

Abstract
Altered microRNA expression patterns are implicated in the formation of many human diseases, including ovarian cancer. Our laboratory previously created Dicerfl/fl/Ptenfl/fl/Amhr2cre/+ mice, which developed high-grade serous carcinomas originating from mouse fallopian tubes, while neither Dicerfl/fl/Amhr2cre/+ nor Ptenfl/fl/Amhr2cre/+ mice developed tumors. To explore miRNAs involved in the tumorigenesis in the double-knockout (DKO) mice, tumor cell lines were established from mouse primary tumors, and the most abundant miRNAs present in mouse normal fallopian tubes, let-7b and miR-34c, were expressed in these cell lines. We found that miR-34c had a more dramatic effect on inhibiting tumor cell viability than let-7b. The action of miR-34c induced tumor cell cycle arrest in G1 phase and apoptosis, and was accompanied with the regulation of key genes involved in cell proliferation and cell cycle G1/S transition. miR-34c suppressed the expression of Ezh2 and Mybl2, which may transcriptionally and functionally activate Cdkn1c. Furthermore, miR-34c levels are extremely low in human serous adenocarcinomas compared with human normal fallopian tubes. Expression of miR-34c in human ovarian cancer cells phenocopied its effects in DKO mouse tumor cells. However, miR-34b/c−/−/Ptenfl/fl/Amhr2cre/+ mice failed to develop high-grade serous carcinomas, implicating a combination of miRNAs in the tumorigenesis process. Thus, while miR-34c is a putative tumor suppressor in high-grade serous ovarian carcinoma with potential therapeutic advantages, screening of additional miRNAs for their effects alone and in combination with miR-34c is highly warranted to uncover miRNAs that synergize with miR-34c against cancer.
Keywords: microRNA, ovarian cancer, Dicer/Pten double knockout, miR-34b/c/Pten double knockout
Introduction
Ovarian cancer is the fifth leading cause of cancer death among women in the United States, with an estimated 22 240 new cases and 14 030 deaths in 2013 [1]. The 5-yr survival rate for women diagnosed with cancer localized to the ovary exceeds 90%. However, nearly 70% of cases are diagnosed at late stages, and the 5-yr survival rate for these patients is only 30%. Approximately 90% of ovarian cancers are epithelial in origin, although the early developmental origins of ovarian cancers remain poorly defined [2]. Although it has been normally proposed that these tumors arise from the surface of the ovary or from inclusion cysts, recent work has brought an alternative hypothesis that a subgroup of these tumors arises from fallopian tube [3, 4]. Histological classification of ovarian cancer results in four major subtypes (i.e., serous [70%], endometrioid [7%], clear cell [10%], and mucinous [3%]) [5]. Although these histotypes may arise from distinct cells of origin and have mutations in different signaling pathways, all ovarian cancers are still treated similarly in the clinical setting. Moreover, cytotoxic chemotherapy is often unsuccessful, due to common resistance to the current chemotherapeutic regimens. The past decade has heralded a new era in the understanding of gene regulation of ovarian cancer, particularly after miRNAs were discovered. We now appreciate that the aberrant expression of miRNAs promotes tumorigenesis, metastasis, and other features of ovarian cancer [6].
miRNAs are aberrantly expressed in a variety of cancers. First described by Calin et al., the loci of miR-15a and -16-1, which are clustered at chromosome 13q14, are frequently deleted in B cell chronic lymphocytic leukemia, followed by an expression reduction of these two miRNAs in cancer samples as compared to normal tissues [7]. Probably the first genome-wide study indicating the importance of miRNAs in ovarian cancer was published in 2006 in which 40% of the miRNAs exhibit altered DNA copy number [8]. Later, impaired miRNA processing was also correlated to ovarian cancer, implicating the potential role of altered miRNA expression in this disease. Analyzed from 111 samples of invasive epithelial ovarian cancer patients, Merritt et al. [9] identified a significant correlation between low DICER and DROSHA expression and advanced tumor stage and suboptimal surgical cytoreduction, while cancer specimens with both high DICER and DROSHA expression were associated with increased median survival (>11 yr vs. 2.66 yr for other subgroups). Using shRNA-mediated knockdown of three factors required for miRNA processing, including DICER, DROSHA, and DGCR8, another group [10] globally repressed miRNA maturation in mice and human cancer cell lines. Their data demonstrated that cancer cells with decreased miRNA levels had a more pronounced transformed phenotype, while, in animals, cells with miRNA processing defects formed tumors with accelerated kinetics. Taken together, these experiments broadly implicate miRNA aberrations in ovarian cancer, probably through important roles as tumor suppressors. However, other studies highlight roles for specific miRNAs as oncogenes (i.e., overexpression accelerates tumorigenesis [11, 12]), clearly indicating that these miRNAs have dual functions in caner [13].
Given that miRNAs may play critical roles in ovarian cancer, our laboratory conditionally deleted Dicer using a conditional knockout (cKO) approach directed by a knockin of the the Cre recombinase gene into the anti-Müllerian hormone receptor type 2 (Amhr2) locus. Amhr2-Cre is expressed embryonically in the mesenchyme of the developing Müllerian ducts and postnatally in ovarian granulosa cells and the smooth muscle and stromal cells of the oviducts and uterus [14–17]. Therefore, tissue-specific recombination of the Dicer floxed allele resulted in the loss of DICER protein in somatic cells of the ovary, oviduct, and uterus, which phenotypically led to female mouse infertility and formation of bilateral tubal diverticuli [18]. However, no tumors were observed in female reproductive tract, although these diverticuli became larger and larger during development. To generate an ovarian cancer mouse model to study the functions of DICER, we later conditionally deleted both Dicer and Pten in mice using the same strategy as our Dicer single cKO mice. Phosphatase and tensin homolog (PTEN) is a tumor suppressor that dephosphorylates phosphatidylinositol 3,4,5-trisphosphate, the product of the lipid kinase phosphatidylinositide 3-kinases (PI3K), therefore serving as a negative regulator of the PI3K signaling pathway [19, 20]. PI3K signaling is a main regulator of cell growth, metabolism, and survival, and overactivity of this signaling has been found in many types of cancers. The Cancer Genome Atlas researchers measured comprehensively genomic and epigenomic abnormalities on clinically annotated high-grade serous ovarian cancer samples and observed mutations of the PI3K/RAS pathway in 45% of all the cases studied [21]. Consistent with this finding, our Dicer/Pten cKO mice developed high-grade serous epithelial cancers that initiated as primary tumors in the fallopian tube and spread to engulf the ovary; these aggressive metastatic cancer cells subsequently spread throughout the abdominal cavity, resulting in ascites formation and death of 100% of the mice by 13 mo [4]. Disabling Pten alone failed to cause a tumor phenotype in the ovary or fallopian tube [22], indicating a synergistic effect of miRNA maturation defects and PI3K signaling overactivity in the onset of ovarian cancer. Therefore, cancer cells isolated from Dicer/Pten double-knockout (DKO) mouse tumors would be a valuable platform to investigate miRNAs and PI3K pathway components in this deadly disease.
Based on our model, we hypothesized that defects in miRNA maturation in Dicer/Pten DKO mice play a critical role in fallopian tube tumorigenesis. Since turning off DICER globally affected miRNAs in mouse reproductive tract, it was still uncertain which miRNA or miRNA combinations functioned as tumor suppressors during the onset of tumor formation in this animal model. Given that DKO tumors originated from fallopian tubes, we hypothesized that loss of most abundant miRNAs in the fallopian tube may be more significant for tumor formation. By profiling miRNAs in mouse fallopian tube by next-generation sequencing, we identified 10 individual miRNAs that make up 92.8% of all fallopian tube miRNAs. Among these 10 miRNAs, 83.3% belonged to the let-7 family. miR-34c, comprising 4.1% of miRNAs, was the most abundant miRNA other than let-7 family members in the mouse fallopian tube (Fig. 1). In addition, the miR-34b and -c locus was frequently lost in high-grade serous ovarian cancers in women [4]. Therefore, we delivered mature miR-34c and let-7b back to DKO mouse tumor cells by in vitro miRNA transfection and investigated their effects on tumor cell viability. We demonstrated that miR-34c had a greater potential effect on inhibiting tumor cell viability than let-7b. The molecular mechanism of miR-34c underlying this effect was further investigated and supported our hypothesis that miR-34c is a putative tumor suppressor in high-grade serous cancers. However, when we deleted miR-34b/c and Pten in mouse reproductive tract, the mice did not phenocopy Dicer/Pten DKO animals, indicating that loss of miR-34b/c was not sufficient to substitute for Dicer in the formation of high-grade serous ovarian cancer in a Pten null background. Our experiments revealed that miR-34c is a putative tumor suppressor in high-grade serous ovarian cancer. However, the formation of this deadly disease is much more complicated where abnormalities of multiple genes must coordinate for its onset.
