

Abstract
The pathology-based classification of Alzheimer’s disease (AD) and other neurodegenerative diseases is a work in progress that is important for both clinicians and basic scientists. Analyses of large autopsy series, biomarker studies, and genomics analyses have provided important insights about AD and shed light on previously unrecognized conditions, enabling a deeper understanding of neurodegenerative diseases in general. After demonstrating the importance of correct disease classification for AD and primary age-related tauopathy, we emphasize the public health impact of an underappreciated AD “mimic,” which has been termed “hippocampal sclerosis of aging” or “hippocampal sclerosis dementia.” This pathology affects >20% of individuals older than 85 years and is strongly associated with cognitive impairment. In this review, we provide an overview of current hypotheses about how genetic risk factors ( GRN , TMEM106B , ABCC9 , and KCNMB2 ), and other pathogenetic influences contribute to TDP-43 pathology and hippocampal sclerosis. Because hippocampal sclerosis of aging affects the “oldest-old” with arteriolosclerosis and TDP-43 pathologies that extend well beyond the hippocampus, more appropriate terminology for this disease is required. We recommend “cerebral age-related TDP-43 and sclerosis” (CARTS). A detailed case report is presented, which includes neuroimaging and longitudinal neurocognitive data. Finally, we suggest a neuropathology-based diagnostic rubric for CARTS.
Keywords: Arteriosclerosis, Cerebrovascular disease, Frontotemporal lobar degeneration, Genome-wide association study, Neurofibrillary tangles, Plaques, VCID.
INTRODUCTION
Prospects for diagnosing and treating neurodegenerative diseases are enhanced when disease classifications reflect the underlying biologic complexity of these conditions. For most neurodegenerative diseases, neuropathologic observations constitute the “gold standard” used in diagnosis and nosology. Yet the pathology-based classifications of neurodegenerative diseases are also dynamic and have evolved recently to capture an increased proportion of the changes in the aged brain that are associated with cognitive impairment (1). These advances have come about with the help of larger and more diverse autopsy cohorts, increasingly robust and quantitative pathological parameters, and greater collaboration among neuropathologists, clinician-scientists, and basic researchers. Here we discuss both recent advances and some areas that merit revision in the study of dementia-related neurodegenerative diseases ( Table 1 ). These studies are directly relevant to the most well-known dementia-inducing disorder, Alzheimer disease (AD).
TABLE 1.
Topics and Organization of the Current Review
| Disease condition | Impact of recent progress and revised pathology-based classification |
|---|---|
| Alzheimer disease (AD) | Increased appreciation of importance of autopsy data and non-AD pathologies |
| Primary age-related tauopathy (PART) | Revised classification is useful for study of non-AD brain disease |
| Hippocampal sclerosis of aging (HS-Aging)/Cerebral age-related TDP-43 and sclerosis | A common age-related brain disease, lacking an accurate classification |
Focusing on AD
AD has an enormous impact on public health and recent studies have substantially revised the classic literature on AD clinicopathologic correlations. For example, we have learned that much of the morbidity attributed to AD as recently as 20 years ago is more accurately associated with non-AD diseases. A basic assumption is that AD is defined by 2 pathologic hallmarks: Aβ amyloid plaques and tau neurofibrillary tangles (NFTs) ( 2 ). The complex but coherent association between AD pathologies and cognitive status was previously addressed ( 3 , 4 ). Here, we focus on 3 basic factors that must be taken into account in disease classifications: autopsy data, non-AD pathologies, and chronological age.
Autopsy-based neuropathological diagnoses are central to AD research. The salience of neuropathologic data and, by extension, pathology-based classification schemes, is illustrated by many prior studies that arrived at correct conclusions only when using the pathological criteria for AD diagnosis rather than using purely clinical criteria for AD diagnosis. One example selected from among many is in testing the association between AD and type 2 diabetes mellitus (T2D), the latter of which affects over a quarter of individuals >65 years of age ( 5 ). Studies analyzing clinical data have reported that T2D is a risk factor for AD ( 6–8 ). By contrast, studies that incorporated autopsy results have consistently arrived at the opposite conclusion, i.e that T2D is not a risk factor for AD pathology. Instead, T2D appears to exert its impact through a different disorder, that is cerebrovascular disease ( 9–14 ). The inclusion of the single study design element of autopsy data is critical to guide all other related work.
The reason for the discrepancy between clinical and pathology-based AD diagnoses relates to the prevalence of non-AD brain diseases, including α-synucleinopathies, non-AD tauopathies, hippocampal sclerosis ([HS] as discussed below), and many subtypes of cerebrovascular disease, all of which can mimic AD clinically ( 4 , 15–17 ). In Table 2 we provide data from the University of Kentucky autopsy series ( 18 , 19 ) listing the frequency of non-AD pathologies by Braak NFT stage ( 20 ). Note that >95% of brains in this cohort have at least 1 brain pathology and most participants had more than 1 pathologic diagnosis. These results are consistent with prior autopsy series ( 4 , 19 , 21–35 ).
TABLE 2.
Even in an Autopsy Cohort That Includes Many Subjects Free of Dementia, Most Individuals Manifest More Than 1 Subtype of Brain Pathology
|
Braak NFT Stages
|
|||||||
|---|---|---|---|---|---|---|---|
| 0 | I | II | III | IV | V | VI | |
| Cases (n) | 20 | 57 | 97 | 58 | 57 | 101 | 188 |
| Cases with B-ASC: moderate or severe | 0% | 12.3% | 21.6% | 12.1% | 21.1% | 19.8% | 14.4% |
| Cases with CAA: moderate or severe | 20.0% | 10.5% | 20.6% | 19.0% | 24.6% | 37.6% | 50.5% |
| Cases with Lacunar and/or microinfarcts | 35.0% | 36.8% | 43.3% | 37.9% | 49.1% | 46.5% | 38.3% |
| Cases with Neocortical Lewy bodies | 5.0% | 14.0% | 9.3% | 10.3% | 7.0% | 10.9% | 19.7% |
| Cases with HS-Aging | 15.0% | 10.5% | 6.2% | 17.2% | 8.8% | 18.8% | 15.4% |
| Cases with >1 non-AD pathology a | 20.0% | 21.1% | 27.0% | 24.1% | 33.3% | 43.6% | 40.1% |
| Cases with PART | n/a | 71.9% | 56.7% | 37.9% | 17.5% | n/a | n/a |
Shown are pathologies among subjects at the University of Kentucky Alzheimer’s Disease Center autopsy cohort who died after age 75 years (total n = 578, of whom 161 subjects had final MMSE score of 28/30 or better) without frontotemporal dementia, stratified by Braak NFT stages. Data are presented as percentages.
B-ASC: brain arteriolosclerosis; CAA: cerebral amyloid angiopathy; HS-Aging: hippocampal sclerosis of aging; PART: primary age-related tauopathy.
a For this purpose, PART is not considered “non-AD” pathology.
Biomarker studies confirm the prevalence of in vivo “suspected non-Alzheimer pathology” (SNAP) ( 36 , 37 ), which refers to neurodegeneration without Aβ amyloidosis according to biomarkers. Approximately 25% of “mild cognitive impairment” (MCI) cases show the SNAP biomarker signature ( 36 ).
This does not indicate that only approximately 25% of MCI subjects show substantial non-AD pathology, but instead, approximately 25% of MCI subjects show impairment that is almost exclusively related to non-AD pathology.
The clinical and biomarker data on patients with cognitive impairments must be guided by pathologic classification to appreciate the heterogeneous diseases underlying these impairments. A first-line biomarker used for diagnosing the cause of cognitive impairment in the elderly is brain MRI ( 38 ). Although MRI-detected hippocampal shrinkage is a relatively strong predictor of subsequent cognitive deterioration ( 39–41 ), this finding has low specificity because multiple separate brain pathologies are associated with hippocampal atrophy. For example, HS of aging (HS-Aging) is a common condition that causes even more severe hippocampal atrophy than AD ( 42–44 ). Distinguishing between causes of hippocampal atrophy is critical because therapies that might impact one condition (eg AD) may not have any effect in another disease (eg HS-Aging). Thus, optimal AD biomarkers would indicate the severity of AD neuropathology rather than cognitive impairment per se , which is not specific to AD ( 27 , 29 , 45 ).
Recent autopsy studies have also shed new light on the interaction between aging and brain disease. The classic AD clinical-pathologic correlation studies of Tomlinson and colleagues were performed on cohorts with average age of death in their early 70s ( 46–49 ). However, the fastest growing population group is persons older than 85 years of age ( 10 ). It is increasingly clear that this “oldest-old” population is affected by conditions that differ from younger cohorts ( 10 , 50–52 ). “Pure” AD cases tend to be younger and to have particular gene variants ( 10 , 29 , 31 , 53–55 ), whereas the prevalence of AD pathology levels off or decreases in advanced old age ( 10 , 15 , 31 , 56 ) ( Figs. 1 and 2 ). By contrast, some non-AD brain pathologies increase in the “oldest-old” ( 10 , 24 , 59 ). Thus, the statement that age is the greatest risk factor for AD should not be taken literally ( 24 , 87 ); a more accurate statement is that age is a risk factor for dementia due to multiple diseases that usually include AD.
FIGURE 1.
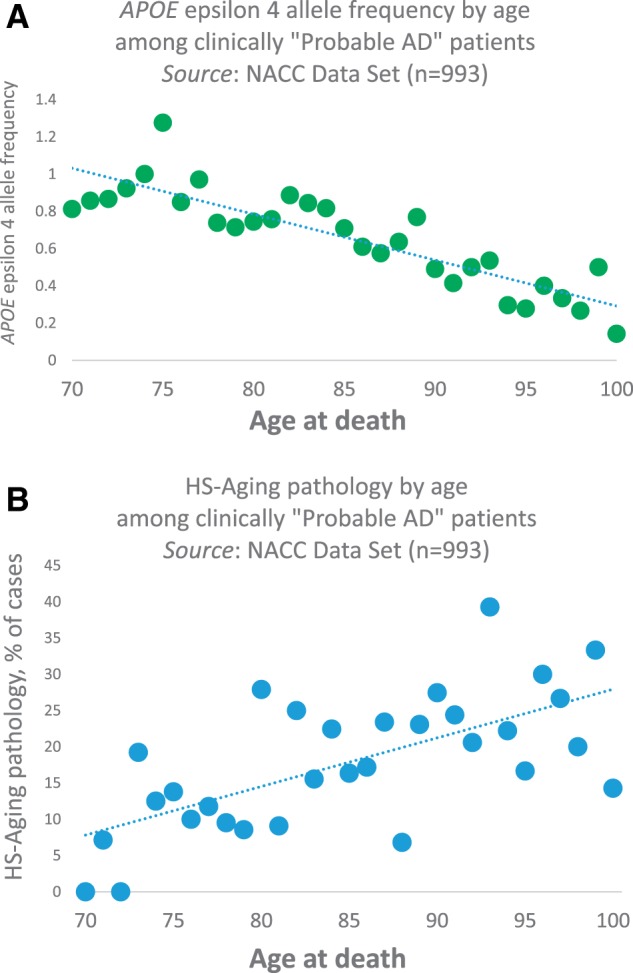
Dementia is associated with risk factors and pathologies that vary across the human aging spectrum. (A, B) Among the “oldest-old” with the clinical diagnosis of “Probable AD,” APOE ϵ4 alleles are decreasingly common after age 75 years (A) , whereas HS-Aging pathology is more prevalent with increasing age (p < 0.001 for both) in the NACC Data Set ( n = 993 of subjects who died between 2005 and 2013) ( 57 ) (B) . Methodology for data analyses was as described previously ( 58 ). These data demonstrate the contribution to dementia of non- APOE genetic risk factors, and common non-AD pathologies in advanced old age.
Although new insights into AD-related brain pathologies have been achieved, many more questions remain. These questions illustrate that there are controversies about disease classifications: What, other than APOE ϵ4, leads to the development of plaques and tangles? Do some cognitively intact individuals tolerate a high burden of plaques and tangles for long periods? Are there environmental and genetic influences that confer protection against the disease? And why do many individuals in advanced old age lack amyloid plaques but still develop hippocampal NFTs?
A Common Pathology With a New Classification: Primary Age-Related Tauopathy (PART)
We consider PART to represent a distinct non-AD pathology ( 59 , 88 ), as indicated in the most recent consensus-based guidelines for the neuropathologic assessment of AD (2) ( Fig. 3 ). However, since tau, α-synuclein, Aβ, and TDP-43 are all pathologically aggregated in multiple diseases ( 1 , 4 , 92–98 ), there are debates about the pathology-based criteria applied to define each disease, including PART ( 99 , 100 ). What is agreed upon is that NFTs are virtually always seen with advanced age, whereas Aβ amyloid plaques are absent in a substantial proportion of elderly brains ( 24 , 59 , 87—91 , 101–103 ). The NFT+/Aβ- pathologic combination (PART) is common, occurring in approximately 20% of all individuals ( Table 2 ).
FIGURE 3.
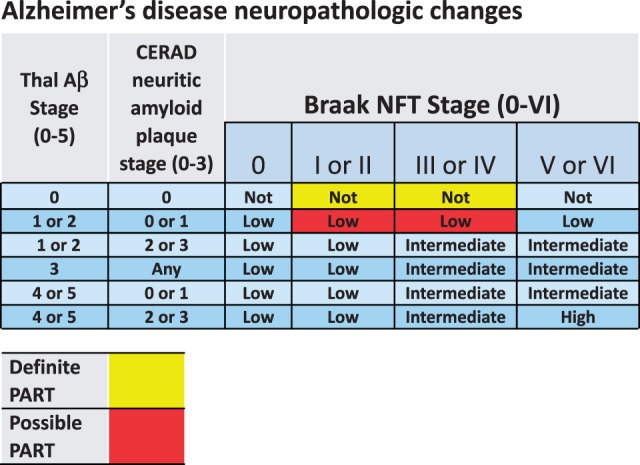
Both Alzheimer disease (AD) and primary age-related tauopathy (PART) are diagnosed based on a grid that incorporates pathologic staging information related to amyloid plaques and neurofibrillary tangles (NFTs). Shown is the grid for determining the level of AD neuropathologic changes at autopsy (2), based on Thal Aβ stages ( 89 ), CERAD neuritic amyloid plaque stages ( 90 ), and Braak NFT stages ( 91 ). Superimposed on the AD classification scheme are criteria for “Definite PART” (red) and “Possible PART” (yellow) ( 59 ) based on the same information.
