

Abstract
Endocrine-disrupting chemicals (EDCs) are found abundantly in the environment, resulting in daily human exposure. This is of concern because many EDCs are known to target the female reproductive system and, more specifically, the ovary. In the female, the ovary is the key organ responsible for reproductive and endocrine functions. Exposure to EDCs is known to cause many reproductive health problems such as infertility, premature ovarian failure, and abnormal sex steroid hormone levels. Some EDCs and their effects on adult ovarian function have been studied extensively over the years, whereas the effects of others remain unclear. This review covers what is currently known about the effects of selected EDCs (bisphenol A, methoxychlor, 2,3,7,8-tetrachlorodibenzo-p-dioxin, phthalates, and genistein) on the adult ovary and the mechanisms by which they act upon the ovary, focusing primarily on their effects on folliculogenesis and steroidogenesis. Furthermore, this review discusses future directions needed to better understand the effects of EDCs, including the need to examine the effects of multiple and more consistent doses and to study different mechanisms of action.
Keywords: 2,3,7,8-tetrachloro-p-dibenzodioxin; bisphenol A; folliculogenesis; genistein; methoxychlor; ovary; phthalates; steroidogenesis
Introduction
Environmental toxicants are chemical compounds found in the environment due to a variety of processes, including manufacturing, combustion, leaching from products, and human contamination (reviewed in [1]). Some environmental toxicants also occur naturally in plants. Many environmental toxicants are known endocrine-disrupting chemicals (EDCs) [2]. This is of concern because women are exposed to EDCs on a daily basis, and some EDCs are known to target the ovary and cause reproductive health problems such as infertility, premature ovarian failure, and abnormal sex steroid hormone levels. Infertility is a public health concern because it affects millions of women worldwide, decreases quality of life, and results in large medical costs. Premature ovarian failure is a public health concern because it leads to early infertility and is associated with an increased risk of osteoporosis, depression, cardiovascular disease, and early death [3, 4]. Abnormal sex steroid hormone levels are of public health concern because they can cause infertility and are associated with other serious conditions including osteoporosis, depression, and cardiovascular disease [1, 5].
The purpose of this minireview is to review the recent research on the impact of selected EDCs on the adult ovary. We focused on the adult ovary because adult humans are exposed to EDCs, and several recent reviews have already discussed developmental exposure to EDCs [6–9]. Furthermore, we chose to review the effects of EDCs on ovarian folliculogenesis, follicle/oocyte health, and steroidogenesis because they are key factors required for normal ovarian function (Figs. 1 and 2). We also present information on effects of the selected EDCs bisphenol A (BPA), methoxychlor (MXC), 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), phthalates, and genistein. We focused on BPA, MXC, and phthalates because they have been identified as contaminants present in human tissue and the environment [10–17]. We selected TCDD because it is widely used as a model EDC, is present in human tissues, and has been shown to be an extremely toxic chemical [18]. We review genistein because women are widely exposed to it, and it is used as a model compound for phytoestrogen studies [19, 20].
Fig. 1.
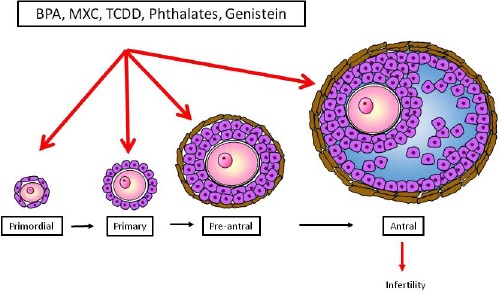
Endocrine-disrupting chemicals can affect folliculogenesis. Females are born with a finite pool of immature primordial follicles, and over time, some will grow and mature to primary, preantral, and then antral follicles. The most mature follicle is the antral follicle. It is the only follicle type capable of ovulating and producing sex steroid hormones. Exposure to EDCs affecting primordial follicles can lead to permanent and early infertility. Effects of EDCs on the antral follicle can result in infertility and altered hormone levels.
Fig. 2.
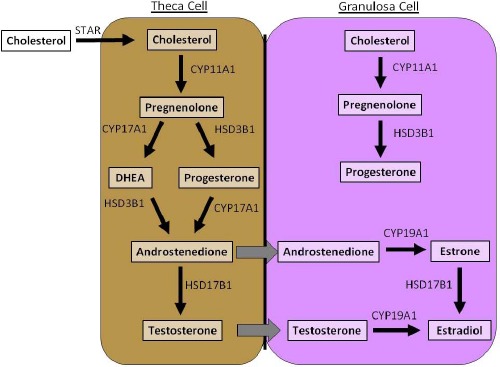
Steroidogenic pathway. The antral follicle is responsible for producing female sex steroid hormones, and it does so by the process known as steroidogenesis. Steroidogenesis requires both theca and granulosa cells. Steroidogenic enzymes present in these cells are responsible for metabolizing cholesterol to 17-β estradiol and other necessary sex steroid hormones. EDCs can alter the expression, protein level, or enzyme activity of steroidogenic enzymes, thus resulting in altered sex steroid hormones.
Bisphenol A
BPA is a plasticizer used commonly in a wide range of consumer products such as food and drink containers, epoxy resins, plastics, baby bottles, thermal receipts, and dental sealants (reviewed in [21]). As a result, humans are constantly exposed to BPA via oral and dermal routes of exposure. BPA has been measured repeatedly in serum [22–24], plasma [25, 26], urine [27], sweat [27], breast milk [28, 29], amniotic fluid [30–32], placental tissue [33], fetal serum [30, 33], and umbilical cords [34]. The currently accepted lowest observed adverse effect level for BPA is 50 mg/kg/day, and the current U.S. Environmental Protection Agency (EPA) reference dose is 50 μg/kg/day, but actual daily human exposure to BPA may be much higher [35]. This raises concern because BPA exposure has been associated with female fertility problems [10], polycystic ovary syndrome [36], and endometriosis [37]. Moreover, in women undergoing fertility treatments, urinary BPA levels have been associated with decreased antral follicle counts and a reduction in the number of oocytes retrieved [38, 39].
BPA has also been shown to reduce fertility [10, 40], reduce the primordial follicle pool [40], lead to premature ovarian failure [40], disturb the estrous cycle [10, 40], and disrupt steroidogenesis in a variety of animal models [10, 40]. Furthermore, studies have shown that adult exposure to BPA (110, 219, and 440 μM) inhibits antral follicle growth in isolated mouse antral follicles in vitro (Fig. 1) [41–43]. It is likely that BPA-induced inhibition of follicle growth is mediated by BPA-induced disruption in steroidogenesis [41], interference with the aryl hydrocarbon receptor (AHR) pathway [42], and dysregulation of cell cycle regulators [43]. It is unlikely, however, that the ability of BPA to inhibit follicle growth is mediated by estrogen receptors because estradiol and ICI 182,780 (a high-affinity estrogen receptor antagonist) co-treatments failed to protect follicles from BPA-induced inhibition of growth [43].
Follicle atresia can also be induced by BPA exposure. In mouse antral follicles, BPA (100 μg/ml) increased the expression of the proapoptotic factor BCL2-associated X protein (Bax) to the antiapoptotic factor B cell lymphoma 2 (Bcl2), and transformation-related protein 53 (Trp53), leading to follicle atresia [43]. In murine granulosa cells, BPA treatment (100 μM) elevated the Bax:Bcl2 ratio at both the protein and the mRNA levels and caused G2-to-M arrest, leading to DNA damage [44]. In an in vivo study in which adult female rats were dosed with BPA for 90 days, BPA treatment (0.001 and 0.1 mg/kg/day) increased follicle atresia and luteal regression by inducing caspase-3-associated apoptosis [45]. Although BPA has been shown to bind to estrogen receptor alpha (ESR1) [46], overexpression of ESR1 did not affect the susceptibility of follicles to BPA-induced atresia, suggesting that BPA-induced follicle atresia is independent of ESR1 [43]. Although some studies have shown that BPA may act independently of ESR1 [43], others have shown that BPA may act through estrogen receptor beta 2 (ESR2) [47] or the nongenomic G protein-coupled receptor (GPCR) [48].
BPA exposure has also been associated with decreased oocyte quality in multiple studies [49–51]. Hunt et al. [49] first reported that BPA exposure (20, 40, and 100 ng/g/day) causes meiotic defects in the oocytes harvested from mice. Subsequently, Trapphoff et al. [50] showed that BPA exposure (3 nM) induces epigenetic changes, which can lead to meiotic errors in cultured mouse follicles. BPA (100 μM) has also been shown to decrease hyaluronic acid in the extracellular matrix of the oocyte cumulus matrix and to adversely affect oocyte meiotic maturation [51].
