

Abstract
The catalytic properties of metallophthalocyanine (MPc) complexes have long been applied to electrochemical sensing of nitric oxide (NO) to amplify sensitivity and reduce the substantial overpotential required for NO oxidation. The latter point has significant ramifications for in situ amperometric detection, as large working potentials oxidize biological interferents (e.g., nitrite, L-ascorbate, and carbon monoxide). Herein, we sought to isolate and quantify, for the first time, the selectivity benefits of MPc modification of glassy carbon electrodes. A series of the most catalytically active MPc complexes towards NO, including Fe(II)Pc, Co(II)Pc, Ni(II)Pc, and Zn(II)Pc, was selected and probed for NO sensing ability under both differential pulse voltammetry (DPV) and constant potential amperometry (CPA). Data from DPV measurements provided information with respect to MPc signal sensitivity amplification (~1.5×) and peak shifting (100-200 mV). Iron-Pc exerted the most specific catalytic activity towards NO over nitrite. Catalyst-enabled reduction of the working potential under CPA was found to improve selectivity for NO over high potential interferents, regardless of MPc. However, impaired selectivity against low potential interferents was also noted.
Keywords: nitric oxide, selective detection, electrocatalyst, metallophthalocyanine, iron-phthalocyanine
TOC image
1. Introduction
Nitric oxide (NO) is an endogenous free radical implicated in a number of essential physiological processes, including inflammation [1], wound healing [2], vasodilation [3], and neurotransmission [4]. Despite a growing understanding of NO’s roles, the ways in which endogenous levels of NO naturally fluctuate and the concentration-dependence of its activity are generally still not agreed upon. Unfortunately, dynamic detection of NO is constrained by NO’s reactive nature and the presence of scavengers, greatly reducing its half-life in biological media [5–7]. Electrochemical sensors enable direct, real-time measurements of NO with fast response, high temporal resolution, low cost, and freedom from exogenous reagents [8,9].
Selectivity remains perhaps the greatest challenge to accurate electrochemical detection of NO in situ [5,10,11]. While electrochemical oxidation of NO is thermodynamically favorable, formation of the initial nitrosyl cation (NO+) is kinetically slow, requiring large overpotentials (Figure 1, adapted from reference 8) that are also capable of oxidizing interferent species present in biological milieu (e.g., nitrite, L-ascorbate, and carbon monoxide) [12]. In this manner, sensor accuracy is diminished greatly. Working electrodes must therefore be chemically modified to differentially enhance the NO signal and/or impede interferent species’ access to the electroactive surface [11]. Selective NO detection is traditionally achieved by the use of either selectively permeable (i.e., permselective) polymer membranes or electrocatalysts [13]. Permselective barriers confer NO selectivity through well-understood size-exclusion, hydrophobic interaction, or charge-repulsion sieving mechanisms—exploiting NO’s small size, lipophilicity, and neutral charge, respectively [14]. In contrast, electrocatalysts do not physically obstruct interferents; rather they enhance the NO signal and/or reduce the overpotential required for oxidation by facilitating electron transfer kinetics.
Figure 1.
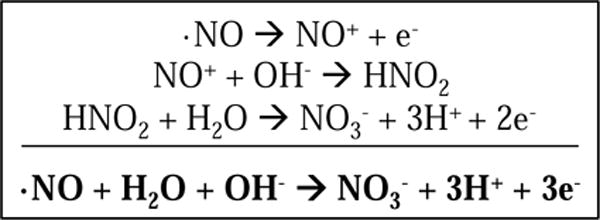
Three-step electrochemical oxidation of nitric oxide.
Metallophthalocyanines (MPc) represent the most common catalysts used in the fabrication of electrochemical NO sensors (Figure S1) [13,15]. Belonging to the same class of transition metal-coordinated macrocycles as metalloporphyrins (MP), MPcs have extended π-systems, which allow them to undergo fast redox processes and thereby facilitate electron transfer to a variety of molecules [16]. Moreover, MPc catalysts are more chemically and thermally stable than their MP counterparts [17]. Coordination of NO with an electrode surface-confined MPc complex (chemisorptive or physisorptive) facilitates initial electron transfer from NO to the MPc, which then relays that electron to the electrode charge sink, generating the current response (Figure S2). The presence of certain MPc complexes is also believed to stabilize the transient NO+ cation before further oxidation to nitrate [16,18]. It is through these stabilizing effects that the NO signal is amplified and voltammetric features are shifted to lower potentials.
