of 21 repeats i.e, 8 repeats more than the Lig4+/+ mouse (Fig. 1A). The difference was also statistically significant when we used another metric of expansion, the standard deviation (SD) of the expanded peak (Fig. 1A and 1C) [23]. Thus, the expanded allele in the Lig4fl/fl mouse shown in Fig. 1A has an SD of 8.4 compared to an SD of 5.8 for the expanded allele in the Lig4+/+ mouse. Notably, the SD for the stable peaks in the Lig4fl/fl profiles are also lower than the SD for the stable peaks in the Lig4+/+ profiles although this difference is not statistically significant (Fig. 1C). While the SD of the stable allele of the Lig4fl/fl mouse shown in Fig. 1A is 2.5, a value characteristic of stable alleles such as heart (Fig. 1A), the SD of the stable allele of the Lig4+/+ mouse is 3.2. This difference arises from the fact that in the Lig4+/+ profile the stable and expanded population overlap, while in the Lig4fl/fl mouse the 2 alleles have diverged enough to reduce or even eliminate any substantial cross-allele contribution. As expected, the observed effect of the loss of LIG4 was confined to the liver as Lig4 is knocked-out specifically in hepatocytes (Fig. 1B and S1). Thus, our data demonstrate that LIG4 and presumably NHEJ, protects against repeat expansions.
3.2. Expansion in the liver only occurs in hepatocytes.
Despite the effect on the size of the expanded allele, the loss of LIG4 does not change the bimodality of the repeat PCR profile. This suggests that all expansion-prone cells are sensitive to the loss of LIG4. Since Lig4 is specifically knocked-out in hepatocytes, we hypothesized that expansions in the liver are limited to the hepatocytes. We thus separated hepatocytes from the non-parenchymal cells (NPCs) that make up the rest of the cells of the liver and performed repeat PCRs on DNA isolated from each cell population. We used 3-month old mice since they are suitable for the liver dissociations needed for the hepatocyte isolation. At this age, the repeat expansions are relatively modest. Nonetheless, expanded alleles can already be distinguished from non-expanded alleles. Consistent with our hypothesis, the repeat PCR profiles of hepatocytes consists of a single distinct peak, corresponding in size to the larger of the 2 peaks seen in the total liver (Fig. 2A). In contrast, the size of the peak in NPCs is similar to that of the stable peak seen in the heart and to the smaller of the 2 allele peaks seen in total liver. Furthermore, the SDs of the NPC peak is 2.5, the same as in heart (Fig. 2A). This is consistent with the repeats being stable in these cells. Thus, expansions in the liver are limited to hepatocytes. We next compared the instability in isolated hepatocytes of 3-months old Lig4fl/fl and Lig4+/+ mice (Fig. 2B-D). While the difference in repeat number added has yet to reach statistical significance (Fig. 2C), the SD metric already shows statistically significant differences between Lig4fl/fl and Lig4+/+ mice (Fig. 2B and 2D). Thus, the SD metric is useful even for analyzing relatively small differences in the extent of repeat expansion. Variability in the propensity to expand is not limited to different cells in the liver. It is also apparent in the variation between different organs in the extent of expansion. For example, testis and liver show extensive expansions, whilst expansions in brain and tail are less extensive (Fig. 1B) and no expansion is seen in heart (Fig. 2A) [18]. This may reflect differences in the levels of proteins that promote or protect against expansions in different cell types.
Fig. 2. Expansions in liver are confined to hepatocytes.
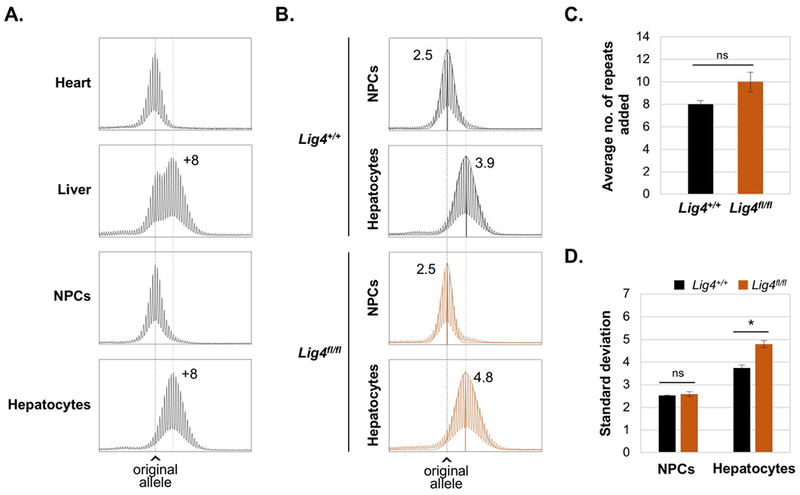
A. and B. Repeat PCR profiles from 3-month old males with 170 repeats. The black dotted line in each panel indicates the size of the original inherited allele, while the gray dotted line indicates the average size of the expanded alleles. A. Representative profiles of heart, liver, NPCs and hepatocytes showing the number of repeats added relative to the original allele. B. Representative profiles of NPCs and hepatocytes isolated from Lig4+/+ and Lig4fl/fl mice livers showing the SD and the calculated normal distribution profile of each allele. C. and D. show the average of repeats added and SDs respectively for 3 Lig4+/+ and 4 Lig4fl/fl animals with ~170 repeats ± SE. * p< 0.005, ns: not significant.
