

Abstract
Background:
Genome-wide association studies (GWAS) are widely used to map genomic regions contributing to lung cancer (LC) susceptibility but they typically do not identify the precise disease-causing genes/variants. To unveil the inherited causal LC variants, we performed focused exome sequencing analyses on genes located in 121 GWAS loci previously implicated in the risk of LC, chronic obstructive pulmonary disease, pulmonary function level and smoking behavior.
Methods:
Germline DNA from 260 LC cases and 318 controls were sequenced utilizing VCRome 2.1 exome capture. Filtering was based upon enrichment of rare and potential deleterious variants in cases (risk alleles) or controls (protective alleles). Allelic association analyses of single variant and gene-based burden tests of multiple variants were performed. Promising candidates were tested in two independent validation studies with a total of 1,773 cases and 1,123 controls.
Results:
We identified 48 rare variants with deleterious effects in the discovery analysis and validated 12 of the 43 candidates that were covered in the validation platforms. The top validated candidates included one well-established truncating variant BRCA2 K3326X (OR 2.36, 95% CI 1.38 - 3.99) and three newly identified variations: LTB p.Leu87Phe (OR 7.52, 95% CI 1.01 - 16.56), P3H2 p.Gln185His (OR 5.39, 95% CI 0.75 - 15.43), and DAAM2 p.Asp762Gly (OR 0.25, 95% CI 0.10 - 0.79). Burden tests revealed strong associations between ZNF93, DAAM2, BRD9, and LTB genes and LC susceptibility.
Conclusion:
Our results extend the catalogue of regions associated with LC and highlight the importance of germline rare coding variants in LC susceptibility.
Keywords: Exome Sequencing, Rare Variants, Lung Cancer (LC)
INTRODUCTION
While over 80% of lung cancers (LC) are attributed to smoking, only about 15% of smokers develop LC. Therefore it remains of great importance to understand the genetic factors that contribute to LC risk. It is well recognized that tobacco-induced chronic obstructive pulmonary disease (COPD) is an important predictor of LC risk. Genome-wide association studies (GWAS) have identified 45 genome-wide significant loci for LC, 22 loci for COPD, 32 loci for smoking behavior (SM), and 63 loci for pulmonary function (PF) levels, totaling 121 unique susceptibility loci (Supplemental Table 1). Interestingly, there is considerable overlap among these susceptibility loci and genes for these phenotypes (LC, COPD, SM, and PF levels). For example, the 6p21-22, 15q24-25.1, and 19q13.2 regions are shared by all four phenotypes, 5p15.33, 6p21.32, 10q23.31, and 10q25 are shared by three phenotypes, and 15 loci shared by two phenotypes (Supplemental Table 1).
While GWAS have been successful in identifying common (minor allele frequency [MAF] > 5%) variants of small effect, the overall amount of LC heritability explained by these known common variants remains small. Further, since the tagSNPs used in GWAS are used to identify genomic regions of interest rather than being selected for causality, identification of the functional variant at a specific locus generally poses a significant challenge. For example, of the 93 common LC-GWAS tophits from the 45 reported susceptibility loci1, only two are protein-coding (CHRNA3 p.Tyr215 and CHRNA5 p.Asp398Asn), and 91 variants fall in non-coding regions (four in UTR, seven in flanking, 70 in intron, and ten in intergenic regions). Alleles that are functionally deleterious will tend to be underrepresented at high frequencies, an assertion supported by the observation of a relationship between putative functionality and MAF. Recent studies suggest that multiple low-frequency (1% < MAF < 5%) or rare (MAF < 1%) variants exhibit stronger effect sizes (odds ratio [OR]) than common variants and contribute to the missing heritability2. Supporting this hypothesis is the observation that several genes containing known low-frequency or rare variants of moderate-to-large effect are associated with LC, for example, PARK2 p.Arg275Trp3, BRCA2 p.Lys3326X and CHEK2 p.Ile157Thr4, CCDC147 p.Arg696Cys and DBH p.Val26Met5.
To unveil the inherited germline rare variants, we employed whole exome sequencing with a focused analysis on the known 121 high priority GWAS susceptibility loci (260 potential target genes). Smoking, family history of LC and COPD are all well-documented risk factors for LC. To efficiently identify the most probable causative variants and genes, we have sequenced selective LC cases with extreme phenotypes (high-risk familial LC patients, sporadic cases reporting heavy smoking histories and or severe COPD) and controls reporting heavy smoking histories but with normal spirometry who are considered resistant to the effects of smoking.
METHODS
Study Population in Discovery
LC cases were derived from four independent case series including sporadic cases from I). Baylor College of Medicine (BCM, n = 68)6–8, II). Harvard School of Public Health (HSPH, n = 101), III). MD Anderson Cancer Center (MDACC, n = 37), and familial cases from IV). Genetic Epidemiology of LC Consortium (GELCC, n = 54, each familial case was chosen from one high-risk LC family that has three or more affected first-degree members)5,9. All cases are white and had histologically confirmed non-small cell LC. Patient clinical and demographic information, such as smoking history (status and pack-years [PY]), was obtained using self-administered questionnaires. For sporadic LC patients, moderate-to-severe COPD phenotype was carefully defined by PF tests (reduced Forced Expiratory Volume in 1 second [FEV1] < 80% predicted, and FEV1/FVC < 0.7). For familial cases, COPD phenotyping data was not available.
The smoking controls were selected from two independent studies: I). Genetic Epidemiology of COPD Study (COPDGene, n = 298) with 10,192 current or ex-smokers, which is a multicenter investigation to examine the genetic epidemiology of COPD and smoking-related lung diseases10; II). BCM COPD and LC study (n = 20), which enrolled current- or former- smokers that was launched in 2002 within the Texas Medical Center in Houston, Texas6–8. All subjects underwent study-related testing that included spirometry, CT scan of the chest, and blood collection. Controls were selected to be white, resistant smokers with normal PF data (defined as post-bronchodilator FEV1 ≥ 80% predicted, FEV1/FVC ≥ 0.7), and with cigarette smoking histories ≥ 10 PY.
DNA was isolated from peripheral blood or saliva from both LC patients and controls. The study was approved by the institutional review board of all sites accruing participants and by the institutional review board at BCM for exome sequencing conducted at the Human Genome Sequencing Center (HGSC).
Library Preparation, Capture Enrichment and Exome Sequencing
DNA samples were constructed into Illumina paired-end pre-capture libraries according to the manufacturer’s protocol. The complete library and capture protocol, as well as oligonucleotide sequences have been described in detail previously11,12. For exome capture, each library pool was hybridized in solution to the BCM-HGSC designed VCRome 2.1 probe set (Roche NimbleGen) according to the manufacturer’s protocol. This exome capture probe set targets the Vertebrate Genome Annotation (Vega), Consensus Coding Sequence project (CCDS), and RefSeq gene models, with 45.2 Mb capture targeting 23,585 genes. Exome sequencing was performed in paired-end mode using the Illumina HiSeq 2000 platform. Sequencing runs generated approximately 300-400 million successful reads on each lane of a flow cell, yielding 7–13 Gb per sample. For exome sequencing yields, samples achieved an average depth of coverage of 200X over exonic regions. Sequence analysis was performed using the BCM-HGSC Mercury analysis pipeline13. All sequence reads were mapped to the GRCh37 Human reference genome using the Burrows-Wheeler aligner (BWA)14. Putative variants, including single nucleotide variants (SNVs), insertions or deletions (Indels), were called using the Atlas2 suite15. Read qualities were recalibrated with GATK and a minimum quality score of 30 was required; also, the variant must have been present in > 15% of the reads that cover the position.