The identity of the transition metal (M) has been reported to influence the site of NO coordination [19], bond orientation [20], electronic structure [21], and oxidation potential [12]. While separate studies have come to different conclusions as to the most catalytically active MPc complex towards NO [12,18,22], no study has yet evaluated the concomitant effects on sensor selectivity. For instance, an electrocatalyst that enhances both the NO and interferent signals will not bear any improvement on sensor selectivity. Signal enhancement from such a catalyst is more likely the result of increased electroactive surface area rather than targeted ligation of NO. This hypothetical catalyst is significant in light of the fact that several MPc complexes do not specifically bind NO at the metal core [19]. For example, nickel-Pc is the most routinely used MPc for NO sensor fabrication due to its substantial NO signal sensitivity amplification, yet several studies have reiterated that the oxidation is not “metal-based” (i.e., mediated through the metal core) [13,18,22,23]. First-principles density functional theory (DFT) calculations demonstrate that nickel- and zinc-Pc only weakly physisorb NO at the metal core, preferring instead to hybridize with the C atoms on the macrocycle’s periphery (Figure S1) [19,20]. Clearly, it is important to determine if these non-metal-based interactions actually furnish selectivity benefits to the detection of NO.
The findings above motivated us to systematically evaluate the selectivity benefits of MPc complexes in isolation (i.e., in the absence of a co-deposited permselective membrane). Based on independent studies carried out by Caro et al. and Vilakazi et al., iron-phthalocyanine (FePc), cobalt-phthalocyanine (CoPc), nickel-phthalocyanine (NiPc), and zinc-phthalocyanine (ZnPc) were identified as the most catalytically active MPc complexes [18,22]. These catalysts were drop-cast on glassy carbon (GC) electrode surfaces and evaluated for selectivity against: nitrite (NO2−), an oxidative product of NO oxidized at similarly high potentials; L-ascorbate (AA), a biologically ubiquitous, redox-active molecule; and carbon monoxide (CO), a molecule of similar size and structure that has been shown to exert similar physiological effects to NO through heme-coordination (e.g., with hemoglobin) [24]. These properties are summarized in Table 1.
Table 1.
Interferents to electrochemical detection of nitric oxide.
In addition to varying the interferent and MPc metal, selectivity was also monitored as a function of electrochemical technique. One of the ways MPcs purportedly improve selectivity is by lowering the required potential for NO oxidation, though actual selectivity benefits have never been rigorously quantified [13]. Herein, NO selectivity was measured using both differential pulse voltammetry (DPV) and constant potential amperometry (CPA). DPV was used to determine shifts, if any, in the NO oxidation potential (Ea,NO) relative to the bare glassy carbon electrode. Measured Ea,NO values of the catalyst-modified electrode were then used as the working potentials for CPA measurements. Comparisons in selectivity when the working potential was set at either the Ea,NO of the bare or MPc-modified electrode allowed for the determination of whether catalyst-enabled reductions of the working potential improved selectivity.
2. Experimental
2.1. Materials and reagents
Iron (II) phthalocyanine (FePc), cobalt (II) phthalocyanine (CoPc), nickel (II) phthalocyanine (NiPc), zinc (II) phthalocyanine (ZnPc), sodium nitrite (NaNO2), and L-ascorbic acid (AA) were purchased from Sigma (St. Louis, MO). Pyridine was obtained from Fischer Scientific (Hampton, NH). Nitric oxide (99.5%), carbon monoxide (99.3%), nitrogen (N2), and argon (Ar) gases were purchased from National Welders Supply (Raleigh, NC). Chemicals and solvents were analytical-reagent grade and used as received without further purification.