In mouse models of two other Repeat Expansion Diseases, myotonic dystrophy type 1 (DM1) and Huntington Disease (HD), expansion has been correlated with increased ploidy [24, 25]. To address the role of ploidy in the FXD mouse we used flow cytometry to isolate diploid (2n), 4n and 8n hepatocytes from Lig4 WT and Lig4fl/fl mice. Since hepatocytes undergo polyploidization with age [26], we again used young animals since that allowed us to isolate sufficient numbers of relatively pure 2n hepatocytes. As can be seen in Fig. 3, the repeat PCR profile of the 2n hepatocytes in both Lig4 WT and Lig4f/flmice was indistinguishable from the PCR profile from the total hepatocyte pool and from the PCR profiles produced from the polyploid hepatocytes. Thus, unlike what is seen in mouse models of other Repeat Expansion Diseases [24, 25], ploidy does not seem to have a role in expansions in the FXD mouse model.
Fig. 3. Hepatocyte ploidy does not affect repeat instability.
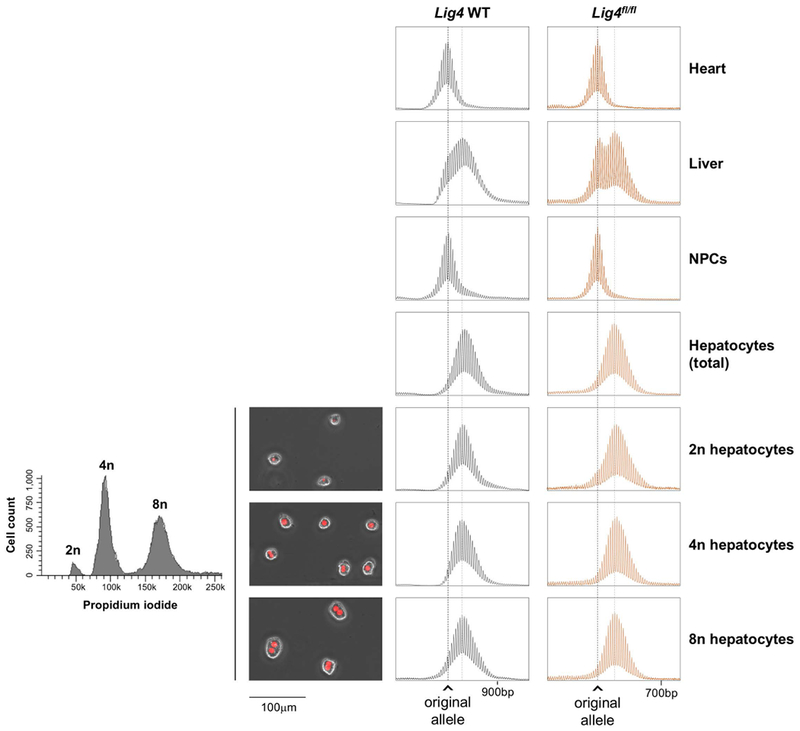
The chart on the left depicts the result of the cell sorting showing the proportion of the diploid and polyploid fractions as assessed by DNA content revealed by PI staining. The adjacent photomicrographs show typical examples of the indicated cell types, with propidium iodide (PI) in red. The right-hand side of the slide shows the Repeat PCR profiles of heart, liver, NPCs, total hepatocytes and diploid and polyploid hepatocyte fractions from a 2-months old Lig4 WT mouse with 260 repeats and a 3-month old Lig4fl/fl mouse with 170 repeats. The black dotted line in each panel indicates the size of the original inherited allele, while the gray dotted line indicates the average size of the expanded alleles.