The update of pathology classification to include PART ( 59 ) provides a universal terminology with both theoretical and clinical implications. To summarize multiple prior studies, human autopsy data indicate the existence of at least 2 common but distinct biologic processes producing NFTs in the hippocampus of elderly persons ( 23 , 60 , 104–106 ). One process (AD) includes Aβ plaques as well as tau NFTs and tends to evolve into dementia. The other (PART) lacks the Aβ plaques and is associated with lesser degree of cognitive impairment and/or other morbidities. Notably, APOE ϵ4 genotype does not appear to be a risk factor for PART, whereas the MAPT “H1” haplotype confers an increased risk for PART ( 59 , 105 , 107 ) and for less common non-AD tauopathies ( 108 , 109 ).
PART differs from AD in terms of overall morbidity and the age range of maximum vulnerability ( 59 ) ( Fig. 2 ). The higher stage PART cases (Braak NFT stages III/IV) tend to show evidence of cognitive impairment ( 59 ). Prior biomarker studies identified neurodegeneration biomarkers in the absence of brain or cerebrospinal fluid Aβ amyloidosis ( 37 , 110–112 ). We also found evidence that PART is a pathologic substrate for individuals who die with subjective memory complaints ( 113 ), which is a very common clinical phenomenon among elderly individuals ( 114 , 115 ).
FIGURE 2.
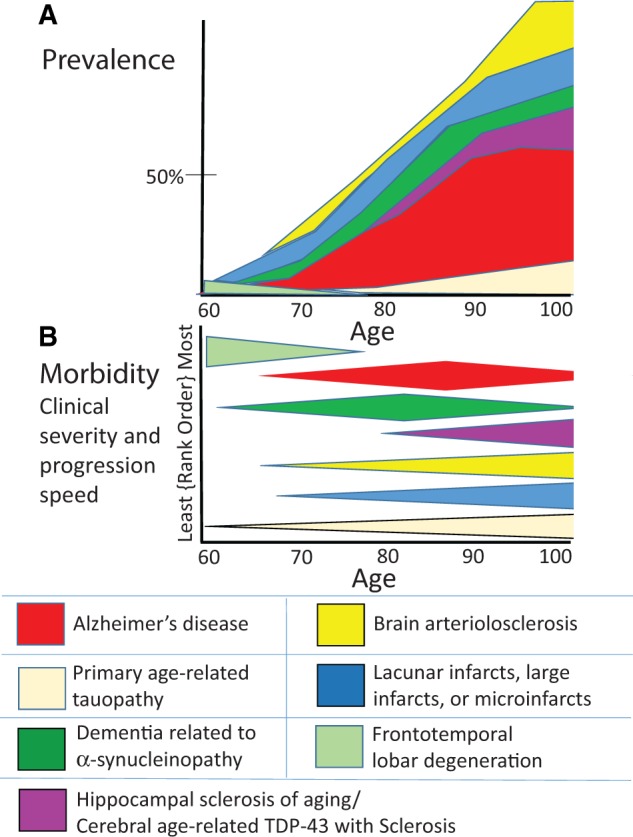
Depiction of prevalence and morbidity of specific pathologies that contribute to dementia. These charts represent the subjective synthesis of the results of multiple published studies ( 19 , 29 , 33 , 52 , 59 , 60 , 61 , 62 , 63 , 58 , 64 , 65–86 ). (A) The prevalence of multiple different specific CNS disease pathologies is depicted (50% prevalence is shown for reference). Note that in large autopsy series the prevalence of advanced AD pathology levels off or decreases in advanced old age whereas other pathologies (HS-Aging, cerebrovascular diseases) increase in that group. Panel (B) shows the same conditions ranked according to the morbidity (neurologic impact and rate of disease progression). Prevalence, morbidity, and age range vary significantly. For example, FTLD is a rare but devastating illness whereas PART is relatively common but lower morbidity, and each mostly afflicts people at separate parts of the human aging spectrum.
Whether PART pathology inevitably progresses to AD is controversial ( 99 , 107 ). Approximately 20% of individuals have PART pathology by their ninth decade 87). The overall proportion of AD and PART cases seems to be stable in centenarians, supporting the hypothesis that PART pathology is not necessarily destined to progress to AD ( 24 , 87 ). A separate issue that remains to be addressed relates to the presence of PART in individuals who satisfy criteria for AD (i.e moderate or frequent neuritic plaques). Using current diagnostic classification ( Fig. 3 ), those cases are not classifiable as PART. However, with the identification of PART, clinicians, pathologists, and basic scientists worldwide can better discriminate amongst various tauopathic diseases, aiding both clinical and basic research. Such studies will set the stage for future preventive or therapeutic strategies.
At the Frontier of Disease Classification: HS-Aging
Despite the recent progress in the field, there are still common and high-morbidity age-related brain diseases that lack a consensus-based classification. Here, we focus on a specific brain disease, with clinical manifestations that overlap with AD, and which has been classified using the term “HS” based on pathologic observations ( 116–126 ). HS in aged individuals is diagnosed at autopsy (according to consensus-based guidelines) when neuron loss and astrocytosis are observed in the hippocampal formation, “out of proportion to AD neuropathologic change in the same structures” (2).
Unfortunately, the current terminology is suboptimal. There is no formal operationalization of what “out of proportion” exactly means. Moreover, the word “sclerosis” (Gr., sklērōsis , “hardness”) lacks a specific connotation in terms of molecular pathogenesis. Pathologists observe what they describe as HS in widely differing conditions including epilepsy, hypoxia and hypoglycemia, frontotemporal lobar degeneration (FTLD), chronic traumatic encephalopathy, and some tauopathies ( 125 , 127–133 ). Recently, a group of experts discussed HS pathologic classification terminology ( 134 ); however, the research subjects in that study were relatively young (most <80 years at death) in comparison to many HS-Aging cases ( 53 , 116 , 135 , 136 ).
The term “HS-Aging” was previously applied to separate this disease from other conditions referred to as HS ( 15 , 56 , 116 , 137–139 ). HS-Aging is distinguished by the advanced age of the affected individuals, by the usual lack of either seizure disorder or frontotemporal dementia symptoms clinically, and by the presence of hippocampal TDP-43 pathology ( 116 , 117 , 140–143 ). Other terms that have been applied include “HpScl” ( 124 , 125 , 144 ) and “HS dementia” ( 125 , 145 , 146 ), with “combined” and “pure” subtypes recognized according to the presence or absence of comorbid pathologies ( 128 ). We note that fewer than 2% of citations returned after a current PubMed search using the words “hippocampal sclerosis” are related to HS-Aging, HpScl, or HS dementia. Thus, whatever final terminology is adopted, it should not be simply “HS” because this is clearly not a disease-defining pathologic endpoint. Here, we describe what is known about this disease including data germane to a useful new terminology.
HS-Aging Is a Common High-Morbidity Disease of Advanced Old Age
The disease referred to as “HS-Aging” affects up to 25% of individuals beyond 85 years of age ( 53 , 56 , 116 , 117 , 135 , 136 , 147 ). The reported prevalence varies across cohorts, perhaps because the “sclerosis” is diagnosed subjectively. Further, 40%–50% of the HS-Aging cases have unilateral HS pathology on hematoxylin and eosin (H&E) stain ( 116 , 147 ). Because this unilateral pathology is associated with cognitive impairment ( 34 ), there would be a large number of false-negatives reported if only one side of the brain were evaluated. Another very important variable is study design. Among research cohorts that are linked to dementia clinics, the samples have tended to be enriched with “pure” AD and FTLD cases, whereas in community-based cohorts, FTLD is quite rare and there are greater numbers of cognitively intact subjects as well as those with dementia due to cerebrovascular disease and HS-Aging ( 22 , 25 , 33 , 61 , 62 , 116 , 117 , 148–153 ).
Research from many centers has found that HS-Aging-type pathology is associated with impaired cognition ( 15 , 34 , 42 , 44 , 53 , 56 , 119 , 122–124 , 136 , 147 , 154–162 ). Lacking more specific biomarkers, some studies have demonstrated associations between HS-Aging pathology and particular cognitive domains ( 116 , 119 , 135 , 136 ). A cognitive profile in patients with autopsy-confirmed HS-Aging was described with relatively impaired Logical Memory Delayed Recall, yet with preserved verbal fluency ( 116 ). These findings were validated in a separate sample to differentiate cognitive impairment with HS-Aging pathology, versus AD or FTLD pathologies, at the group level ( 15 ).
HS-Aging is generally misdiagnosed in living individuals as AD because of the presence of a memory impairment and cognitive deterioration ( 15 , 124 ). Put another way, a relatively large proportion (>10%, increased in advanced age) of “clinical AD” is actually HS-Aging. This may improve as some individuals with SNAP ( 36 ), based on clinical biomarkers, are removed from clinical AD cohorts. However, identification of HS-Aging cases will remain challenging in the large group of individuals with comorbid AD and HS-Aging pathologies because they are not SNAP ( 15 , 53 , 116 , 163 ). Indeed, HS-Aging was found in 2 (9%) of the first 22 subjects followed to autopsy among intensely longitudinally studied subjects with clinical AD in the AD Neuroimaging Initiative (ADNI) cohort ( 164 ), whereas only 4 (18%) of these cases had pure AD pathology ( 165 ).
HS-Aging: Clues About Pathogenesis and the Preclinical State
Although the pathogenesis of HS-Aging is incompletely understood, some prior findings suggest that ischemia or other vascular dysfunction may contribute to the disease phenotype. As stated by Zarow et al ( 117 ), “HS has long been hypothesized to result from ischemic–hypoxic insult to the brain”. The CA1 sector is fed by small end-arterioles from the anterior choroidal and posterior cerebral arteries and is known to be susceptible to hypoxic injury ( 166 ). In a study of 13 aged individuals with HS, Dickson et al reported severe “arteriosclerosis” in 12 of the 13 cases ( 121 ), and others have also published data compatible with a link between HS-Aging and cerebrovascular disease ( 117 , 123 , 157 , 167 , 168 ). Subsequent studies have provided a more specific focus. We found that among vascular neuropathologies, only arteriolosclerosis is associated with HS-Aging pathology ( 169 ). The association between HS-Aging and arteriolosclerosis pathology throughout the brain was confirmed in a subsequent study ( 24 ) and is discussed further below.
In apparent contradiction to the hypothesis that HS-Aging is purely associated with cerebrovascular disease, HS-Aging brains also demonstrate characteristics that are indicative of a neurodegenerative condition. The clinical course of HS-Aging tends to follow the trajectory of a neurodegenerative disease ( 53 , 116 ). A key pathologic biomarker for HS-Aging is aberrant hippocampal TDP-43 pathology that often resembles the pathologic pattern observed in FTLD-TDP ( 140 , 141 , 146 , 170–172 ). In both HS-Aging and FTLD-TDP, slender TDP-43-immunoreactive neurites are observed in hippocampus, subiculum, and amygdala (termed “Type A” TDP-43 pathology) ( 86 , 140 , 170 ). Some gene variants that are associated with increased risk for HS-Aging ( 53 , 124 , 144 , 173 , 174 ) were previously associated with increased risk for FTLD ( 175 , 176 ).
Thus, HS-Aging has been suggested to reflect both cerebrovascular dysfunction and abnormal proteostasis leading to protein misfolding, a mechanistic leitmotif of neurodegenerative processes ( 177 , 178 ). Although these dual vascular and proteostasis mechanisms may seem contradictory, there is increasing evidence of synergistic “mixed” mechanisms that could lead to neurodegeneration ( 179–181 ). Analyzing this requires insights into TDP-43 pathology, which is the most specific marker for HS-Aging ( 116 , 117 , 140 ).
TDP-43 pathology was discovered in the context of brains along the clinical/genetic/pathologic spectrum that includes both amyotrophic lateral sclerosis (ALS) and FTLD ( 141 ); however, TDP-43 pathology is not specific for ALS/FTLD spectrum disorders. TDP-43 pathology has been reported in Alexander disease, Cockayne syndrome, Down syndrome, Guam ALS-parkinsonism-dementia, a subset of Lewy body disorders, low-grade glial neoplasms, inclusion body myositis, and chronic traumatic encephalopathy ( 97 , 120 , 142 , 182–187 ). As such, TDP-43 pathology in some cases must represent a secondary (“downstream”) manifestation of diverse neurodegenerative, developmental, and “reactive” influences. Moreover, monogenic, early onset familial AD is frequently comorbid with hippocampal TDP-43 pathology ( 120 , 188 ), which indicates molecular synergy for specific misfolded proteins, likely due to abnormal proteostasis. These observations argue strongly that conditions outside the ALS/FTLD spectrum may include TDP-43 pathology. A staging schema has been proposed to describe how TDP-43 pathology is distributed in brains with comorbid AD pathology, many of which also had HS-Aging ( 189 , 190 ). Notably, in multiple cohorts of aged persons, TDP-43 pathology is far more strongly linked to HS-Aging than early AD pathology ( 15 , 53 , 116 , 136 , 191 ). However, within the amygdala of subjects with advanced AD, protein misfolding of tau, Aβ, α-synuclein, and TDP-43 pathologies tends to occur ( 61 , 192 , 193 ). Accounting for the existing uncertainties, the above findings collectively indicate that multiple mechanisms can result in hippocampal TDP-43 pathology. The challenge is to define a unique condition with both TDP-43 (TDP[+]) and HS (HS[+]) pathologies.
For determining “boundary zones” that are meaningful for pathology-based classification, a key goal is to understand the disease in its preclinical stage(s). Current knowledge derives predominantly from cross-sectional data, and here we are referring mostly to autopsy series that include subjects over 85 years of age. The term “pre-HpScl” was used to describe hippocampal pathology characterized by none to minimal neuronal loss yet with abundant TDP-43 pathology ( 170 ). This partially overlaps with “segmental” HS-Aging, where only select portions of the TDP[+] hippocampal formation showed evidence of neuronal loss on H&E despite extensive additional sampling ( 194 ). Notably, in cases with unilateral HS according to H&E evaluation, the contralateral side is almost always positive for TDP-43 pathology ( 116 ).