Distilled water was purified to a final resistivity of 18.2 MΩ·cm and a total organic content of ≤6 ppb using a Millipore Milli-Q UV Gradient A10 System (Bedford, MA). Saturated solutions of gaseous NO (1.9 mM) and CO (0.9 mM) were prepared by purging ~20 mL of phosphate buffered saline (PBS; 10 mM, pH 7.4) on ice with Ar for 30 min to remove oxygen, followed by purging with NO or CO gas for 30 min [28]. Saturated solutions were prepared on the same day as use and stored at 4 °C between calibrations. All electrochemical experiments were carried out in 20 mL of deoxygenated PBS at room temperature (23 °C) using a CH Instruments 1030 8-channel Electrochemical Analyzer (Austin, TX). The electrode configuration consisted of glassy carbon (GC; 3 mm dia.) inlaid disc working electrodes sealed in Kel-F (6 mm total dia.; CH Instruments), a silver-silver chloride (Ag|AgCl) reference electrode (3.0 M KCl; CH Instruments), and a coiled Pt wire counter electrode. All potentials are reported versus the Ag|AgCl reference electrode.
2.2. Preparation of catalyst-modified glassy carbon electrodes
Glassy carbon disc working electrodes were sequentially polished with 1.0, 0.3, and 0.05 μm alumina slurries to achieve a mirror finish. To avoid surface cracking and loss of electrical contact, glassy carbon electrodes were wiped lightly with tissue and rinsed copiously with DI water (in place of sonication) to remove imbedded alumina. After drying under a N2 stream, catalysts were drop-cast onto the GC electrode surface using 30 μL of 1.5 mM MPc solution dissolved in pyridine. After evaporation of the pyridine in ambient (~20 min), excess catalyst not physi- or chemisorbed to the surface was removed by rinsing with ethanol and then pyridine. Catalyst-modified electrodes were dried under a N2 stream before immediate use. Adsorption of the catalyst was confirmed by X-ray electron spectroscopy (XPS). Briefly, a Kratos Axis Ultra DLD X-ray photoelectron spectrometer with a monochromatic Al Kα X-ray source (base pressure = 6 × 10−9 torr) was used to collect both survey (80 eV pass energy) and high-resolution scans (20 eV pass energy) of the signature peaks of the transition metal centers. All data were corrected to the carbon 1s peak at 284.6 eV. Plates of glassy carbon (10 mm × 30 mm × 1 mm; SPI Supplies; West Chester, PA) were used in place of disk electrodes to facilitate surface analysis.
2.3. Electrochemical measurements via differential pulse voltammetry
Bare and MPc-modified GC electrodes were swept repetitively from 0 to +1100 mV for eight scans via differential pulse voltammetry (DPV) to achieve a constant background trace before measurements. Anodic peak potentials (Ea,NO) were measured and calibration curves collected for NO via DPV with aliquot concentrations ranging from 0.475 to 47.5 μM. Peak current trended linearly with NO concentration, the slope representing the sensitivity. Similar DPV calibration plots were generated for nitrite and AA over a concentration range between 0.1 – 3.0 mM. Carbon monoxide calibrations were generated over 9 – 180 μM. Of note, signal related to CO concentration was never observed, even at the saturation limit of 0.9 mM. All DPV traces were collected by sweeping from 0 to +1100 mV with an amplitude of 50 mV, step increase of 4 mV, period of 0.5 s, pulse width of 0.2 s, and sample width of 0.0167 s. Voltammograms herein are presented in the polarographic convention.
2.4. Electrochemical measurements via constant potential amperometry
The Ea,NO peak potential measured for the bare GC electrodes under DPV was used as the working potential for constant potential amperometric (CPA) measurements (Ea,NO = +1022 mV). For MPc-modified electrodes, the working potential was set at the Ea,NO of the bare electrode with measurements repeated at the specific Ea,NO of the MPc-modified electrode for comparison. In all cases, electrodes were polarized for 10 min prior to measurement at the working potential being tested. Staircase calibration plots were collected with consecutive 0.475 μM injections of saturated NO solution. Similar plots were generated with 0.250 mM injections of nitrite and AA and 1.125 μM injections of saturated CO solution.