3.3. Mre11ATLD1/ATLD1 mice do not show altered expansion dynamics.
Since Mre11 null mice are not viable, we crossed our FXD mice with mice carrying an allele of Mre11 that has a premature termination codon at amino acid 633 [12]. In humans this mutation results in the DNA repair deficiency, ataxia-telangiectasialike disorder-1 (ATLD1) (MIM#604391) [10]. In mice, this mutation is associated with significantly reduced levels of MRE11 protein, as well as cell checkpoint defects and increased chromosomal instability [12]. As can be seen in Fig. 4A-C, Mre11ATLD1/ATLD1 mice have no evidence of altered instability in any of the organs tested suggesting that this mutation does not affect somatic instability. Single sperm PCR also showed no significant difference between Mre11ATLD1/ATLD1 and Mre11+/+ mice (Fig. 4D), suggesting that this mutation also does not affect expansion in the germ line either.
Fig. 4. The Mre11ATLD1/ATLD1 mutation does not affect repeat expansion.
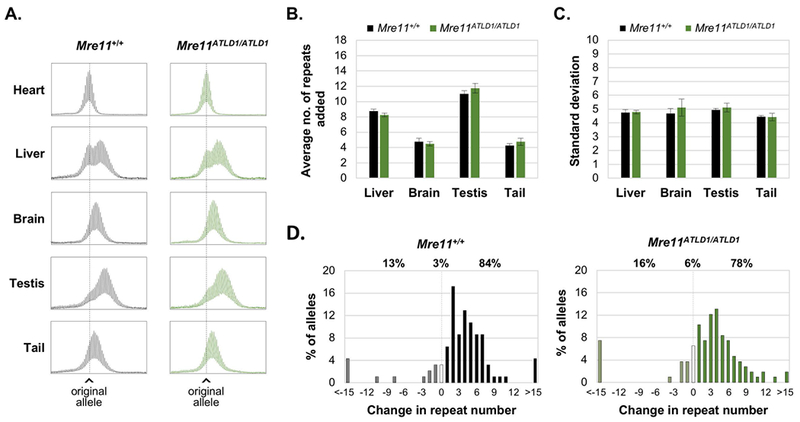
A. Representative repeat PCR profiles for different organs of 6-month old Mre11+/+ and Mre11ATLD1/ATLD1 mice with 160 repeats. The dotted line in each panel indicates the size of the original inherited allele. B. and C. show the average of repeats added and SDs respectively for four 6-month old animals with ~160 repeats. The error bars show the standard error. D. Distribution of the change in the repeat number in sperm collected from 3 Mre11+/+ and 3 Mre11ATLD1/ATLD1 3-month old mice with ~158 repeats. The percentage of alleles that were larger, identical or smaller than the parental allele of each genotype is shown above the graph.
4. Discussion
In this study we have identified LIG4, and thus NHEJ, as protecting against expansion in a mouse model of the FXDs since loss of Lig4 in hepatocytes resulted in an increase in repeat expansions (Fig. 1 and 2). In contrast, a hypomorphic Mre11 mutation (Mre11ATLD1/ATLD1) had no effect on repeat expansion (Fig. 4). While it is possible that the lack of an effect of MRE11 reflects redundancy with other exonucleases, our demonstration that one such nuclease, EXO1, protects against expansion [4], suggests that this is not the case. The effect of Lig4 and Mre11 mutations in the FXD mouse contrasts with what is seen in yeast where rather than increasing expansions, a mutation in Lig4 results in significantly more contractions and a mutation in Mre11, rather than having no effect, actually causes more expansions [27]. In a mouse model of DM1, the loss of DNA-PKcs, another component of the NHEJ pathway, was shown to have no significant effect on expansion [28]. However, this result does not conclusively rule out an effect of NHEJ, since not all end-joining reactions are DNA-PKcs-dependent [29].