Single Rare Variant Filtering and Functional Annotation
Our analysis was restricted to rare variants mapping within the exonic regions of the 121 known GWAS loci (Supplemental Table 1 for genomic coordinates and 260 target genes). Variants were annotated for effect on the protein and predicted function using the SNP & Variation Suite (SVS, Golden Helix, Inc). To identify pathogenic variants, a three-step filtering protocol was designed utilizing automated filtering followed by manual review (Figure 1)
Figure 1.
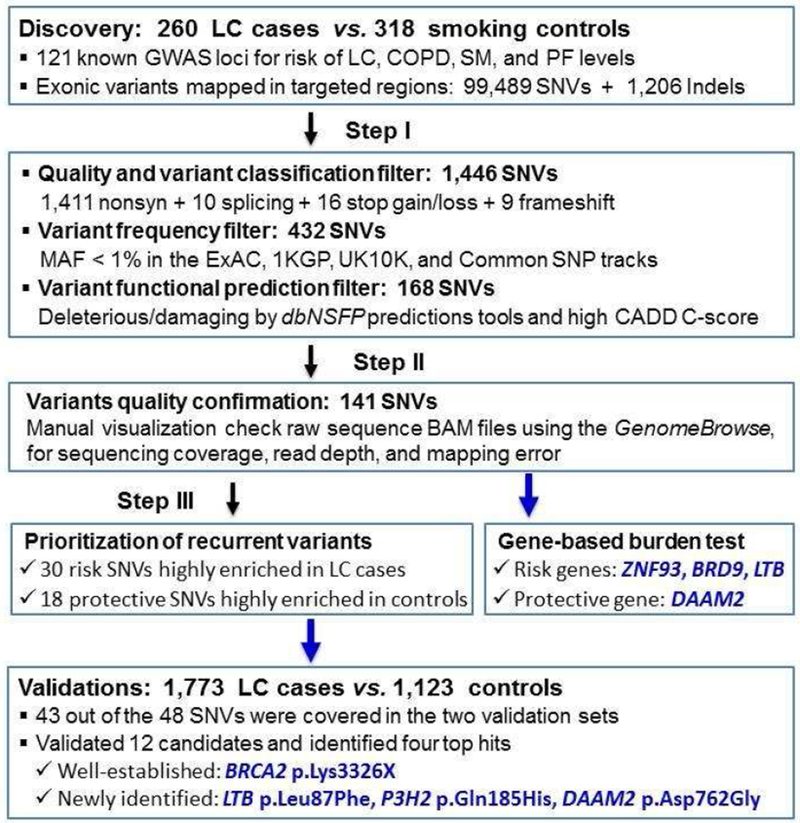
Workflow and Annotation Pipeline for the Identification of Candidate Variants
Abbreviations: GWAS, Genome-wide association studies; LC, lung cancer; SNV, Single nucleotide variants; Indels, Insertions or deletions; MAF, minor allele frequency; ExAC, Exome Aggregation Consortium; 1KGP, The 1000 Genomes Project; dbNSFP, database for non-synonymous SNPs functional predictions; CADD, Combined Annotation Dependent Depletion
- Automated filtering identified variants that fulfilled the following four criteria:
- Mutation type, including missense and disruptive (defined as nonsense, stop-gain/loss, splice site destructions and frame-shift Indels, which severely disrupt protein structure);
- Mutation effects, i.e., the variant is predicted to result in truncation of the protein, or it is predicted to be damaging/deleterious (not to be benign/tolerated) to the protein using SIFT, PolyPhen-2, Mutation taster and scaled C-scores from the Combined Annotation-Dependent Depletion (CADD) method 16 that strongly correlates with both molecular functionality and pathogenicity;
- The MAF < 1% in the Europeans in the reference databases including Exome Aggregation Consortium (ExAC), 1000 Genomes Project (1KGP), UK10K project, and UCSC Common SNPs tracks. Novel variants were defined as never having been reported in a publicly available database and UCSC All SNPs 135/137/141 tracks.
After implementing the above automated filtering schema, manual review of the raw BAM files were then performed using the GenomeBrowse (Golden Helix, Inc). This filter was used to remove the false-positive events that result from mapping errors, and mutations found in a “noisy” background (multiple mismatches or Indels in flanking sequences). These highly rare and predicted deleterious mutations were used to perform the gene-based burden analysis.
We further prioritized candidate variants that are highly enriched in the case group (risk alleles) or the control group (protective alleles).
Gene-based Burden Analysis of Multiple Rare Deleterious Variants
To have greater power to detect significant associations to rare variants, we performed gene-based collapsing tests for those genes that included ≥ 2 rare and predicted deleterious variants (from filtering steps I and II), including the Combined Multivariate and Collapsing (CMC) test and the Kernel-Based Adaptive Cluster (KBAC) test17,18. To measure their cumulative effect, the CMC first bins variants according to MAF criterion (thresholds at 1%, 0.1% and 0.01%) based on the observed data, then collapses the multiple variants within each bin and finally uses Hotelling’s T2 to perform multivariate testing on the counts across the various bins. The KBAC test first counts multi-marker genotypes within a given gene based on the variant data, and then performs a special case/control test based on the weighted sum of these allele counts. To account for multiple comparisons, we calculated False Discovery Rate (FDR) 19 adjusted P-values.
Study Population in Validation
To discover robust associations and validate the promising candidates, we analyzed two independent sets: I). Wayne State University (WSU) study which enrolled at Karmanos Cancer Institute or Henry Ford Health System. Study participants either underwent spirometry or had PF test data abstracted from medical records20. We carefully selected LC cases with ≥ 10 PY, and smoking controls with normal PF (FEV1 ≥ 80% predicted, and FEV1/FVC ≥ 0.7) and ≥ 10 PY. Genotyping was performed using the Illumina MEGA panel which includes > 1.7 million variants. II). Transdisciplinary Research in Cancer of the Lung team of the International Lung Cancer Consortium (TRICL-ILCCO) study. Subjects were selected from four sites in the TRICL-ILCCO: HSPH, International Agency for Research on Cancer (IARC), University of Liverpool, and Mount Sinai Hospital and Princess Margaret Hospital (MSH-PMH) in Toronto. Exome capture (Agilent SureSelect XT Custom ELID and Whole Exome v5) and sequencing were performed at the Center for Inherited Disease Research (CIDR). Both validation studies were approved by the institute ethics review committees, and all participants provided written informed consent.
Allelic Association Analysis in the Combined Datasets
We then tabulated the minor allele and the reference allele counts per candidate in the combined discovery and validation datasets, and performed allelic association analysis which compares frequencies of alleles in LC cases vs. controls, and LC cases vs. ExAC reference population (non-Finnish Europeans, n = 33,370). We note that the individuals in the reference set are not necessarily healthy – many have adult-onset diseases such as type 2 diabetes and schizophrenia. Since the numbers of mutation carriers are small (< 5), Fisher exact tests were used for the allelic association analysis. ORs, 95% confidence intervals (CIs) and FDR adjusted P values were calculated.
RESULTS
Demographic and clinical information including age, gender, smoking history, histology and PF data are summarized in Table 1. The discovery set included 260 LC cases (54 familial and 206 sporadic cases; 75/206 sporadic cases also had moderate-to-severe COPD and 318 smoking controls with ≥ 15 PY and normal PF data. The validation populations (WSU and TRICL-ILCCO) included 1,773 cases and 1,123 controls. Among the combined 2,033 cases and 1,441 controls of European descent, 129 cases (6%) and 303 (21%) controls were nonsmokers, mostly from the TRICL-ILCCO study. In terms of smoking intensity (mean PY), in the discovery, lower PY was reported in LC cases than in controls (mean 46 vs 54, P < 0.001), whereas much higher PY were reported in cases than controls in the two validation studies (mean 52 vs. 35 in WSU, and 43 vs 23 in TRICL-ILCCO, respectively; both P < 0.001). Regarding LC histology, adenocarcinoma was the most common type across three datasets, with 52% in discovery, 51% and 44% in two validations, respectively.