2.5. Calculations and statistical analysis
With the data collected from both DPV and CPA, selectivity coefficients for NO against interferent species were calculated according to Eq. 1, where Sj is the sensitivity towards interferent j, and SNO is the NO sensitivity. Of note, more negative coefficients are indicative of greater selectivity.
| (Eq. 1) |
Sensitivity amplification factors were calculated according to Eq. 2, where SNO,MPc represents the NO sensitivity of the MPc-modified electrode and SNO,bare that of the bare GC electrode.
| (Eq. 2) |
As a comparative metric for MPc-modified electrodes, theoretical selectivity coefficients were calculated according to Eq. 3, assuming perfectly selective amplification of solely the NO signal (i.e., no amplification of the interferent response), where Sj,bare is the bare GC electrode sensitivity towards interferent j.
| (Eq. 3) |
An experimental selectivity coefficient (from Eq. 1) smaller (i.e., less negative) than its corresponding perfectly selective value (Eq. 3) was thus indicative of partial non-selective catalytic behavior. Sensitivity to NO, NO oxidation potentials, sensitivity amplification factors, and selectivity coefficients are presented (either numerically or with error bars) as the mean ± the standard error of the mean. Comparisons between data sets were performed using two-tailed t-tests.
3. Results and Discussion
The outer aromatic ring of MPc macrocycles coordinates strongly with parallel carbon planes due to the overlap in π-orbitals, which facilitates functionalization of glassy carbon electrodes [29–31]. Drop-casting was selected as the method of surface modification due to its simplicity and reproducibility [16,32]. Although practically all NO sensors that incorporate MPc complexes do so in conjunction with a permselective membrane (either co-deposited or as a discrete secondary layer) [17,32–35], the GC electrodes herein were modified solely with the MPc complexes to observe their selectivity and catalytic effects in isolation [12]. Electrode surface modification was confirmed with XPS by identifying signature transition metal peaks (Figure S3). The metal/carbon ratio was calculated for relative comparisons of the extent of surface modification, though some literature suggests catalytic enhancements are not dependent on surface concentration [36].
3.1. Differential pulse voltammetry
Metallophthalocyanines are known to exert catalytic effects by shifting voltammetric features to lower overpotentials and by amplifying current responses [13]. To observe both of these effects, differential pulse voltammograms were collected in the presence of NO on bare and MPc-modified GC electrodes. Incorporation of MPc catalyst elicited a marked shift in NO’s anodic peak towards lower potentials (ΔEa,NO = 100-200 mV), confirming successful facilitation of electron transfer (Figure 2A). Although the influence of the metal center identity was not strong, the order in peak shift was ZnPc ≈ NiPc > FePc > CoPc as shown in Figure 2B. This sequence is in agreement with the ordering of catalytic activity reported by Caro et al. based on the kinetic current measured on a rotating disc electrode [18]. The second peak feature in the trace of NiPc (ca. +1050 mV) was also observed in the absence of NO and was attributed to the Ni(III)|Ni(II) couple [22,37]. In order to precisely quantify amplifications to the current response, calibration curves were generated by plotting peak anodic current versus NO concentration (Figure 2C displays an example calibration for the FePc-modified GC electrode). Representative overlaid DPV traces and their corresponding calibration curves for modified and bare GC electrodes are provide in Figure S4. Signal sensitivity amplifications with MPc-incorporation were then calculated, normalized to the bare GC performance (Figure 2D; Eq. 2). In addition to lowering the peak anodic potential, all catalysts increased sensor sensitivity towards NO by a factor of ~1.5. Although the identity of the metal center did not significantly impact the degree of amplification, the same ordering as with respect to peak shifting persisted.
Figure 2.