Prior autopsy studies that reported a relatively high proportion of cases with HS[−]TDP[+] pathology ( 136 , 61 , 195 ), in contrast to studies with a higher proportion of HS[+]TDP[+] cases ( 116 , 147 ), reflect the lack of universally applied diagnostic criteria for aging-related TDP-43 or HS-Aging pathologies. Thus, what one group may call HS[+]TDP[+], another would categorize as HS[−]TDP[+]. Yet taken together, the published studies are compatible with a progressive disease with limbic TDP-43 pathology in the early stages and HS in later stages.
Do TDP[+] cases in advanced age represent a subtype of FTLD-TDP? This is debatable, but there are reasons to consider TDP[+] in older subjects to be separate from FTLD-TDP. TDP-43 pathology seen in HS-Aging cases does not appear to progress to full-blown frontotemporal dementia (clinically) or FTLD-TDP (pathologically) when evaluated in community-based cohorts ( 25 , 26 , 135 , 151 , 61 ). TDP-43 pathology is rare before 65 years but allocortical TDP-43 pathology (i.e within amygdala, hippocampus, entorhinal cortex) is common in octogenarians and nonagenarians ( 136 , 196 , 197 ). In a high-quality community-based cohort ( n = 544, average age of death 89.0 years), 52% had limbic TDP-43 pathology but none had FTLD-TDP ( 198 ). Likewise, in the largest neuropathologic study to date of centenarians, there was not a single example of FTLD-TDP ( 24 ). The enormous difference in prevalence ( Fig. 2 ) is a key point: FTLD-TDP affects approximately 20 000 individuals in United States ( 62 ), whereas HS-Aging afflicts well over 10 times as many if one extrapolates from large autopsy series ( 15 , 56 , 116 , 117 , 120 , 135 , 157 , 199 ). In Figure 4 , we provide cognitive, neuroimaging, and neuropathologic data on a patient with clinical “probable AD” yet whose autopsy showed HS[+]TDP[+] pathology by our criteria, and with very minimal Aβ deposition. To make progress in studying this common disease phenotype, it is critical to generate consensus about the disease-defining criteria. These efforts will be aided by clinical studies because there currently is no validated animal model.
FIGURE 4.

Case study illustrates clinical and neuropathologic features of common comorbid diseases. The female subject was followed from 77 years until death at age 102 years. Detailed neurocognitive tests were performed until age 98 years; MCI was diagnosed clinically at age 93, “Probable Alzheimer Disease” at age 95. APOE genotype was ϵ3/ϵ3. (A) Panel shows results of MMSE (global cognition) and animal naming (verbal fluency) results; note that the verbal fluency was relatively stable even after global cognitive status was impaired. A brain MRI (horizontal plane) 10 years before death (B) showed hippocampal atrophy (arrows). Immunohistochemistry demonstrated extremely sparse Aβ amyloid pathology in temporal neocortex (C) and no neuritic plaques. Aβ amyloid in AD brain (D) is shown for comparison. In the hippocampal formation there was Braak NFT stage II tauopathy (E) and PART and HS-Aging were both diagnosed. Note that the hippocampal sclerosis is diagnosed according to consensus-based criteria (2): “cell loss and gliosis out of proportion to plaques and tangles,” rather than complete destruction of the structure. TDP-43 pathology was present in the hippocampus; (F) shows dentate gyrus with inclusions (arrows) and (G) shows subiculum with neuronal inclusion (arrow) and slender nontapering TDP-43 neurites (arrowheads). Widespread brain arteriolosclerosis pathology was also observed (Fig. 5). Panel (H) is a low-power photomicrograph showing the hippocampal formation including CA1, dentate granule (dg), and subiculum (Sub) regions. Adjacent sections were stained for phospho-Tau (P-Tau) and P-TDP-43 and the pathology was depicted schematically (inset I) using an Aperio ScanScope as described previously ( 56 ): red dots for NFTs, green dots for P-TDP-43 inclusions, and cyan region shows area with P-TDP-43 neurites. As noted previously ( 56 ), the tauopathic distribution in TDP[+]HS[+] cases is slightly different from “classic” early Braak NFT stages. Scale bars: C , 100 μm; D , 80 μm; E , 40 μm; F , 30 μm; G , 70 μm; H , 2 mm.
In a recent neuroimaging study, Kotrotsou et al found that in elderly individuals dying with eventual autopsy-proven HS-Aging, premortem MRI studies showed extensive brain atrophy outside of the hippocampal formation, particularly in the frontal lobes ( 200 ). Furthermore, in the AD Neuroimaging Initiative (ADNI) data set ( 164 ), the HS-Aging risk alleles (described below) were associated with widespread MRI-detected brain atrophy outside of the hippocampus ( 201 ). Previous pathologic observations are compatible with the hypothesis that HS-Aging is actually a generalized disease that is often comorbid with pan-cerebral arteriolosclerosis pathology, rather than one localized to medial temporal lobe structures ( 15 , 56 , 136 , 158 , 169 , 194 ).
We interpret these prior findings to indicate that there is a common cerebral disease, affecting persons in advanced age, characterized by a spectrum of pathologies:
In other words, the signal feature of the disease is TDP-43 pathology, rather than HS. Yet, there are more complexities. Perhaps analogous to the many non-AD diseases that are associated with hippocampal NFTs ( 202–204 ), some rare diseases, fundamentally different from HS-Aging, also show the HS[+]TDP[+] pattern ( 127 , 132 , 205 , 206 ). To understand what makes HS-Aging unique, further knowledge is required about its specific pathogenesis.
Genetics of HS-Aging
Genetic risk factors can provide insights into disease-specific mechanisms. For example, APOE gene variants are not associated with altered risk for either TDP[+] or HS[+] neuropathology ( 15 , 116 , 124 , 135 , 207 ). This supports the hypothesis that HS-Aging is a separate disease entity from AD.
Genotypes linked to HS-Aging pathology have now been identified ( Table 3 ). Potential risk alleles were first analyzed in 2 specific genes ( GRN and TMEM106B ), in line with the hypothesis that HS-Aging is related pathogenetically to FTLD-TDP. The first gene variant linked to HS-Aging pathology was rs5848, a single nucleotide polymorphism (SNP) located in the 3′UTR of GRN ( 173 ). This SNP is also associated with altered expression of GRN ( 144 , 211 ). Whereas many different GRN mutations cause FTLD-TDP ( 212–216 ), the HS-Aging SNP (rs5848) is apparently a disease-modifying allele that impacts the manifestation of multiple different diseases rather than specifically of HS-Aging. For example, rs5848 has been linked to AD, Parkinson disease, C9ORF72 neurodegeneration, and bipolar disorder ( 173 , 217–221 ), whereas several groups have reported that rs5848 is not linked to FTLD ( 208 , 222 ).
TABLE 3.
Genes and Single Nucleotide Polymorphisms Associated With Risk for Hippocampal Sclerosis of Aging
| Gene | SNP | Experiment that Linked the Gene to HS-Aging: SNP-Focused or GWAS | Replicated? | References |
|---|---|---|---|---|
| GRN | rs5848 | SNP-focused | Yes | ( 124 , 144 , 173 , 174 ) |
| TMEM106B | rs1990622 | SNP-focused | Yes | ( 170 , 174 , 194 , 196 , 208 , 209 ) |
| ABCC9 | rs704180 | GWAS | Yes | ( 24 , 151 , 174 , 58 ) a |
| KCNMB2 | rs9637454 | GWAS | No | ( 210 ) |
a Reference ( 58 ) refers only to ABCC9 association with brain arteriolosclerosis.
The TMEM106B SNP rs1990622 is a risk allele for FTLD-TDP, as determined using a genome-wide association study (GWAS) ( 223 ), and the same SNP is linked to a coding variant and altered protein expression ( 224 ). TMEM106B polypeptide is a lysosomal protein that apparently affects GRN expression ( 225–227 ). The risk allele is associated with increased vulnerability to ALS and for neurodegeneration linked to C9ORF72 repeat expansions ( 209 , 228 ). Further, in SNP-focused studies, rs1990622 status was found to be associated with HS pathology ( 53 , 151 , 229 ), altered AD phenotype ( 210 , 229 , 230 ), and cognition independent of AD or HS pathologies ( 196 ).
The studies on specific GRN and TMEM106B gene variants due to their link with other diseases left unaddressed the possibility of genotypes associated specifically with HS-Aging. A complementary experimental approach is GWAS, which is unbiased by prior mechanistic hypotheses. GWASs have now identified 2 putative HS-Aging risk genes, both of which encode potassium channel regulators: ABCC9 and KCNMB2 ( 151 , 231 ).
The association between ABCC9 SNP rs704180 and HS-Aging pathology attained genome-wide significance in the GWAS paper ( 151 ), and has since been replicated ( 174 ), although not as extensively as GRN and TMEM106B . This intronic SNP is associated with altered ABCC9 mRNA expression ( 232 ), and nearby ABCC9 gene variants are also linked to other neurologic diseases such as sleep disorder and depression ( 233–235 ). The ABCC9 gene encodes proteins that regulate potassium channels ( 233 , 234 ), serving as a metabolic “sensor” relevant to vascular responses to hypoxia, ischemia, and inflammation ( 236 ). There are published studies that support direct connections between ABCC9 and neurodegenerative disease mechanisms ( 237–244 ).
A second gene linked through GWAS to HS-Aging pathology is KCNMB2 ( 231 ). Intriguingly, Zarei et al found that the KCNMB2 gene product could be relevant to hippocampal physiology ( 245 ). As yet, the finding of association between the KCNMB2 SNP rs9637454 and HS-Aging remains to be replicated.
In summary, genomic studies provided meaningful indications about what may cause or protect against HS-Aging. The associations for both GRN and TMEM106B SNPs with HS-Aging risk have now been replicated, providing strong support for a mechanism relevant to both HS-Aging and FTLD-TDP. However, these HS-Aging risk SNPs are risk-modifying alleles in multiple diseases, thereby begging the question about the disease-specific “upstream” factors. The impact of these particular gene variants may be analogous to the MAPT H1 haplotype that confers increased risk for PART, progressive supranuclear palsy, and other “sporadic” tauopathies ( 57 , 105 , 108 , 246 , 247 ), as opposed to MAPT mutations that directly cause familial FTLD-MAPT ( 248–250 ). As with PART in comparison to FTLD-MAPT, the ultimate manifestations of HS-Aging and FTLD-TDP are also profoundly different from each other in terms of clinical (i.e course and age range) and pathological features ( 15 , 56 ). Although it is an important insight that particular GRN and TMEM106B SNPs can increase risk for TDP-43 pathology in multiple diseases, the experiments that discovered these phenomena were blind to the disease-specific “upstream” mechanisms involved in a disease that is far more common than FTLD-TDP. Published genome-wide analyses have implicated ABCC9 gene variants specific to HS-Aging. Because genomics information provides insights into pathogenesis, this may help in the delineation of the disease phenotype, shifting the focus to another brain pathology, namely aging-related brain arteriolosclerosis (B-ASC).
Previously Unsuspected Pathologic Synergies: B-ASC and HS-Aging
B-ASC describes pathologic thickening of the walls of brain arterioles not due to brain amyloid (58, 63, 169 , 251–255 ). This subtype of small vessel pathology is very common in the brains of older individuals and is associated with impaired cognitive status, independent of other pathologies ( 255 ). In prior studies by us and others, it was implied that B-ASC represents a well-defined and classifiable subtype of vascular pathology in the CNS.
However, we have much to learn about brain arterioles in healthy and disease states. A recent review made trenchant points: “The term arteriolosclerosis actually does not define a lesion at all. It is a generic term meaning ‘hardening of small arteries’. In fact, the term encompasses 2 distinct lesions: (1) a fibromuscular proliferation of the intima, the ‘hyperplastic type’ and (2) a deposition of amorphous material in the arteriolar wall, the ‘hyaline type’” ( 256 ). Moreover, the current classification of arteriosclerosis is not based on a consensus document by a major cerebrovascular, cardiovascular, or pathology organization ( 256 ). Despite progress in the field, relatively little is known about the neuropathology of elements that comprise brain arterioles, a complicated arrangement of endothelial cells, pericytes, smooth muscle cells, basement membrane, astrocyte end-feet, and extracellular matrix ( 64 , 255 , 257–261 ). Small blood vessels participate in energy exchange, removal of waste, blood pressure regulation, neuroglial activity, and neuroimmune functions and it seems clear that we still have much to learn.
Photomicrographs to convey some of the heterogeneity of vascular changes that may be diagnosed as B-ASC at autopsy are shown in Figure 5 . In aged brains, arteriolar morphologies include pathologic variants other than “hyperplastic-” and “hyaline”-type changes. For example, some arterioles show degenerative changes in smooth muscle cells, whereas other arteriolar structures comprise multiple lumens ( 262 , 263 ). Future work may better capture the heterogeneity of arteriolar disease phenotypes and provide the basis for robust clinical-pathologic correlations.
FIGURE 5.

Brain arteriolosclerosis (B-ASC) pathology is a complex phenotype. These panels show B-ASC vascular profiles in brains from different aged individuals to provide a small sampling of the heterogeneity of B-ASC. (A-C) Panels show hematoxylin and eosin staining. Panel (A) is a low-power photomicrograph depicting a vessel in the amygdala of a person with advanced AD and cerebrovascular disease. Note the large expanse of hyalinized material (*) that extends from the vessel wall, along with a patch of lymphocytic inflammation (arrow). By contrast, in the hippocampus of the case study ( Fig. 4 ) there is a smaller blood vessel (boxed in B , magnified in C ) that shows a vascular profile with apparent fibrinoid necrosis and/or microcalcifications in the paucicellular vessel wall. Another pattern we have seen in many cases is multiple vascular profiles in the same vessel bed, as shown in panel (D) (arterioles are here visualized using α-SMA immunohistochemistry). Collagen can be visualized using a trichrome stain (panels E, F are separate HS-Aging cases); a B-ASC profile is shown in E with the collagen labeled green. Cases with hippocampal TDP-43 in our experience often show neocortical B-ASC as visualized by the green-staining arterioles (arrows) in this low-power photomicrograph near the pia (*) of frontal neocortex, Brodmann Area 9. Scale bars: A , 200 μm; B , 90 μm; C , 10 μm; D , 25 μm; E , 70 μm; F , 100 μm.