Table 1.
Basic Characteristics of LC Cases and Controls in the Discovery and Validations
| Characteristics | Discovery | Validation: WSU Study | Validation: TRICL-ILCCO Study | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Case (n = 260) * |
Smoking control (n = 318) # |
P value & | Case (n = 831) |
Smoking control (n = 266) # |
P value & | Case (n = 942) |
Control (n = 857) # |
P value & | |
| Age, year | |||||||||
| Mean (SD) | 64 (6.3) | 62.6 (4.9) | 0.258 | 63.6 (9.8) | 59.5 (9.1) | <0.001 | 62.4 (12.3) | 60.8 (11.8) | 0.006 |
| Range | 30 - 87 | 55 - 80 | 31 - 88 | 35 - 86 | 23.8 - 91 | 19.8 - 90 | |||
| Sex | |||||||||
| Male (%) | 165 (63.4) | 172 (54.1) | 0.039 | 398 (47.9) | 129 (48.5) | 0.920 | 515 (54.7) | 498 (58.1) | 0.15 |
| Female (%) | 95 (39.7) | 146 (45.9) | 433 (52.1) | 137 (51.5) | 427 (45.3) | 359 (41.9) | |||
| Smoking status | |||||||||
| Never (%) | 21 (8.1) | - | <0.001 | - | - | <0.001 | 108 (11.5) | 303 (35.6) | <0.001 |
| Former (%) | 163 (62.7) | 156 (49.1) | 372 (44.8) | 153 (57.5) | 380 (40.4) | 357 (41.9) | |||
| Current (%) | 76 (29.2) | 162 (50.9) | 459 (55.2) | 113 (42.5) | 450 (47.9) | 194 (22.5) | |||
| Smoking pack-years | <0.001 | ||||||||
| Mean (SD) | 45.5 (32.9) | 53.6 (18.4) | <0.001 | 51.8 (30.0) | 35.3 (20.5) | <0.001 | 42.6 (28.0) | 22.8 (18.6) | - |
| Range | 0-165 | 10 - 97 | 10 - 216 | 10 – 124 | 0-196 | 0-105 | |||
| FEV1 (% pred) # | |||||||||
| Mean (SD) | 69.1 | 93.9 (10.5) | <0.001 | 68.6 (20.4) | 94.5 (10.3) | <0.001 | - | - | - |
| Range | 22-124 | 80 - 129.1 | 15 - 135.1 | 80 - 123.2 | - | - | |||
| FEV1/FVC # | |||||||||
| Mean (SD) | 0.59 | 0.77 (0.05) | <0.001 | 0.66 (0.12) | 0.79 (0.04) | < 0.001 | - | - | - |
| Range | 0.27-0.94 | 0.70 - 0.9 | 0.27-0.97 | 0.70-0.94 | - | - | |||
| Histology | |||||||||
| Adenocarcinoma | 136 (52.3) | - | - | 415 (51.3) | - | - | 325 (44.3) | - | - |
| Squamous | 82 (31.5) | - | 176 (21.8) | - | 248 (33.8) | - | |||
| Other | 42 (16.2) | - | 218 (26.9) | - | 161 (21.9) | - | |||
Of the 260 LC cases, 54 were unrelated familial cases; and 75 out of the 206 sporadic LC cases also had severe COPD.
Controls with normal pulmonary function are defined as FEV1 > 80% and FEV1/FVC > 0.7 predicted. These data are not available for familial cases in the discovery and the TRICL-ILCCO study subjects.
P value from the two-sided chi-square test (for categorical variables) and Student’s t test (for continuous variables).
Analysis of Recurrent Rare and Deleterious Variants
In the discovery set, of 99,489 SNVs and 1,206 Indels mapped in the exons of the target 121 known loci (260 genes), 1,446 were functional mutation types (1,411 nonsynonymous SNVs [nsSNVs], 10 splice-sites, 16 stop gain/loss, and nine frameshifts), 432 of these were rare, and 168 were further predicted to be potential deleterious. Our stepwise filtering strategy (Figure 1 and Table 2) identified 48 recurrent candidate variants of which 30 were highly enriched in LC patients (risk-conferring) and 18 enriched in controls (protective), including three stop-gains, three splice-sites, and 42 nsSNVs. These 48 candidates were located in 33 genes at 25 of the risk loci, and presented in total 68 smoking controls and 85 patients (17 familial and 68 sporadic cases; 8 sporadic carriers had severe COPD; 70% were adenocarcinoma histology). Among the candidates carriers, 13 cases (two familial and 11 sporadic patients; none of them had severe COPD) and eight controls were multi-carriers whom had carried ≥ 2 candidates (Supplemental Table 2). It is interesting to note, four out of the 13 multi-carriers patients had p.Gly337Glu in ZNF93 (Zinc-finger 93, OMIM # 603975). In particular, one adenocarcinoma patient (age 60, male, 30 PY) was a carrier of four candidates, including two candidates from ZNF93. For the controls, six out of eight multi-carriers carried 1~2 candidates from DAAM2 (Disheveled-associated activator of morphogenesis 2, OMIM # 606627).
Table 2.