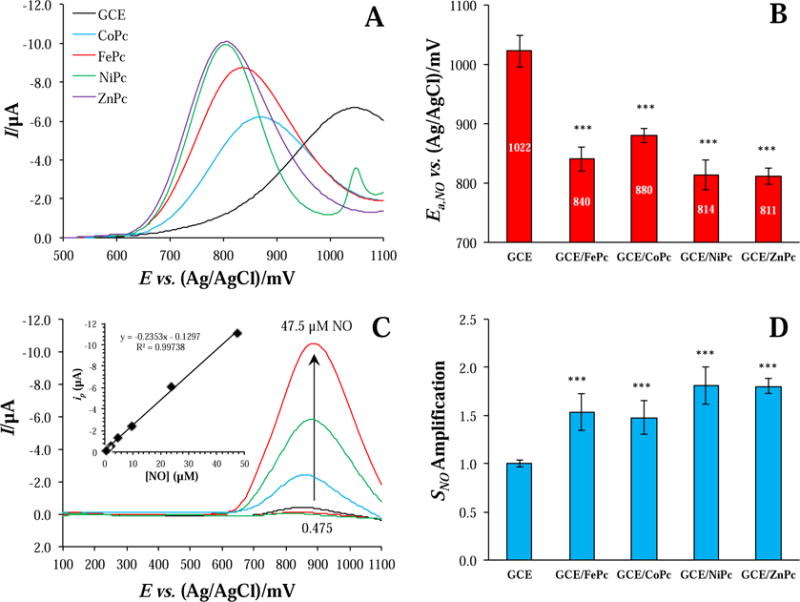
(A) Differential pulse voltammograms of bare and MPc-modified GC electrodes in the presence of 23.75 uM NO in pH 7.4 PBS with (B) corresponding peak potentials (n ≥ 3). (C) Overlay of DPV traces collected in the presence of different NO concentrations on a FePc-modified GC electrode (Inset: calibration curve from the peak currents as a function of concentration). (D) Nitric oxide sensitivity amplification of MPc-modified electrodes relative to bare GC (n ≥ 3). * = p < 0.05, ** = p < 0.01, and *** = p < 0.001 with respect to GCE.
3.2. NO selectivity under DPV
Whereas permselective barriers impart selectivity by impeding interferent diffusion to the working electrode surface, a selective electrocatalyst (ideally) enhances the signal of solely the target analyte and not interferents. In order to assess the catalytic selectivity of different MPc complexes towards NO, potential off-target signal sensitivity amplifications were studied for relevant biological interferents: nitrite, L-ascorbate, and carbon monoxide (Table 1). Experimental selectivity coefficients were calculated for both MPc-modified and bare GC electrodes as a logarithmic ratio of sensitivities (Eq. 1). As a comparative metric to the experimental selectivity coefficients, theoretical coefficients were calculated for each MPc complex assuming perfectly selective amplification of just the NO signal (Eq. 3). Any undesirable increases in the sensitivity towards an interferent will yield an experimental selectivity coefficient that is smaller (i.e., less negative) than the predicted coefficient that assumes perfectly selective sensitivity amplification.
As indicated by the discrepancies in experimental and theoretical selectivity coefficients in Figure 3A, it is clear that signal sensitivity amplifications from CoPc, NiPc, and ZnPc are not perfectly selective for NO against nitrite—most severely in the case of NiPc. Metallophthalocyanines have been shown to mediate electron transfers to/from a variety of molecules and ions, including nitrite [16,36,38]. Moreover, in the reaction to oxidize NO, nitrous acid (i.e., protonated nitrite) is formed before complete oxidation to nitrate (Figure 1). The considerable overlap in redox chemistry thus makes nitrite a potent interferent. Of all the MPc complexes investigated in this study, only FePc demonstrated a statistically insignificant difference between experimental and theoretical selectivity performance versus nitrite, indicating the most specific catalytic amplification of NO signal.
Figure 3.
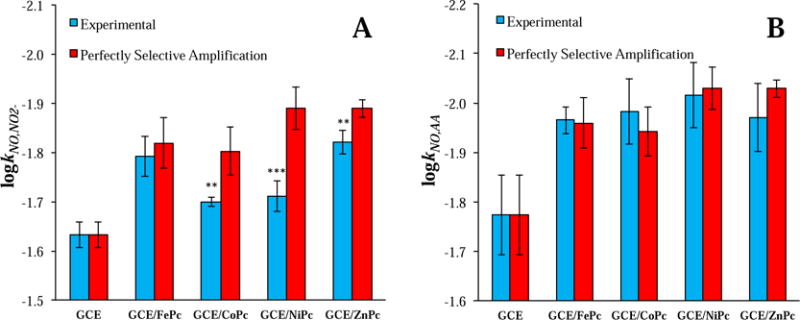
Experimental and theoretical (Eq. 3) selectivity coefficients for NO versus (A) nitrite and (B) L-ascorbate measured via DPV on MPc-modified and bare GC electrodes (n ≥ 3). Statistical information is with respect to perfectly selective amplification. * = p < 0.05, ** = p < 0.01, and *** = p < 0.001.