BPA exposure also adversely affects sex steroid hormone levels by interfering with steroidogenesis in several animal models. However, the specific effects of BPA on steroidogenesis differ by species and doses. In rat ovarian theca interstitial cells and granulosa cell cultures, BPA exposure (0.1–10 μM) significantly increased testosterone and progesterone levels by increasing the expression of several key cytochrome p450 steroidogenic enzymes such as 17-α hydroxylase (Cyp17a1), cholesterol side chain cleavage enzyme (Cyp11a1), and steroidogenic acute regulatory protein (Star) [52] (Fig. 2). Conversely, BPA exposure (1–100 μM) decreased estradiol levels by decreasing the expression of aromatase (Cyp19a1) [52]. In porcine granulosa cells, BPA exposure (0.01–10 μM) increased the basal levels of progesterone but inhibited follicle-stimulating hormone (FSH)-induced estradiol production [53]. In contrast, another study conducted with porcine ovarian granulosa cell cultures showed that exposure to BPA for 48 h decreased progesterone production (0.1–10 μM) and stimulated estradiol production at low BPA concentrations (0.1 μM) but inhibited estradiol production at higher BPA concentrations (1 and 10 μM) [54]. In the two studies using porcine granulosa cell cultures, the dose ranges were similar, but the culture time lengths were different. Thus, differences in results obtained from the two studies may be due to the different lengths of the cell culture time. In mouse antral follicle cultures, BPA (10 μg/ml and 100 μg/ml) also inhibited steroid hormone production by disrupting expression of steroidogenic enzymes [55]. Specifically, BPA (10 μg/ml and 100 μg/ml) acutely decreased Cyp11a1 expression as early as 18 h, leading to a decrease in progesterone levels at 24 h and further leading to decreases in androstenedione, testosterone, and estradiol, as well as the expression of Star at 72 h [55]. Interestingly, some of the effects of BPA on steroidogenesis in mice are reversible with removal of BPA [55] or supplementation with pregnenolone, a precursor hormone in the ovarian steroidogenesis pathway [41]. In vivo studies also indicate that BPA can disrupt steroidogenesis in the adult ovary. In rats, BPA (0.001 mg/kg/day and 0.1 mg/kg/day) significantly decreased serum estradiol levels by decreasing the protein levels of CYP19A1 and StAR in granulosa cells and theca interstitial cells [45]. Together, these studies show that BPA exposure disrupts normal steroidogenesis in the ovary by affecting several steroidogenic enzymes and upstream hormone levels in both in vitro and in vivo settings.
The mechanisms by which BPA exerts ovotoxic effects are still not fully understood. Oxidative stress, glucose metabolism, and insulin signaling have been shown to impair testicular functions, induce toxicity, and lead to infertility in adult male animals [56–59], but limited information is available on whether such processes are involved in BPA-induced ovarian toxicity. In one study, BPA at 25 mg/kg/day caused oxidative damage in ovarian tissues [60], but the study only used one dose of BPA and thus, there is a need for additional studies using a wide range of BPA doses. Such studies will be helpful in understanding how BPA disrupts female reproduction.
Methoxychlor
MXC is an organochlorine pesticide used in many countries against insects that attack fruits, vegetables, and home gardens. It is present in food and water samples [11–13], and as a result, humans are exposed to MXC. Studies have shown that 35% of agricultural commodities contain pesticide residues including MXC [13]. Badach et al. [12] showed that MXC levels ranged from 0.0165 to 1.1507 μg/L in well water and from 0.0177 to 0.9660 μg/L in samples from water mains. Although MXC production was stopped in the United States due to the failure of the manufacturer to properly register its production with the EPA, it is still an important chemical to study because it is currently used on agricultural products in many other countries that are also imported into the United States and other countries, resulting in global human exposure. Furthermore, Golovleva et al. [14] showed that MXC is persistent in soil and that its residues are present even 18 mo after soil treatment using microorganisms that biodegrade MXC, and Shegunova et al. [15] showed that MXC residues are present even in areas where the chemical has not been used, and they propose this is due to atmospheric redistribution.
MXC is metabolized predominantly to 1,1,1-trichloro-2-(4-hydroxyphenyl)-2-(4-methoxyphenyl) ethane (MOH) and the bisphenolic compound 1,1,1-trichloro-2,2-bis(4-hydroxyphenyl) ethane (HPTE) by cytochrome p450 enzymes in the body [61]. Several studies have consistently shown that MXC, as well as its metabolites, interferes with folliculogenesis. For example, studies have shown that treatment with MXC (100 mg/kg) in vivo inhibits folliculogenesis (Fig. 1), likely by reducing the expression of Esr2 and by increasing the expression of anti-Müllerian hormone (Amh) in preantral and early antral follicles [16, 62]. Studies have also shown that treatment with MXC (1–100 μg/ml) or its major metabolites in vitro inhibits growth of isolated antral follicles, likely by reducing the expression of G1-S phase cell cycle regulators such as cyclin D2 (Ccnd2) and cyclin-dependent kinase 4 (Cdk4), by decreasing the antiapoptotic factor Bcl2 and increasing the proapoptotic factor Bax [63, 64]. In addition, MXC exposure in vivo (32 and 64 mg/kg/day) induces oxidative stress in mouse ovaries [65, 66].
Multiple studies have also shown consistently that MXC and its metabolites inhibit ovarian steroidogenesis. For example, MXC (1–100 μg/ml) inhibits the production of estradiol, testosterone, and androstenedione in isolated mouse antral follicles [67]. The effects of MXC (1–100 μg/ml) on steroid hormone levels likely stem from its ability to inhibit the expression of key factors in the estradiol biosynthesis pathway (i.e., Cyp19a1, Cyp17a1, Cyp11a1, Star, 17β-hydroxysteroid dehydrogenase-1 [Hsd17b1], and 3β-hydroxysteroid dehydrogenase-1 [Hsd3b1]) (Fig. 2), along with its ability to induce expression of cytochrome P450 1b1 (Cyp1b1), an enzyme that metabolizes estradiol [67]. The MXC metabolite HTPE (500 nM) inhibits Cyp11a1 activity, leading to decreased progesterone production by cultured rat ovarian follicular cells [68]. Similarly, HPTE (1–10 μM) reduces FSH-stimulated synthesis of progesterone and estrogen in cultured rat granulosa cells by decreasing the expression levels of Cyp11a1 and Cyp19a1 [69]. The MXC metabolite MOH inhibits steroidogenesis both by reducing the availability of pregnenolone [70] and by inhibiting the expression levels of Cyp11a1, Cyp17a1, and Cyp19a1 mRNA in mouse antral follicles in vitro [71].
The mechanisms by which MXC and its metabolites interact with ovarian cells to inhibit folliculogenesis and steroidogenesis are unclear. Some studies show that MXC and its metabolites may bind to estrogen receptors and exert estrogenic, antiestrogenic, and antiandrogenic properties, depending on the receptor subtype with which it interacts [72, 73]. This possibility is supported by studies that have shown that fetal and neonatal exposure to MXC (20 μg/kg and 100 mg/kg) alters methylation on the promoter region of Esr1, suppressing the expression of Esr1 and causing ovarian dysfunction in the rat [16, 74]. It is also supported by studies by Paulose et al. [75] that show that ESR1 overexpression in antral follicles was more sensitive to toxicity induced by MXC and its metabolites than control antral follicles, suggesting that disruption in the equilibrium between ESR1 and ESR2 in the ovary may alter the response of the ovary to estrogenic chemicals. In addition, some studies suggest that MXC may interact with pathways that cross-talk with estrogen receptors. Specifically, evidence provided by Basavarajappa et al. [76] suggests that MXC may act through the AHR pathway to inhibit follicle growth and induce atresia in mouse antral follicles.
Although studies consistently show that MXC inhibits follicle growth and steroidogenesis, little is known about the effects of MXC on female fertility. Thus, future studies should examine whether MXC exposure during adulthood causes subfertility or infertility. Furthermore, little is known about whether other organochlorine pesticides affect folliculogenesis and steroidogenesis. Therefore, future studies should examine the effects of organochlorine pesticides as well as mixtures of organochlorine pesticides on the ovary and female reproductive system.
2,3,7,8-Tetrachlorodibenzo-p-Dioxin
TCDD is a persistent environmental contaminant inadvertently produced as a by-product of herbicide and pesticide manufacturing, bleaching process at tree pulp and paper mills, and burning of municipal solid waste [77–79]. TCDD is the most toxic member of the dioxin class of chemicals. It also has a long environmental half-life, accumulates in the food chain [18], and is found in human fat tissue [18], blood serum [18, 80], breast milk [81], and ovarian follicular fluid [82].