Although many unknowns remain in the study of B-ASC, insights have been gained. Studies of organs outside the brain indicate that arteriolosclerosis is associated with metabolic or cardiovascular disorders such as diabetes and hypertension ( 57 , 256 , 264–267 ). Ighodaro et al recently tested risk factors of B-ASC among 2390 persons who had come to autopsy with known B-ASC status using the National Alzheimer’s Coordinating Center data set ( 255 , 268 ). These analyses indicated that advanced age at death was associated with B-ASC severity. Self-reported hypertension was only associated with B-ASC in the <80 years age at death group.
Interestingly, in the ≥80 years age at death group, the ABCC9 gene variant rs704180, previously associated with HS-Aging, was also associated with B-ASC ( 255 ). By contrast, neither GRN nor TMEM106B SNPs were associated with B-ASC ( 255 ). The hypothesis that ABCC9 is associated with arteriolosclerosis pathology throughout many different brain regions was supported in analyses of centenarians’ brains ( 24 ). Intriguingly, Lim et al recently reported that B-ASC is associated with “sleep fragmentation,” ( 269 ) and ABCC9 gene variants have been linked to sleep problems ( 235 ,, 270 , 271 ).
The observation that the same ABCC9 gene variant is associated with risk for both B-ASC and HS-Aging pathologies in old age provides the basis for a novel hypothesis combining cerebrovascular and neurodegenerative paradigms ( 255 ). One exciting aspect of implicating ABCC9 in disease pathogenesis is that ABCC9 gene product-modifying drugs (both agonists and antagonists) are widely used in the human pharmacopeia ( 272–275 ). We emphasize that the findings of ABCC9 require further validation; this is still a new hypothesis that requires more study.
Any hypothesis to conceptualize the aging-related disease that was previously labeled HS-Aging or HS dementia must include TDP-43 pathology. In each of the many conditions associated with TDP-43 pathology there is a chronic genetic and/or environmental insult to the brain. It is possible that a subtype of chronic vascular insult(s) could induce TDP-43 phosphorylation and misfolding. Numerous studies indicate that TDP-43 pathology does not appear to arise following acute hypoxic/ischemic neuronal injury ( 116 , 117 , 140 , 142 ). This would be directly analogous to brain trauma, where a single traumatic event is not associated with TDP-43 pathology ( 276 ), yet approximately 80% of brains with chronic traumatic encephalopathy are positive for TDP-43 pathology ( 127 ). If chronic vascular dysfunction can lead to TDP[+] disease phenotype, then that disease may constitute a novel targetable cause of dementia. An intriguing possibility is that epidemiologic phenomena previously attributed to AD ( 65 , 66 ) are related instead to this common, high-morbidity, but hitherto largely ignored disease.
Revised Terminology: A Recommendation
Whatever the pathogenetic mechanisms are, new terminology is required. Neither “HS-Aging,” nor any other extant term, is truly applicable. A terminology that focuses on FTLD, TDP-43, or HS in isolation would not be accurate based on the experimental data. Because the disease preferentially affects the “oldest-old,” often includes TDP-43 pathology and arteriolosclerosis well beyond the hippocampus, and may evolve to HS, we recommend the term “cerebral age-related TDP-43 and sclerosis” (CARTS). Features seen in CARTS are illustrated in Figure 6 .
FIGURE 6.

The disease we have referred to as hippocampal sclerosis of aging (HS-Aging) is a complex phenotype that includes hippocampal sclerosis (A-C) , TDP-43 pathology (D-F) , and brain arteriolosclerosis (G-I) . Features of pathologically unaffected compartments are shown ( A, hippocampus; D, neurons; and G, arterioles) in contrast to the compartments (B, E, H) and brain areas (C, F, I) affected in this disease. Because the extant classification is suboptimal, we propose a new terminology to classify this disease: cerebral age-related TDP-43 and sclerosis, CARTS.
A diagnosis of CARTS indicates robust TDP-43 pathology in the hippocampus of persons aged >85 years at death and would otherwise incorporate the current TDP[+] cases termed HS-Aging, HpScl, and HS dementia. As stated above, it is relevant that both HS and arteriolosclerosis are often parts of the phenotype. We recommend that HS is not necessary for the diagnosis of CARTS because HS is ill-defined, nonspecific, and often segmental and, therefore, sampling bias would be considerable. Although B-ASC may well play a role in pathogenesis, the current diagnosis of B-ASC is too inconsistent to be practical as part of the diagnostic rubric at this time. The TDP-43 pathology frequently extends outside the hippocampus, but CARTS should not have the extremely dense subcortical TDP-43 pathology that occurs in FTLD-TDP. A hypothetical staging schema is presented in Table 4 . Some older FTLD-TDP cases may be challenging to discriminate from CARTS in the absence of other biomarkers, but CARTS affects a virtually nonoverlapping age group in comparison with FTLD-TDP. AD and CARTS pathologies are both common pathologies and thus are expected to be comorbid frequently, although distinct ‘boundary zones’ will require further research.
TABLE 4.
Proposed Staging of Disease Referred to as HS-Aging or “CARTS”

|
|---|

|
Inclusion:
o >85 years of age at death.
o TDP-43 pathology in limbic structures, preferably in more than one region.
Amygdala, subiculum, hippocampus proper, parahippocampal gyrus.
o Hippocampal sclerosis and arteriolosclerosis should be evaluated but are not
required for diagnosis.
Exclusion:
o Exclude cases with frontotemporal dementia (FTD)-spectrum, including behavioral variant FTD, semantic dementia, progressive nonfluent aphasia, and logopenic aphasia (on clinical grounds and optimally on neuropsychological testing).
o Exclude cases with predominantly amygdala TDP-43 in the context of advanced Alzheimer disease pathology (Braak NFT stages V/VI).
CONCLUSION
High-quality, large autopsy cohorts and collaborative efforts among neuropathologists have enabled recent advances in the field of disease classification relevant to dementia. There are diagnostic “border zones” that need to be clarified and a better understanding of the “mixed” pathologies that are typical in advanced old age is needed. As these challenges are addressed, the diagnoses increasingly reflect the biologic complexity and should help in efforts to identify appropriate patient groups for clinical trials. Particular diagnostic categories are in different stages of scientific “maturity,” with some having been studied in thousands of published papers, whereas others will require substantial additional work to achieve an accurate nosology. TDP-43 pathologies appear to be analogous to tau tangles, “upstream” factors and comorbid pathologies can disturb protein homeostasis, especially with the added influence of gene variants that increase risk across different diseases ( Fig. 7 ). Categorizing the “downstream” pathology is complex because of the overlapping pathologic phenotypes. This is particularly true for CARTS because TDP-43, HS, and B-ASC pathologies all occur in multiple conditions. The recently revised pathologic concepts are not all truly novel. On the contrary, investigators had previously reported many of the manifestations of the brain diseases but lacked adequate contextual data. It is safe to assert that for all prior advances, current data are imperfect and skepticism should be sustained in considering the current diagnostic terms and criteria. Categorization of brain diseases of aging is still a work in progress.
FIGURE 7.
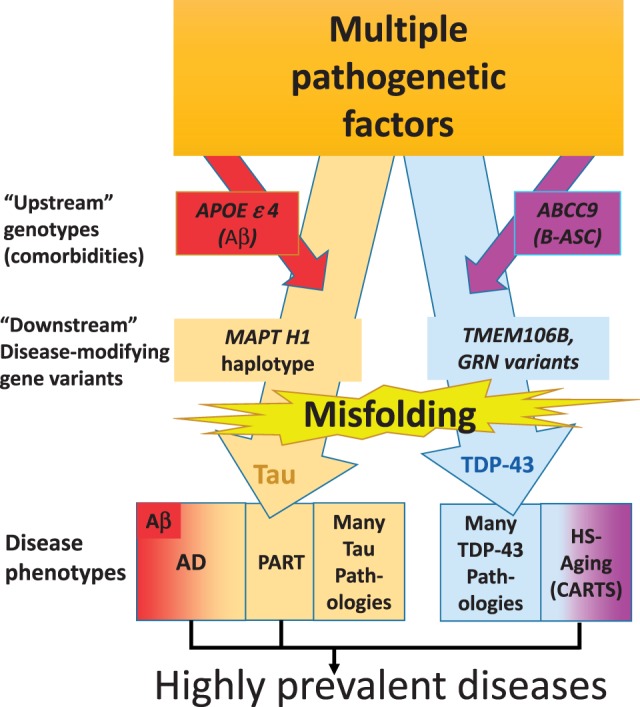
Pathogenesis helps guide classification of neurodegenerative diseases. This schematic depicts hypotheses about the multiple genetic and environmental factors that promote tau and TDP-43 pathologies. These may include comorbid pathologies such as amyloid plaques (associated with APOE ϵ4 allele) or brain arteriolosclerosis (B-ASC, associated with ABCC9 gene variant). There also are “downstream” genetic risk modifiers, such as GRN (rs5848), TMEM106B (rs1990662), and MAPT H1 haplotype, which appear to influence many different disease phenotypes each. Ultimately, protein misfolding contributes to symptomatic manifestations. While there are large number of rarer conditions, the common tau and TDP-43 diseases linked to cognitive impairment in advanced old age are AD, PART, and HS-Aging/CARTS.
ACKNOWLEDGMENTS
We are deeply grateful to all of the study volunteers. We thank Sonya Anderson and Ela Patel for technical support, Greg Cooper, MD, Nancy Stiles, MD, Stephen Scheff PhD, and Allison Caban-Holt, PhD for the clinical evaluations.
REFERENCES
- 1. Kovacs GG. Molecular pathological classification of neurodegenerative diseases: Turning towards precision medicine . Int J Mol Sci 2016. ; 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Montine TJ, Phelps CH, Beach TG , et al. . National Institute on Aging-Alzheimer's association guidelines for the neuropathologic assessment of Alzheimer's disease: A practical approach . Acta Neuropathol 2012. ; 123 : 1 – 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: A complex but coherent relationship . J Neuropathol Exp Neurol 2009. ; 68 : 1 – 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nelson PT, Alafuzoff I, Bigio EH , et al. . Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature (Review) . J Neuropathol Exp Neurol 2012. ; 71 : 362 – 81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Caspersen CJ, Thomas GD, Boseman LA , et al. . Aging, diabetes, and the public health system in the United States . Am J Public Health 2012. ; 102 : 1482 – 97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chatterjee S, Peters SA, Woodward M , et al. . Type 2 diabetes as a risk factor for dementia in women compared with men: A pooled analysis of 2.3 million people comprising more than 100,000 cases of dementia . Diab Care 2016. ; 39 : 300 – 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Profenno LA, Porsteinsson AP, Faraone SV. Meta-analysis of Alzheimer's disease risk with obesity, diabetes, and related disorders . Biol Psychiatry 2010. ; 67 : 505 – 12 [DOI] [PubMed] [Google Scholar]
- 8. Lu FP, Lin KP, Kuo HK. Diabetes and the risk of multi-system aging phenotypes: A systematic review and meta-analysis . PLoS One 2009. ; 4 : e4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abner EL, Nelson PT, Kryscio RJ , et al. . Diabetes is associated with cerebrovascular but not Alzheimer neuropathology . Alzheimers Dement 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nelson PT, Head E, Schmitt FA , et al. . Alzheimer's disease is not “brain aging”: Neuropathological, genetic, and epidemiological human studies . Acta Neuropathol 2011. ; 121 : 571 – 87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nelson PT, Smith CD, Abner EA , et al. . Human cerebral neuropathology of Type 2 diabetes mellitus . Biochim Biophys Acta 2009. ; 1792 : 454 – 69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dugger BN, Malek-Ahmadi M, Monsell SE , et al. . A cross-sectional analysis of late-life cardiovascular factors and their relation to clinically defined neurodegenerative diseases . Alzheimer Dis Assoc Disord 2016. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Beeri MS, Silverman JM, Davis KL , et al. . Type 2 diabetes is negatively associated with Alzheimer's disease neuropathology . J Gerontol A Biol Sci Med Sci 2005. ; 60 : 471 – 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ahtiluoto S, Polvikoski T, Peltonen M , et al. . Diabetes, Alzheimer disease, and vascular dementia: A population-based neuropathologic study . Neurology 2010. ; 75 : 1195 – 202 [DOI] [PubMed] [Google Scholar]
- 15. Brenowitz WD, Monsell SE, Schmitt FA , et al. . Hippocampal sclerosis of aging is a key Alzheimer's disease mimic: Clinical-pathologic correlations and comparisons with both Alzheimer's disease and non-tauopathic frontotemporal lobar degeneration . J Alzheimers Dis 2014. ; 39 : 691 – 702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kovacs GG, Ferrer I, Grinberg LT , et al. . Aging-related tau astrogliopathy (ARTAG): Harmonized evaluation strategy . Acta Neuropathol 2016. ; 131 : 87 – 102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dickson DW. Neuropathology of non-Alzheimer degenerative disorders . Int J Clin Exp Pathol 2009. ; 3 : 1 – 23 [PMC free article] [PubMed] [Google Scholar]
- 18. Schmitt FA, Nelson PT, Abner E , et al. . University of Kentucky Sanders-Brown Healthy Brain Aging Volunteers: Donor characteristics, procedures, and neuropathology . Curr Alzheimer Res 2012. ; 9 : 724 – 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nelson PT, Jicha GA, Schmitt FA , et al. . Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: Neuritic plaques and neurofibrillary tangles “do count” when staging disease severity . J Neuropathol Exp Neurol 2007. ; 66 : 1136 – 46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes . Acta Neuropathol 1991. ; 82 : 239 – 59 [DOI] [PubMed] [Google Scholar]
- 21. Jicha GA, Abner EL, Schmitt FA , et al. . Preclinical AD workgroup staging: Pathological correlates and potential challenges . Neurobiol Aging 2012. ; 33 : 622 e1 – e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jicha GA, Parisi JE, Dickson DW , et al. . Neuropathologic outcome of mild cognitive impairment following progression to clinical dementia . Arch Neurol 2006. ; 63 : 674 – 81 [DOI] [PubMed] [Google Scholar]