Candidate Rare Deleterious Variants Identified in the Discovery and Tested in the Validations
| Known Association (25 loci) |
Gene (33 genes) |
Variant (48 SNVs) |
Identifier RS ID |
Ref/ Alt |
CADD C* |
MAF% | N. carriers in Case / Control & | |||
|---|---|---|---|---|---|---|---|---|---|---|
| ExAC # (33,370) |
Case / Control (2,033 / 1,441) |
Discovery ‡ (260 / 318) |
WSU Study (831 / 266) |
TRICL-ILCCO Study (942 / 857) |
||||||
| 1q23.2 (LC) | DUSP23 | Cys95Ser | rs147728803 | G/C | 31 | 0.07 | 0.10 / 0.31 | 0 / 4 C1 | 0 / 1 | 4 / 4 |
| 2q36.3 (PF) | COL4A4 | Pro1587Arg | rs190148408 | G/C | 20 | 0.27 | 0.32 / 0.38 | 0 / 5 | 6 / 1 | 7 / 5 |
| COL4A3 | Pro1109Ser | rs55816283 | C/T | 17 | 0.56 | 0.52 / 0.48 | 0 / 6 C2,7 | 8 / 3 | 13 / 5 | |
| 3p24.1 (LC_PF) | ZCWPW2 | Asn23Ser | rs148504648 | A/G | 13 | 0.33 | 0.19 / 0.45 | 0 / 4 C3 | 1 / 2 | 7 / 7 |
| DPPA2 | Ala157Ser | rs144052288 | C/A | 17 | 0.22 | 0.37 / 0.34 | 5 S8 / 1 | - | 4 / 7 | |
| 3q13.13 (COPD_SM) | DZIP3 | Gly67Cys | rs745923043 | G/T | 29 | 0.003 | 0.08 / 0.04 | 2 S1,2 / 0 | - | 0 / 1 |
| Pro990Leu | rs140068430 | C/T | 26 | 0.03 | 0.07 / 0 | 2 S3 / 0 | 0 / 0 | 1 / 0 | ||
| 3q28 (LC_SM) | P3H2 | Gln185His | rs117688924 | C/A | 27 | 0.06 | 0.19 / 0.03 | 3 / 0 | 2 / 1 | 3 / 0 |
| 4p16.1 (LC) | DRD5 | Met75Thr | rs151282040 | T/C | 19 | 0.18 | 0.12 / 0.14 | 4 S5 / 1C4 | 0 / 0 | 1 / 3 |
| Cys335X | rs145497708 | C/A | 36 | 0.23 | 0.23 / 0.09 | 4 / 1 | 1 / 0 | - | ||
| 5p15.33 | BRD9 | Splice 3’ | rs201402002 | T/C | 16 | 0.17 | 0.33 / 0.09 | 5 S1 / 0 | - | 3 / 2 |
| (LC_COPD_SM_PF) | SLC12A7 | Splice 5’ | rs150315797 | G/A | 13 | 0.06 | 0.12 / 0 | 3 S6 / 0 | - | 0/0 |
| DAAM2 | Arg172His | rs200589550 | G/A | 31 | 0.31 | 0.22 / 0.45 | 1 S3 / 6 C1 | 4 / 3 | 4 / 4 | |
| 6p21.2 (PF) | Pro555Leu | rs201570348 | C/T | 25 | 0.25 | 0.15 0.14 | 0 / 3 C4 | 3 / 0 | 3 / 1 | |
| Asp762Gly | rs200287086 | A/G | 24 | 0.28 | 0.10 / 0.38 | 0 / 6 C2,3,5,6 | 3 / 1 | 1 / 4 | ||
| 6p21.33 | LTB | Leu87Phe | rs4647187 | G/A | 23 | 0.09 | 0.27 / 0.03 | 3 / 0 | 2 / 0 | 6 / 1 |
| (LC_COPD_SM_PF) | SAPCD1 | Gln76X | rs139815351 | C/T | 35 | 0.15 | 0.24 / 0.14 | 4 S5 / 1 | 3 / 1 | 3 / 2 |
| 8q11.21 (PF) | SNTG1 | Splice 5’ | rs201831443 | G/T | 14 | 0.11 | 0.08 / 0.13 | 2 S1,3 / 0 | - | 0 / 3 |
| Val121Leu | rs138262840 | G/C | 27 | 0.20 | 0.42 / 0.24 | 3 S6 / 0 | 5 / 4 | 9 / 3 | ||
| 9q22.32 (PF) | PTCH1 | Asp436Asn | rs142274954 | C/T | 23 | 0.11 | 0.15 / 0.03 | 2 / 0 | 2 / 0 | 2 / 1 |
| 9q34.2 (SM) | DBH | Val195Met | rs145059403 | G/A | 28 | 0.09 | 0.05 / 0.10 | 0 / 3 | 1 / 0 | 1 / 0 |
| IFIT3 | Leu390Arg | rs116926108 | T/G | 24 | 0.12 | 0.17 / 0.34 | 3 S2,7 / 0 | 1 / 1 | 3 / 4 | |
| 10q23 | CEP55 | Arg191Gln | rs368583889 | G/A | 34 | 0.002 | 0.38 / 0 | 2 / 0 | - | - |
| (LC_COPD_SM) | Glu321Lys | rs146992036 | G/A | 28 | 0.26 | 0.15 / 0.03 | 3 S11 / 0 | 1 / 1 | 2 / 0 | |
| PLCE1 | Thr467Ile | rs192219615 | C/T | 25 | 0.16 | 0 / 0.34 | 0 / 4C5 | 0 / 1 | 0 / 0 | |
| CCDC147 | Arg696Cys | rs41291850 | C/T | 28 | 1.09 | 0.84 / 0.59 | 3 S7 / 0 | 14 /1 | 17 / 16 | |
| 10q25.1 (LC_SM_PF) | ITPRIP | Arg181Trp | rs151176986 | G/A | 27 | 0.15 | 0.12 / 0.55 | 2 / 0 | 1 / 1 | 2 / 7 |
| Asp236Tyr | rs372849615 | C/A | 23 | 0.003 | 0.12 / 0 | 2 S11 / 0 | - | 1 / 0 | ||
| 11p14.1 (SM) | BDNF | Thr2Ile | rs8192466 | G/A | 24 | 0.15 | 0.15 / 0.10 | 3 F1 / 0 | 1 / 0 | 2 / 3 |
| 12p13.33 (LC) | RAD52 | Arg396Cys | rs112677599 | G/A | 15 | 0.15 | 0.17 / 0.55 | 3 / 0 | 1 / 1 | 3 / 7 |
| 13q12.12 (LC) | MIPEP | Leu197Pro | rs150167906 | A/G | 32 | 0.11 | 0.27 / 0.10 | 3 / 1 | 3 / 1 | 5 / 1 |
| BRCA2 | Phe12Ser | rs587782872 | T/C | 23 | novel | 0 / 0.31 | 0 / 2 | - | - | |
| Tyr42Cys | rs4987046 | A/G | 15 | 0.22 | 0.12 / 0.55 | 4 F2 / 1 C6 | 1 / 2 | 0 / 5 | ||
| 13q13.1 (LC) | Gly1433Trp | rs1036091086 | G/T | 24 | 0.003 | 0 / 0.31 | 0 / 2 | - | - | |
| Lys3326X | rs11571833 | A/T | 38 | 0.90 | 1.47 / 0.62 | 7 / 4 | 22 / 3 | 31 / 11 | ||
| 16q21 (PF) | CCDC113 | Lys100Asn | rs144246110 | A/T | 21 | 0.34 | 0.27 / 0.24 | 1 / 5 C8 | 1 / 0 | 9 / 2 |
| ADAMTS18 | Gln146His | rs151326659 | C/G | 23 | 0.17 | 0.25 / 0.34 | 0 / 3 | - | 6 / 5 | |
| 16q23.1 (PF) | Arg1053Trp | rs148703569 | G/A | 28 | 0.23 | 0.22 / 0.42 | 0 / 4 C5,8 | 3 / 2 | 6 / 6 | |
| CLEC3A | Ala66Pro | rs150149068 | G/C | 24 | 0.29 | 0.54 / 0.13 | 3/0 | - | 10 / 3 | |
| 18p11.3 (LC) | LAMA1 | Gly967Asp | rs141851670 | C/T | 26 | 0.25 | 0.24 / 0.14 | 2 / 0 | 5 / 0 | 3 / 4 |
| Gly1227Arg | rs776158943 | C/T | 25 | 0.008 | 0.38 / 0 | 2 F2 / 0 | - | - | ||
| 19p12 (SM) | ZNF93 | Gly337Glu | rs 145491369 | G/A | 26 | 0.61 | 1.08 / 0.42 | 6 S4,8,9,10 / 1 | - | 20 / 9 |
| Lys388Asn | rs140935689 | G/C | 24 | 0.08 | 0.17 / 0.10 | 3 S8,9,10 / 0 | 0 / 1 | 4 / 2 | ||
| 20q13.33 ( LC) | RTEL1 | Gln397Glu | rs150285674 | C/G | 24 | 0.06 | 0.17 / 0.07 | 2 F1,S8 / 0 | 3 / 1 | 2 / 1 |
| Met652Thr | rs148080505 | T/C | 16 | 0.03 | 0.10 / 0.10 | 0 / 3 C7 | 0 / 0 | 4 / 0 | ||
| 22q12.1 (LC) | CHEK2 | Ile157Thr | rs17879961 | A/G | 21 | 0.47 | 0.49 / 0.62 | 1 / 4 | 5 / 0 | 14 / 14 |
| Met424Val | rs375130261 | T/C | 26 | 0.005 | 0 / 0.31 | 0 / 2 | - | - | ||
| 22q12.2 (LC) | MTMR3 | Pro1192His | rs773098171 | C/A | 29 | 0.006 | 0.08 / 0 | 2 / 0 | - | 0 / 0 |
The CADD (combined annotation-dependent depletion) C-score is the overall measure of deleteriousness, ≥ 20 indicates the top 1%, and ≥ 30 indicates the top 0.1% in the human genome.