Studies consistently show that TCDD exposure targets the ovary and leads to altered folliculogenesis (Fig. 1). However, the effects of TCDD on folliculogenesis differ by species. In mice, TCDD (0.1–100 nM) exposure does not affect the growth of antral follicles in vitro, suggesting that it does not affect proliferation of granulosa cells [83]. In pigs, however, TCDD (0.1 and 10 nM) reduces the percentage of proliferating cells in follicles over time [84]. Similarly, in rats, TCDD exposure decreases the number of antral follicles without increasing in atresia, suggesting that TCDD has an antiproliferative effect on the rat ovary [85]. Reasons for species differences in response to TCDD are unclear but may stem from differences in the abilities of species to metabolize TCDD as well as species differences in expression of the AHR, the primary receptor for TCDD [86].
TCDD exposure has also been shown to reduce or block ovulation in rodents in vivo [87–89]. The mechanism by which TCDD (32 μg/kg body weight) blocks ovulation likely involves the ability of TCDD to reduce the numbers of granulosa cells in S phase and inhibit the levels of cyclin dependent kinase 2 (Cdk2) and Ccnd2 following equine chorionic gonadotropin treatment in immature rats [90].
In addition to interfering with folliculogenesis and ovulation, TCDD can interfere with ovarian steroidogenesis (Fig. 2). Karman et al. [83, 91] showed that TCDD exposure (0.1–100 nM) decreased progesterone (1 nM), androstenedione (0.1 and 1 nM), testosterone (0.1 and 1 nM), and estradiol (0.1–10 nM) levels in a nonmonotonic dose response manner in isolated mouse antral follicles. Furthermore, Karman et al. [83] showed that the addition of pregnenolone substrate (10 μM) restored hormone levels to control levels, suggesting that TCDD may act prior to pregnenolone formation to decrease hormone levels in mice. The effects of TCDD on ovarian steroidogenesis appear to be due to the ability of TCDD to inhibit key steroidogenic enzymes (Hsd17b1 and Cyp19a1), leading to reduced steroidogenic capacity of antral follicles [91].
These data are consistent with those in other studies in other species and culture systems, which also show that TCDD inhibits ovarian steroidogenesis and that it often does so in a nonmonotonic fashion [84, 92–94]. Specifically, TCDD inhibits estradiol levels in female mice in vivo [95], and oral administration of TCDD (20, 50, and 125 ng/kg/day) weekly for 29 wk reduces estradiol levels in Sprague-Dawley rats [96]. In addition, TCDD exposure (90 ng/kg/day) increases the testosterone-to-estradiol ratio in juvenile mice [97], and TCDD decreases estradiol levels in the chicken ovary [98]. Furthermore, TCDD exposures of 0.1 nM or 10 nM reduce estradiol levels, whereas only 10 nM exposure significantly reduces progesterone levels in isolated porcine thecal and granulosa cell co-cultures [99]. These results suggest that, like hormones and other environmental contaminants that act as endocrine disruptors, TCDD may have multiple modes of action in antral follicles, depending on the dose.
Phthalates
Phthalates are synthetic chemicals found ubiquitously in the environment and are known to have endocrine-disrupting characteristics (reviewed in [17]). More than 18 billion pounds of phthalates are used each year, predominantly as plasticizers in polyvinyl chloride (PVC) products such as upholstery, table cloths, shower curtains, pesticides, solvents, and infant toys [100]. Phthalates are used to impart flexibility to plastics but leach from plastic products into the environment over time due to their noncovalent bonds [17]. Phthalates can also be found in air, sediments, agricultural and urban soil, wastewater, and natural bodies of water [101–103].
Di-(2-ethylhexyl) phthalate (DEHP) is the plasticizer most commonly used for PVC and is approved for use in medical devices such as tubing, blood bags, and dialysis equipment [104]. It is also used to manufacture disposable medical examination and sterile surgical vinyl gloves [104]. Dibutyl phthalate (DBP), butyl benzyl phthalate (BBP), and diethyl phthalate (DEP) are also produced in high volumes and are commonly used in consumer products [17]. The estimated range of daily human exposure to DEHP is 0.71–4.6 μg/kg/day, 0.84–5.22 μg/kg/day to DBP, 0.26–0.88 μg/kg/day to BBP, and 2.32–12 μg/kg/day to DEP [105].
Phthalates have been shown to alter follicle development and growth and impair follicle functionality (Fig. 1). Studies in mice have shown that exposure to phthalates may lead to acceleration of primordial follicle recruitment (Fig. 1). Specifically, exposure to DEHP (20 μg–750 mg/kg/day) in vivo for 10 to 30 days accelerates primordial follicle recruitment through a mechanism that involves overactivation of the phosphatidylinositol 3-kinase (PI3K) pathway [106]. In contrast, exposure to DEHP in vitro did not alter folliculogenesis, but exposure to mono-(2ethylhexyl) phthalate (MEHP), the bioactive metabolite of DEHP, in vitro, accelerates primordial follicle recruitment [107]; thereby indicating that DEHP accelerates folliculogenesis through its metabolite MEHP. Exposure to DBP (1000 μg/ml) in vitro inhibits growth of antral follicles by disrupting the cell cycle [108]. Specifically, DBP inhibits the expression of Ccnd2, cyclin E1 (Ccne1), cyclin A2 (Ccna2), and cyclin B1 (Ccnb1) and increases expression of cyclin-dependent kinase inhibitor 1A (Cdkn1a) [108]. These changes indicate DBP exposure may result in cell cycle arrest, thereby inhibiting antral follicle growth in vitro [108]. Collectively, these studies provide evidence that exposure to certain phthalates affects ovarian folliculogenesis, but additional studies are required to determine if other phthalates inhibit follicle growth and to determine the mechanisms by which they do so.
Phthalates also adversely affect the health of follicles. Exposure to DEHP (600 mg/kg) by oral gavage for 60 consecutive days decreases primary and secondary follicle numbers and increases atretic follicles [109]. This may have been caused by a DEHP-induced increase in apoptotic granulosa cells [109]. MEHP has been shown to affect follicle health by inducing oxidative stress by increasing reactive oxygen species levels and disrupting the expression and activity of the antioxidants superoxide dismutase 1 (SOD1) and glutathione peroxidase (GPX), thereby, resulting in MEHP-induced inhibition of antral follicle growth [110]. BBP (1 μM) has been shown to reduce human granulosa cell viability most likely by increasing mRNA and protein levels of the AHR, aryl hydrocarbon receptor nuclear translocator (ARNT), and CYP1B1 [111].
Although these studies collectively indicate that phthalates directly affect follicle health, future studies are needed to determine whether the effects of phthalates on the ovary significantly impact fertility and the timing of reproductive senescence. In addition, future studies are needed to examine the impact of other phthalates on follicle and oocyte health and to determine the mechanisms by which phthalates induce ovarian toxicity.
Like BPA, MXC, and TCDD, phthalates have been shown to alter steroidogenesis (Fig. 2). In vivo studies have shown that DEHP exposure (300 and 600 mg/kg) in rats decreases serum estradiol levels [109]. This is likely the result of decreased Cyp19a1 expression [109]. Similar results are seen in vitro as well. Studies showed that exposure to DEHP (10 and 100 μg/ml) decreased estradiol levels in mouse antral follicles [112, 113]. The same studies showed that DEHP exposure (100 μg/ml) reduced Cyp19a1 expression [112, 113]. Exposure to MEHP (0.1–10 μg/ml) decreased testosterone, estrone, and estradiol levels in cultured mouse antral follicles [107]. These effects are likely due to the ability of MEHP to inhibit the expression of Cyp17a1, Hsd17b1, and Cyp19a1 [107]. Similar to the in vitro mouse study, a study by Reinsberg et al. [114] showed that increasing MEHP exposure decreases estradiol levels and aromatase activity in human granulosa cells. In contrast, Inada et al. [115] found that exposure to MEHP (10, 30, and 100 μg/ml) increased the combined levels of progesterone, androstenedione, testosterone, and estradiol in cultured rat antral follicles. The increases in steroid hormones are likely attributable to overactivation of progesterone and testosterone synthesis. Reasons for differences between the effects of MEHP on rat and those on mouse follicles are unknown but could stem from species differences in metabolism of MEHP. Studies also show that exposure to DBP (1000 μg/ml) significantly reduced the amount of accumulated estradiol in cultured mouse antral follicles, although the exact mechanism by which this occurs is not fully understood [108].