- 23. Nelson PT, Abner EL, Schmitt FA , et al. . Brains with medial temporal lobe neurofibrillary tangles but no neuritic amyloid plaques are a diagnostic dilemma but may have pathogenetic aspects distinct from Alzheimer disease . J Neuropathol Exp Neurol 2009. ; 68 : 774 – 84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Neltner JH, Abner EL, Jicha GA , et al. . Brain pathologies in extreme old age . Neurobiol Aging 2016. ; 37 : 1 – 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rahimi J, Kovacs GG. Prevalence of mixed pathologies in the aging brain . Alzheimers Res Ther 2014. ; 6 : 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bennett DA, Schneider JA, Arvanitakis Z , et al. . Neuropathology of older persons without cognitive impairment from two community-based studies . Neurology 2006. ; 66 : 1837 – 44 [DOI] [PubMed] [Google Scholar]
- 27. Schneider JA, Arvanitakis Z, Bang W , et al. . Mixed brain pathologies account for most dementia cases in community-dwelling older persons . Neurology 2007. ; 69 : 2197 – 204 [DOI] [PubMed] [Google Scholar]
- 28. Robinson JL, Molina-Porcel L, Corrada MM , et al. . Perforant path synaptic loss correlates with cognitive impairment and Alzheimer's disease in the oldest-old . Brain 2014. ; 137 : 2578 – 87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barker WW, Luis CA, Kashuba A , et al. . Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the state of Florida Brain Bank . Alzheimer Dis Assoc Disord 2002. ; 16 : 203 – 12 [DOI] [PubMed] [Google Scholar]
- 30. Jellinger KA. Clinicopathological analysis of dementia disorders in the elderly—An update . J Alzheimers Dis 2006. ; 9 : 61 – 70 [DOI] [PubMed] [Google Scholar]
- 31. Brenowitz WD, Nelson PT, Besser LM , et al. . Cerebral amyloid angiopathy and its co-occurrence with Alzheimer's disease and other cerebrovascular neuropathologic changes . Neurobiol Aging 2015. ; 36 : 2702 – 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yarchoan M, Xie SX, Kling MA , et al. . Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias . Brain 2012. ; 135 : 3749 – 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schneider JA, Aggarwal NT, Barnes L , et al. . The neuropathology of older persons with and without dementia from community versus clinic cohorts . J Alzheimers Dis 2009. ; 18 : 691 – 701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nelson PT, Abner EL, Schmitt FA , et al. . Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons . Brain Pathol 2010. ; 20 : 66 – 79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Petrovitch H, Ross GW, He Q , et al. . Characterization of Japanese-American men with a single neocortical AD lesion type . Neurobiol Aging 2008. ; 29 : 1448 – 55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jack CR, Jr, Knopman DS, Chetelat G , et al. . Suspected non-Alzheimer disease pathophysiology—Concept and controversy . Nat Rev Neurol 2016. ; 12 : 117 – 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Knopman DS, Jack CR, Jr., Wiste HJ , et al. . Brain injury biomarkers are not dependent on beta-amyloid in normal elderly . Ann Neurol 2013. ; 73 : 472 – 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bocchetta M, Galluzzi S, Kehoe PG , et al. . The use of biomarkers for the etiologic diagnosis of MCI in Europe: An EADC survey . Alzheimers Dement 2015. ; 11 : 195 – 206 [DOI] [PubMed] [Google Scholar]
- 39. Ewers M, Walsh C, Trojanowski JQ , et al. . Prediction of conversion from mild cognitive impairment to Alzheimer's disease dementia based upon biomarkers and neuropsychological test performance . Neurobiol Aging 2012. ; 33 : 1203 – 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jack CR, Jr, Lowe VJ, Weigand SD , et al. . Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: Implications for sequence of pathological events in Alzheimer's disease . Brain 2009. ; 132 : 1355 – 65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Risacher SL, Shen L, West JD , et al. . Longitudinal MRI atrophy biomarkers: Relationship to conversion in the ADNI cohort . Neurobiol Aging 2010. ; 31 : 1401 – 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dawe RJ, Bennett DA, Schneider JA , et al. . Neuropathologic correlates of hippocampal atrophy in the elderly: A clinical, pathologic, postmortem MRI study . PLoS One 2011. ; 6 : e26286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zarow C, Wang L, Chui HC , et al. . MRI shows more severe hippocampal atrophy and shape deformation in hippocampal sclerosis than in Alzheimer's disease . Int J Alzheimers Dis 2011. ; 2011 : 483972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kaur B, Himali JJ, Seshadri S , et al. . Association between neuropathology and brain volume in the Framingham heart study . Alzheimer Dis Assoc Disord 2014. ; 28 : 219 – 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fernando MS, Ince PG. Vascular pathologies and cognition in a population-based cohort of elderly people . J Neurol Sci 2004. ; 226 : 13 – 7 [DOI] [PubMed] [Google Scholar]
- 46. Blessed G, Tomlinson BE, Roth M. The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects . Br J Psychiatry 1968. ; 114 : 797 – 811 [DOI] [PubMed] [Google Scholar]
- 47. Roth M, Tomlinson BE, Blessed G. Correlation between scores for dementia and counts of `senile plaques' in cerebral grey matter of elderly subjects . Nature 1966. ; 209 : 109 – 10 [DOI] [PubMed] [Google Scholar]
- 48. Tomlinson BE, Blessed G, Roth M. Observations on the brains of non-demented old people . J Neurol Sci 1968. ; 7 : 331 – 56 [DOI] [PubMed] [Google Scholar]
- 49. Tomlinson BE, Blessed G, Roth M. Observations on the brains of demented old people . J Neurol Sci 1970. ; 11 : 205 – 42 [DOI] [PubMed] [Google Scholar]
- 50. von Gunten A, Ebbing K, Imhof A , et al. . Brain aging in the oldest-old . Curr Gerontol Geriatr Res 2010. ; pii: 358531. doi: 10.1155/2010/358531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bancher C, Jellinger KA. Neurofibrillary tangle predominant form of senile dementia of Alzheimer type: A rare subtype in very old subjects . Acta Neuropathol 1994. ; 88 : 565 – 70 [DOI] [PubMed] [Google Scholar]
- 52. Imhof A, Kovari E, von Gunten A , et al. . Morphological substrates of cognitive decline in nonagenarians and centenarians: A new paradigm? J Neurol Sci 2007. Jun 15; 257 ( 1-2 ): 72 – 9 [DOI] [PubMed] [Google Scholar]
- 53. Murray ME, Cannon A, Graff-Radford NR , et al. . Differential clinicopathologic and genetic features of late-onset amnestic dementias . Acta Neuropathol 2014. ; 128 : 411 – 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Naj AC, Jun G, Reitz C , et al. . Effects of multiple genetic loci on age at onset in late-onset Alzheimer disease: A genome-wide association study . JAMA Neurol 2014. ; 71 : 1394 – 404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wingo TS, Lah JJ, Levey AI , et al. . Autosomal recessive causes likely in early-onset Alzheimer disease . Arch Neurol 2012. ; 69 : 59 – 64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nelson PT, Smith CD, Abner EL , et al. . Hippocampal sclerosis of aging, a prevalent and high-morbidity brain disease . Acta Neuropathol 2013. ; 126 : 161 – 77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Baker AB. Structure of the small cerebral arteries in hypertension . Am J Pathol 1941. ; 17 : 39 – 46 [PMC free article] [PubMed] [Google Scholar]
- 58. Kril JJ, Patel S, Harding AJ , et al. . Patients with vascular dementia due to microvascular pathology have significant hippocampal neuronal loss . J Neurol Neurosurg Psychiatry 2002. ; 72 : 747 – 51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Crary JF, Trojanowski JQ, Schneider JA , et al. . Primary age-related tauopathy (PART): A common pathology associated with human aging . Acta Neuropathol 2014. ; 128 : 755 – 66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Noda K, Sasaki K, Fujimi K , et al. . Quantitative analysis of neurofibrillary pathology in a general population to reappraise neuropathological criteria for senile dementia of the neurofibrillary tangle type (tangle-only dementia): The Hisayama Study . Neuropathology 2006. ; 26 : 508 – 18 [DOI] [PubMed] [Google Scholar]
- 61. Kovacs GG, Milenkovic I, Wohrer A , et al. . Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: A community-based autopsy series . Acta Neuropathol 2013. ; 126 : 365 – 84 [DOI] [PubMed] [Google Scholar]
- 62. Knopman DS, Roberts RO. Estimating the number of persons with frontotemporal lobar degeneration in the US population . J Mol Neurosci 2011. ; 45 : 330 – 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Snyder HM, Corriveau RA, Craft S , et al. . Vascular contributions to cognitive impairment and dementia including Alzheimer's disease . Alzheimers Dement 2015. ; 11 : 710 – 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chou SH, Shulman JM, Keenan BT , et al. . Genetic susceptibility for ischemic infarction and arteriolosclerosis based on neuropathologic evaluations . Cerebrovasc Dis 2013. ; 36 : 181 – 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jones DS, Greene JA. Is Dementia in decline? Historical trends and future trajectories . N Engl J Med 2016. ; 374 : 507 – 9 [DOI] [PubMed] [Google Scholar]
- 66. Satizabal CL, Beiser AS, Chouraki V , et al. . Incidence of dementia over three decades in the Framingham heart study . N Engl J Med 2016. ; 374 : 523 – 32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mizutani T, Shimada H. Neuropathological background of twenty-seven centenarian brains . J Neurol Sci 1992. ; 108 : 168 – 77 [DOI] [PubMed] [Google Scholar]
- 68. Petrovitch H, Ross GW, Steinhorn SC , et al. . AD lesions and infarcts in demented and non-demented Japanese-American men . Ann Neurol 2005. ; 57 : 98 – 103 [DOI] [PubMed] [Google Scholar]
- 69. Wilson RS, Leurgans SE, Boyle PA , et al. . Neurodegenerative basis of age-related cognitive decline . Neurology 2010. ; 75 : 1070 – 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Beach TG, Sue LI, Walker DG , et al. . The sun health research institute brain donation program: Description and experience, 1987-2007 . Cell Tissue Bank 2008. ; 9 : 229 – 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sonnen JA, Larson EB, Crane PK , et al. . Pathological correlates of dementia in a longitudinal, population-based sample of aging . Ann Neurol 2007. ; 62 : 406 – 13 [DOI] [PubMed] [Google Scholar]
- 72. Riley KP, Snowdon DA, Markesbery WR. Alzheimer's neurofibrillary pathology and the spectrum of cognitive function: Findings from the Nun Study . Ann Neurol 2002. ; 51 : 567 – 77 [DOI] [PubMed] [Google Scholar]
- 73. O'Brien RJ, Resnick SM, Zonderman AB , et al. . Neuropathologic studies of the baltimore longitudinal study of Aging (BLSA) . J Alzheimers Dis 2009. ; 18 : 665 – 75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jellinger KA, Attems J. Prevalence and pathology of vascular dementia in the oldest-old . J Alzheimers Dis 2010. ; 21 : 1283 – 93 [DOI] [PubMed] [Google Scholar]
- 75. Nagy Z, Hindley NJ, Braak H , et al. . The progression of Alzheimer's disease from limbic regions to the neocortex: Clinical, radiological and pathological relationships . Dement Geriatr Cogn Disord 1999. ; 10 : 115 – 20 [DOI] [PubMed] [Google Scholar]
- 76. Knopman DS, Parisi JE, Salviati A , et al. . Neuropathology of cognitively normal elderly . J Neuropathol Exp Neurol 2003. ; 62 : 1087 – 95 [DOI] [PubMed] [Google Scholar]
- 77. Thal DR, Del Tredici K, Braak H. Neurodegeneration in normal brain aging and disease . Sci Aging Knowledge Environ 2004. ; 2004 : pe26. [DOI] [PubMed] [Google Scholar]
- 78. Del Tredici K, Braak H. Neurofibrillary changes of the Alzheimer type in very elderly individuals: Neither inevitable nor benign: Commentary on “No disease in the brain of a 115-year-old woman” . Neurobiol Aging 2008. ; 29 : 1133 – 6 [DOI] [PubMed] [Google Scholar]
- 79. Jellinger KA, Attems J. Prevalence of dementia disorders in the oldest-old: An autopsy study . Acta Neuropathol 2010. ; 119 : 421 – 33 [DOI] [PubMed] [Google Scholar]
- 80. Gold G, Bouras C, Kovari E , et al. . Clinical validity of Braak neuropathological staging in the oldest-old . Acta Neuropathol 2000. ; 99 : 579 – 82 discussion 83-4 [DOI] [PubMed] [Google Scholar]
- 81. Savva GM, Wharton SB, Ince PG , et al. . Age, neuropathology, and dementia . N Engl J Med 2009. ; 360 : 2302 – 9 [DOI] [PubMed] [Google Scholar]
- 82. Giannakopoulos P, Gold G, Kovari E , et al. . Assessing the cognitive impact of Alzheimer disease pathology and vascular burden in the aging brain: The Geneva experience . Acta Neuropathol 2007. ; 113 : 1 – 12 [DOI] [PubMed] [Google Scholar]
- 83. Prohovnik I, Perl DP, Davis KL , et al. . Dissociation of neuropathology from severity of dementia in late-onset Alzheimer disease . Neurology 2006. ; 66 : 49 – 55 [DOI] [PubMed] [Google Scholar]
- 84. Xuereb JH, Brayne C, Dufouil C , et al. . Neuropathological findings in the very old. Results from the first 101 brains of a population-based longitudinal study of dementing disorders . Ann N Y Acad Sci 2000. ; 903 : 490 – 6 [DOI] [PubMed] [Google Scholar]
- 85. Gold G, Giannakopoulos P, Herrmann FR , et al. . Identification of Alzheimer and vascular lesion thresholds for mixed dementia . Brain 2007. ; 130 : 2830 – 6 [DOI] [PubMed] [Google Scholar]