Density functional theory first-principles calculations of the optimized MPc-NO structures have demonstrated that NO binds strongly to FePc and CoPc at the metal center [19,20]. Iron-Pc in particular has the greatest theoretical change in adsorption energy upon hybridization, accounting for its high specificity towards NO [39,40]. Calculations via DFT carried out by Nguyen et al. have shown that MPc-NO adsorption energy decreases with d-orbital occupation, suggesting the need for unoccupied d-orbitals for proper interaction with the half-occupied π*-orbital of NO (resulting from its unpaired electron) [19]. The fact that FePc and CoPc strongly bind NO may also account for their higher NO oxidation potentials relative to NiPc and ZnPc (Figure 2B) as binding energy has been used as a reactivity descriptor to explain why Fe N4 macrocycles that strongly bind oxygen have lower catalytic activity [41]. Durable FePc-NO adduct formation may also lead to surface consumption of active electrocatalytic sites [18].
Transition metals further across the periodic table have more highly occupied d-orbitals; thus NO only weakly physisorbs to aromatic carbons along the periphery of NiPc and ZnPc macrocycles. It has also long been understood that the catalytic effects observed for NiPc are distinctly not “metal-based” [23], and lack of targeted ligation of NO may account for poorer selectivity against nitrite relative to the other MPc complexes studied herein (Figure 3A).
L-ascorbate (AA) is an equally relevant interferent capable of being oxidized at lower potentials than NO (Ea,AA = 100-300 mV). Of note, AA is present at greater concentrations than nitrite physiologically (Table 1). Comparisons between experimental and theoretical selectivity coefficients for NO against AA revealed no statistically significant difference or off-target amplification of the AA signal occurring with MPc modification (Figure 3B). The specificity of all MPc catalysts for NO over AA is attributed to AA not sharing any part of its intrinsic redox chemistry with NO (in contrast to nitrite) [37]. The lack of AA signal enhancement indicates that these MPc complexes do not amplify the NO signal through non-specific means (e.g., as with an increase in the electrode’s electroactive surface area) regardless of M-NO binding ability.
Lastly, neither the bare nor MPc-modified GC electrodes exhibited measureable activity towards CO (Table 2). Heterogeneous electrochemical oxidation of CO requires direct coordination with a transition metal electrode, such as tin or platinum [27,42]. As such, no activity was anticipated on bare GC. The continued lack of activity upon modification with the MPc catalysts suggests that either CO does not bind to the metal centers or such binding does not meaningfully facilitate electron transfer from CO to the GC electrode sink. Pre-treatment of MPc-modified electrodes in CO-spiked solution neither decreased sensitivity towards NO nor altered peak position, demonstrating that the catalytic effects towards NO are maintained even in the presence of CO.
Table 2.
Summary of nitric oxide sensitivity amplification and selectivity performance of MPc-modified and bare GC electrodes as measured by differential pulse voltammetry (n ≥ 3).
| Electrode | Ea,NO (mV) | SNO(pA/nM) | AMPca | logkNO,NO2−b | logkNO,AAb | logkNO,CO |
|---|---|---|---|---|---|---|
| GCE (bare) | 1022 ± 27 | −292 ± 10 | 1 | −1.63 ± 0.03 | −1.77 ± 0.08 | < −4 |
| GCE/FePc | 840 ± 20 | −448 ± 56 | 1.53 ± 0.19 | −1.79 ± 0.04 | −1.98 ± 0.07 | < −4 |
| GCE/CoPc | 880 ± 12 | −431 ± 52 | 1.48 ± 0.18 | −1.70 ± 0.01 | −1.97 ± 0.03 | < −4 |
| GCE/NiPc | 814 ± 25 | −527 ± 56 | 1.81 ± 0.19 | −1.71 ± 0.03 | −2.02 ± 0.07 | < −4 |
| GCE/ZnPc | 811 ± 14 | −526 ± 22 | 1.80 ± 0.08 | −1.82 ± 0.02 | −1.97 ± 0.07 | < −4 |
3.3. Constant potential amperometry
Differential pulse voltammetry measures current across a range of potentials, the output of which may be used to ascertain electrochemical properties of the redox system under observation. Potentiodynamic techniques of this kind, however, are constrained by temporal resolution and the potentiostat’s ability to apply complex waveforms. Potentiostatic techniques, such as CPA, are better suited for continuous measurement, since a constant potential is applied while sampling the current at a set rate. Though faster and simpler than potentiodynamic techniques, CPA suffers from an inherent lack of specificity [11,33]. For example, a DPV trace may show two peaks for two analytes, while CPA will return the sum of each analyte’s contribution as a single current. We thus set out to determine if and how the selectivity benefits of MPc complexes observed with DPV could be transferred to CPA, a more widely employed technique.