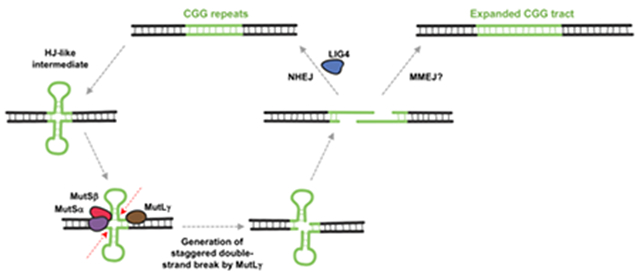
A protective role for NHEJ has important implications for the expansion mechanism. Specifically, it suggests that expansion may proceed via a DSB intermediate that can be either processed via NHEJ to generate a non-expanded allele or processed via a second pathway that results in expansion. While we cannot rule out the possibility that NHEJ is protecting against a form of expansion that is only apparent in the absence of LIG4, the simplest interpretation of our data is that LIG4 is acting downstream of the MMR proteins that we have previously shown to be essential for expansion [2–4, 30]. We thus suggest a model for expansion that accommodates a DSB intermediate as illustrated in Fig. 5. We hypothesize that MutLγ, a protein required for HJ resolution during meiosis that is also essential for expansions [4], recognizes an HJ-like structure formed by the repeat, perhaps a double-hairpin or loop-out structure. Such a structure could arise in a number of different ways, perhaps during repair synthesis or transcription. During transcription for example, R-loop formation [31–34] may provide an opportunity for hairpin formation by the non-template strand. Resolution of the R-loop would then leave the template strand free to form a hairpin of its own. This would explain the dependence on transcription/open chromatin for expansions to occur [35]. However the HJ-like structure is generated, it’s processing results in a staggered DSB with overhangs containing CGG-repeats. As such, the two ends are able to anneal directly without further resectioning. This product could then be processed by NHEJ to complete error-free repair by simple ligation of the staggered break. Alternatively, if the repeats anneal out of register, the resultant gaps may be processed by an alternate pathway that leads to expansions. This alternate pathway may be a simple gap-filling, perhaps similar to the final steps of microhomology-mediated end-joining (MMEJ). The number of repeats added to the expanded allele would correspond to the size of the gap generated by out of register alignment of the 3’ overhangs. In this regard, it is interesting that Polβ, a DNA polymerase important for expansion [15], is very effective at such gap-filling on a synthetic substrate [36]. Since the Polβ-mediated gap-filling can be accompanied by the incorporation of additional repeats [36], this could add to the size of the expanded allele. Since NHEJ and the expansion pathway compete for a common substrate, LIG4 would act to reduce the number of expansions. Since the two ends of the DSB generated by MutLγ within the repeat do not require resectioning in order to anneal, the loss of MRE11 would not be expected to impact these events.
Fig. 5. Model for repeat instability at the FX locus.
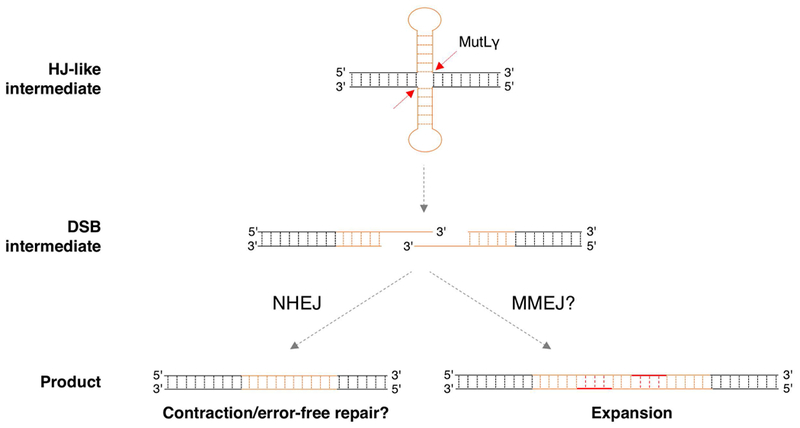
Repair synthesis or transcription could provide an opportunity for secondary structure formation by both strands of the FX repeat. The resultant “double hairpin” may resemble a Holliday junction (HJ) that can be processed by MutLγ to generate a staggered DSB within the repeat. Since the staggered ends each contain CGG repeats they are complementary and can thus anneal without need for end-resectioning. This product could then be processed by NHEJ to generate contractions or to restore the original repeat tract. Out of register annealing of the staggered ends could be processed by an alternative pathway such as gap filling to generate expansions.
In summary, our data suggest that the expansion involves the MRE11- and NHEJ-independent processing of a DSB intermediate. A better understanding of the steps involved may help us identify other factors that may modify expansion and disease risk in humans.
Supplementary Material
{kind=link}
{kind=link}
Highlights.
DNA Ligase IV and thus non-homologous end-joining protects against repeat expansion.
A mutation in MRE11 does not affect repeat expansion.
Repeat expansion likely involves repair of a double-strand break that does not need end resection.