Collectively, these studies provide evidence that phthalate exposure during adulthood alters ovarian steroidogenesis, but future studies need to examine whether similar effects of phthalates occur in humans and whether the ability of phthalates to alter steroid levels leads to infertility and other adverse outcomes such as cardiovascular disease and osteoporosis. Furthermore, future studies should focus on biologically relevant dose ranges, and attempt to mimic normal human exposure by examining the effects of exposure to phthalate mixtures as well as mixtures of other ubiquitous toxicants on the ovary.
Genistein
Genistein is an isoflavone phytoestrogen found naturally in plant structures such as soy beans, chickpeas, sunflower seeds, and lentils [116]. Genistein, like other phytoestrogens, is also consumed as a supplement to treat numerous ailments including cancer, kidney disease, neuronal injury, sexual dysfunction, depression, and menopausal symptoms [117, 118]. Humans are exposed to genistein primarily through consumption of soy-based dietary products such as soy milk, tofu, and soy flour, and thus, genistein is considered an important environmental estrogen in the human diet [117, 119]. Genistein, like other phytoestrogens, has been shown to bind to and signal through estrogen receptors [120–122] with a greater affinity to ESR2 than to ESR1 [121], thereby making the estrogen receptor-rich ovary a major target tissue for genistein.
Genistein exposure has been shown to alter folliculogenesis in rats in vivo [123, 124] (Fig. 1), but the results have varied depending on age, strain, and dose. Genistein exposure (50 mg/kg) decreases healthy primordial, primary, and secondary follicles but increases the amount of antral follicles in 18-day-old Wistar rats, suggesting that genistein exposure accelerates follicle recruitment [123]. Additionally, the same study showed that genistein exposure (50 mg/kg) increases the number of atretic secondary and antral follicles [123], suggesting that genistein-induced accelerated follicles may not survive to produce viable oocytes. Conversely, other studies indicate that genistein exposure (160 mg/kg) increases primordial follicles and decreases antral follicles in 3-mo-old Sprague-Dawley rats [124], suggesting that genistein inhibits follicle growth. Studies using a mixture of soy isoflavones (SIF) with a 60% concentration of genistein have shown that SIF at 200 mg/kg increases the number of apoptotic cells in primary and antral follicles and corpora lutea in rats [125]. Furthermore, 100 mg/kg SIF also increases the number of apoptotic cells in antral follicles and corpora lutea of rats [125]. This phenomenon is likely due to increased protein levels of the apoptotic factors caspase 3, FAS, and BAX combined with decreased protein levels of antiapoptotic factor BCL2 [125]. The reasons for differences in the ability of genistein to accelerate or inhibit follicle growth are unclear. Future studies should be conducted with a wider range of doses, various dosing periods, and ages and should examine the possible mechanisms by which genistein increases or decreases folliculogenesis in vivo.
The effects of genistein exposure on steroidogenesis have been studied extensively in vitro. Studies showed that exposure to genistein alters progesterone levels, but the actual effect depends on the dose and species. One study showed that genistein (45 μM) decreases prolactin-induced progesterone in porcine theca cells [126]. In another study, exposure to genistein (1–50 μM) decreased progesterone levels in human granulosa cells [127]. Furthermore, genistein exposure (50 μM and 100 μM) decreased production of progesterone from pregnenolone in human granulosa cells [127, 128]. Studies done with rat granulosa cells have also provided results that varied by dose. In one study, genistein exposure (0.5–50 μM) reduced basal levels of progesterone and FSH-induced progesterone levels (10 μM) [129]. Another study showed that low concentrations of genistein (0.1–3 μM) increased FSH-induced progesterone levels, but high concentrations of genistein (30–100 μM) decreased FSH-induced progesterone levels [130]. Genistein exposure (30 and 100 μM) also reduced FSH-induced estradiol levels in rat granulosa cells [130]. Similarly, genistein (50 and 100 μM) decreased estradiol levels in human granulosa cells, possibly due to altered levels in precursor hormones or changes in steroidogenic enzymes [127, 128].
It is possible that the effects of genistein on sex steroid hormone levels may be due to dysregulation of steroidogenic enzymes. For example, genistein exposure (10 and 50 μM) reduces mRNA expression and activity of CYP19A1 in human granulosa cells [131]. Because Cyp19a1 is the key enzyme responsible for transforming precursor hormones to estradiol (Fig. 2), down-regulation of this enzyme may explain the decreased estradiol levels seen in many studies. Genistein exposure (1–50 μM) also decreases CYP11A1 protein levels and Hsd3b1 expression in porcine granulosa cells [132]. These two enzymes are responsible for transforming cholesterol and pregnenolone to progesterone (Fig. 2). Thus, inhibition of these enzymes may cause the genistein-induced decrease in progesterone levels.
The reasons for the different effects of genistein on sex steroid hormone levels are unclear. Future studies should examine the potential mechanisms by which genistein affects estradiol levels. In addition, studies should be conducted using whole antral follicles because steroid hormone production relies heavily on both the theca and granulosa cells and most previous studies have been conducted solely on isolated theca or granulosa cells.
Conclusions
Adult women are exposed to several EDCs, including BPA, phthalates, MXC, TCDD, and genistein. These EDCs have been shown to target the ovary and adversely affect folliculogenesis, follicle/oocyte health, and steroidogenesis (Figs. 1 and 2). This is of concern for public health because the selected EDCs are used extensively in a wide variety of commonly used items or they are present as contaminants in the environment, resulting in ubiquitous human exposure. Furthermore, the effects of EDCs on folliculogenesis, follicle/oocyte health, and steroidogenesis can have lasting effects on reproductive and nonreproductive health because folliculogenesis, follicle/oocyte health, and steroidogenesis are essential for fertility, maintenance of appropriately timed reproductive senescence, and regulation of skeletal, cardiovascular, and brain health [1–5].
Although recent studies indicate that EDCs are ovarian toxicants, we still have limited information about the impact of many EDCs in women and in animal models. Furthermore, we have limited information on the mechanisms by which EDCs exert ovarian toxicity. Thus, future studies are needed to fully understand the impact of EDCs on the ovary and to determine the mechanisms by which they exert ovarian toxicity. Additionally, future studies should examine the effects of EDCs on the hypothalamic-pituitary-gonadal axis, as a whole, to see if EDCs indirectly affect the ovary by affecting the hypothalamus or pituitary. By better understanding the impact of EDCs on the adult ovary, we may be able to develop better policies for preventing EDC-induced ovarian toxicity. Increased comprehension of the mechanisms by which EDCs inhibit folliculogenesis, decrease follicle/oocyte health, and alter steroidogenesis may help us develop novel treatments for EDC-induced ovarian damage and thus, develop novel treatments for EDC-induced abnormalities in reproductive and nonreproductive health.