Fig. 1.
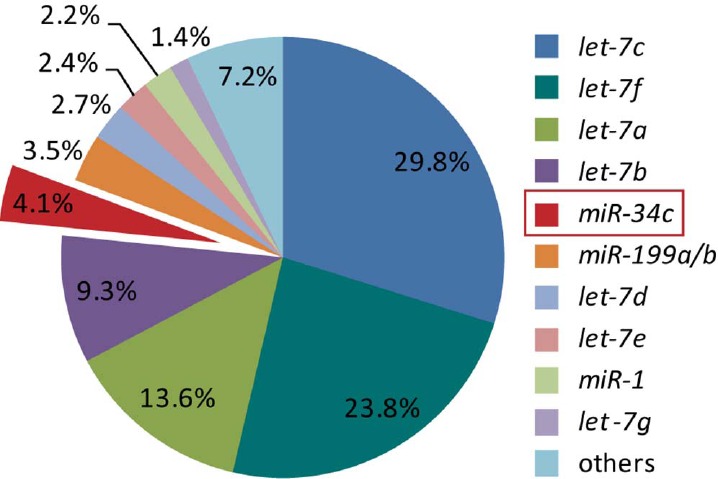
let-7 and miR-34c are the most abundant miRNAs in mouse normal fallopian tubes. Illumina deep sequencing uncovered miRNAs in mouse normal fallopian tubes. Level of individual miRNA was expressed as percentage of total miRNAs reads.
Materials and Methods
miR-34b/c/Pten DKO Mice
Mice were maintained in the vivarium at Baylor College of Medicine (BCM) according to the institutional guidelines for the care and use of laboratory animals. miR-34b/c+/−/Ptenfl/+/Amhr2cre/+ mice were generated by mating female miR-34b/c−/− mice with male Ptenfl/fl/Amhr2cre/+ mice. After a series of intercrosses, male miR-34b/c−/−/Ptenfl/fl/Amhr2cre/+ mice and female miR-34b/c−/−/Ptenfl/fl mice were mated to efficiently generate female miR-34b/c−/−/Ptenfl/fl/Amhr2cre/+ and miR-34b/c−/−/Ptenfl/fl (control) mice.
Dicer/Pten DKO Mouse Tumor Cells and Human Serous Ovarian Cancer Cell Lines
Dicer/Pten DKO mouse tumor cell lines were established by culturing primary fallopian tube serous tumors that engulfed the ovaries in Dicer/Pten DKO mice [4]. The cells were cultured in Dulbecco modified Eagle medium/F12 (Gibco) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Invitrogen). Human serous ovarian cancer cell lines OVCAR8 and OVCAR5 were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Invitrogen).
Small RNA Sequencing
Total RNA was extracted from mouse normal fallopian tubes using the mirVana miRNA Isolation Kit (Applied Biosystems). RNA quality and the presence of small RNAs were determined on a 2100 Bioanalyzer (Agilent Technologies). Small RNA library construction was performed using the DGE-Small RNA Sample Prep Kit (Illumina). Purified cDNA was quantified with the Quant-iT PicoGreen dsDNA Kit (Invitrogen) and diluted to 3 pM for sequencing on the Illumina 1G Genome Analyzer (University of Houston). The raw sequence data were passed through a series of quality-control filters to discard reads without a 3′-adapter, reads with copy number less than 4, reads that were less than 10 nucleotides, and reads with more than 10 consecutive identical nucleotides. The remaining usable sequence reads were aligned to mouse miRNA hairpin precursor sequences in miRBase, with up to three mismatches tolerated in an alignment. If a read aligned equally well to multiple precursors, its copy number was equally apportioned. For each mature miRNA sequence, we counted the number of reads that were exact matches, allowing a four-nucleotide extension into the precursor sequence on each side of the mature miRNA. Each miRNA profile was scale normalized using the total number of usable sequence reads, after which 40 units were added to each miRNA expression value.
Immunofluorescence
The expression of keratin 8 and keratin 14 in cultured DKO mouse tumor cells were studied by immunofluorescence. DKO mouse tumor cell lines were seeded onto Lab-Tek chamber slides (Nunc) 24 hours prior to the staining. Tumor cells were rinsed briefly in PBS and fixed in ice-cold methanol for 15 min at room temperature. Tumor cells were washed twice in ice-cold PBS and then permeabilized with PBS containing 0.25% Triton X-100 for 10 min. After gentle washing three times with PBS each for 5 min, tumor cells were incubated in 1% bovine serum albumin (BSA) in PBS containing 0.1% Tween (PBST) for 30 min to block unspecific binding of the antibodies. Tumor cells were then stained with primary antibodies to keratin 8 (1:50; Abcam) or keratin 14 (1:1000; Covance) in PBST containing 1% BSA in a humidified chamber overnight at 4°C. After primary antibody staining, tumor cells were washed three times with PBS each for 10 min, then stained with Alexa Fluor 488 donkey anti-rabbit IgG (1:1000; Invitrogen) for 1 h at room temperature in the dark. Finally, tumor cells were washed with PBS three times and then examined under an inverted fluorescence microscope to detect the expression of keratin 8 and keratin 14.
Semiquantitative PCR
Genomic DNA was isolated from Dicer/Pten DKO mouse tumor cell lines using DNeasy Blood & Tissue kit (Qiagen). Deletion of Dicer and Pten in Dicer/Pten DKO mouse tumor cell lines were validated using genomic DNA PCR with primers as follows: Dicer_Primer_1: 5′-GGTTACATGGCTAGACTCAAAGC-3′, Dicer_Primer_2: 5′-AGGTGCCTTTCGTTTAGGAAC-3′, Dicer_Primer_3: 5′-AAAGCAGAACTCTAATGCCCC-3′; Pten_Primer_1: 5′-ACTCAAGGCAGGGATGAGC-3′, Pten_Primer_2: 5′-AATCTAGGGCCTCTTGTGCC-3′, Pten_Primer_3: 5′-GCTTGATATCGAATTCCTGCAGC-3′. Genomic DNA of kidney and tumor from Dicer/Pten DKO mouse were used as controls.
miRNA Mimic Transfection on Dicer/Pten DKO Mouse Tumor Cells
miRNA mimic transient transfection was performed using Lipofectamine RNAiMAX (Invitrogen) following the manufacturer's instructions. To achieve a maximum transfection efficiency and lower toxicity, the transfection conditions were optimized in preliminary experiments by transfecting DKO mouse tumor cells with Dye547-labeled miRNA mimic negative control (Dharmacon). To evaluate the effect of miR-34c or let-7b on DKO tumor cells, tumor cells were reverse transfected with 30 nM mmu-miR-34c or mmu-let-7b miRIDINA mimics (Dharmacon) in 96- or 6-well culture plates. Briefly, miRNA duplex and RNAiMAX diluted in Opti-MEM I medium (Invitrogen) were gently mixed and incubated in the well of culture plates for 15 min. The appropriate number of DKO tumor cells in complete growth medium without antibiotics were plated onto the well to give 30%–50% confluence 24 h after plating. The cells were mixed gently by rocking the plate back and forth, and then let sit in the culture hood for 5 min. Transfected cells were then incubated at 37°C in a CO2 incubator for 48 h before various assays. In all transfection experiments, a miRIDIAN miRNA mimic negative control #1 (Dharmacon) was applied in parallel at the same concentration as miR-34c or let-7b, serving as a control.
Cell Viability and Apoptosis Assay
The viability of DKO tumor cells transfected with miR-34c or let-7b mimics was determined by ATP level quantitation-based Promega CellTiter-Glo Luminescent Cell Viability Assay. Cellular apoptosis was determined by Promega Caspase-Glo 3/7 assay. At 48 h after transfection, cells were moved out of the incubator and equilibrated to room temperature for approximately 30 min. Cell viability and apoptosis were then measured following the manufacturer's instructions. An Omega microplate luminescence reader was used to record luminescence. Data were plotted as mean ± SEM of three determinations. Statistical differences were tested using one-way ANOVA followed by Tukey multiple comparison test for viability and two-tailed paired t-test for apoptosis (Prism; GraphPad). P < 0.05 was considered statistically significant.
Gene Expression Profiling
Total RNA was extracted from DKO tumor cell lines transfected with miR-34c (n = 3) and control miRNA (n = 3) using the RNeasy mini RNA isolation kit (Qiagen). RNA quality was determined on a 2100 Bioanalyzer. Gene expression profiles were generated on Mouse WG-6 version 2.0 Beadchips (Illumina) at University of Texas Health Science Center Microarray Core Laboratory. The array platform consists of 45 281 transcript probes representing 30 854 unique genes. Expression data were quantile normalized. Array datasets have been deposited into the Gene Expression Omnibus (GSE57493). Differentially expressed genes were identified using t-test and fold change on log-transformed data. Predicted targets were identified using TargetScanMouse (release 5.0) [23] and miRanda (August 2010) [24].