MAF% were reported for the non-Finnish Europeans in ExAC database, n = 33,370.
Of the 48 SNVs, 15 were not covered in the WSU Study, six were not covered in TRICL-ILCCO Study, and five were not covered by both validation sets, shown as “-”. The MAF% of these SNVs was based on the available cases and controls.
In the discovery, 13 LC cases and 8 controls carrying multiple candidates (see details in Supplemental Table 2); Entries followed by superscript “C” refers to the same control subject, “F” to the same familial cases, and “S” to the same sporadic cases.
In the validation sets, of the 48 candidates, five (10%) were not covered by both validation studies. Specifically, 15 (31%) were not covered by the WSU study, while six (13%) were not covered in the TRICL-ILCCO study. As shown in Table 3, the top most risk-conferring variant from the allelic association analysis is a known stop codon, p.Lys3326X (K3326X) in BRCA2 (OMIM # 600185). This stop gain results from A > T transversion in the 27th exon that leads to the loss of the final 93 amino acids (AAs) of the BRCA2 protein (UniProt # P51587). The MAF of K3326X in cases is significantly higher than controls (MAF 1.47% vs. 0.62%; OR 2.36, 95% CI 1.38 - 3.99) and ExAC population (MAF 1.47% vs. 0.9%; OR 1.68, 95% CI 1.29 - 2.20). This truncating variant has a highest scaled CADD C-score of 38 and predicted to be in the top 0.1% most deleterious substitutions in the human genome. The K3326X occurred in the highly conserved COOH-terminal domain which plays a critical role in the homology-directed repair of DNA double strand breaks.
Table 3.
Top Hits from Allelic Association Analysis of Combined Discovery and Validation Sets
| Candidate Variants | Case / Control / ExAC * (N = 2,033 / 1,441 / 33,370) |
Allelic OR (95% CI) and FDR adjusted P value † | |||||
|---|---|---|---|---|---|---|---|
| N. Minor allele | MAF% | Case vs. Control | Case vs. ExAC | ||||
| Strong association | |||||||
| BRCA2 | Lys3326X | 60 / 18 / 602 | 1.47 / 0.62 / 0.90 | 2.36 (1.38-3.99) | 0.0004 | 1.68 (1.29-2.20) | 0.0002 |
| LTB | Leu87Phe | 11 / 1 / 57 | 0.27 / 0.03 / 0.09 | 7.52 (1.01-16.56) | 0.008 | 3.07 (1.61-5.85) | 0.001 |
| P3H2 | Gln185His | 8 / 1 / 40 | 0.19 / 0.03 / 0.06 | 5.39 (0.75-15.43) | 0.032 | 3.33 (1.56-7.12) | 0.003 |
| DAAM2 | Asp762Gly | 4 / 11 / 34 | 0.10 / 0.38 / 0.27 | 0.25 (0.10-0.79) | 0.007 | 0.34 (0.11-0.94) | 0.011 |
| ZNF93 | Gly337Glu # | 26 / 10 / 404 | 1.08 / 0.42 / 0.61 | 2.51 (1.22-5.20) | 0.005 | 1.82 (1.22-2.72) | 0.003 |
| CLEC3A | Ala66Pro # | 13 / 3 / 195 | 0.54 / 0.13 / 0.29 | 4.18 (1.19-11.69) | 0.008 | 1.89 (1.08-3.31) | 0.021 |
| BRD9 | Splice acceptor # | 8 / 2 / 14 | 0.33 / 0.09 / 0.17 | 3.77 (0.95-10.41) | 0.041 | 2.28 (0.97-4.93) | 0.039 |
| Suggestive signal | |||||||
| PLCE1 | Thr467Ile ‡ | 0 / 5 / 109 | 0 / 0.34 / 0.16 | 0.15 (0.01-1.02) | 0.053 | 0.15 (0.10-0.75) | 0.013 |
| MIPEP | Leu197Pro | 11 / 3 / 76 | 0.27 / 0.10 / 0.11 | 2.58 (0.87-9.22) | 0.059 | 2.41 (1.28-4.53) | 0.007 |
| RTEL1 | Gln397Glu | 7 / 2 / 41 | 0.17 / 0.07 / 0.06 | 2.45 (0.52-11.76) | 0.065 | 2.83 (1.27-6.27) | 0.009 |
| SNTG1 | Val121Leu | 17 / 7 / 126 | 0.42 / 0.24 / 0.20 | 1.72 (0.71-4.11) | 0.118 | 2.13 (1.28-3.54) | 0.004 |
| ZNF93 | Lys388Asn | 7 / 3 / 50 | 0.17 / 0.10 / 0.08 | 1.63 (0.43-6.27) | 0.152 | 2.34 (1.06-5.16) | 0.028 |
The non-Finnish Europeans in ExAC database, n = 33,370.
These variants were not covered in the WSU Study, the allele counts were based on the discovery and TRICL-ILCCO validation sets, including 1,202 LC cases and 1,175 controls.
This variant was absented in LC case, we thus added 0.5 to each cell in the analysis.
P values were calculated by Fisher’s exact test and bolded if significant after FDR adjustment.
Another two top candidates were missense variants, p.Leu87Phe in LTB (Lymphotoxin Beta, OMIM # 600978) and p.Gln185His in P3H2 (Prolyl 3-Hydroxylase 2, also known as LEPREL1; OMIM # 610341). Both variants were carried by only one control (MAF 0.034%), but occurred in 11 and eight cases (MAF 0.27% and 0.19%), respectively, with effect size of 7.52 (95% CI 1.01 - 16.56) and 5.39 (95% CI 0.75 - 15.43), respectively. Likewise, these two variants were exceedingly rare in ExAC (MAF 0.09% and 0.06, respectively) and predicted to be in the 1% most deleterious (CADD scores 23 and 27, respectively). The LTB p.Leu87Phe occurred at the 3rd exon of the gene and a remarkably conserved β-strand structure which links the Transmembrane and TNF domains of the protein (UniProt # Q06643, Figure 2A); The P3H2 p.Gln185His is located in the 2nd exon of the gene, and between the 2nd and 3rd Tetratricopeptide-like helical repeats of the protein (UniProt # Q8IVL5, Figure 2B).
Figure 2.
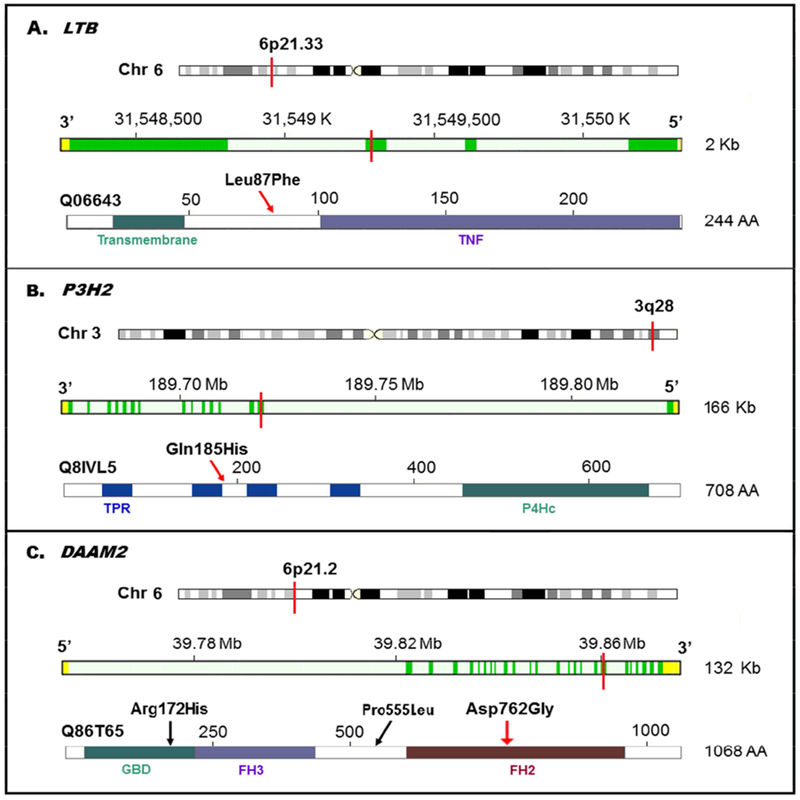
Chromosomal Position, Gene Exon, Protein Domain(s), and the Top Candidates
A. LTB p.Leu87Phe located in the 3rd exon, the β-strand which links the Transmembrane and TNF domains;
B. P3H2 p.Gln185His located in the 2nd exon, between the 2nd and 3rd Tetratricopeptide-like helical repeat (TPR) domains;