References
- 1. Silbergeld EK, Flaws JA.. Environmental exposures and women's health. Clin Obstet Gynecol 2002; 45:1119–1128. [DOI] [PubMed] [Google Scholar]
- 2. Craig ZR, Wang W, Flaws JA.. Endocrine-disrupting chemicals in ovarian function: effects on steroidogenesis, metabolism and nuclear receptor signaling. Reproduction 2011; 142:633–646. [DOI] [PubMed] [Google Scholar]
- 3. Sowers MR, La Pietra MT.. Menopause: its epidemiology and potential association with chronic diseases. Epidemiol Rev 1995; 17:287–302. [DOI] [PubMed] [Google Scholar]
- 4. Bagur AC, Mautalen CA.. Risk for developing osteoporosis in untreated premature menopause. Calcif Tissue Int 1992; 51:4–7. [DOI] [PubMed] [Google Scholar]
- 5. Sharara FI, Seifer DB, Flaws JA.. Environmental toxicants and female reproduction. Fertil Steril 1998; 70:613–622. [DOI] [PubMed] [Google Scholar]
- 6. Zama AM, Uzumcu M.. Epigenetic effects of endocrine-disrupting chemicals on female reproduction: an ovarian perspective. Front Neuroendocrinol 2010; 31:420–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Padmanabhan V, Sarma HN, Savabieasfahani M, Steckler TL, Veiga-Lopez A.. Developmental reprogramming of reproductive and metabolic dysfunction in sheep: native steroids vs. environmental steroid receptor modulators. Int J Androl 2010; 33:394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jefferson WN, Padilla-Banks E, Newbold RR.. Disruption of the developing female reproductive system by phytoestrogens: genistein as an example. Mol Nutr Food Res 2007; 51:832–844. [DOI] [PubMed] [Google Scholar]
- 9. Uzumcu M, Zachow R.. Developmental exposure to environmental endocrine disruptors: consequences within the ovary and on female reproductive function. Reprod Toxicol 2007; 23:337–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peretz J, Vrooman L, Ricke WA, Hunt PA, Ehrlich S, Hauser R, Padmanabhan V, Taylor HS, Swan SH, VandeVoort CA, Flaws JA.. Bisphenol a and reproductive health: update of experimental and human evidence, 2007-2013. Environ Health Perspect 2014; 122:775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Badach H, Nazimek T, Kaminski R, Turski W.. Organochlorine pesticides concentration in the drinking water from regions of extensive agriculture in Poland. Ann Agric Environ Med 2000; 7:25–28. [PubMed] [Google Scholar]
- 12. Badach H, Nazimek T, Kaminska IA.. Pesticide content in drinking water samples collected from orchard areas in central Poland. Ann Agric Environ Med 2007; 14:109–114. [PubMed] [Google Scholar]
- 13. Krol WJ, Arsenault TL, Pylypiw HM Jr, . Incorvia Mattina MJ. Reduction of pesticide residues on produce by rinsing. J Agric Food Chem 2000; 48:4666–4670. [DOI] [PubMed] [Google Scholar]
- 14. Golovleva LA, Polyakova AB, Pertsova RN, Finkelshtein ZI.. The fate of methoxychlor in soils and transformation by soil microorganisms. J Environ Sci Health B 1984; 19:523–538. [DOI] [PubMed] [Google Scholar]
- 15. Shegunova P, Klanova J, Holoubek I.. Residues of organochlorinated pesticides in soils from the Czech Republic. Environ Pollut 2007; 146:257–261. [DOI] [PubMed] [Google Scholar]
- 16. Armenti AE, Zama AM, Passantino L, Uzumcu M.. Developmental methoxychlor exposure affects multiple reproductive parameters and ovarian folliculogenesis and gene expression in adult rats. Toxicol Appl Pharmacol 2008; 233:286–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hannon PR, Flaws JA.. The effects of phthalates on the ovary. Front Endocrinol (Lausanne) 2015; 6:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schecter A, Birnbaum L, Ryan JJ, Constable JD.. Dioxins: an overview. Environ Res 2006; 101:419–428. [DOI] [PubMed] [Google Scholar]
- 19. Polkowski K, Mazurek AP.. Biological properties of genistein. A review of in vitro and in vivo data. Acta Pol Pharm 2000; 57:135–155. [PubMed] [Google Scholar]
- 20. Jefferson WN. Adult ovarian function can be affected by high levels of soy. J Nutr 2010; 140:2322s–2325s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV.. Human exposure to bisphenol A (BPA). Reprod Toxicol 2007; 24:139–177. [DOI] [PubMed] [Google Scholar]
- 22. Sajiki J, Takahashi K, Yonekubo J.. Sensitive method for the determination of bisphenol-A in serum using two systems of high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl 1999; 736:255–261. [DOI] [PubMed] [Google Scholar]
- 23. Inoue K, Kato K, Yoshimura Y, Makino T, Nakazawa H.. Determination of bisphenol A in human serum by high-performance liquid chromatography with multi-electrode electrochemical detection. J Chromatogr B Biomed Sci Appl 2000; 749:17–23. [DOI] [PubMed] [Google Scholar]
- 24. Takeuchi T, Tsutsumi O.. Serum bisphenol a concentrations showed gender differences, possibly linked to androgen levels. Biochem Biophys Res Commun 2002; 291:76–78. [DOI] [PubMed] [Google Scholar]
- 25. Inoue K, Yamaguchi A, Wada M, Yoshimura Y, Makino T, Nakazaw H.. Quantitative detection of bisphenol A and bisphenol A diglycidyl ether metabolites in human plasma by liquid chromatography-electrospray mass spectrometry. J Chromatogr B Biomed Sci Appl 2001; 765:121–126. [DOI] [PubMed] [Google Scholar]
- 26. Volkel W, Bittner N, Dekant W.. Quantitation of bisphenol A and bisphenol A glucuronide in biological samples by high performance liquid chromatography-tandem mass spectrometry. Drug Metab Dispos 2005; 33:1748–1757. [DOI] [PubMed] [Google Scholar]
- 27. Genuis SJ, Beesoon S, Birkholz D, Lobo RA.. Human excretion of bisphenol A: blood, urine, and sweat (BUS) study. J Environ Public Health 2012; 2012:185731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Otaka H, Yasuhara A, Morita M.. Determination of bisphenol A and 4-nonylphenol in human milk using alkaline digestion and cleanup by solid-phase extraction. Anal Sci 2003; 19:1663–1666. [DOI] [PubMed] [Google Scholar]
- 29. Sun Y, Irie M, Kishikawa N, Wada M, Kuroda N, Nakashima K.. Determination of bisphenol A in human breast milk by HPLC with column-switching and fluorescence detection. Biomed Chromatogr 2004; 18:501–507. [DOI] [PubMed] [Google Scholar]
- 30. Ikezuki Y, Tsutsumi O, Takai Y, Kamei Y, Taketani Y.. Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Hum Reprod 2002; 17:2839–2841. [DOI] [PubMed] [Google Scholar]
- 31. Yamada H, Furuta I, Kato EH, Kataoka S, Usuki Y, Kobashi G, Sata F, Kishi R, Fujimoto S.. Maternal serum and amniotic fluid bisphenol A concentrations in the early second trimester. Reprod Toxicol 2002; 16:735–739. [DOI] [PubMed] [Google Scholar]
- 32. Engel SM, Levy B, Liu Z, Kaplan D, Wolff MS.. Xenobiotic phenols in early pregnancy amniotic fluid. Reprod Toxicol 2006; 21:110–112. [DOI] [PubMed] [Google Scholar]
- 33. Schonfelder G, Wittfoht W, Hopp H, Talsness CE, Paul M, Chahoud I.. Parent bisphenol A accumulation in the human maternal-fetal-placental unit. Environ Health Perspect 2002; 110:A703–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Todaka E, Mori C.. Necessity to establish new risk assessment and risk communication for human fetal exposure to multiple endocrine disruptors in Japan. Congenit Anom (Kyoto) 2002; 42:87–93. [DOI] [PubMed] [Google Scholar]
- 35. Taylor JA. Vom Saal FS, Welshons WV, Drury B, Rottinghaus G, Hunt PA, Toutain PL, Laffont CM, VandeVoort CA. Similarity of bisphenol A pharmacokinetics in rhesus monkeys and mice: relevance for human exposure. Environ Health Perspect 2011; 119:422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kandaraki E, Chatzigeorgiou A, Livadas S, Palioura E, Economou F, Koutsilieris M, Palimeri S, Panidis D, Diamanti-Kandarakis E.. Endocrine disruptors and polycystic ovary syndrome (PCOS): elevated serum levels of bisphenol A in women with PCOS. J Clin Endocrinol Metab 2011; 96:E480–484. [DOI] [PubMed] [Google Scholar]