Quantitative PCR
Quantitative PCR (QPCR) validation of microarray gene expression profiling was performed on samples independent of those in the microarray experiment. Total RNA (1 μg) was reverse transcribed using Superscript III reverse transcriptase and random hexamers (Invitrogen). Complementary DNA was diluted 50-fold and 2.5 μl was used for each 10-μl QPCR reaction. Custom primers were designed using Integrated DNA Technologies SciTools (http://www.idtdna.com/scitools/Applications/RealTimePCR/) and primer sequences are listed in Supplemental Table S1 (Supplemental Data are available online at www.biolreprod.org). QPCR was performed on ABI Prism 7500 Fast Real-Time PCR system and β-actin severed as an endogenous control. Each sample was analyzed in triplicate. The relative quantity of target transcripts were calculated using the 2−ΔΔCt method [25] and normalized to the average 2−ΔΔCt of miRNA mimic negative control. Statistical differences were calculated using Student t-test, and P < 0.05 was considered statistically significant.
The level of miR-34c in human ovarian serous adenocarcinoma was measured using Taqman QPCR. Permission to perform all experiments was obtained from the Institutional Review Board (IRB) for BCM and its affiliated institutions. Human normal fallopian tubes were collected with BCM, IRB approval, and written, informed consent from patients undergoing surgery at Ben Taub General Hospital (Houston, TX) for benign gynecologic conditions, as reported previously [26]. Anonymized human high-grade serous ovarian cancer samples were obtained from the Tissue Acquisition and Distribution Core of the Dan L. Duncan Cancer Center and Department of Pathology and Immunology under an approved BCM IRB protocol. Total RNA was extracted from human normal fallopian tubes (n = 6), human serous ovarian cancer cell lines (n = 2), and human serous ovarian adenocarcinomas (n = 12) using the mirVana miRNA isolation kit. RNA quality and the presence of small RNAs were inspected on a 2100 Bioanalyzer. Total RNA (25 ng) was reverse transcribed with Taqman microRNA RT primers and Multiscribe Reverse Transcriptase (Invitrogen). Levels of mature miR-34c were then determined by QPCR using commercially available Taqman probes (assay ID 000428; Applied Biosystems) per the manufacturer's instructions with U6 small nuclear RNA (assay ID 001973; Applied Biosystems) used as an internal standard for normalization. Relative fold changes were calculated using the 2−ΔΔCt method. Statistical analysis was performed using two-tail paired t-test (Prism, GraphPad). P < 0.05 were taken as statistically significant.
Flow Cytometry
Cell cycle profile of tumor cells overexpressing miR-34c was analyzed by propidium iodide (PI) staining followed by a flow cytometry analysis. Briefly, tumor cells were trypsinized and resuspended in cold PBS. Cells were then fixed with cold 70% ethanol overnight. After removing 70% ethanol from fixed cells, tumor cell nucleic acids were stained with 40 μg/ml PI and 100 μg/ml RNase in PBS. Cells were incubated at 37°C for 30 min in the dark and then analyzed in a FACSCanto II analyzer (BD Biosciences). Cell cycle profiles were generated using flowjo software.
miR-34c Cloning
Genomic DNA was isolated from a p53 wild-type human ovarian cancer cell line HEY using Gentra DNA purification kit (Qiagen). DNA quality was determined by calculating the ratio of absorbance at 260 nm to absorbance at 280 nm and agarose gel electrophoresis. The primary miR-34c sequence was amplified from genomic DNA using Red Taq DNA polymerase (Sigma) and miR-34c-specific primers: forward, 5′-ACTCGAGAAAGCAAAATCCCTGGAGGTG-3′; and reverse, 5′-ATACGCGTTGGTTAAGAGCCTGAAGG-3′. Pri-miR-34c PCR products were separated by gel electrophoresis and extracted using Zymoclean Gel DNA Recovery Kit (ZYMO Research Corp.), and then cloned into pGEM-T Easy Vector (Promega) and sequencing confirmed. To functionally study miR-34c over-expression in human ovarian cancer cells, pri-miR-34c was subcloned from pGEM-T Easy Vector to a lentiviral vector driven by a CMV promoter (pLenti-miR; Open Biosystems). The pri-miR-34c was coexpressed with TurboRed fluorescent protein (TurboRFP) for easy tracking of miRNA expression in human cancer cells.
Lentivirus Production and Transduction
CMV-TurboRFP-miR-34c-IRES-puroR vector was packaged and transduced into OVCAR8 and OVCAR5 cells. A similar vector with nontargeting sequence served as the negative control. Briefly, miR-34c or control vector, as well as helper virus DNA plasmids carrying gag, pol, VSVG, and tat, were transfected into 293T cells using Lipofectamine 2000 (Invitrogen). Cell medium containing virus was syringe filtered (0.45 μm) 48 h after transfection and added to OVCAR8 and OVCAR5 cells at an appropriate dilution. The virus was replaced with fresh medium 6 h after infection. Transduction efficiency was measured by examining the cells under fluorescence microscope.
Human ovarian cancer cells transduced with miR-34c lentivirus or control virus were seeded onto 96-well plates on Day 5. Cell proliferation was evaluated by measuring cell viability at different time points after cell seeding using CellTiter-Glo assay. Data are expressed as relative cell number normalized to control virus transduced cells, and each data point represents an average of triplicate measurements. Statistical analysis was performed using two-tail paired t-test (Prism; GraphPad). P < 0.05 was taken as statistically significant.
Results
Repertoire of miRNAs in Mouse Fallopian Tubes
Using Amhr2-Cre, mice lacking Dicer and Pten in the female reproductive tract generated high-grade serous carcinomas arising from the fallopian tube [4]. Given that DICER biologically encodes a ribonuclease essential for the production of mature miRNAs, Dicer deletion in the DKO mouse model resulted in hindered miRNA maturation, which may be responsible for tumor formation. Therefore, we explored the repertoire of approximately 18- to 30-nucleotide small RNAs expressed in mouse fallopian tubes using Illumina next-generation sequencing technology. The raw sequencing data were passed through a series of quality-control filters, and the usable small RNA reads generated were mapped to known mature miRNAs in miRBase. A total of 10 individual miRNAs were found to make up 92.8% of all fallopian tube miRNAs, where 83.3% belong to the let-7 family. let-7c, let-7f, let-7a, and let-7b are the most abundant four miRNAs. miR-34c, comprising 4.1% of miRNAs, is the most abundant miRNA other than let-7 family members in the mouse fallopian tube (Fig. 1 and Supplemental Table S2)
Establishment of Dicer/Pten DKO Mouse Tumor Cell Lines
We isolated cells from Dicer/Pten DKO mouse primary oviductal serous tumors that engulfed the ovaries. Once adapted to in vitro culture conditions, these cells were maintained for growth for at least 15 passages without entering senescence. When we injected these cells back to severe combined immunodeficiency (SCID) mice, these cells formed xenografts and induced ascites in the abdomen [4]. The unlimited number of cell division and the xenograft-forming capacity indicated the cancerous nature of these cells, and, therefore, these cell lines could be used to study functions of different miRNAs in mouse cancer. We randomly picked up three established DKO tumor cell lines, and cells at early passages were thawed for further experiments. To make sure that these early passaged cells are predominantly tumor cells and have minimum stromal cell contamination, we stained the cells fluorescently with epithelial markers keratin 8 and keratin 14. All the three cell lines studied, DKO-1, DKO-2, and DKO-3, showed typical epithelial-like cell morphology with polygonal shape, more regular dimensions, and growth in discrete patches (Supplemental Fig. S1). As shown, more than 90% of cells had an intense cytoplasmic staining of keratin 8 and keratin 14, further validating that the cancer cell lines established are epithelial-like cells. Deletion of Dicer and Pten in the three cell lines was confirmed by PCR using the genomic DNA isolated from cultured cell lines (Supplemental Fig. S2).
miR-34c Delivery Inhibited DKO Cell Viability by Blocking Cell Division and Inducing Apoptosis
The DKO cancer cell lines are a unique model to study the functions of miRNAs in serous ovarian cancer, since these cancerous cells lack synthesis of mature miRNAs. We therefore delivered individual mature miRNA into these cells by transfecting them with Dharmacon miRNA mimics. let-7b or miR-34c mimics were transfected into DKO tumor cell lines using Lipofectamine RNAiMAX. At 2 days after transfection, miR-34c was discovered to inhibit 50%–60% of cell viability of all the three DKO tumor cell lines as compared to control cells. In contrast, let-7b did not significantly inhibit tumor cell viability (Fig. 2A), indicating a more important role of miR-34c in Dicer/Pten DKO mouse tumorigenesis. Upon inspection of DKO tumor cell lines transfected with miR-34c under a microscope, we did not observe many dead cells; however, we noted that cells were slightly enlarged and cell numbers were apparently lower than control cells, suggesting that miR-34c may induce cell cycle arrest. We therefore studied cell cycle profiles of miR-34c and control miRNA transfected cells using PI staining. Flow cytometric data showed a robust cell division of control cells with high percentage of cells entering S and G2 phase. However, miR-34c dramatically inhibited cell division with much lower percentages of cells entering S and G2 phase. Analyzed by flowjo software, miR-34c transfection resulted in a decrease of S- and G2-phase cells, from 38.2% to 8.1% in DKO-1, a decrease from 30.3% to 15.2% in DKO-2, and a decrease from 52.2% to 16.1% in DKO-3 (Fig. 2B).