- 86. Knopman DS, Rocca WA, Cha RH , et al. . Survival study of vascular dementia in Rochester, Minnesota . Archives of Neurology 2003. ; 60 : 85 – 90 [DOI] [PubMed] [Google Scholar]
- 87. Braak H, Thal DR, Ghebremedhin E , et al. . Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years . J Neuropathol Exp Neurol 2011. ; 70 : 960 – 9 [DOI] [PubMed] [Google Scholar]
- 88. Jack CR, Jr , PART, SNAP . Acta Neuropathol 2014. ; 128 : 773 – 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Thal DR, Capetillo-Zarate E, Del Tredici K , et al. . The development of amyloid beta protein deposits in the aged brain . Sci Aging Knowledge Environ 2006. ; 2006 : re1. [DOI] [PubMed] [Google Scholar]
- 90. Mirra SS. The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer's disease: A commentary . Neurobiol Aging 1997. ; 18 : S91 – 4 [DOI] [PubMed] [Google Scholar]
- 91. Braak H, Braak E, Bohl J. Staging of Alzheimer-related cortical destruction . Eur Neurol 1993. ; 33 : 403 – 8 [DOI] [PubMed] [Google Scholar]
- 92. Lippa CF, Duda JE, Grossman M , et al. . DLB and PDD boundary issues: Diagnosis, treatment, molecular pathology, and biomarkers . Neurology 2007. ; 68 : 812 – 9 [DOI] [PubMed] [Google Scholar]
- 93. Taniguchi-Watanabe S, Arai T, Kametani F , et al. . Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau . Acta Neuropathol 2016. ; 131 : 267 – 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bech S, Hjermind LE, Salvesen L , et al. . Amyloid-related biomarkers and axonal damage proteins in parkinsonian syndromes . Parkinsonism Relat Disord 2012. ; 18 : 69 – 72 [DOI] [PubMed] [Google Scholar]
- 95. Nogalska A, D'Agostino C, Engel WK , et al. . Novel demonstration of amyloid-beta oligomers in sporadic inclusion-body myositis muscle fibers . Acta Neuropathol 2010. ; 120 : 661 – 6 [DOI] [PubMed] [Google Scholar]
- 96. Van Broeckhoven C, Haan J, Bakker E , et al. . Amyloid beta protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch) . Science 1990. ; 248 : 1120 – 2 [DOI] [PubMed] [Google Scholar]
- 97. Weihl CC, Temiz P, Miller SE , et al. . TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia . J Neurol Neurosurg Psychiatry 2008. ; 79 : 1186 – 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Tan RH, Kril JJ, Fatima M , et al. . TDP-43 proteinopathies: Pathological identification of brain regions differentiating clinical phenotypes . Brain 2015. ; 138 : 3110 – 22 [DOI] [PubMed] [Google Scholar]
- 99. Duyckaerts C, Braak H, Brion JP , et al. . PART is part of Alzheimer disease . Acta Neuropathol 2015. ; 129 : 749 – 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Braak H, Del Tredici K. Are cases with tau pathology occurring in the absence of Abeta deposits part of the AD-related pathological process? Acta Neuropathol 2014. ; 128 : 767 – 72 [DOI] [PubMed] [Google Scholar]
- 101. Santacruz KS, Sonnen JA, Pezhouh MK , et al. . Alzheimer disease pathology in subjects without dementia in 2 studies of aging: The nun study and the adult changes in thought study . J Neuropathol Exp Neurol 2011. ; 70 : 832 – 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Bouras C, Hof PR, Morrison JH. Neurofibrillary tangle densities in the hippocampal formation in a non-demented population define subgroups of patients with differential early pathologic changes . Neurosci Lett 1993. ; 153 : 131 – 5 [DOI] [PubMed] [Google Scholar]
- 103. Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories . Neurobiol Aging 1997. ; 18 : 351 – 7 [DOI] [PubMed] [Google Scholar]
- 104. Mungas D, Tractenberg R, Schneider JA , et al. . A 2-process model for neuropathology of Alzheimer's disease . Neurobiol Aging 2014. ; 35 : 301 – 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Santa-Maria I, Haggiagi A, Liu X , et al. . The MAPT H1 haplotype is associated with tangle-predominant dementia . Acta Neuropathol 2012. ; 124 : 693 – 704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Jellinger KA, Attems J. Neurofibrillary tangle-predominant dementia: Comparison with classical Alzheimer disease . Acta Neuropathol 2007. ; 113 : 107 – 17 [DOI] [PubMed] [Google Scholar]
- 107. Jellinger KA, Alafuzoff I, Attems J , et al. . PART, a distinct tauopathy, different from classical sporadic Alzheimer disease . Acta Neuropathol 2015. ; 129 : 757 – 62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bonifati V, Joosse M, Nicholl DJ , et al. . The tau gene in progressive supranuclear palsy: Exclusion of mutations in coding exons and exon 10 splice sites, and identification of a new intronic variant of the disease-associated H1 haplotype in Italian cases . Neurosci Lett 1999. ; 274 : 61 – 5 [DOI] [PubMed] [Google Scholar]
- 109. Pastor P, Ezquerra M, Tolosa E , et al. . Further extension of the H1 haplotype associated with progressive supranuclear palsy . Mov Disord 2002. ; 17 : 550 – 6 [DOI] [PubMed] [Google Scholar]
- 110. Knopman DS, Caselli RJ. Appraisal of cognition in preclinical Alzheimer's disease: A conceptual review . Neurodegener Dis Manag 2012. ; 2 : 183 – 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Vos SJ, Xiong C, Visser PJ , et al. . Preclinical Alzheimer's disease and its outcome: A longitudinal cohort study . Lancet Neurol 2013. ; 12 : 957 – 65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wirth M, Villeneuve S, Haase CM , et al. . Associations between Alzheimer disease biomarkers, neurodegeneration, and cognition in cognitively normal older people . JAMA Neurol 2013. ; 70 : 1512 – 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kryscio RJ, Abner EL, Jicha GA , et al. . Self-reported memory complaints: A comparison of demented and unimpaired outcomes . J Prev Alzheimers Dis 2016. ; 3 : 13 – 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Abner EL, Kryscio RJ, Caban-Holt AM , et al. . Baseline subjective memory complaints associate with increased risk of incident dementia: The PREADVISE trial . J Prev Alzheimers Dis 2015. ; 2 : 11 – 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kryscio RJ, Abner EL, Cooper GE , et al. . Self-reported memory complaints: Implications from a longitudinal cohort with autopsies . Neurology 2014. ; 83 : 1359 – 65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Nelson PT, Schmitt FA, Lin Y , et al. . Hippocampal sclerosis in advanced age: Clinical and pathological features . Brain 2011. ; 134 : 1506 – 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Zarow C, Sitzer TE, Chui HC. Understanding hippocampal sclerosis in the elderly: Epidemiology, characterization, and diagnostic issues . Curr Neurol Neurosci Rep 2008. ; 8 : 363 – 70 [DOI] [PubMed] [Google Scholar]
- 118. Attems J, Jellinger KA. Hippocampal sclerosis in Alzheimer disease and other dementias . Neurology 2006. ; 66 : 775. [DOI] [PubMed] [Google Scholar]
- 119. Corey-Bloom J, Sabbagh MN, Bondi MW , et al. . Hippocampal sclerosis contributes to dementia in the elderly . Neurology 1997. ; 48 : 154 – 60 [DOI] [PubMed] [Google Scholar]
- 120. Davidson YS, Raby S, Foulds PG , et al. . TDP-43 pathological changes in early onset familial and sporadic Alzheimer's disease, late onset Alzheimer's disease and Down's syndrome: association with age, hippocampal sclerosis and clinical phenotype . Acta Neuropathol 2011. ; 122 : 703 – 13 [DOI] [PubMed] [Google Scholar]
- 121. Dickson DW, Davies P, Bevona C , et al. . Hippocampal sclerosis: A common pathological feature of dementia in very old (> or = 80 years of age) humans . Acta Neuropathol 1994. ; 88 : 212 – 21 [DOI] [PubMed] [Google Scholar]
- 122. Jellinger KA. Hippocampal sclerosis: A common pathological feature of dementia in very old humans . Acta Neuropathol 1994. ; 88 : 599. [DOI] [PubMed] [Google Scholar]
- 123. Leverenz JB, Lipton AM. Clinical aspects of hippocampal sclerosis . Handb Clin Neurol 2008. ; 89 : 565 – 7 [DOI] [PubMed] [Google Scholar]
- 124. Pao WC, Dickson DW, Crook JE , et al. . Hippocampal sclerosis in the elderly: Genetic and pathologic findings, some mimicking Alzheimer disease clinically . Alzheimer Dis Assoc Disord 2011. ; 25 : 364 – 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Probst A, Taylor KI, Tolnay M. Hippocampal sclerosis dementia: A reappraisal . Acta Neuropathol 2007. ; 114 : 335 – 45 [DOI] [PubMed] [Google Scholar]
- 126. Thom M, D'Arrigo C, Scaravilli F. Hippocampal sclerosis with hypertrophy of end folium pyramidal cells . Acta Neuropathol 1999. ; 98 : 107 – 10 [DOI] [PubMed] [Google Scholar]
- 127. McKee AC, Cairns NJ, Dickson DW , et al. . The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy . Acta Neuropathol 2016. ; 131 : 75 – 86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Dutra JR, Cortes EP, Vonsattel JP. Update on hippocampal sclerosis . Curr Neurol Neurosci Rep 2015. ; 15 : 592. [DOI] [PubMed] [Google Scholar]
- 129. Hatanpaa KJ, Raisanen JM, Herndon E , et al. . Hippocampal sclerosis in dementia, epilepsy, and ischemic injury: Differential vulnerability of hippocampal subfields . J Neuropathol Exp Neurol 2014. ; 73 : 136 – 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Miller LA, Munoz DG, Finmore M. Hippocampal sclerosis and human memory . Arch Neurol 1993. ; 50 : 391 – 4 [DOI] [PubMed] [Google Scholar]
- 131. Thom M. Hippocampal sclerosis: Progress since Sommer . Brain Pathol 2009. ; 19 : 565 – 72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Yokota O, Davidson Y, Bigio EH , et al. . Phosphorylated TDP-43 pathology and hippocampal sclerosis in progressive supranuclear palsy . Acta Neuropathol 2010. ; 120 : 55 – 66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Beach TG, Sue L, Scott S , et al. . Hippocampal sclerosis dementia with tauopathy . Brain Pathol 2003. ; 13 : 263 – 78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Rauramaa T, Pikkarainen M, Englund E , et al. . Consensus recommendations on pathologic changes in the hippocampus: A postmortem multicenter inter-rater study . J Neuropathol Exp Neurol 2013. ; 72 : 452 – 61 [DOI] [PubMed] [Google Scholar]
- 135. Leverenz JB, Agustin CM, Tsuang D , et al. . Clinical and neuropathological characteristics of hippocampal sclerosis: A community-based study . Arch Neurol 2002. ; 59 : 1099 – 106 [DOI] [PubMed] [Google Scholar]
- 136. Nag S, Yu L, Capuano AW , et al. . Hippocampal sclerosis and TDP-43 pathology in aging and Alzheimer disease . Ann Neurol 2015. ; 77 : 942 – 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Bachstetter AD, Van Eldik LJ, Schmitt FA , et al. . Disease-related microglia heterogeneity in the hippocampus of Alzheimer's disease, dementia with Lewy bodies, and hippocampal sclerosis of aging . Acta Neuropathol Commun 2015. ; 3 : 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Hebert SS, Wang WX, Zhu Q , et al. . A study of small RNAs from cerebral neocortex of pathology-verified Alzheimer's disease, dementia with Lewy bodies, hippocampal sclerosis, frontotemporal lobar dementia, and non-demented human controls . J Alzheimers Dis 2013. ; 35 : 335 – 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Scheff SW, Neltner JH, Nelson PT. Is synaptic loss a unique hallmark of Alzheimer's disease? Biochem Pharmacol 2014. ; 88 : 517 – 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Amador-Ortiz C, Lin WL, Ahmed Z , et al. . TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease . Ann Neurol 2007. ; 61 : 435 – 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Neumann M, Sampathu DM, Kwong LK , et al. . Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis . Science 2006. ; 314 : 130 – 3 [DOI] [PubMed] [Google Scholar]
- 142. Lee EB, Lee VM, Trojanowski JQ , et al. . TDP-43 immunoreactivity in anoxic, ischemic and neoplastic lesions of the central nervous system . Acta Neuropathol 2008. ; 115 : 305 – 11 [DOI] [PubMed] [Google Scholar]
- 143. Wilson RS, Yu L, Trojanowski JQ , et al. . TDP-43 pathology, cognitive decline, and dementia in old age . JAMA Neurol 2013. ; 70 : 1418 – 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Dickson DW, Baker M, Rademakers R. Common variant in GRN is a genetic risk factor for hippocampal sclerosis in the elderly . Neurodegener Dis 2010. ; 7 : 170 – 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Blass DM, Hatanpaa KJ, Brandt J , et al. . Dementia in hippocampal sclerosis resembles frontotemporal dementia more than Alzheimer disease . Neurology 2004. ; 63 : 492 – 7 [DOI] [PubMed] [Google Scholar]
- 146. Amador-Ortiz C, Ahmed Z, Zehr C , et al. . Hippocampal sclerosis dementia differs from hippocampal sclerosis in frontal lobe degeneration . Acta Neuropathol 2007. ; 113 : 245 – 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Zarow C, Weiner MW, Ellis WG , et al. . Prevalence, laterality, and comorbidity of hippocampal sclerosis in an autopsy sample . Brain Behav 2012. ; 2 : 435 – 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Mortimer JA. The Nun Study: Risk factors for pathology and clinical-pathologic correlations . Curr Alzheimer Res 2012. ; 9 : 621 – 7 [DOI] [PubMed] [Google Scholar]
- 149. Bennett DA, Schneider JA, Aggarwal NT , et al. . Decision rules guiding the clinical diagnosis of Alzheimer's disease in two community-based cohort studies compared to standard practice in a clinic-based cohort study . Neuroepidemiology 2006. ; 27 : 169 – 76 [DOI] [PubMed] [Google Scholar]