- 37. Caserta D, Di Segni N, Mallozzi M, Giovanale V, Mantovani A, Marci R, Moscarini M., Bisphenol A. and the female reproductive tract: an overview of recent laboratory evidence and epidemiological studies. Reprod Biol Endocrinol 2014; 12:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Souter I, Smith KW, Dimitriadis I, Ehrlich S, Williams PL, Calafat AM, Hauser R.. The association of bisphenol-A urinary concentrations with antral follicle counts and other measures of ovarian reserve in women undergoing infertility treatments. Reprod Toxicol 2013; 42:224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mok-Lin E, Ehrlich S, Williams PL, Petrozza J, Wright DL, Calafat AM, Ye X, Hauser R.. Urinary bisphenol A concentrations and ovarian response among women undergoing IVF. Int J Androl 2010; 33:385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang W, Hafner KS, Flaws JA.. In utero bisphenol A exposure disrupts germ cell nest breakdown and reduces fertility with age in the mouse. Toxicol Appl Pharmacol 2014; 276:157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peretz J, Gupta RK, Singh J, Hernandez-Ochoa I, Flaws JA.. Bisphenol A impairs follicle growth, inhibits steroidogenesis, and downregulates rate-limiting enzymes in the estradiol biosynthesis pathway. Toxicol Sci 2011; 119:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ziv-Gal A, Craig ZR, Wang W, Flaws JA.. Bisphenol A inhibits cultured mouse ovarian follicle growth partially via the aryl hydrocarbon receptor signaling pathway. Reprod Toxicol 2013; 42:58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peretz J, Craig ZR, Flaws JA.. Bisphenol A inhibits follicle growth and induces atresia in cultured mouse antral follicles independently of the genomic estrogenic pathway. Biol Reprod 2012; 87:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xu J, Osuga Y, Yano T, Morita Y, Tang X, Fujiwara T, Takai Y, Matsumi H, Koga K, Taketani Y, Tsutsumi O.. Bisphenol A induces apoptosis and G2-to-M arrest of ovarian granulosa cells. Biochem Biophys Res Commun 2002; 292:456–462. [DOI] [PubMed] [Google Scholar]
- 45. Lee SG, Kim JY, Chung JY, Kim YJ, Park JE, Oh S, Yoon YD, Yoo KS, Yoo YH, Kim JM.. Bisphenol A exposure during adulthood causes augmentation of follicular atresia and luteal regression by decreasing 17beta-estradiol synthesis via downregulation of aromatase in rat ovary. Environ Health Perspect 2013; 121:663–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wetherill YB, Akingbemi BT, Kanno J, McLachlan JA, Nadal A, Sonnenschein C, Watson CS, Zoeller RT, Belcher SM.. In vitro molecular mechanisms of bisphenol A action. Reprod Toxicol 2007; 24:178–198. [DOI] [PubMed] [Google Scholar]
- 47. Susiarjo M, Hassold TJ, Freeman E, Hunt PA.. Bisphenol A exposure in utero disrupts early oogenesis in the mouse. PLoS Genet 2007; 3:e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bouskine A, Nebout M, Brucker-Davis F, Benahmed M, Fenichel P.. Low doses of bisphenol A promote human seminoma cell proliferation by activating PKA and PKG via a membrane G-protein-coupled estrogen receptor. Environ Health Perspect 2009; 117:1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hunt PA, Koehler KE, Susiarjo M, Hodges CA, Ilagan A, Voigt RC, Thomas S, Thomas BF, Hassold TJ.. Bisphenol a exposure causes meiotic aneuploidy in the female mouse. Curr Biol 2003; 13:546–553. [DOI] [PubMed] [Google Scholar]
- 50. Trapphoff T, Heiligentag M, El Hajj N, Haaf T, Eichenlaub-Ritter U.. Chronic exposure to a low concentration of bisphenol A during follicle culture affects the epigenetic status of germinal vesicles and metaphase II oocytes. Fertil Steril 2013; 100:1758–1767.e1751. [DOI] [PubMed] [Google Scholar]
- 51. Mlynarcikova A, Nagyova E, Fickova M, Scsukova S.. Effects of selected endocrine disruptors on meiotic maturation, cumulus expansion, synthesis of hyaluronan and progesterone by porcine oocyte-cumulus complexes. Toxicol in Vitro 2009; 23:371–377. [DOI] [PubMed] [Google Scholar]
- 52. Zhou W, Liu J, Liao L, Han S, Liu J.. Effect of bisphenol A on steroid hormone production in rat ovarian theca-interstitial and granulosa cells. Mol Cell Endocrinol 2008; 283:12–18. [DOI] [PubMed] [Google Scholar]
- 53. Mlynarcikova A, Kolena J, Fickova M, Scsukova S.. Alterations in steroid hormone production by porcine ovarian granulosa cells caused by bisphenol A and bisphenol A dimethacrylate. Mol Cell Endocrinol 2005; 244:57–62. [DOI] [PubMed] [Google Scholar]
- 54. Grasselli F, Baratta L, Baioni L, Bussolati S, Ramoni R, Grolli S, Basini G.. Bisphenol A disrupts granulosa cell function. Domest Anim Endocrinol 2010; 39:34–39. [DOI] [PubMed] [Google Scholar]
- 55. Peretz J, Flaws JA.. Bisphenol A down-regulates rate-limiting Cyp11a1 to acutely inhibit steroidogenesis in cultured mouse antral follicles. Toxicol Appl Pharmacol 2013; 271:249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kabuto H, Hasuike S, Minagawa N, Shishibori T.. Effects of bisphenol A on the metabolisms of active oxygen species in mouse tissues. Environ Res 2003; 93:31–35. [DOI] [PubMed] [Google Scholar]
- 57. El-Beshbishy HA, Aly HA, El-Shafey M.. Lipoic acid mitigates bisphenol A-induced testicular mitochondrial toxicity in rats. Toxicol Ind Health 2013; 29:875–887. [DOI] [PubMed] [Google Scholar]
- 58. D'Cruz SC, Jubendradass R, Jayakanthan M, Rani SJ, Mathur PP.. Bisphenol A impairs insulin signaling and glucose homeostasis and decreases steroidogenesis in rat testis: an in vivo and in silico study. Food Chem Toxicol 2012; 50:1124–1133. [DOI] [PubMed] [Google Scholar]
- 59. Chouhan S, Yadav SK, Prakash J, Westfall S, Ghosh A, Agarwal NK, Singh SP.. Increase in the expression of inducible nitric oxide synthase on exposure to bisphenol A: a possible cause for decline in steroidogenesis in male mice. Environ Toxicol Pharmacol 2015; 39:405–416. [DOI] [PubMed] [Google Scholar]
- 60. Avci B, Bahadir A, Tuncel OK, Bilgici B.. Influence of alpha-tocopherol and alpha-lipoic acid on bisphenol-A-induced oxidative damage in liver and ovarian tissue of rats. Toxicol Ind Health 2014; [DOI] [PubMed] [Google Scholar]
- 61. Stresser DM, Kupfer D.. Human cytochrome P450-catalyzed conversion of the proestrogenic pesticide methoxychlor into an estrogen. Role of CYP2C19 and CYP1A2 in O-demethylation. Drug Metab Dispos 1998; 26:868–874. [PubMed] [Google Scholar]
- 62. Uzumcu M, Kuhn PE, Marano JE, Armenti AE, Passantino L.. Early postnatal methoxychlor exposure inhibits folliculogenesis and stimulates anti-Müllerian hormone production in the rat ovary. J Endocrinol 2006; 191:549–558. [DOI] [PubMed] [Google Scholar]
- 63. Gupta RK, Meachum S, Hernandez-Ochoa I, Peretz J, Yao HH, Flaws JA.. Methoxychlor inhibits growth of antral follicles by altering cell cycle regulators. Toxicol Appl Pharmacol 2009; 240:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Miller KP, Gupta RK, Greenfeld CR, Babus JK, Flaws JA.. Methoxychlor directly affects ovarian antral follicle growth and atresia through Bcl-2- and Bax-mediated pathways. Toxicol Sci 2005; 88:213–221. [DOI] [PubMed] [Google Scholar]
- 65. Gupta RK, Schuh RA, Fiskum G, Flaws JA.. Methoxychlor causes mitochondrial dysfunction and oxidative damage in the mouse ovary. Toxicol Appl Pharmacol 2006; 216:436–445. [DOI] [PubMed] [Google Scholar]
- 66. Symonds DA, Merchenthaler I, Flaws JA.. Methoxychlor and estradiol induce oxidative stress DNA damage in the mouse ovarian surface epithelium. Toxicol Sci 2008; 105:182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Basavarajappa MS, Craig ZR, Hernandez-Ochoa I, Paulose T, Leslie TC, Flaws JA.. Methoxychlor reduces estradiol levels by altering steroidogenesis and metabolism in mouse antral follicles in vitro. Toxicol Appl Pharmacol 2011; 253:161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Akgul Y, Derk RC, Meighan T, Rao KM, Murono EP.. The methoxychlor metabolite, HPTE, inhibits rat luteal cell progesterone production. Reprod Toxicol 2011; 32:77–84. [DOI] [PubMed] [Google Scholar]
- 69. Zachow R, Uzumcu M.. The methoxychlor metabolite, 2,2–bis-(p-hydroxyphenyl)-1,1,1-trichloroethane, inhibits steroidogenesis in rat ovarian granulosa cells in vitro. Reprod Toxicol 2006; 22:659–665. [DOI] [PubMed] [Google Scholar]