Fig. 2.
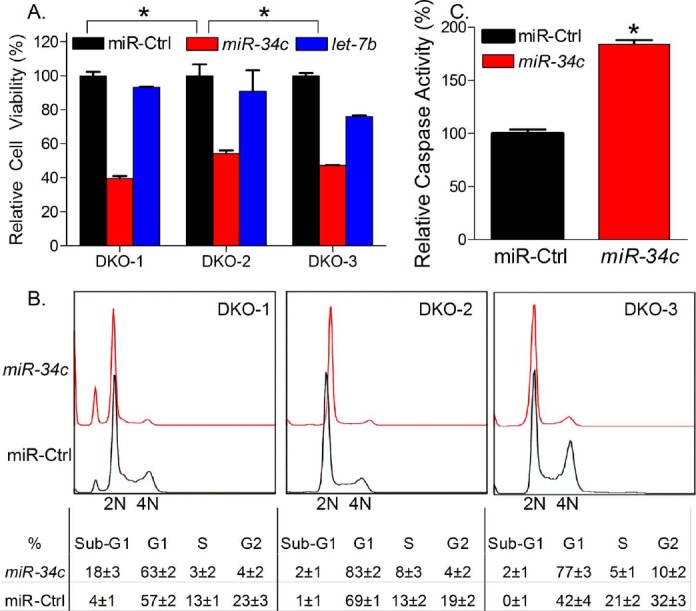
miR-34c inhibited Dicer/Pten DKO cancer cell growth by inducing cell cycle arrest in G1 phase and apoptosis. Dicer/Pten cancer cell lines were transfected with mmu-miR-34c, mmu-let-7b, or miRNA mimic negative control using Lipofectamine RNAiMAX. After 2 days, viability of treated Dicer/Pten DKO cancer cells was measured by Promega CellTiter-Glo assay (A). Representative cell cycle profile of miR-34c-treated cells was determined by flow cytometry using PI staining and analyzed by flowjo software (B). Caspase activities of miR-34c-treated DKO-1 were measured by Promega caspase-Glo 3/7 assay (C). Data of cell viability, cell cycle profile, and caspase activity are expressed as mean values ± SEM for three determinations (*P < 0.05).
An inhibition of cell division to block cells entering S and G2 phase indicated a cell cycle arrest in G1 phase. In DKO-2 and DKO-3, the percentage of G1-phase cells was significantly increased by miR-34c transfection, from 69.2% to 83.2% in DKO-2, and 42.1% to 77.2% in DKO-3. However, we did not see a dramatic increase of G1-phase cells in DKO-1 transfected with miR-34c, which only resulted in an increase from 57.2% to 63.4%. Instead, 18.3% of DKO-1 transfected with miR-34c were in sub-G1 phase, indicating DNA fragmentation in these cells, while, in control DKO-1, only 4.2% cells were in sub-G1 phase. In DKO-2 and DKO-3, although small, an miR-34c-mediated increase of sub-G1 phase cells could also be noted, from 1.1% to 2.4% in DKO-2 and from 0.1% to 2.2% in DKO-3. DNA fragmentation is one of the hallmarks of apoptosis. Therefore, using Promega Caspase 3/7 apoptosis assay, a significant increase of caspase activities was observed in DKO-1 cells transfected with miR-34c, implicating the induction of apoptosis (Fig. 2C). Thus, our functional studies demonstrated that miR-34c induced cell cycle arrest in G1 phase and induced apoptosis in DKO tumor cell lines.
miR-34c-Regulated Genes Were Associated with G1/S-Phase Transition
One mechanism of miRNA-mediated gene regulation is through degradation of miRNA-bound mRNA. Therefore, we studied miR-34c-regulated genes by generating gene expression profiles of miR-34c and control transfected DKO tumor cells. We identified 300 genes (364 probes) downregulated and 385 genes (439 probes) upregulated in miR-34c transfected cells compared with control cells (fold change, ≥2; P < 0.05). To identify putative miR-34c direct targets, we used two algorithms for miRNA target prediction, including TargetScan and miRanda. Of 300 genes that were downregulated in miR-34c transfected cells, 96 genes (32.0% of downregulated genes) were identified as miR-34c predicted targets. Furthermore, of 385 genes that were upregulated in miR-34c transfected cells, 37 genes were also identified as miR-34c predicted targets and accounted for 9.6% of the upregulated genes. Thus, our gene expression profiling data identified a panel of genes that were directly or indirectly regulated by miR-34c.
Biologically, miR-34c actions resulted in cell cycle arrest in G1 phase of DKO tumor cells. Therefore, we explored our gene expression profiling data for genes associated with G1/S-phase transition. A subset of miR-34c-regulated genes was identified as key mediators of DNA replication and cell cycle G1/S progression. Based on their biological functions, these genes could be subgrouped into several complexes, including prereplication complex, DNA polymerase complex, and cyclin/cyclin-dependent kinase complex (Table 1). We validated miR-34c-mediated regulations by QPCR and compared these regulations with gene-level changes in DKO mouse early tumors versus normal mouse fallopian tubes [4]. A reverse correlation was identified where miR-34c upregulated genes were downregulated in DKO mouse tumors or miR-34c downregulated genes were upregulated in DKO mouse tumors (Table 1). Thus, our data indicate a concomitant reversal of a subset of cancer gene signatures when miR-34c was delivered back to DKO mouse tumor cells. Taken that these genes are key mediators of DNA replication and cell cycle control, miR-34c mediated partial restoration of gene expression to a more “normal” state and, to some extent, explains the biological consequence of miR-34c transfection.
Table 1.
miR-34c regulated key mediators in cell proliferation and cell cycle G1/S transition.
| Genea | Complex | Fold change (miR-34c/miR-Ctrl) | Fold change (DKO mouse early tumor/normal FL) | Algorithmsb | |
|---|---|---|---|---|---|
| mRNA microarray | QPCR validation | ||||
| Orc6l* | Prereplication complex | −2.4 | −5.9 | 1.8 | M |
| Cdt1 | −3.2 | −6.3 | 6.3 | ||
| Cdc6* | −2.6 | −5.5 | 5.8 | M | |
| Mcm2 | −2.3 | −3.0 | 4.7 | ||
| Mcm3 | −2.4 | −4.9 | 4.4 | ||
| Mcm5* | −9.7 | −18.7 | 12.3 | M | |
| Mcm6 | −2.4 | −3.5 | 8.2 | ||
| Mcm7 | −2.8 | −4.2 | 5.1 | ||
| Mcm10 | −3.4 | −6.0 | 13.3 | ||
| Rfc1* | DNA polymerase complex | −3.3 | −3.3 | 1.4 | M |
| Rfc2* | −2.0 | −2.4 | 1.2 | M | |
| Rfc4 | −2.2 | −4.3 | 3.4 | ||
| Rfc5 | −2.3 | −5.3 | 2.2 | ||
| Top2a | −3.7 | −11.7 | 21.3 | ||
| Pold1 | −2.1 | −3.4 | 2.4 | ||
| Pold2 | −2.9 | −4.6 | 1.3 | ||
| Ccne2* | Cyclin/cyclin-depedent kinase complex | −2.6 | −4.6 | 3.5 | M, T |
| Ccnd3 | −2.5 | −2.9 | 2.9 | ||
| Cdk2 | −2.1 | −3.4 | 1.9 | ||
| Cdkn1c | 8.9 | 18.3 | −2.4 | ||
Of 20 genes, 19 were downregulated, of which 6 are predicted miR-34c targets.
Prediction algorithms: FL, fallopian tube; M, miRanda; T, TargetScan.