C. DAMM2 p.Asp762Gly located the 18th exon, the 2nd Formin Homology (FH) domain.
The top candidate mutations were indicated with red lines in the chromosome and gene exons (genomic location, assembly GRCh37), and red arrows in the protein. The gene annotation also shows forward (DAMM2) or reverse (LTB and P3H2) strand of the chromosome.
In contrast to the above three risk-conferring variants, we also identified one protective variant, p.Asp762Gly in DAAM2. The LC risk for DAAM2 p.Asp762Gly carriers decreased 4-fold compared to study controls (OR 0.25, 95% CI 0.10 - 0.79) and 3-fold compared to ExAC population (OR 0.34, 95% CI 0.11 - 0.94). The p.Asp762Gly is located in the 18th exon of the gene, close to two acetylation sites, Lys765 and Lys766, and lies in the 2nd formin homology domain of the protein (UniProt # Q86T65; Figure 2C). In the same gene, another candidate p.Arg172His, although not statistically significant, was also enriched in controls, with MAF 0.45% in smoker controls and 0.31% in ExAC, comparing to 0.22% for cases.
Other promising candidates with consistent allelic associations include p.Gly337Glu in ZNF93, p.Ala66Pro in CLEC3A (C-type Lectin family 3 member A, OMIM # 613588), and a splice acceptor (rs201402002) in BDR9 (Bromodomain Containing 9). Unfortunately, these three variants were not covered in the WSU study. In addition, five SNVs showed suggestive evidence (only significant in LC case vs. ExAC population): MIPEP p.Leu197Pro, RTEL1 p.Gln397Glu, PLCE1 p.Thr467Ile, SNTG1 p.Val121Leu, and ZNF93 p.Lys388Asn (Table 3).
Gene-based Burden Analysis of Rare Variants
Table 4 summarizes the burden test results from the gene-based multiple rare and predicted deleterious SNVs. Among the 21 candidate genes with multiple rare deleterious SNVs, four genes showed strong association, ZNF93, DAAM2, BRD9, and LTB, with FDR adjusted P < 0.05 in both CMC and KBAC tests.
Table 4.
Gene Based Association Collapsing Tests in the Discovery Data
| Genes * (21 genes) | N. rare deleterious SNVs per gene # | N. SNVs MAF% distribution | N. carriers in Case / Control (n = 260 / 318) | FDR adjusted P value † | |||
|---|---|---|---|---|---|---|---|
| Bin 1: 0.1 - 1 | Bin 2: 0.01 - 0.1 | Bin 3: < 0.01 | CMC test | KBAC test | |||
| Risk genes | |||||||
| ZNF93 | 4 | 1 | 1 | 2 | 10 / 2 | 0.011 | 0.009 |
| BRD9 | 2 | 0 | 1 | 1 | 6 / 1 | 0.046 | 0.039 |
| LTB | 4 | 0 | 2 | 2 | 6 / 1 | 0.048 | 0.039 |
| DRD5 | 3 | 2 | 1 | 0 | 8 / 3 | 0.090 | 0.077 |
| SNTG1 | 3 | 2 | 1 | 0 | 5 / 1 | 0.094 | 0.079 |
| LAMA1 | 4 | 1 | 1 | 2 | 5 / 1 | 0.095 | 0.079 |
| SLC12A7 | 4 | 1 | 2 | 1 | 5 / 1 | 0.095 | 0.079 |
| IFIT3 | 3 | 1 | 1 | 1 | 6 / 2 | 0.140 | 0.115 |
| CEP55 | 4 | 1 | 2 | 1 | 6 / 2 | 0.141 | 0.115 |
| BDNF | 2 | 1 | 0 | 1 | 4 / 1 | 0.204 | 0.152 |
| RAD52 | 3 | 1 | 1 | 1 | 4 / 1 | 0.205 | 0.154 |
| ITPRIP | 3 | 1 | 1 | 1 | 4 / 1 | 0.205 | 0.154 |
| P3H2 | 4 | 1 | 1 | 2 | 5 / 2 | 0.266 | 0.189 |
| DZIP3 | 4 | 0 | 2 | 2 | 5 / 2 | 0.267 | 0.189 |
| BRCA2 | 6 | 2 | 1 | 3 | 13 / 9 | 0.275 | 0.193 |
| CCDC147 | 2 | 1 | 1 | 0 | 4 / 2 | 0.415 | 0.329 |
| RTEL1 | 3 | 0 | 2 | 1 | 3 / 3 | 0.992 | 0.998 |
| Protective genes | |||||||
| DAAM2 | 4 | 3 | 1 | 0 | 3 / 15 | 0.019 | 0.013 |
| ADAMTS18 | 4 | 2 | 1 | 1 | 2/8 | 0.186 | 0.125 |
| CHEK2 | 4 | 1 | 2 | 1 | 2/7 | 0.265 | 0.188 |
| DBH | 2 | 0 | 1 | 1 | 1 / 3 | 0.692 | 0.486 |
Only genes with two or more rare deleterious variants are included in the analysis from the Discovery.
N of rare deleterious SNVs (after filtering steps I-II) within the genes.
Significant P values are bolded.
DISCUSSION
Despite previous family-based linkage studies and intensive population-based GWAS analyses and candidate gene screening, a large proportion of the heritability of LC remains unexplained. Our focused analyses led to identification of four rare and deleterious inherited variants associated with LC susceptibility, including one well established truncating variant BRCA2 K3326X and three newly identified missense variations: LTB p.Leu87Phe, P3H2 p.Gln185His, and DAAM2 p.Asp762Gly. It should be noted that none of the candidate rare variants we have identified in the present study were in linkage disequilibrium (LD) with the known LC-GWAS common SNPs. The limits of LD between common and rare variants were quantified by Wray21 and supported by previous studies22,23.
This study confirms a robust association between a known rare truncating variant, K3326X in 13q13.1 BRCA2 and LC risk. The effect size in the current study (OR 2.36) is nearly identical to the previously identified association with squamous cell LC risk (OR 2.47)24, upper aero-digestive tract cancer (OR 2.53)25, and far exceeds the small increase in breast and ovarian cancer risk (OR 1.26)26,27. The molecular mechanisms that underpin this finding are unknown. In relation to the effect on cellular and biochemical properties of this variant, cancer cell lines show that mice and cells with the exon 27 truncated protein are hypersensitive to ionizing radiation28, cross-linking agents29,30, and exhibited increased susceptibility to various types of solid tumors31. It has been demonstrated that ovarian cancer patients with BRCA1/BRCA2 germline mutations respond favorably to PARP inhibitors in clinical trials32–34. Therefore, it is possible LC patients with BRCA2 K3326X may also similarly respond favorably to PARP inhibition and benefit from treatment.