Expansions may arise from simple gap filling of a staggered break.
Acknowledgements
We would like to acknowledge all the hard work done by the staff who take care of our mice and without whom this work would not have been possible. Thank you also to Pradeep Dagur and staff from the NHLBI Flow Cytometry Core for help with the flow sorting of hepatocytes and to Andre Nussenzweig and John Petrini for mice. We also thank all the Usdin lab members for ideas, encouragement and support.
Funding.
This work was supported by the Intramural Program of the NIDDK to KU (DK057808).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
References
- [1].Lozano R, Rosero CA, Hagerman RJ, Fragile X spectrum disorders, Intractable Rare Dis Res, 3 (2014) 134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zhao XN, Lokanga R, Allette K, Gazy I, Wu D, Usdin K, A MutSbeta-Dependent Contribution of MutSalpha to Repeat Expansions in Fragile X Premutation Mice?, PLoS Genet, 12 (2016) e1006190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lokanga RA, Zhao XN, Usdin K, The mismatch repair protein MSH2 is rate limiting for repeat expansion in a fragile X premutation mouse model, Hum. Mutat, 35 (2014) 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhao X-N, Zhang Y, Wilkins K, Edelmann W, Usdin K, Mlh3 promotes while Exo1 protects against repeat expansion in mouse model for the Fragile X-related disorders, PLoS Genet., (2018) In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ranjha L, Anand R, Cejka P, The Saccharomyces cerevisiae Mlh1-Mlh3 heterodimer is an endonuclease that preferentially binds to Holliday junctions, J. Biol. Chem, 289 (2014) 5674–5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Usdin K, Woodford KJ, CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro, Nucleic Acids Res, 23 (1995) 4202–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yu A, Barron MD, Romero RM, Christy M, Gold B, Dai J, Gray DM, Haworth IS, Mitas M, At physiological pH, d(CCG)15 forms a hairpin containing protonated cytosines and a distorted helix, Biochemistry, 36 (1997) 3687–3699. [DOI] [PubMed] [Google Scholar]
- [8].Mitas M, Yu A, Dill J, Haworth IS, The trinucleotide repeat sequence d(CGG)15 forms a heat-stable hairpin containing Gsyn. Ganti base pairs, Biochemistry, 34 (1995) 12803–12811. [DOI] [PubMed] [Google Scholar]
- [9].Nadel Y, Weisman-Shomer P, Fry M, The fragile X syndrome single strand d(CGG)n nucleotide repeats readily fold back to form unimolecular hairpin structures, J Biol Chem, 270 (1995) 28970–28977. [DOI] [PubMed] [Google Scholar]
- [10].Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM, The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder, Cell, 99 (1999) 577–587. [DOI] [PubMed] [Google Scholar]
- [11].Entezam A, Biacsi R, Orrison B, Saha T, Hoffman GE, Grabczyk E, Nussbaum RL, Usdin K, Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model, Gene, 395 (2007) 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Theunissen JW, Kaplan MI, Hunt PA, Williams BR, Ferguson DO, Alt FW, Petrini JH, Checkpoint failure and chromosomal instability without lymphomagenesis in Mre11(ATLD1/ATLD1) mice, Mol. Cell, 12 (2003) 1511–1523. [DOI] [PubMed] [Google Scholar]
- [13].Shull ER, Lee Y, Nakane H, Stracker TH, Zhao J, Russell HR, Petrini JH, McKinnon PJ, Differential DNA damage signaling accounts for distinct neural apoptotic responses in ATLD and NBS, Genes Dev, 23 (2009) 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA, Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase, J Biol Chem, 274 (1999) 305–315. [DOI] [PubMed] [Google Scholar]
- [15].Lokanga RA, Senejani AG, Sweasy JB, Usdin K, Heterozygosity for a hypomorphic Polbeta mutation reduces the expansion frequency in a mouse model of the Fragile X-related disorders, PLoS Genet, 11 (2015) e1005181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hayward BE, Zhou Y, Kumari D, Usdin K, A Set of Assays for the Comprehensive Analysis of FMR1 Alleles in the Fragile X-Related Disorders, J. Mol. Diagn, 18 (2016) 762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhao X, Zhang Y, Wilkins K, Edelmann W, Usdin K, MutLgamma promotes repeat expansion in a Fragile X mouse model while EXO1 is protective, PLoS Genet, 14 (2018) e1007719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lokanga RA, Entezam A, Kumari D, Yudkin D, Qin M, Smith CB, Usdin K, Somatic expansion in mouse and human carriers of fragile X premutation alleles, Hum Mutat, 34 (2013) 