- 150. Jicha GA, Abner E, Schmitt FA , et al. . Clinical features of mild cognitive impairment differ in the research and tertiary clinic settings . Dement Geriatr Cogn Disord 2008. ; 26 : 187 – 92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Nelson PT, Estus S, Abner EL , et al. . ABCC9 gene polymorphism is associated with hippocampal sclerosis of aging pathology . Acta Neuropathol 2014. ; 127 : 825 – 43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Whitwell JL, Wiste HJ, Weigand SD , et al. . Comparison of imaging biomarkers in the Alzheimer Disease Neuroimaging Initiative and the Mayo Clinic Study of Aging . Arch Neurol 2012. ; 69 : 614 – 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Jack CR, Jr, Therneau TM, Wiste HJ , et al. . Transition rates between amyloid and neurodegeneration biomarker states and to dementia: a population-based, longitudinal cohort study . Lancet Neurol 2016. ; 15 : 56 – 64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Kawas CH, Kim RC, Sonnen JA , et al. . Multiple pathologies are common and related to dementia in the oldest-old: The 90+ study . Neurology 2015. ; 85 : 535 – 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Zabar Y, Carson KA, Troncoso JC , et al. . Dementia due to hippocampal sclerosis: Clinical features and comparison to Alzheimer's disease . Neurology 1998. ; 50 : A59 – 60 [Google Scholar]
- 156. Crystal HA, Dickson DW, Sliwinski MJ , et al. . Pathological markers associated with normal aging and dementia in the elderly . Ann Neurol 1993. ; 34 : 566 – 73 [DOI] [PubMed] [Google Scholar]
- 157. White L. Brain lesions at autopsy in older Japanese-American men as related to cognitive impairment and dementia in the final years of life: A summary report from the Honolulu-Asia aging study . J Alzheimers Dis 2009. ; 18 : 713 – 25 [DOI] [PubMed] [Google Scholar]
- 158. Keage HA, Hunter S, Matthews FE , et al. . TDP-43 pathology in the population: Prevalence and associations with dementia and age . J Alzheimers Dis 2014. ; 42 : 641 – 50 [DOI] [PubMed] [Google Scholar]
- 159. Crystal HA, Dickson D, Davies P , et al. . The relative frequency of “dementia of unknown etiology” increases with age and is nearly 50% in nonagenarians . Arch Neurol 2000. ; 57 : 713 – 9 [DOI] [PubMed] [Google Scholar]
- 160. Clark AW, White CL, III , et al. . Primary degenerative dementia without Alzheimer pathology . Can J Neurol Sci 1986. ; 13 : 462 – 70 [DOI] [PubMed] [Google Scholar]
- 161. Corrada MM, Berlau DJ, Kawas CH. A population-based clinicopathological study in the oldest-old: The 90+ study . Curr Alzheimer Res 2012. ; 9 : 709 – 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Robinson JL, Geser F, Corrada MM , et al. . Neocortical and hippocampal amyloid-beta and tau measures associate with dementia in the oldest-old . Brain 2011. ; 134 : 3708 – 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Mok W, Chow TW, Zheng L , et al. . Clinicopathological concordance of dementia diagnoses by community versus tertiary care clinicians . Am J Alzheimers Dis Other Demen 2004. ; 19 : 161 – 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Weiner MW, Aisen PS, Jack CR, Jr , et al. . The Alzheimer's disease neuroimaging initiative: Progress report and future plans . Alzheimers Dement 2010. ; 6 : 202 – 11 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Toledo JB, Cairns NJ, Da X , et al. . Clinical and multimodal biomarker correlates of ADNI neuropathological findings . Acta Neuropathologica Commun 2013. ; 1 : 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Duvernoy HM. The Human Hippocampus: Functional Anatomy, Vascularization and Serial Sections With MRI . New York: : Srpinger-Verlag; 2005 [Google Scholar]
- 167. White L, Petrovitch H, Hardman J , et al. . Cerebrovascular pathology and dementia in autopsied Honolulu-Asia Aging Study participants . Ann N Y Acad Sci 2002. ; 977 : 9 – 23 [DOI] [PubMed] [Google Scholar]
- 168. Reed BR, Mungas DM, Kramer JH , et al. . Profiles of neuropsychological impairment in autopsy-defined Alzheimer's disease and cerebrovascular disease . Brain 2007. ; 130 : 731 – 9 [DOI] [PubMed] [Google Scholar]
- 169. Neltner JH, Abner EL, Baker S , et al. . Arteriolosclerosis that affects multiple brain regions is linked to hippocampal sclerosis of ageing . Brain 2014. ; 137 : 255 – 67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Aoki N, Murray ME, Ogaki K , et al. . Hippocampal sclerosis in Lewy body disease is a TDP-43 proteinopathy similar to FTLD-TDP Type A . Acta Neuropathol 2015. ; 129 : 53 – 64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Arai T, Hasegawa M, Akiyama H , et al. . TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis . Biochem Biophys Res Commun 2006. ; 351 : 602 – 11 [DOI] [PubMed] [Google Scholar]
- 172. Davidson Y, Kelley T, Mackenzie IR , et al. . Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43 . Acta Neuropathol 2007. ; 113 : 521 – 33 [DOI] [PubMed] [Google Scholar]
- 173. Rademakers R, Eriksen JL, Baker M , et al. . Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia . Hum Mol Genet 2008. ; 17 : 3631 – 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Nelson PT, Wang WX, Partch AB , et al. . Reassessment of risk genotypes (GRN, TMEM106B, and ABCC9 variants) associated with hippocampal sclerosis of aging pathology . J Neuropathol Exp Neurol 2015. ; 74 : 75 – 84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Deming Y, Cruchaga C. TMEM106B: A strong FTLD disease modifier . Acta Neuropathol 2014. ; 127 : 419 – 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Van Deerlin VM, Wood EM, Moore P , et al. . Clinical, genetic, and pathologic characteristics of patients with frontotemporal dementia and progranulin mutations . Arch Neurol 2007. ; 64 : 1148 – 53 [DOI] [PubMed] [Google Scholar]
- 177. Trojanowski JQ, Lee VM. “Fatal attractions” of proteins. A comprehensive hypothetical mechanism underlying Alzheimer's disease and other neurodegenerative disorders . Ann N Y Acad Sci 2000. ; 924 : 62 – 7 [DOI] [PubMed] [Google Scholar]
- 178. Trojanowski JQ, Goedert M, Iwatsubo T , et al. . Fatal attractions: Abnormal protein aggregation and neuron death in Parkinson's disease and Lewy body dementia . Cell Death Differ 1998. ; 5 : 832 – 7 [DOI] [PubMed] [Google Scholar]
- 179. Snyder HM, Corriveau RA, Craft S , et al. . Vascular contributions to cognitive impairment and dementia including Alzheimer's disease . Alzheimers Dement 2015. ; 11 ( 6 ): 710 – 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Montine TJ, Koroshetz WJ, Babcock D , et al. . Recommendations of the Alzheimer's disease-related dementias conference . Neurology 2014. ; 83 : 851 – 60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Weller RO, Hawkes CA, Carare RO , et al. . Does the difference between PART and Alzheimer's disease lie in the age-related changes in cerebral arteries that trigger the accumulation of Abeta and propagation of tau? Acta Neuropathol 2015. ; 129 : 763 – 6 [DOI] [PubMed] [Google Scholar]
- 182. Walker AK, Daniels CM, Goldman JE , et al. . Astrocytic TDP-43 pathology in Alexander Disease . J Neurosci 2014. ; 34 : 6448 – 58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Ling H, Holton JL, Lees AJ , et al. . TDP-43 pathology is present in most post-encephalitic parkinsonism brains . Neuropathol Appl Neurobiol 2014. ; 40 : 654 – 7 [DOI] [PubMed] [Google Scholar]
- 184. McKee AC, Gavett BE, Stern RA , et al. . TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy . J Neuropathol Exp Neurol 2010. ; 69 : 918 – 29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Sakurai A, Makioka K, Fukuda T , et al. . Accumulation of phosphorylated TDP-43 in the CNS of a patient with Cockayne syndrome . Neuropathology 2013. ; 33 : 673 – 7 [DOI] [PubMed] [Google Scholar]
- 186. Geser F, Winton MJ, Kwong LK , et al. . Pathological TDP-43 in parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam . Acta Neuropathol 2008. ; 115 : 133 – 45 [DOI] [PubMed] [Google Scholar]
- 187. Nakashima-Yasuda H, Uryu K, Robinson J , et al. . Co-morbidity of TDP-43 proteinopathy in lewy body related diseases . Acta Neuropathol 2007. ; 114 : 221 – 9 [DOI] [PubMed] [Google Scholar]
- 188. Lippa CF, Rosso AL, Stutzbach LD , et al. . Transactive response DNA-binding protein 43 burden in familial Alzheimer disease and Down syndrome . Arch Neurol 2009. ; 66 : 1483 – 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Josephs KA, Murray ME, Whitwell JL , et al. . Staging TDP-43 pathology in Alzheimer's disease . Acta Neuropathol 2014. ; 127 : 441 – 50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Josephs KA, Murray ME, Whitwell JL , et al. . Updated TDP-43 in Alzheimer's disease staging scheme . Acta Neuropathol 2016. Apr; 131 ( 4 ): 571 – 85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Cykowski MD, Takei H, Van Eldik LJ , et al. . Hippocampal sclerosis but not normal aging or Alzheimer's disease is associated with TDP-43 pathology in aged persons' basal forebrain . J Neuropathol Exp Neurol 2016. ; 75 : 397 – 407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Uchikado H, Lin WL, DeLucia MW , et al. . Alzheimer disease with amygdala lewy bodies: A distinct form of alpha-synucleinopathy . J Neuropathol Exp Neurol 2006. ; 65 : 685 – 97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Uchino A, Takao M, Hatsuta H , et al. . Incidence and extent of TDP-43 accumulation in aging human brain . Acta Neuropathol Commun 2015. ; 3 : 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Ighodaro ET, Jicha GA, Schmitt FA , et al. . Hippocampal sclerosis of aging can be segmental: Two cases and review of the literature . J Neuropathol Exp Neurol 2015. ; 74 : 642 – 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Nascimento C, Suemoto CK, Rodriguez RD , et al. . Higher Prevalence of TDP-43 proteinopathy in cognitively normal Asians: A clinicopathological study on a multiethnic sample . Brain Pathol 2016. ; 26 : 177 – 85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Yu L, De Jager PL, Yang J , et al. . The TMEM106B locus and TDP-43 pathology in older persons without FTLD . Neurology 2015. ; 84 : 927 – 34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Geser F, Robinson JL, Malunda JA , et al. . Pathological 43-kDa transactivation response DNA-binding protein in older adults with and without severe mental illness . Arch Neurol 2010. ; 67 : 1238 – 50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Dickson DW, Rademakers R, Nicholson AM , et al. . The TMEM106B locus and TDP-43 pathology in older persons without FTLD . Neurology 2015. ; 85 : 1354 – 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Amador-Ortiz C, Dickson DW. Neuropathology of hippocampal sclerosis . Handb Clin Neurol 2008. ; 89 : 569 – 72 [DOI] [PubMed] [Google Scholar]
- 200. Kotrotsou A, Schneider JA, Bennett DA , et al. . Neuropathologic correlates of regional brain volumes in a community cohort of older adults . Neurobiol Aging 2015. ; 36 : 2798 – 805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Nho K, Saykin AJ , Disease Neuroimaging Initiative Alzheimer’s , et al. . Hippocampal sclerosis of aging, a common Alzheimer's disease 'mimic': Risk genotypes are associated with brain atrophy outside the temporal lobe . J Alzheimers Dis 2016. Mar 15; 52 ( 1 ): 373 – 83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Wang Y, Mandelkow E. Tau in physiology and pathology . Nat Rev Neurosci 2016. ; 17 : 22 – 35 [DOI] [PubMed] [Google Scholar]
- 203. Lewis J, Dickson DW. Propagation of tau pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies . Acta Neuropathol 2016. ; 131 : 27 – 48 [DOI] [PubMed] [Google Scholar]
- 204. Abisambra JF, Scheff S. Brain injury in the context of tauopathies . J Alzheimers Dis 2014. ; 40 : 495 – 518 [DOI] [PubMed] [Google Scholar]
- 205. Miklossy J, Steele JC, Yu S , et al. . Enduring involvement of tau, beta-amyloid, alpha-synuclein, ubiquitin and TDP-43 pathology in the amyotrophic lateral sclerosis/parkinsonism-dementia complex of Guam (ALS/PDC) . Acta Neuropathol 2008. ; 116 : 625 – 37 [DOI] [PubMed] [Google Scholar]
- 206. Murray ME, Bieniek KF, Banks Greenberg M , et al. . Progressive amnestic dementia, hippocampal sclerosis, and mutation in C9ORF72 . Acta Neuropathol 2013. ; 126 : 545 – 54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Troncoso JC, Kawas CH, Chang CK , et al. . Lack of association of the apoE4 allele with hippocampal sclerosis dementia . Neurosci Lett 1996. ; 204 : 138 – 40 [DOI] [PubMed] [Google Scholar]
- 208. Simon-Sanchez J, Seelaar H, Bochdanovits Z , et al. . Variation at GRN 3'-UTR rs5848 is not associated with a risk of frontotemporal lobar degeneration in Dutch population . PLoS One 2009. ; 4 : e7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Vass R, Ashbridge E, Geser F , et al. . Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis . Acta Neuropathol 2011. ; 121 : 373 – 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Satoh JI, Kino Y, Kawana N , et al. . TMEM106B expression is reduced in Alzheimer's disease brains . Alzheimers Res Ther 2014. ; 6 : 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Fenoglio C, Galimberti D, Cortini F , et al. . Rs5848 variant influences GRN mRNA levels in brain and peripheral mononuclear cells in patients with Alzheimer's disease . J Alzheimers Dis 2009. ; 18 : 603 – 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Baker M, Mackenzie IR, Pickering-Brown SM , et al. . Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17 . Nature 2006. ; 442 : 916 – 9 [DOI] [PubMed] [Google Scholar]
- 213. Boeve BF, Baker M, Dickson DW , et al. . Frontotemporal dementia and parkinsonism associated with the IVS1 + 1G->A mutation in progranulin: A clinicopathologic study . Brain 2006. ; 129 : 3103 – 14 [DOI] [PubMed] [Google Scholar]