An interesting finding is the association with immunity-related gene LTB, localized to the 6p21.33 MHC region which has been previously implicated in risk of LC, COPD, SM, and PF levels35–38. Functional studies in gene-knockout and transgenic mice systems have shown that LTB is of fundamental importance in fibrogenesis and carcinogenesis due to its action through a distinct receptor LTβR and NFκB39. The LTB protein plays a key role in innate immunity and inflammation which has been the focus of intensive basic science and translational research40. Another finding in the chromosome 6p region was the protective effects of DAAM2. This 6p21.2 loci is known to multiple disease susceptibility including PF levels35, smoking cessation41, renal cancer42, schizophrenia43, and hypospadias44. The DAAM2 gene is involved in the regulation of actin cytoskeleton in several different tissues, including the tracheal airways system45, and abnormally regulated in nasopharyngeal carcinoma46 and COPD47. The DAAM2 protein is one of the key WNT/plantar cell polarity signaling pathway proteins which has been documented to promote oncogenesis, stem cell renewal and tumor proliferation48,49.
The 3q28 P3H2 plays a critical role in collagen metabolic processes and oxidation reduction, and inhibits cell proliferation50. Previous work has shown that P3H2 are novel targets for epigenetic silencing in breast cancer51. The pathogenic mutations p.Gly508Val was associated with high myopia 52–54. Moreover, P3H2 expression from TISSUES database shows the highest levels in Lung55.
A main strength of this study is the accurate PF data and smoking exposure data. In the discovery where only a small number of individuals were exome sequenced, the inclusion of even a small proportion of misclassified individuals could affect the analysis. On the other hand, extreme phenotypes increase statistical power. Our discovery set (260 cases) included the 102 cases from our previous study5, 48 sporadic cases reporting heavy smoking histories and severe COPD and 54 familial cases who are likely enriched for disease-associated genetic signals. In the WSU validation study, the smoking controls were strictly selected in terms of normal PF despite heavy smoking history, and thus considered to be resistant to the effects of smoking; whereas for LC cases, 75 sporadic LC cases had severe COPD in whom the tobacco exposure would be considered quite substantial. It should be noted however that our study is still underpowered for the association analysis of each rare variant in a limited number of 2,033 cases and 1,441 controls. Although the association of CCDC147 p.Arg696Cys and DBH p.Val26Met between the LC cases and ExAC reference population did not attain statistical significance, our result is in line with our previous work5. Another limitation is that we have focused on missense, nonsense, stop-gain/loss, splice sites, and frame-shift variants in our variant filtration strategies, and we have not evaluated certain classes of variants, such as large gene-disrupting duplications and noncoding variants, like flanking intronic and UTR regions that may disrupt gene expression. Whilst larger studies and/or whole genome sequencing analysis might identify more rare variants with deleterious effects, the paucity of findings of recurrent rare variants impacting LC risk is intriguing.
In conclusion, our results provide evidence that rare deleterious germline variants, BRCA2 p.Lys3326X, LTB p.Leu87Phe, P3H2 p.Gln185His, and DAAM2 p.Asp762Gly, contributes to LC susceptibility. However, further in-depth functional follow-up studies are still needed to evaluate the pathogenicity of each of the strong candidates reported in this study.
Supplementary Material
ACKNOWLEDGEMENT
We would like to thank the patients and their families for participating in this research. We thank Dr. Richard Gibbs, Donna Muzny, Xiaoyun Liao, Van Le, Sandra Lee, and Margi Sheth from the Human Genome Sequencing Center at Baylor for performing the exome sequencing for all the samples in the discovery phase. The authors declared no competing financial interests.
Funding Support: This work was supported by grants from the National Institutes of Health (R01CA127219, R01CA141769, R01CA060691, R01CA87895, R01CA80127, R01CA84354, R01CA134682, R01CA134433, R01CA074386, R01CA092824, R01HL089856, R01HL089897, R01HL113264, R01HL082487, R01HL110883, R03CA77118, P20GM103534, P30CA125123, P30CA023108, P30CA022453, P30ES006096, P50CA090578, U01CA76293, U19CA148127, K07CA181480, N01-HG-65404, HHSN261201300011I, and HHSN268201 200007C), Intramural Research Program of the National Human Genome Research Institute (JEB-W), Herrick Foundation. The MSH-PMH study is supported by The Canadian Cancer Society Research Institute (020214) to R. H., Ontario Institute of Cancer and the Alan Brown Chair to G. L. and Lusi Wong Programs at the Princess Margaret Hospital Foundation.
WEB RESOURCES AND ABBREVIATION
- GWAS
Genome-wide association studies Catalog, www.genome.gov/gwastudies/
- ExAC
Exome Aggregation Consortium, http://exac.broadinstitute.org
- 1KGP
The 1000 Genomes Project, http://www.1000genomes.org
- CADD
Combined Annotation Dependent Depletion, cadd.gs.washington.edu/
- DbNSFP
annotation database for non-synonymous SNPs functional predictions
- GELCC
Genetic Epidemiology of Lung Cancer Consortium
- COPDGene
Chronic Obstructive Pulmonary Disease Genetic Epidemiology
- WSU
Wayne State University
- TRICL
Transdisciplinary Research in Cancer of the Lung
- ILCCO
International Lung Cancer Consortium
- LC
lung cancer
- COPD
chronic obstructive pulmonary disease
- PF
pulmonary function
- SM
smoking behavior
- PY
pack-year
- FEV1
forced expiratory volume in one second
- FVC
forced vital capacity
- SNV
Single nucleotide variants; Indels, Insertions or deletions
- MAF
minor allele frequency
- FDR
false discovery rate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: None
The authors declare no conflict of interest.