Genes with predicted miR-34c binding sites in their 3′ UTR sequences.
miR-34c Was Downregulated in Human Serous Adenocarcinoma as Compared to Human Normal Fallopian Tubes
Our in vitro data in DKO mouse tumor cells demonstrate a potential tumor-suppressive effect of miR-34c, which may account for the onset of mouse fallopian tube tumors. To translate our mouse work to women, we determined whether miR-34c level was also down-regulated in human ovarian cancer as compared to human normal fallopian tubes. Using Taqman QPCR, miR-34c levels were determined to be 83-fold decreased (Fig. 3A) in human serous adenocarcinomas compared with fallopian tube, consistent with the miR-34b/c allele loss [4] and previously reported epigenetic changes in miR-34b/c [27].
Fig. 3.
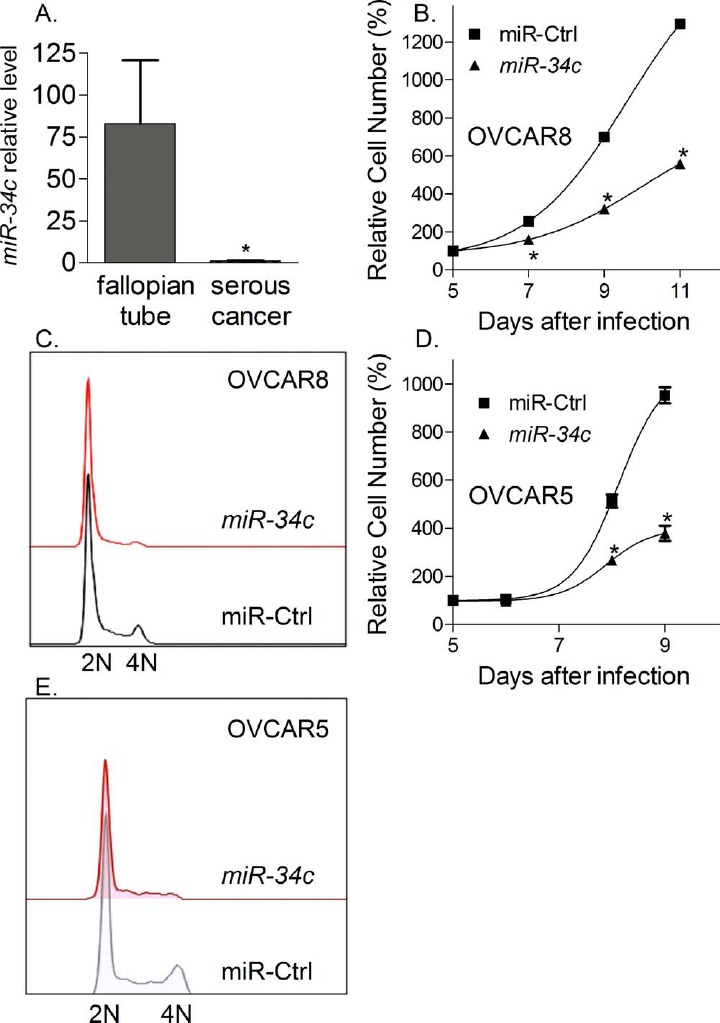
miR-34c in human ovarian cancer. A) Human normal fallopian tubes have a much higher miR-34c level than human serous ovarian cancer (P = 0.0029). Taqman QPCR identified relative miR-34c levels in human normal fallopian tubes (n = 6), human serous ovarian cancer cell lines (n = 2), and human serous ovarian adenocarcinomas (n = 12). B and D) miR-34c overexpression inhibited the proliferation of human ovarian cancer cell OVCAR8 and OVCAR5. Cell viability was measured by Promega CellTiter-Glo assay and expressed as mean values ± SEM for three determinations (*P < 0.05). C and E) miR-34c overexpression arrested OVCAR8 and OVCAR5 in G1 phase. Cell cycle profiles were generated by flow cytometry using PI staining and analyzed by flowjo software.
miR-34c Overexpression Slowed Human Serous Ovarian Cancer Cell Proliferation and Induced Cell Cycle Arrest at G1 Phase
To evaluate the effect of miR-34c in human serous ovarian cancer, we overexpressed miR-34c in OVCAR8 and OVCAR5 cells using lentivirus. OVCAR8 and OVCAR5 cells are serous ovarian cancer cell lines, and have a mutated p53. Overexpression of miR-34c significantly slowed cancer cell proliferation using a CellTiter Glo cell viability assay (Fig. 3, B and D). Cell cycle analysis demonstrated a G1-phase arrest in miR-34c infected cells as compared to miR control (miRCtrl) infected cells (Fig. 3, C and E), consistent with the effect of miR-34c in DKO mouse tumor cells. Thus, our data indicate a potential tumor-suppressive effect of miR-34c in both mouse and human cancer cells.
miR-34b/c/Pten DKO Mice Did Not Develop Any Tumors
To test in vivo whether the loss of miR-34c can substitute for Dicer deficiency in the tumorigenesis of high-grade serous ovarian cancer, we generated conditional DKO mice for miR-34b/c and Pten (miR34-b/c−/−/Ptenfl/fl/Amhr2cre/+). However, unlike Dicer/Pten DKO mice, these miR34-b/c/Pten DKO mice did not develop any tumors in the fallopian tube, nor in the ovary. The ovaries, oviducts, and uteri from miR34-b/c/Pten DKO mice looked grossly normal at the age of 12–13 mo (Fig. 4, A and B), a time at which all Dicer/Pten DKO mice had prominent metastatic cancers and ascites, and had died. Histology data showed normal morphology of ovaries with preantral and antral follicles at different stages, as well as corpora lutea (Fig. 4, C and D). Cross-sectional views of DKO mouse oviducts demonstrated a typical normal histology of mouse oviducts (Fig. 4, E and F), indicating that loss of miR-34b/c did not drive tumor formation in the Pten-deficient female reproductive tract. Thus, our data show that development of tumors in Dicer/Pten DKO mice is not attributed to loss of only the miR-34b/c locus. A screening of additional miRNAs in the mouse miRNome is highly warranted.
Fig. 4.
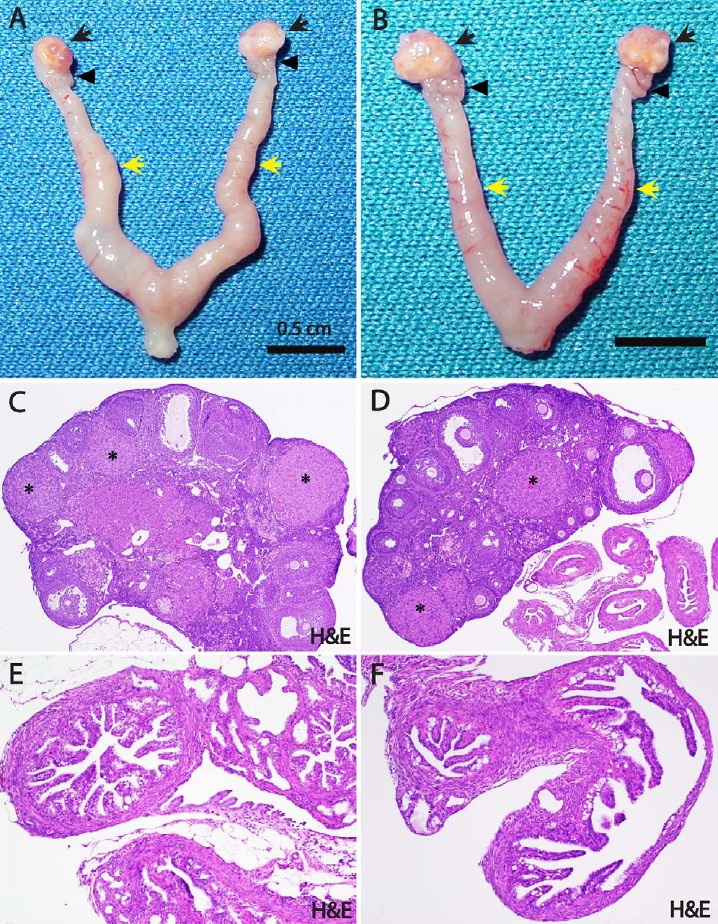
The normal reproductive morphology of miR-34b/c/Pten DKO mice. A and B) The ovaries (black arrows), oviducts (black arrowheads), and uteri (yellow arrows) from miR-34b/c−/−/Ptenfl/fl/Amhr2cre/+ mice look grossly normal (12–13 mo old). C and D) Histologically, the ovaries of these mice show a normal morphology with preantral and antral follicles displayed at different stages, as well as with corpora lutea (asterisks) (H&E; original magnification ×4). E and F) Cross-sectional views of miR-34b/c−/−/Ptenfl/fl/Amhr2cre/+ mouse oviducts displaying a typical normal histology of the mouse oviduct (H&E; original magnification ×10).