- 70. Craig ZR, Hannon PR, Flaws JA.. Pregnenolone co-treatment partially restores steroidogenesis, but does not prevent growth inhibition and increased atresia in mouse ovarian antral follicles treated with mono-hydroxy methoxychlor. Toxicol Appl Pharmacol 2013; 272:780–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Craig ZR, Leslie TC, Hatfield KP, Gupta RK, Flaws JA.. Mono-hydroxy methoxychlor alters levels of key sex steroids and steroidogenic enzymes in cultured mouse antral follicles. Toxicol Appl Pharmacol 2010; 249:107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gaido KW, Maness SC, McDonnell DP, Dehal SS, Kupfer D, Safe S.. Interaction of methoxychlor and related compounds with estrogen receptor alpha and beta, and androgen receptor: structure-activity studies. Mol Pharmacol 2000; 58:852–858. [PubMed] [Google Scholar]
- 73. Waters KM, Safe S, Gaido KW.. Differential gene expression in response to methoxychlor and estradiol through ERalpha, ERbeta, and AR in reproductive tissues of female mice. Toxicol Sci 2001; 63:47–56. [DOI] [PubMed] [Google Scholar]
- 74. Zama AM, Uzumcu M.. Fetal and neonatal exposure to the endocrine disruptor methoxychlor causes epigenetic alterations in adult ovarian genes. Endocrinology 2009; 150:4681–4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Paulose T, Hernandez-Ochoa I, Basavarajappa MS, Peretz J, Flaws JA.. Increased sensitivity of estrogen receptor alpha overexpressing antral follicles to methoxychlor and its metabolites. Toxicol Sci 2011; 120:447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Basavarajappa MS, Hernandez-Ochoa I, Wang W, Flaws JA.. Methoxychlor inhibits growth and induces atresia through the aryl hydrocarbon receptor pathway in mouse ovarian antral follicles. Reprod Toxicol 2012; 34:16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hites RA. Dioxins: an overview and history. Environ Sci Technol 2011; 45:16–20. [DOI] [PubMed] [Google Scholar]
- 78. Frakes RA, Zeeman CQ, Mower B.. Bioaccumulation of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) by fish downstream of pulp and paper mills in Maine. Ecotoxicol Environ Saf 1993; 25:244–252. [DOI] [PubMed] [Google Scholar]
- 79. Tuppurainen K, Asikainen A, Ruokojarvi P, Ruuskanen J.. Perspectives on the formation of polychlorinated dibenzo-p-dioxins and dibenzofurans during municipal solid waste (MSW) incineration, and other combustion processes. Acc Chem Res 2003; 36:652–658. [DOI] [PubMed] [Google Scholar]
- 80. Humblet O, Sergeyev O, Altshul L, Korrick SA, Williams PL, Emond C, Birnbaum LS, Burns JS, Lee MM, Revich B, Shelepchikov A, Feshin D et al. Temporal trends in serum concentrations of polychlorinated dioxins, furans, and PCBs among adult women living in Chapaevsk, Russia: a longitudinal study from 2000 to 2009. Environ Health 2011; 10:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ulaszewska MM, Zuccato E, Davoli E.. PCDD/Fs and dioxin-like PCBs in human milk and estimation of infants' daily intake: a review. Chemosphere 2011; 83:774–782. [DOI] [PubMed] [Google Scholar]
- 82. Tsutsumi O, Uechi H, Sone H, Yonemoto J, Takai Y, Momoeda M, Tohyama C, Hashimoto S, Morita M, Taketani Y.. Presence of dioxins in human follicular fluid: their possible stage-specific action on the development of preimplantation mouse embryos. Biochem Biophys Res Commun 1998; 250:498–501. [DOI] [PubMed] [Google Scholar]
- 83. Karman BN, Basavarajappa MS, Craig ZR, Flaws JA.. 2,3,7,8-Tetrachlorodibenzo-p-dioxin activates the aryl hydrocarbon receptor and alters sex steroid hormone secretion without affecting growth of mouse antral follicles in vitro. Toxicol Appl Pharmacol 2012; 261:88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Grochowalski A, Pieklo R, Gasinska A, Chrzaszcz R, Gregoraszczuk EL.. Accumulation of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in porcine preovulatory follicles after in vitro exposure to TCDD: effects on steroid secretion and cell proliferation. Cytobios 2000; 102:21–31. [PubMed] [Google Scholar]
- 85. Heimler I, Trewin AL, Chaffin CL, Rawlins RG, Hutz RJ.. Modulation of ovarian follicle maturation and effects on apoptotic cell death in Holtzman rats exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in utero and lactationally. Reprod Toxicol 1998; 12:69–73. [DOI] [PubMed] [Google Scholar]
- 86. Valdez KE, Shi Z, Ting AY, Petroff BK.. Effect of chronic exposure to the aryl hydrocarbon receptor agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin in female rats on ovarian gene expression. Reprod Toxicol 2009; 28:32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hernandez-Ochoa I, Karman BN, Flaws JA.. The role of the aryl hydrocarbon receptor in the female reproductive system. Biochem Pharmacol 2009; 77:547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gao X, Petroff BK, Rozman KK, Terranova PF.. Gonadotropin-releasing hormone (GnRH) partially reverses the inhibitory effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on ovulation in the immature gonadotropin-treated rat. Toxicology 2000; 147:15–22. [DOI] [PubMed] [Google Scholar]
- 89. Petroff BK, Gao X, Rozman KK, Terranova PF.. Interaction of estradiol and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in an ovulation model: evidence for systemic potentiation and local ovarian effects. Reprod Toxicol 2000; 14:247–255. [DOI] [PubMed] [Google Scholar]
- 90. Jung NK, Park JY, Park JH, Kim SY, Park JK, Chang WK, Lee HC, Kim SW, Chun SY.. Attenuation of cell cycle progression by 2,3,7,8-tetrachlorodibenzo-p-dioxin eliciting ovulatory blockade in gonadotropin-primed immature rats. Endocr J 2010; 57:863–871. [DOI] [PubMed] [Google Scholar]
- 91. Karman BN, Basavarajappa MS, Hannon P, Flaws JA.. Dioxin exposure reduces the steroidogenic capacity of mouse antral follicles mainly at the level of HSD17B1 without altering atresia. Toxicol Appl Pharmacol 2012; 264:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Dasmahapatra AK, Wimpee BA, Trewin AL, Wimpee CF, Ghorai JK, Hutz RJ.. Demonstration of 2,3,7,8-tetrachlorodibenzo-p-dioxin attenuation of P450 steroidogenic enzyme mRNAs in rat granulosa cell in vitro by competitive reverse transcriptase-polymerase chain reaction assay. Mol Cell Endocrinol 2000; 164:5–18. [DOI] [PubMed] [Google Scholar]
- 93. Gregoraszczuk EL. Dioxin exposure and porcine reproductive hormonal activity. Cad Saude Publica 2002; 18:453–462. [DOI] [PubMed] [Google Scholar]
- 94. Heimler I, Rawlins RG, Owen H, Hutz RJ.. Dioxin perturbs, in a dose- and time-dependent fashion, steroid secretion, and induces apoptosis of human luteinized granulosa cells. Endocrinology 1998; 139:4373–4379. [DOI] [PubMed] [Google Scholar]
- 95. Huang L, Huang R, Ran XR, Liu HY, Zhang Y, Dai LJ, Li B.. Three-generation experiment showed female C57BL/6J mice drink drainage canal water containing low level of TCDD-like activity causing high pup mortality. J Toxicol Sci 2011; 36:713–724. [DOI] [PubMed] [Google Scholar]
- 96. Chen X, Ma XM, Ma SW, Coenraads PJ, Zhang CM, Liu J, Zhao LJ, Sun M, Tang NJ.. Proteomic analysis of the rat ovary following chronic low-dose exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). J Toxicol Environ Health A 2009; 72:717–726. [DOI] [PubMed] [Google Scholar]
- 97. Maranghi F, Tassinari R, Moracci G, Altieri I, Rasinger JD, Carroll TS, Hogstrand C, Lundebye AK, Mantovani A.. Dietary exposure of juvenile female mice to polyhalogenated seafood contaminants (HBCD, BDE-47, PCB-153, TCDD): comparative assessment of effects in potential target tissues. Food Chem Toxicol 2013; 56:443–449. [DOI] [PubMed] [Google Scholar]
- 98. Sechman A, Antos P, Katarzynska D, Grzegorzewska A, Wojtysiak D, Hrabia A.. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on secretion of steroids and STAR, HSD3B and CYP19A1 mRNA expression in chicken ovarian follicles. Toxicol Lett 2014; 225:264–274. [DOI] [PubMed] [Google Scholar]
- 99. Grochowalski A, Chrzaszcz R, Pieklo R, Gregoraszczuk EL.. Estrogenic and antiestrogenic effect of in vitro treatment of follicular cells with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Chemosphere 2001; 43:823–827. [DOI] [PubMed] [Google Scholar]