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rossi M, Zhu L, McMillin SM, Pydi SP, Jain S, Wang L, Cui Y, Lee RJ, Cohen AH, Kaneto H, Birnbaum MJ, Ma Y, Rotman Y, Liu J, Cyphert TJ, Finkel T, McGuinness OP, Wess J, Hepatic Gi signaling regulates whole-body glucose homeostasis, J Clin Invest, 128 (2018) 746–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bale SS, Geerts S, Jindal R, Yarmush ML, Isolation and co-culture of rat parenchymal and non-parenchymal liver cells to evaluate cellular interactions and response, Sci Rep, 6 (2016) 25329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kegel V, Deharde D, Pfeiffer E, Zeilinger K, Seehofer D, Damm G, Protocol for Isolation of Primary Human Hepatocytes and Corresponding Major Populations of Non-parenchymal Liver Cells, J Vis Exp, (2016) e53069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T, Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice, Curr Biol, 8 (1998) 1395–1398. [DOI] [PubMed] [Google Scholar]
- [23].Mollersen L, Rowe AD, Larsen E, Rognes T, Klungland A, Continuous and periodic expansion of CAG repeats in Huntington’s disease R6/1 mice, PLoS Genet, 6 (2010) e1001242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].van den Broek WJ, Wansink DG, Wieringa B, Somatic CTG*CAG repeat instability in a mouse model for myotonic dystrophy type 1 is associated with changes in cell nuclearity and DNA ploidy, BMC Mol. Biol, 8 (2007) 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lee JM, Pinto RM, Gillis T, St Claire JC, Wheeler VC, Quantification of age-dependent somatic CAG repeat instability in Hdh CAG knock-in mice reveals different expansion dynamics in striatum and liver, PLoS One, 6 (2011) e23647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gentric G, Desdouets C, Celton-Morizur S, Hepatocytes polyploidization and cell cycle control in liver physiopathology, Int J Hepatol, 2012 (2012) 282430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sundararajan R, Gellon L, Zunder RM, Freudenreich CH, Double-strand break repair pathways protect against CAG/CTG repeat expansions, contractions and repeat-mediated chromosomal fragility in Saccharomyces cerevisiae, Genetics, 184 (2010) 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Savouret C, Brisson E, Essers J, Kanaar R, Pastink A, te Riele H, Junien C, Gourdon G, CTG repeat instability and size variation timing in DNA repair-deficient mice, EMBO J, 22 (2003) 2264–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chang HH, Watanabe G, Gerodimos CA, Ochi T, Blundell TL, Jackson SP, Lieber MR, Different DNA End Configurations Dictate Which NHEJ Components Are Most Important for Joining Efficiency, J Biol Chem, 291 (2016) 24377–24389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhao XN, Kumari D, Gupta S, Wu D, Evanitsky M, Yang W, Usdin K, Mutsbeta generates both expansions and contractions in a mouse model of the Fragile X-associated disorders, Hum Mol Genet, 24 (2015) 7087–7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Loomis EW, Sanz LA, Chedin F, Hagerman PJ, Transcription-associated R-loop formation across the human FMR1 CGG-repeat region, PLoS Genet, 10 (2014) e1004294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Groh M, Lufino MM, Wade-Martins R, Gromak N, R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome, PLoS Genet, 10 (2014) e1004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kumari D, Usdin K, Sustained expression of FMR1 mRNA from reactivated fragile X syndrome alleles after treatment with small molecules that prevent trimethylation of H3K27, Hum. Mol. Genet, 25 (2016) 3689–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Abu Diab M, Mor-Shaked H, Cohen E, Cohen-Hadad Y, Ram O, Epsztejn-Litman S, Eiges R, The G-rich Repeats in FMR1 and C9orf72 Loci Are Hotspots for Local Unpairing of DNA, Genetics, 210 (2018) 1239–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Adihe Lokanga R, Zhao XN, Entezam A, Usdin K, X inactivation plays a major role in the gender bias in somatic expansion in a mouse model of the fragile X-related disorders: implications for the mechanism of repeat expansion, Hum Mol Genet, 23 (2014) 4985–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Crespan E, Czabany T, Maga G, Hubscher U, Microhomology-mediated DNA strand annealing and elongation by human DNA polymerases lambda and beta on normal and repetitive DNA sequences, Nucleic Acids Res, 40 (2012) 5577–5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.