Discussion
During the last decade, putative roles of miRNAs in cancer have been widely described. Many researchers, including those in our laboratory, have profiled global miRNA expression levels in ovarian cancer [28–35]. These miRNA expression analyses suggest tumor-suppressive or oncogenic roles of miRNAs. However, the patterns of deregulated miRNA expression in different studies do not overlap well. The great inconsistency is probably due to the different methodologies applied (e.g., next-generation deep sequencing and miRNA microarray [36]) and various normal controls used by different groups. Consequently, there is no simple conclusion as to which miRNA(s) are more important in ovarian cancer tumorigenesis. In human studies investigating miRNA biological functions in ovarian cancer, there are difficulties in finding ovarian cancer cell lines where these miRNAs are deregulated. However, different cancer cell lines have different miRNA expression patterns and different cellular contexts unique to their division and proliferation. It is impossible to compare the relative significance of deregulated miRNAs in ovarian cancer by using these human cancer cell lines. In the present study, we established cancer cell lines derived from primary fallopian tube tumors developed in Dicer/Pten DKO mice. Histologically, these primary tumors and human high-grade serous ovarian carcinomas are very much alike [4]. In addition, Dicer deletion is indispensable for the onset of tumor formation, indicating that impaired maturation of certain miRNA(s) played critical roles therein. Therefore, the Dicer/Pten DKO cancer cell lines that we established would become a valuable platform to study the relative significance of various miRNAs in high-grade serous ovarian carcinomas.
The cancer cell lines established from primary fallopian tube tumors developed in Dicer/Pten DKO mice are morphologically epithelial cells and express epithelial cell markers (Supplemental Fig. S1). This is consistent with our previously published Dicer/Pten DKO mouse tumor histology data, where serous epithelial cancer was identified [4]. However, genomic DNA PCR of the three mouse cancer cell lines showed a deletion of Dicer and Pten, indicating that these DKO cells are originally derived from Amhr2-positive stromal cells (Supplemental Fig. S2). Therefore, mouse cancer cell lines in the present study are in line with previous findings, where a potential cancer mechanism of stromal-to-epithelial transition was suggested in the Dicer/Pten DKO mouse model [4]. Histological analysis of the fallopian tubes from young Dicer/Pten DKO mice showed an initiation and expansion of mitotically active cancer cells in the stromal compartment, but not the epithelial layer, and these cancer cells expressed keratin 8, keratin 14, keratin 17, and E-cadherin, and gradually filled the stromal compartment of the fallopian tube and compressed the lumen [4].
One potential drawback of this study is that we picked up the most abundant miRNAs present in normal fallopian tubes to be studied in Dicer/Pten DKO cancer cells, rather than screening the whole set of miRNAs identified in normal fallopian tubes. Although miRNA abundance may not generally correlate with its relevance in Dicer/Pten DKO mouse cancer, the loss of abundant miRNAs might have a greater chance of contributing to tumorigenesis as compared to low-level miRNAs. Using next-generation sequencing, we identified that the most abundant five miRNAs in mouse normal fallopian tubes are let-7c, let-7f, let-7a, let-7b, and miR-34c, where let-7 family members are the most abundant miRNA, accounting for 83.3% of the total miRNAs in fallopian tube. Other than let-7 family members, miR-34c is the most abundant miRNA, accounting for 4.1% of the total reads. Therefore, our studies focused on let-7 and miR-34c, since both have been demonstrated to be tumor suppressors in many cancers. let-7 and its family members are highly conserved across species in sequence and function. Deregulation of let-7 has been implicated in a less-differentiated cellular state and the development of cancer [37]. let-7 miRNAs are downregulated in numerous cancers, including cancers of the ovary, lung, prostate, breast, and thyroid [29, 38–41]. Low levels of let-7 have been correlated with poor prognosis, while overexpression of let-7 inhibits the growth of lung cancer and breast cancer mouse models [42]. Significance of miR-34c in cancers has also been well documented, most notably as part of the TP53 stress and DNA damage response pathway [43]. miR-34c is a member of the miR-34 family, which consists of three highly conserved miRNAs, miR-34a, miR-34b, and miR-34c, in vertebrates encoded at two different gene loci [44]. The miR-34 family has been implicated in playing a role in ovarian cancer [4, 45, 46]. Several targets of the miR-34 family mediate cell progression and block apoptosis. Most of these targets, like CDK4, CDK6, HMGA2, c-Met, and AKT, have been validated for miR-34a, while the other two family members, miR-34b and miR-34c, have been less well studied [43, 44, 47]. In the current study, we delivered mature let-7b or miR-34c to Dicer/Pten DKO cancer cells. Given that Dicer is genetically deleted in these cells, there is no way to express miRNA precursors using plasmids. We observed that miR-34c mimics significantly inhibited DKO cancer cell viability 2 days after transfection, while the viability of let-7b transfected cells remained basically unchanged or slightly decreased. Our data thus suggest that miR-34c is more related to PI3K/mTOR pathway-mediated cell division and proliferation than let-7, since one of the casual effects in Dicer/Pten DKO mouse tumorigenesis is the overactivation of PI3K/mTOR pathway [4].
We observed a cell proliferation inhibition by miR-34c in DKO mouse cancer cells and human ovarian cancer cell line, which was due to cell cycle arrest in the G1 phase. However, in one DKO cancer cell line, G1 cell cycle arrest was not as dramatic as the other two cells and the human ovarian cancer cell line; instead, an increase of sub-G1 cells was observed, which indicates an induction of apoptosis. The different outcome of miR-34c actions presumably depends on different expression levels of miR-34c in these cells. In addition, the slight difference of cellular context in the three DKO mouse cancer cells might affect the balance of miR-34c action between apoptosis and cell cycle arrest. Our data, therefore, are consistent with previous publications showing that ectopic miR-34 caused a cell cycle arrest in G1 phase [48, 49] or an induction in apoptosis [50, 51]. The mechanism of miR-34c-induced apoptosis in mouse DKO cancer cells, however, is unknown. BCL2 has been demonstrated to be a target of miR-34a, and miR-34a-defective MEFs show a decrease in spontaneous apoptosis [52]. However, our gene expression data show that Bcl2 level was very low in DKO mouse cancer cells, and miR-34c increased Bcl2 by 1.3 fold, although this increase was not statistically significant. Chang et al. [50] has showed that miR-34a-induced apoptosis is, at least in part, due to the presence of wild-type TP53 and a feedback of miR-34a to TP53. Given that our mouse DKO cancer cell lines have a wild-type Tp53, although at a very low level [4], miR-34c-induced apoptosis in DKO-1 is likely Tp53 dependent, but Bcl2 independent. Our gene expression profiling of miR-34c-treated cells identified miR-34c-regulated genes that are critical for G1-phase cell cycle progression. We excluded some genes, such as Aurkb, Incenp, Birc5, Cdca3, and Cdca8, the levels of which were also downregulated by miR-34c. The expression levels of these genes oscillate during the cell cycle and have a lower expression in G1 phase; therefore, the downregulation of these genes might only be a cell cycle-nonspecific effect. However, we could not rule out the possibility that these genes may also be regulated by miR-34c. In addition, Cdca8, a component of a chromosomal passenger complex required for stability of the bipolar mitotic spindle, is predicted to be a miR-34c direct target by miRanda algorithm. In Table 1, miR-34c-regulated key mediators for DNA replication and cell cycle G1/S transition could be further subgrouped into several complexes, including prereplication complex, DNA polymerase complex, and cyclin/cyclin-dependent kinase complex. A total of 6 out of 20 genes downregulated by miR-34c are predicted to be miR-34c targets by either miRanda or TargetScan, implicating a direct effect of miR-34c on cell cycle arrest.