- 214. Cruts M, Gijselinck I, van der Zee J , et al. . Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21 . Nature 2006. ; 442 : 920 – 4 [DOI] [PubMed] [Google Scholar]
- 215. Cruts M, Kumar-Singh S, Van Broeckhoven C. Progranulin mutations in ubiquitin-positive frontotemporal dementia linked to chromosome 17q21 . Curr Alzheimer Res 2006. ; 3 : 485 – 91 [DOI] [PubMed] [Google Scholar]
- 216. Snowden JS, Pickering-Brown SM, Mackenzie IR , et al. . Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia . Brain 2006. ; 129 : 3091 – 102 [DOI] [PubMed] [Google Scholar]
- 217. van Blitterswijk M, Mullen B, Wojtas A , et al. . Genetic modifiers in carriers of repeat expansions in the C9ORF72 gene . Mo Neurodegener 2014. ; 9 : 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Kamalainen A, Viswanathan J, Natunen T , et al. . GRN variant rs5848 reduces plasma and brain levels of granulin in Alzheimer's disease patients . J Alzheimers Dis 2013. ; 33 : 23 – 7 [DOI] [PubMed] [Google Scholar]
- 219. Chang KH, Chen CM, Chen YC , et al. . Association between GRN rs5848 polymorphism and Parkinson's disease in Taiwanese population . PloS One 2013. ; 8 : e54448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Galimberti D, Dell'Osso B, Fenoglio C , et al. . Progranulin gene variability and plasma levels in bipolar disorder and schizophrenia . PloS One 2012. ; 7 : e32164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Pickering-Brown SM, Rollinson S, Du Plessis D , et al. . Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: Comparison with patients with MAPT and no known mutations . Brain 2008. ; 131 : 721 – 31 [DOI] [PubMed] [Google Scholar]
- 222. Rollinson S, Rohrer JD, van der Zee J , et al. . No association of PGRN 3'UTR rs5848 in frontotemporal lobar degeneration . Neurobiol Aging 2011. ; 32 : 754 – 5 [DOI] [PubMed] [Google Scholar]
- 223. Van Deerlin VM, Sleiman PM, Martinez-Lage M , et al. . Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions . Nat Genet 2010. ; 42 : 234 – 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Nicholson AM, Finch NA, Wojtas A , et al. . TMEM106B p.T185S regulates TMEM106B protein levels: implications for frontotemporal dementia . J Neurochem 2013. ; 126 : 781 – 91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Brady OA, Zheng Y, Murphy K , et al. . The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function . Hum Mol Genet 2013. ; 22 : 685 – 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Lang CM, Fellerer K, Schwenk BM , et al. . Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration . J Biol Chem 2012. ; 287 : 19355 – 65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Finch N, Carrasquillo MM, Baker M , et al. . TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers . Neurology 2011. ; 76 : 467 – 74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Wood HB. TMEM106B is a susceptibility locus for Ftld . Nat Rev Neurol 2010. ; 6 : 184. [DOI] [PubMed] [Google Scholar]
- 229. Rutherford NJ, Carrasquillo MM, Li M , et al. . TMEM106B risk variant is implicated in the pathologic presentation of Alzheimer disease . Neurology 2012. ; 79 : 717 – 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Lu RC, Wang H, Tan MS , et al. . TMEM106B and APOE polymorphisms interact to confer risk for late-onset Alzheimer's disease in Han Chinese . J Neural Transm 2014. ; 121 : 283 – 7 [DOI] [PubMed] [Google Scholar]
- 231. Beecham GW, Hamilton K, Naj AC , et al. . Genome-wide association meta-analysis of neuropathologic features of Alzheimer's disease and related dementias . PLoS Genet 2014. ; 10 : e1004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Nelson PT, Wang WX, Wilfred BR , et al. . Novel human ABCC9/SUR2 brain-expressed transcripts and an eQTL relevant to hippocampal sclerosis of aging . J Neurochem 2015. ; 134 : 1026 – 39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Nichols CG, Singh GK, Grange DK. KATP channels and cardiovascular disease: Suddenly a syndrome . Circ Res 2013. ; 112 : 1059 – 72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Shi WW, Yang Y, Shi Y , et al. . K(ATP) channel action in vascular tone regulation: From genetics to diseases . Sheng Li Xue Bao 2012. ; 64 : 1 – 13 [PMC free article] [PubMed] [Google Scholar]
- 235. Parsons MJ, Lester KJ, Barclay NL , et al. . Replication of genome-wide association studies (GWAS) loci for sleep in the British G1219 cohort . Am J Med Genet B Neuropsychiatr Genet 2013. ; 162B : 431 – 8 [DOI] [PubMed] [Google Scholar]
- 236. Nelson PT, Jicha GA, Wang WX , et al. . ABCC9/SUR2 in the brain: Implications for hippocampal sclerosis of aging and a potential therapeutic target . Ageing Res Rev 2015. ; 24 : 111 – 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Gao J, Xu D, Sabat G , et al. . Disrupting KATP channels diminishes the estrogen-mediated protection in female mutant mice during ischemia-reperfusion . Clin Proteomics 2014. ; 11 : 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Kong M, Ba M. Protective effects of diazoxide against Abeta(2)(5)(-)(3)(5)-induced PC12 cell apoptosis due to prevention of endoplasmic reticulum stress . Neuroreport 2012. ; 23 : 493 – 7 [DOI] [PubMed] [Google Scholar]
- 239. Liu D, Pitta M, Lee JH , et al. . The KATP channel activator diazoxide ameliorates amyloid-beta and tau pathologies and improves memory in the 3xTgAD mouse model of Alzheimer's disease . J Alzheimers Dis 2010. ; 22 : 443 – 57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Heurteaux C, Bertaina V, Widmann C , et al. . K+ channel openers prevent global ischemia-induced expression of c-fos, c-jun, heat shock protein, and amyloid beta-protein precursor genes and neuronal death in rat hippocampus . Proc Natl Acad Sci U S A 1993. ; 90 : 9431 – 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Liu D, Slevin JR, Lu C , et al. . Involvement of mitochondrial K+ release and cellular efflux in ischemic and apoptotic neuronal death . J Neurochem 2003. ; 86 : 966 – 79 [DOI] [PubMed] [Google Scholar]
- 242. Liu D, Lu C, Wan R , et al. . Activation of mitochondrial ATP-dependent potassium channels protects neurons against ischemia-induced death by a mechanism involving suppression of Bax translocation and cytochrome c release . J Cereb Blood Flow Metab 2002. ; 22 : 431 – 43 [DOI] [PubMed] [Google Scholar]
- 243. Goodman Y, Mattson MP. K+ channel openers protect hippocampal neurons against oxidative injury and amyloid beta-peptide toxicity . Brain Res 1996. ; 706 : 328 – 32 [DOI] [PubMed] [Google Scholar]
- 244. Macauley SL, Stanley M, Caesar EE , et al. . Hyperglycemia modulates extracellular amyloid-beta concentrations and neuronal activity in vivo . J Clin Invest 2015. ; 125 : 2463 – 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Zarei MM, Song M, Wilson RJ , et al. . Endocytic trafficking signals in KCNMB2 regulate surface expression of a large conductance voltage and Ca(2+)-activated K+ channel . Neuroscience 2007. ; 147 : 80 – 9 [DOI] [PubMed] [Google Scholar]
- 246. Ezquerra M, Pastor P, Gaig C , et al. . Different MAPT haplotypes are associated with Parkinson's disease and progressive supranuclear palsy . Neurobiol Aging 2011. ; 32 : 547 e11 – 6 [DOI] [PubMed] [Google Scholar]
- 247. Myers AJ, Pittman AM, Zhao AS , et al. . The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts . Neurobiol Dis 2007. ; 25 : 561 – 70 [DOI] [PubMed] [Google Scholar]
- 248. Cairns NJ, Bigio EH, Mackenzie IR , et al. . Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the consortium for frontotemporal lobar degeneration . Acta Neuropathol 2007. ; 114 : 5 – 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Kaivorinne AL, Kruger J, Kuivaniemi K , et al. . Role of MAPT mutations and haplotype in frontotemporal lobar degeneration in Northern Finland . BMC Neurol 2008. ; 8 : 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Goedert M, Spillantini MG. Pathogenesis of the tauopathies . J Mol Neurosci 2011. ; 45 : 425 – 31 [DOI] [PubMed] [Google Scholar]
- 251. Esiri MM, Wilcock GK, Morris JH. Neuropathological assessment of the lesions of significance in vascular dementia (Comparative Study) . J Neurol Neurosurg Psychiatry 1997. ; 63 : 749 – 53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Back SA, Kroenke CD, Sherman LS , et al. . White matter lesions defined by diffusion tensor imaging in older adults . Ann Neurol 2011. ; 70 : 465 – 76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Jellinger KA. The pathology of “vascular dementia”: A critical update . J Alzheimers Dis 2008. ; 14 : 107 – 23 [DOI] [PubMed] [Google Scholar]
- 254. Smith EE, Schneider JA, Wardlaw JM , et al. . Cerebral microinfarcts: The invisible lesions . Lancet Neurol 2012. ; 11 : 272 – 82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Ighodaro ET, Abner EL, Fardo DW , et al. . Risk factors and global cognitive status related to brain arteriolosclerosis in elderly individuals . J Cereb Blood Flow Metab 2016; doi: 10.1177/0271678X15621574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Fishbein MC, Fishbein GA. Arteriosclerosis: Facts and fancy . Cardiovasc Pathol 2015. ; 24 : 335 – 42 [DOI] [PubMed] [Google Scholar]
- 257. Vinters HV, Ellis WG, Zarow C , et al. . Neuropathologic substrates of ischemic vascular dementia . J Neuropathol Exp Neurol 2000. ; 59 : 931 – 45 [DOI] [PubMed] [Google Scholar]
- 258. Smallwood A, Oulhaj A, Joachim C , et al. . Cerebral subcortical small vessel disease and its relation to cognition in elderly subjects: A pathological study in the oxford project to investigate memory and ageing (OPTIMA) cohort . Neuropathol Appl Neurobiol 2012. ; 38 : 337 – 43 [DOI] [PubMed] [Google Scholar]
- 259. Buchman AS, Yu L, Boyle PA , et al. . Microvascular brain pathology and late-life motor impairment . Neurology 2013. ; 80 : 712 – 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Bridges LR, Andoh J, Lawrence AJ , et al. . Blood-brain barrier dysfunction and cerebral small vessel disease (arteriolosclerosis) in brains of older people . J Neuropathol Exp Neurol 2014. ; 73 : 1026 – 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Toledo JB, Arnold SE, Raible K , et al. . Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the national Alzheimer's coordinating centre . Brain 2013. ; 136 : 2697 – 706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Arsene D, Ardeleanu C. Vascular convolutes in the brain: A peculiar entity . Rom J Morphol Embryol 2006. ; 47 : 37 – 41 [PubMed] [Google Scholar]
- 263. Hassler O. Vascular changes in senile brains. A micro-angiographic study . Acta Neuropathol 1965. ; 5 : 40 – 53 [DOI] [PubMed] [Google Scholar]
- 264. Rizzoni D, Porteri E, Guelfi D , et al. . Structural alterations in subcutaneous small arteries of normotensive and hypertensive patients with non-insulin-dependent diabetes mellitus . Circulation 2001. ; 103 : 1238 – 44 [DOI] [PubMed] [Google Scholar]
- 265. Schofield I, Malik R, Izzard A , et al. . Vascular structural and functional changes in type 2 diabetes mellitus: Evidence for the roles of abnormal myogenic responsiveness and dyslipidemia . Circulation 2002. ; 106 : 3037 – 43 [DOI] [PubMed] [Google Scholar]
- 266. Moritz AR, Oldt MR. Arteriolar sclerosis in hypertensive and non-hypertensive individuals . Am J Pathol 1937. ; 13 : 679 – 728.7 [PMC free article] [PubMed] [Google Scholar]
- 267. Fishbein GA, Fishbein MC. Arteriosclerosis: Rethinking the current classification . Arch Pathol Lab Med 2009. ; 133 : 1309 – 16 [DOI] [PubMed] [Google Scholar]
- 268. Beekly DL, Ramos EM, Lee WW , et al. . The national Alzheimer's coordinating center (NACC) database: The uniform data set . Alzheimer Dis Assoc Disord 2007. ; 21 : 249 – 58 [DOI] [PubMed] [Google Scholar]
- 269. Lim AS, Yu L, Schneider JA , et al. . Sleep fragmentation, cerebral arteriolosclerosis, and brain infarct pathology in community-dwelling older people . Stroke 2016. ; 47 : 516 – 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Allebrandt KV, Amin N, Muller-Myhsok B , et al. . A K(ATP) channel gene effect on sleep duration: From genome-wide association studies to function in Drosophila . Mol Psychiatry 2013. ; 18 : 122 – 32 [DOI] [PubMed] [Google Scholar]
- 271. Scheinfeldt LB, Gharani N, Kasper RS , et al. . Using the coriell personalized medicine collaborative data to conduct a genome-wide association study of sleep duration . Am J Med Genet B Neuropsychiatr Genet 2015. ; 168 : 697 – 705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Edwards G, Weston AH. Pharmacology of the potassium channel openers . Cardiovasc Drugs Ther 1995. ; 9 (Suppl 2) : 185 – 93 [DOI] [PubMed] [Google Scholar]
- 273. Horinaka S. Use of nicorandil in cardiovascular disease and its optimization . Drugs 2011. ; 71 : 1105 – 19 [DOI] [PubMed] [Google Scholar]
- 274. Lawson K. Potassium channel openers as potential therapeutic weapons in ion channel disease . Kidney Int 2000. ; 57 : 838 – 45 [DOI] [PubMed] [Google Scholar]
- 275. Gribble FM, Reimann F. Pharmacological modulation of K(ATP) channels . Biochem Soc Trans 2002. ; 30 : 333 – 9 [DOI] [PubMed] [Google Scholar]
- 276. Johnson VE, Stewart W, Trojanowski JQ , et al. . Acute and chronically increased immunoreactivity to phosphorylation-independent but not pathological TDP-43 after a single traumatic brain injury in humans . Acta Neuropathol 2011. ; 122 : 715 – 26 [DOI] [PMC free article] [PubMed] [Google Scholar]