- 100. Blount BC, Milgram KE, Silva MJ, Malek NA, Reidy JA, Needham LL, Brock JW.. Quantitative detection of eight phthalate metabolites in human urine using HPLC-APCI-MS/MS. Anal Chem 2000; 72:4127–4134. [DOI] [PubMed] [Google Scholar]
- 101. Berge A, Cladiere M, Gasperi J, Coursimault A, Tassin B, Moilleron R.. Meta-analysis of environmental contamination by phthalates. Environ Sci Pollut Res Int 2013; 20:8057–8076. [DOI] [PubMed] [Google Scholar]
- 102. Hongjun Y, Wenjun X, Qing L, Jingtao L, Hongwen Y, Zhaohua L.. Distribution of phthalate esters in topsoil: a case study in the Yellow River Delta, China. Environ Monit Assess 2013; 185:8489–8500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Martine B, Marie-Jeanne T, Cendrine D, Fabrice A, Marc C.. Assessment of adult human exposure to phthalate esters in the urban centre of Paris (France). Bull Environ Contam Toxicol 2013; 90:91–96. [DOI] [PubMed] [Google Scholar]
- 104. National Toxicology Program.Di(2-ethylhexyl) phthalate. Rep Carcinog 2011; 12:156–159. [PubMed] [Google Scholar]
- 105. Koch HM, Calafat AM.. Human body burdens of chemicals used in plastic manufacture. Philos Trans R Soc Lond B Biol Sci 2009; 364:2063–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hannon PR, Peretz J, Flaws JA.. Daily exposure to Di(2-ethylhexyl) phthalate alters estrous cyclicity and accelerates primordial follicle recruitment potentially via dysregulation of the phosphatidylinositol 3-kinase signaling pathway in adult mice. Biol Reprod 2014; 90:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hannon PR, Brannick KE, Wang W, Flaws JA.. Mono(2-ethylhexyl) phthalate accelerates early folliculogenesis and inhibits steroidogenesis in cultured mouse whole ovaries and antral follicles. Biol Reprod 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Craig ZR, Hannon PR, Wang W, Ziv-Gal A, Flaws JA.. Di-n-butyl phthalate disrupts the expression of genes involved in cell cycle and apoptotic pathways in mouse ovarian antral follicles. Biol Reprod 2013; 88:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Xu C, Chen JA, Qiu Z, Zhao Q, Luo J, Yang L, Zeng H, Huang Y, Zhang L, Cao J, Shu W.. Ovotoxicity and PPAR-mediated aromatase downregulation in female Sprague-Dawley rats following combined oral exposure to benzo[a]pyrene and di-(2-ethylhexyl) phthalate. Toxicol Lett 2010; 199:323–332. [DOI] [PubMed] [Google Scholar]
- 110. Wang W, Craig ZR, Basavarajappa MS, Hafner KS, Flaws JA.. Mono-(2-ethylhexyl) phthalate induces oxidative stress and inhibits growth of mouse ovarian antral follicles. Biol Reprod 2012; 87:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Chen HS, Chiang PH, Wang YC, Kao MC, Shieh TH, Tsai CF, Tsai EM.. Benzyl butyl phthalate induces necrosis by AhR mediation of CYP1B1 expression in human granulosa cells. Reprod Toxicol 2012; 33:67–75. [DOI] [PubMed] [Google Scholar]
- 112. Gupta RK, Singh JM, Leslie TC, Meachum S, Flaws JA, Yao HH.. Di-(2-ethylhexyl) phthalate and mono-(2-ethylhexyl) phthalate inhibit growth and reduce estradiol levels of antral follicles in vitro. Toxicol Appl Pharmacol 2010; 242:224–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hannon PR, Brannick KE, Wang W, Gupta RK, Flaws JA.. Di(2-ethylhexyl) phthalate inhibits antral follicle growth, induces atresia, and inhibits steroid hormone production in cultured mouse antral follicles. Toxicol Appl Pharmacol 2015; 284:42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Reinsberg J, Wegener-Toper P, van der Ven K, van der Ven H, Klingmueller D.. Effect of mono-(2-ethylhexyl) phthalate on steroid production of human granulosa cells. Toxicol Appl Pharmacol 2009; 239:116–123. [DOI] [PubMed] [Google Scholar]
- 115. Inada H, Chihara K, Yamashita A, Miyawaki I, Fukuda C, Tateishi Y, Kunimatsu T, Kimura J, Funabashi H, Miyano T.. Evaluation of ovarian toxicity of mono-(2-ethylhexyl) phthalate (MEHP) using cultured rat ovarian follicles. J Toxicol Sci 2012; 37:483–490. [DOI] [PubMed] [Google Scholar]
- 116. Mazur W. Phytoestrogen content in foods. Baillieres Clin Endocrinol Metab 1998; 12:729–742. [DOI] [PubMed] [Google Scholar]
- 117. Khan SI, Zhao J, Khan IA, Walker LA, Dasmahapatra AK.. Potential utility of natural products as regulators of breast cancer-associated aromatase promoters. Reprod Biol Endocrinol 2011; 9:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Andres S, Abraham K, Appel KE, Lampen A.. Risks and benefits of dietary isoflavones for cancer. Crit Rev Toxicol 2011; 41:463–506. [DOI] [PubMed] [Google Scholar]
- 119. Reinli K, Block G.. Phytoestrogen content of foods—a compendium of literature values. Nutr Cancer 1996; 26:123–148. [DOI] [PubMed] [Google Scholar]
- 120. Yoon K, Kwack SJ, Kim HS, Lee BM.. Estrogenic endocrine-disrupting chemicals: molecular mechanisms of actions on putative human diseases. J Toxicol Environ Health B Crit Rev 2014; 17:127–174. [DOI] [PubMed] [Google Scholar]
- 121. Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B, Gustafsson JA.. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998; 139:4252–4263. [DOI] [PubMed] [Google Scholar]
- 122. Bialesova L, Brtko J, Lenko V, Macejova D.. Nuclear receptors—target molecules for isoflavones in cancer chemoprevention. Gen Physiol Biophys 2013; 32:467–478. [DOI] [PubMed] [Google Scholar]
- 123. Medigovic I, Ristic N, Trifunovic S, Manojlovic-Stojanoski M, Milosevic V, Zikic D, Nestorovic N.. Genistein affects ovarian folliculogenesis: a stereological study. Microsc Res Tech 2012; 75:1691–1699. [DOI] [PubMed] [Google Scholar]
- 124. Zhuang XL, Fu YC, Xu JJ, Kong XX, Chen ZG, Luo LL.. Effects of genistein on ovarian follicular development and ovarian life span in rats. Fitoterapia 2010; 81:998–1002. [DOI] [PubMed] [Google Scholar]
- 125. Wang W, Sun Y, Liu J, Li Y, Li H, Xiao S, Weng S, Zhang W.. Soy isoflavones administered to rats from weaning until sexual maturity affect ovarian follicle development by inducing apoptosis. Food Chem Toxicol 2014; 72:51–60. [DOI] [PubMed] [Google Scholar]
- 126. Gregoraszczuk E, Slomczynska M, Stoklosowa S.. Effect of genistein, tyrphostin and herbimycin on prolactin-stimulated progesterone production by porcine theca and luteal cells. J Physiol Pharmacol 1999; 50:477–484. [PubMed] [Google Scholar]
- 127. Whitehead SA, Cross JE, Burden C, Acute Lacey M.. and chronic effects of genistein, tyrphostin and lavendustin A on steroid synthesis in luteinized human granulosa cells. Hum Reprod 2002; 17:589–594. [DOI] [PubMed] [Google Scholar]
- 128. Lacey M, Bohday J, Fonseka SM, Ullah AI, Whitehead SA.. Dose-response effects of phytoestrogens on the activity and expression of 3beta-hydroxysteroid dehydrogenase and aromatase in human granulosa-luteal cells. J Steroid Biochem Mol Biol 2005; 96:279–286. [DOI] [PubMed] [Google Scholar]
- 129. Whitehead SA, Lacey M.. Protein tyrosine kinase activity of lavendustin A and the phytoestrogen genistein on progesterone synthesis in cultured rat ovarian cells. Fertil Steril 2000; 73:613–619. [DOI] [PubMed] [Google Scholar]
- 130. Haynes-Johnson D, Lai MT, Campen C, Palmer S.. Diverse effects of tyrosine kinase inhibitors on follicle-stimulating hormone-stimulated estradiol and progesterone production from rat granulosa cells in serum-containing medium and serum-free medium containing epidermal growth factor. Biol Reprod 1999; 61:147–153. [DOI] [PubMed] [Google Scholar]
- 131. Rice S, Mason HD, Whitehead SA.. Phytoestrogens and their low dose combinations inhibit mRNA expression and activity of aromatase in human granulosa-luteal cells. J Steroid Biochem Mol Biol 2006; 101:216–225. [DOI] [PubMed] [Google Scholar]
- 132. Tiemann U, Schneider F, Vanselow J, Tomek W.. In vitro exposure of porcine granulosa cells to the phytoestrogens genistein and daidzein: effects on the biosynthesis of reproductive steroid hormones. Reprod Toxicol 2007; 24:317–325. [DOI] [PubMed] [Google Scholar]