Cdkn1c was found to be dramatically upregulated by miR-34c (8.9-fold up by gene expression profiling, and 18.3-fold up by QPCR). Cdkn1c encodes p57Kip2, which is a cyclin-dependent kinase inhibitor with a broader spectrum. Compared to the other two Cip/Kip family members, p21Cip1 and p27Kip1, p57Kip2 is the least studied. However, as reviewed by Borriello et al. [53], downregulation of CDKN1C has been observed in various cancers, including ovarian cancer, hepatocellular cancer, lung cancer, breast cancer, pancreatic cancer, and colorectal cancer, indicating an underrated role of this gene in cancer. So far, no CDKN1C mutations have been detected in cancer specimens. Most frequently, p57Kip2 suppression in cancer is due to CDKN1C methylation and diminished transcription, although post-transcriptional events have been described in some instances [53]. Epigenetic mechanisms have suggested that histone methyltransferase EZH2-mediated H3K27 trimethylation is likely the key mark associated with the transcriptional repression of CDKN1C in breast cancer cells [54]. Consistent with this observation, overexpression of EZH2 in breast cancer is associated with a downregulated CDKN1C level [54]. In our DKO mouse model, Ezh2 mRNA level is upregulated by 6.1-fold in early tumors versus normal fallopian tubes, while Cdkn1c is 2.4-fold downregulated in early tumors (data not shown). We found that miR-34c transfection significantly downregulates Ezh2 in DKO mouse cancer cells (Fig. 5A), leading to a hypothesis that miR-34c-induced upregulation of Cdkn1c may be due to its suppression on Ezh2. On the other hand, Mybl2 is found to be 2.9-fold upregulated in DKO mouse tumors (data not shown) and 5.2-fold downregulated by miR-34c treatment in DKO cancer cells (Fig. 5B). MYBL2 encodes a regulatory protein B-Myb, which has critical roles in cell proliferation and differentiation. In cell cycle regulation, B-Myb competes with cyclin A2 for binding to p57Kip2, resulting in release of active cyclin/CDK2 kinase, indicating an inhibitory effect of B-Myb on p57Kip2 function [55]. Therefore, our data implicate a miR-34c-mediated transcriptional and post-transcriptional activation of p57Kip2, which would partially explain miR-34c-induced cell cycle arrest in mouse DKO cancer cells and, to some extent, get an insight into tumorigenesis of Dicer/Pten DKO mouse. However, how miR-34c regulates Ezh2 and Mybl2 requires further investigation.
Fig. 5.
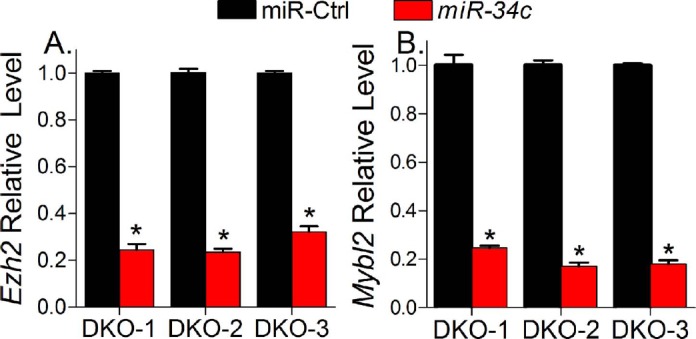
miR-34c suppressed the expression of Ezh2 and Mybl2. QPCR analysis shows relative quantity of Ezh2 (A) and Mybl2 (B) after miR-34c transfection in DKO-1, DKO-2, and DKO-3 cells. Data are expressed as mean values ± SEM for three determinations (*P < 0.05).
To obtain in vivo evidence that miR-34c is a tumor suppressor in Dicer/Pten DKO tumors, we generated conditional DKO mice for miR-34b/c and Pten (miR-34b/c−/−/Ptenfl/fl/Amhr2cre/+). However, the loss of miR-34b/c loci cannot substitute for Dicer deficiency in the tumorigenesis of high-grade serous ovarian cancer. Although this finding cannot discredit our conclusion that miR-34c is a potential tumor suppressor in Dicer/Pten DKO ovarian cancer, miR-34c is not likely the sole tumor suppressor miRNA in mouse fallopian tube tumorigenesis. miRNAs are a class of regulatory molecules, mainly modulating the expression of protein-encoding genes. An individual miRNA can simultaneously modulate the expression of dozens—and in some cases even hundreds—of different mRNA targets. An individual protein-encoding gene can also be regulated by more than one miRNA. miR-34c-mediated partial restoration of gene expression to a more “normal” state in miR-34c-treated Dicer/Pten DKO cancer cells does indicate the involvement of miR-34c loss in the formation of Dicer/Pten DKO cancer gene signature. However, it does not mean that miR-34b/c deletion would substitute for Dicer deletion in generating the same cancer gene signature, since other miRNAs in miR-34b/c/Pten DKO mice may also regulate miR-34b/c targets so as to compensate for the effect of miR-34b/c loss. One of these miRNAs might be miR-34a. The high similarity among mmu-miR-34a, mmu-miR-34b, and mmu-miR-34c suggested that they may have the same targets. An expression analysis after separate transfection of three human miR-34s showed that the affected mRNAs were almost identical [48]. Moreover, a recent publication identified a significant overlap between miR-34a- and miR-34c-regulated protein synthesis [47]. On the other hand, miRNAs other than miR-34s may regulate miR-34 targets. A validated miR-34 target, CCNE2, is also targeted by miR-449 in silico (TargetScan) or experimentally [56]. Taken together, all these observations indicate complicated, redundant regulations of miRNAs on genes, which might be involved in tumorigenesis. miR-34c is involved in this process; however, it cannot be the only miRNA of importance, and should be more relevant to tumor proliferation than tumor initiation.
Supplementary Material
Footnotes
Supported by grants from the Ovarian Cancer Research Fund, the Marsha Rivkin Center for Ovarian Cancer, the Baylor Partnership, and the Dan L. Duncan Cancer Center.
References
- 1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013.CA Cancer J Clin 2013;63:11–30. [DOI] [PubMed] [Google Scholar]
- 2. Birrer MJ. The origin of ovarian cancer-is it getting clearer? N Engl J Med 2010;363:1574–1575. [DOI] [PubMed] [Google Scholar]
- 3. Levanon K, Crum C, Drapkin R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact.J Clin Oncol 2008;26:5284–5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim J, Coffey DM, Creighton CJ, Yu Z, Hawkins SM, Matzuk MM. High-grade serous ovarian cancer arises from fallopian tube in a mouse model.Proc Natl Acad Sci U S A 2012;109:3921–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seidman JD, Horkayne-Szakaly I, Haiba M, Boice CR, Kurman RJ, Ronnett BM. The histologic type and stage distribution of ovarian carcinomas of surface epithelial origin.Int J Gynecol Pathol 2004;23:41–44. [DOI] [PubMed] [Google Scholar]
- 6. Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer.Nat Rev Cancer 2006;6:259–269. [DOI] [PubMed] [Google Scholar]
- 7. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia.Proc Natl Acad Sci U S A 2002;99:15524–15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang L, Huang J, Yang N, Greshock J, Megraw MS, Giannakakis A, Liang S, Naylor TL, Barchetti A, Ward MR, Yao G, Medina A, et al. microRNAs exhibit high frequency genomic alterations in human cancer.Proc Natl Acad Sci U S A 2006;103:9136–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Merritt WM, Lin YG, Han LY, Kamat AA, Spannuth WA, Schmandt R, Urbauer D, Pennacchio LA, Cheng JF, Nick AM, Deavers MT, Mourad-Zeidan A, et al. Dicer, Drosha, and outcomes in patients with ovarian cancer.N Engl J Med 2008;359:2641–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis.Nat Genet 2007;39:673–677. [DOI] [PubMed] [Google Scholar]
- 11. Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B, Mazzaferro V, Lowe SW, Croce CM. Dejean A. miR-221 overexpression contributes to liver tumorigenesis.Proc Natl Acad Sci U S A 2010;107:264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Song SJ, Ito K, Ala U, Kats L, Webster K, Sun SM, Jongen-Lavrencic M, Manova-Todorova K, Teruya-Feldstein J, Avigan DE, Delwel R, Pandolfi PP. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation.Cell Stem Cell 2013;13:87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Edson MA, Nagaraja AK, Matzuk MM. The mammalian ovary from genesis to revelation.Endocr Rev 2009;30:624–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jamin SP, Arango NA, Mishina Y, Hanks MC, Behringer RR. Requirement of Bmpr1a for Müllerian duct regression during male sexual development.Nat Genet 2002;32:408–410. [DOI] [PubMed] [Google Scholar]
- 15. Jeyasuria P, Ikeda Y, Jamin SP, Zhao L, De Rooij DG, Themmen AP, Behringer RR, Parker KL. Cell-specific knockout of steroidogenic factor 1 reveals its essential roles in gonadal function.Mol Endocrinol 2004;18:1610–1619. [DOI] [PubMed] [Google Scholar]