REFERENCE
- 1.Bosse Y, Amos CI. A Decade of GWAS Results in Lung Cancer. Cancer Epidemiol Biomarkers Prev. 2018;27(4):363–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorlov IP, Gorlova OY, Sunyaev SR, Spitz MR, Amos CI. Shifting paradigm of association studies: value of rare single-nucleotide polymorphisms. Am J Hum Genet. 2008;82(1):100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiong D, Wang Y, Kupert E, et al. A recurrent mutation in PARK2 is associated with familial lung cancer. Am J Hum Genet. 2015;96(2):301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, McKay JD, Rafnar T, et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat Genet. 2014;46(7):736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y, Kheradmand F, Davis CF, et al. Focused Analysis of Exome Sequencing Data for Rare Germline Mutations in Familial and Sporadic Lung Cancer. J Thorac Oncol. 2016;11(1):52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee SH, Goswami S, Grudo A, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nature medicine. 2007;13(5):567–569. [DOI] [PubMed] [Google Scholar]
- 7.Grumelli S, Corry DB, Song LZ, et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS medicine. 2004;1(1):e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shan M, Cheng HF, Song LZ, et al. Lung myeloid dendritic cells coordinately induce TH1 and TH17 responses in human emphysema. Science translational medicine. 2009;1(4):4ra10. [DOI] [PubMed] [Google Scholar]
- 9.Liu P, Vikis HG, Wang D, et al. Familial aggregation of common sequence variants on 15q24-25.1 in lung cancer. Journal of the National Cancer Institute. 2008;100(18):1326–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Regan EA, Hokanson JE, Murphy JR, et al. Genetic epidemiology of COPD (COPDGene) study design. COPD. 2010;7(1):32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bainbridge MN, Wang M, Wu Y, et al. Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol. 2011;12(7):R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lupski JR, Gonzaga-Jauregui C, Yang Y, et al. Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot-Marie-Tooth neuropathy. Genome medicine. 2013;5(6):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reid JG, Carroll A, Veeraraghavan N, et al. Launching genomics into the cloud: deployment of Mercury, a next generation sequence analysis pipeline. BMC bioinformatics. 2014;15:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Challis D, Yu J, Evani US, et al. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC bioinformatics. 2012;13:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nature genetics. 2014;46(3):310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83(3):311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu DJ, Leal SM. A novel adaptive method for the analysis of next-generation sequencing data to detect complex trait associations with rare variants due to gene main effects and interactions. PLoS Genet. 2010;6(10):e1001156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc Ser B. 1995;57(1):289–300. [Google Scholar]
- 20.Schwartz AG, Lusk CM, Wenzlaff AS, et al. Risk of Lung Cancer Associated with COPD Phenotype Based on Quantitative Image Analysis. Cancer Epidemiol Biomarkers Prev. 2016;25(9):1341–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wray NR. Allele frequencies and the r2 measure of linkage disequilibrium: impact on design and interpretation of association studies. Twin Res Hum Genet. 2005;8(2):87–94. [DOI] [PubMed] [Google Scholar]
- 22.Lopez de Maturana E, Ibanez-Escriche N, Gonzalez-Recio O, et al. Next generation modeling in GWAS: comparing different genetic architectures. Hum Genet. 2014;133(10):1235–1253. [DOI] [PubMed] [Google Scholar]
- 23.de Los Campos G, Sorensen D, Gianola D. Genomic heritability: what is it? PLoS Genet. 2015;11(5):e1005048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, McKay JD, Rafnar T, et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nature genetics. 2014;46(7):736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delahaye-Sourdeix M, Anantharaman D, Timofeeva MN, et al. A rare truncating BRCA2 variant and genetic susceptibility to upper aerodigestive tract cancer. Journal of the National Cancer Institute. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michailidou K, Hall P, Gonzalez-Neira A, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nature genetics. 2013;45(4):353–361, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meeks HD, Song H, Michailidou K, et al. BRCA2 Polymorphic Stop Codon K3326X and the Risk of Breast, Prostate, and Ovarian Cancers. J Natl Cancer Inst. 2016;108(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morimatsu M, Donoho G, Hasty P. Cells deleted for Brca2 COOH terminus exhibit hypersensitivity to gamma-radiation and premature senescence. Cancer research. 1998;58(15):3441–3447. [PubMed] [Google Scholar]
- 29.Atanassov BS, Barrett JC, Davis BJ. Homozygous germ line mutation in exon 27 of murine Brca2 disrupts the Fancd2-Brca2 pathway in the homologous recombination-mediated DNA interstrand cross-links’ repair but does not affect meiosis. Genes, chromosomes & cancer. 2005;44(4):429–437. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Andreassen PR, D’Andrea AD. Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Molecular and cellular biology. 2004;24(13):5850–5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McAllister KA, Bennett LM, Houle CD, et al. Cancer susceptibility of mice with a homozygous deletion in the COOH-terminal domain of the Brca2 gene. Cancer research. 2002;62(4):990–994. [PubMed] [Google Scholar]
- 32.Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–251. [DOI] [PubMed] [Google Scholar]
- 33.Fong PC, Yap TA, Boss DS, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–2519. [DOI] [PubMed] [Google Scholar]
- 34.Ledermann JA, Harter P, Gourley C, et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol. 2016;17(11):1579–1589. [DOI] [PubMed] [Google Scholar]
- 35.Repapi E, Sayers I, Wain LV, et al. Genome-wide association study identifies five loci associated with lung function. Nat Genet. 2010;42(1):36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Broderick P, Webb E, et al. Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nature genetics. 2008;40(12):1407–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Broderick P, Wang Y, Vijayakrishnan J, et al. Deciphering the impact of common genetic variation on lung cancer risk: a genome-wide association study. Cancer Res. 2009;69(16):6633–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hancock DB, Eijgelsheim M, Wilk JB, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet. 2010;42(1):45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drutskaya MS, Efimov GA, Kruglov AA, Kuprash DV, Nedospasov SA. Tumor necrosis factor, lymphotoxin and cancer. IUBMB Life. 2010;62(4):283–289. [DOI] [PubMed] [Google Scholar]
- 40.Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3(9):745–756. [DOI] [PubMed] [Google Scholar]
- 41.Uhl GR, Liu QR, Drgon T, et al. Molecular genetics of successful smoking cessation: convergent genome-wide association study results. Arch Gen Psychiatry. 2008;65(6):683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hirata H, Hinoda Y, Nakajima K, et al. Wnt antagonist gene polymorphisms and renal cancer. Cancer. 2009;115(19):4488–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meda SA, Ruano G, Windemuth A, et al. Multivariate analysis reveals genetic associations of the resting default mode network in psychotic bipolar disorder and schizophrenia. Proc Natl Acad Sci U S A. 2014;111(19):E2066–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geller F, Feenstra B, Carstensen L, et al. Genome-wide association analyses identify variants in developmental genes associated with hypospadias. Nat Genet. 2014;46(9):957–963. [DOI] [PubMed] [Google Scholar]
- 45.Matusek T, Djiane A, Jankovics F, Brunner D, Mlodzik M, Mihaly J. The Drosophila formin DAAM regulates the tracheal cuticle pattern through organizing the actin cytoskeleton. Development. 2006;133(5):957–966. [DOI] [PubMed] [Google Scholar]
- 46.Zeng ZY, Zhou YH, Zhang WL, et al. Gene expression profiling of nasopharyngeal carcinoma reveals the abnormally regulated Wnt signaling pathway. Hum Pathol. 2007;38(1):120–133. [DOI] [PubMed] [Google Scholar]
- 47.Wu X, Sun X, Chen C, Bai C, Wang X. Dynamic gene expressions of peripheral blood mononuclear cells in patients with acute exacerbation of chronic obstructive pulmonary disease: a preliminary study. Crit Care. 2014;18(6):508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barrow JR. Wnt/PCP signaling: a veritable polar star in establishing patterns of polarity in embryonic tissues. Semin Cell Dev Biol. 2006;17(2):185–193. [DOI] [PubMed] [Google Scholar]
- 49.Tanaka K Formin family proteins in cytoskeletal control. Biochem Biophys Res Commun. 2000;267(2):479–481. [DOI] [PubMed] [Google Scholar]
- 50.Pokidysheva E, Boudko S, Vranka J, et al. Biological role of prolyl 3-hydroxylation in type IV collagen. Proc Natl Acad Sci U S A. 2014;111(1):161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shah R, Smith P, Purdie C, et al. The prolyl 3-hydroxylases P3H2 and P3H3 are novel targets for epigenetic silencing in breast cancer. Br J Cancer. 2009;100(10):1687–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mordechai S, Gradstein L, Pasanen A, et al. High myopia caused by a mutation in LEPREL1, encoding prolyl 3-hydroxylase 2. Am J Hum Genet. 2011;89(3):438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo H, Tong P, Peng Y, et al. Homozygous loss-of-function mutation of the LEPREL1 gene causes severe non-syndromic high myopia with early-onset cataract. Clin Genet. 2014;86(6):575–579. [DOI] [PubMed] [Google Scholar]
- 54.Feng CY, Huang XQ, Cheng XW, Wu RH, Lu F, Jin ZB. Mutational screening of SLC39A5, LEPREL1 and LRPAP1 in a cohort of 187 high myopia patients. Sci Rep. 2017;7(1):1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Santos A, Tsafou K, Stolte C, Pletscher-Frankild S, O’Donoghue SI, Jensen LJ. Comprehensive comparison of large-scale tissue expression datasets. PeerJ. 2015;3:e1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.