- 16. Deutscher E. Hung-Chang Yao H. Essential roles of mesenchyme-derived beta-catenin in mouse Müllerian duct morphogenesis.Dev Biol 2007;307:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arango NA, Kobayashi A, Wang Y, Jamin SP, Lee HH, Orvis GD, Behringer RR. A mesenchymal perspective of Müllerian duct differentiation and regression in Amhr2-lacZ mice.Mol Reprod Dev 2008;75:1154–1162. [DOI] [PubMed] [Google Scholar]
- 18. Nagaraja AK, Andreu-Vieyra C, Franco HL, Ma L, Chen R, Han DY, Zhu H, Agno JE, Gunaratne PH, DeMayo FJ, Matzuk MM. Deletion of Dicer in somatic cells of the female reproductive tract causes sterility.Mol Endocrinol 2008;22:2336–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate.J Biol Chem 1998;273:13375–13378. [DOI] [PubMed] [Google Scholar]
- 20. Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN.Cell 1998;95:29–39. [DOI] [PubMed] [Google Scholar]
- 21. Cancer Genome Atlas Research Network.Integrated genomic analyses of ovarian carcinoma.Nature 2011;474:609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fan HY, Liu Z, Paquet M, Wang J, Lydon JP, DeMayo FJ, Richards JS. Cell type-specific targeted mutations of Kras and Pten document proliferation arrest in granulosa cells versus oncogenic insult to ovarian surface epithelial cells.Cancer Res 2009;69:6463–6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs.Genome Res 2009;19:92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression.Nucleic Acids Res 2008;36:D149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method.Methods 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 26. Hawkins SM, Creighton CJ, Han DY, Zariff A, Anderson ML, Gunaratne PH, Matzuk MM. Functional microRNA involved in endometriosis.Mol Endocrinol 2011;25:821–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Corney DC, Hwang CI, Matoso A, Vogt M, Flesken-Nikitin A, Godwin AK, Kamat AA, Sood AK, Ellenson LH, Hermeking H, Nikitin AY. Frequent downregulation of miR-34 family in human ovarian cancers.Clin Cancer Res 2010;16:1119–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iorio MV, Visone R, Di Leva G, Donati V, Petrocca F, Casalini P, Taccioli C, Volinia S, Liu CG, Alder H, Calin GA, Menard S, et al. MicroRNA signatures in human ovarian cancer.Cancer Res 2007;67:8699–8707. [DOI] [PubMed] [Google Scholar]
- 29. Dahiya N, Sherman-Baust CA, Wang TL, Davidson B, Shih Ie M, Zhang Y, Wood W, 3rd, , Becker KG, Morin PJ. MicroRNA expression and identification of putative miRNA targets in ovarian cancer.PLoS One 2008;3:e2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang L, Volinia S, Bonome T, Calin GA, Greshock J, Yang N, Liu CG, Giannakakis A, Alexiou P, Hasegawa K, Johnstone CN, Megraw MS, et al. Genomic and epigenetic alterations deregulate microRNA expression in human epithelial ovarian cancer.Proc Natl Acad Sci U S A 2008;105:7004–7009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee CH, Subramanian S, Beck AH, Espinosa I, Senz J, Zhu SX, Huntsman D, van de Rijn M, Gilks CB. MicroRNA profiling of BRCA1/2 mutation-carrying and non-mutation-carrying high-grade serous carcinomas of ovary.PLoS One 2009;4:e7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang H, Kong W, He L, Zhao JJ, O'Donnell JD, Wang J, Wenham RM, Coppola D, Kruk PA, Nicosia SV, Cheng JQ. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN.Cancer Res 2008;68:425–433. [DOI] [PubMed] [Google Scholar]
- 33. Nam EJ, Yoon H, Kim SW, Kim H, Kim YT, Kim JH, Kim JW, Kim S. MicroRNA expression profiles in serous ovarian carcinoma.Clin Cancer Res 2008;14:2690–2695. [DOI] [PubMed] [Google Scholar]
- 34. Wyman SK, Parkin RK, Mitchell PS, Fritz BR, O'Briant K, Godwin AK, Urban N, Drescher CW, Knudsen BS, Tewari M. Repertoire of microRNAs in epithelial ovarian cancer as determined by next generation sequencing of small RNA cDNA libraries.PLoS One 2009;4:e5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Creighton CJ, Fountain MD, Yu Z, Nagaraja AK, Zhu H, Khan M, Olokpa E, Zariff A, Gunaratne PH, Matzuk MM, Anderson ML. Molecular profiling uncovers a p53-associated role for microRNA-31 in inhibiting the proliferation of serous ovarian carcinomas and other cancers.Cancer Res 2010;70:1906–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Creighton CJ, Reid JG, Gunaratne PH. Expression profiling of microRNAs by deep sequencing.Brief Bioinform 2009;10:490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roush S, Slack FJ. The let-7 family of microRNAs.Trends Cell Biol 2008;18:505–516. [DOI] [PubMed] [Google Scholar]
- 38. Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, Mitsudomi T, Takahashi T. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival.Cancer Res 2004;64:3753–3756. [DOI] [PubMed] [Google Scholar]
- 39. Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Menard S, Palazzo JP, et al. MicroRNA gene expression deregulation in human breast cancer.Cancer Res 2005;65:7065–7070. [DOI] [PubMed] [Google Scholar]
- 40. Visone R, Pallante P, Vecchione A, Cirombella R, Ferracin M, Ferraro A, Volinia S, Coluzzi S, Leone V, Borbone E, Liu CG, Petrocca F, et al. Specific microRNAs are downregulated in human thyroid anaplastic carcinomas.Oncogene 2007;26:7590–7595. [DOI] [PubMed] [Google Scholar]
- 41. Nadiminty N, Tummala R, Lou W, Zhu Y, Shi XB, Zou JX, Chen H, Zhang J, Chen X, Luo J, deVere White RW, Kung HJ, et al. MicroRNA let-7c is downregulated in prostate cancer and suppresses prostate cancer growth.PLoS One 2012;7:e32832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME. The role of let-7 in cell differentiation and cancer.Endocr Relat Cancer 2010;17:F19–36. [DOI] [PubMed] [Google Scholar]
- 43. Hermeking H. The miR-34 family in cancer and apoptosis.Cell Death Differ 2010;17:193–199. [DOI] [PubMed] [Google Scholar]
- 44. He X, He L, Hannon GJ. The guardian's little helper: microRNAs in the p53 tumor suppressor network.Cancer Res 2007;67:11099–11101. [DOI] [PubMed] [Google Scholar]
- 45. Mullany LK, Fan HY, Liu Z, White LD, Marshall A, Gunaratne P, Anderson ML, Creighton CJ, Xin L, Deavers M, Wong KK, Richards JS. Molecular and functional characteristics of ovarian surface epithelial cells transformed by KrasG12D and loss of Pten in a mouse model in vivo.Oncogene 2011;30:3522–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Corney DC, Hwang CI, Matoso A, Vogt M, Flesken-Nikitin A, Godwin AK, Kamat AA, Sood AK, Ellenson LH, Hermeking H, Nikitin AY. Frequent downregulation of miR-34 family in human ovarian cancers.Clin Cancer Res 2010;16:1119–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ebner OA, Selbach M. Quantitative proteomic analysis of gene regulation by miR-34a and miR-34c.PLoS One 2014;9:e92166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, et al. A microRNA component of the p53 tumour suppressor network.Nature 2007;447:1130–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, Meister G, Hermeking H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest.Cell Cycle 2007;6:1586–1593. [DOI] [PubMed] [Google Scholar]
- 50. Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, Arking DE, Beer MA, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis.Mol Cell 2007;26:745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis.Mol Cell 2007;26:731–743. [DOI] [PubMed] [Google Scholar]
- 52. Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, Zhai Y, Giordano TJ, Qin ZS, Moore BB, MacDougald OA, Cho KR, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes.Curr Biol 2007;17:1298–1307. [DOI] [PubMed] [Google Scholar]
- 53. Borriello A, Caldarelli I, Bencivenga D, Criscuolo M, Cucciolla V, Tramontano A, Oliva A, Perrotta S, Della Ragione F. p57(Kip2) and cancer: time for a critical appraisal.Mol Cancer Res 2011;9:1269–1284. [DOI] [PubMed] [Google Scholar]
- 54. Yang X, Karuturi RK, Sun F, Aau M, Yu K, Shao R, Miller LD, Tan PB, Yu Q. CDKN1C (p57) is a direct target of EZH2 and suppressed by multiple epigenetic mechanisms in breast cancer cells.PLoS One 2009;4:e5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Joaquin M, Watson RJ. The cell cycle-regulated B-Myb transcription factor overcomes cyclin-dependent kinase inhibitory activity of p57(KIP2) by interacting with its cyclin-binding domain.J Biol Chem 2003;278:44255–44264. [DOI] [PubMed] [Google Scholar]
- 56. Bou Kheir T, Futoma-Kazmierczak E, Jacobsen A, Krogh A, Bardram L, Hother C, Gronbaek K, Federspiel B, Lund AH, Friis-Hansen L. miR-449 inhibits cell proliferation and is down-regulated in gastric cancer.Mol Cancer 2011;10:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.