With the potential set to the oxidation potential of NO on bare GC (+1022 mV), current response was measured with successive injections of NO to generate staircase calibration plots for bare and MPc-modified GC electrodes (Figure 4A). Catalyst inclusion increased the sensitivity 2-3-fold with the greatest amplifications observed for FePc and NiPc (Figure 4B). Under DPV, by comparison, the MPc-derived signal increases were comparatively milder (~1.5×). The applied potential of +1022 mV under CPA represented a significant overpotential to MPc-catalyzed NO oxidation (Figure 2B). In order to determine if the resulting 2-3× signal sensitivity amplification was specific to NO, experimental and theoretical selectivity coefficients were compared (Figure 4C; Eq. 3). The large discrepancies observed across all MPcs point to off-target amplification of the nitrite signal, even in the case of FePc, the most selective catalyst under DPV. Of note, CPA and DPV experimental selectivity coefficients against nitrite were congruous (Figure 4C), revealing that only signal sensitivity amplification in excess of ~1.5× under CPA was non-specific to NO. The only exception was ZnPc, for which the NO sensitivity diminished with successive CPA trials, possibly indicative of breakdown/stripping at the continuously-maintained, high overpotential.
Figure 4.
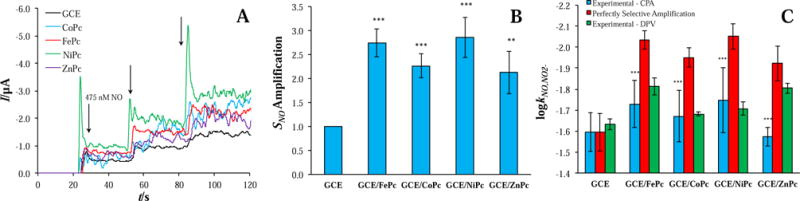
(A) Staircase amperograms of bare and MPc-modified GC electrodes with successive NO injections in pH 7.4 PBS and an applied potential of +1022 mV; (B) corresponding NO sensitivity amplifications relative to bare GC (n ≥ 3). Statistical information is with respect to GCE. (C) Experimental and theoretical (Eq. 3) selectivity coefficients for NO versus nitrite (n ≥ 3). Statistical information is with respect to perfectly selective amplification. * = p < 0.05, ** = p < 0.01, and *** = p < 0.001.
Due to the fact that the CPA current is the sum of all redox species’ current contributions, increasing the applied potential extends the range of potential interferents included in the sum. Nitric oxide and nitrite share similar redox chemistry with oxidation potentials that closely mirror each other, even in the presence of an electrocatalyst (ΔEa < 80 mV) [43]. As a result, an excessive overpotential (e.g., +1022 mV compared with the reduced Ea,NO values of MPc-modified electrodes) will amplify both signals as opposed to selectively amplifying NO. To potentially eliminate this non-specific signal sensitivity amplification, the CPA applied potential was reduced to the specific NO oxidation potential of each MPc catalyst (Figure 2B). Selectivity coefficients were then re-measured (Table 3). As anticipated, the NO sensitivity fell substantially without the large overpotential; however, selectivity versus nitrite improved slightly (except for NiPc, for which the NO signal fell precipitously). Of note, amplifications were less than 1.1 for MPc-modified electrodes, indicating that selectivity improvements against nitrite were a result of reducing the nitrite signal to a greater degree than the NO signal (as opposed to selectively amplifying the NO signal).
Table 3.
Summary of nitric oxide sensitivity amplification and selectivity of MPc-modified and bare GC electrodes measured under constant potential amperometry with different applied potentials (n ≥ 3).
| Eapplied(mV)a | Electrode | SNO(pA/nM) | AMPcb | logkNO,NO2−c | logkNO,AAb | logkNO,CO |
|---|---|---|---|---|---|---|
| 1022 | GCE (bare) | −895 ± 74 | 1 | −1.60 ± 0.09 | −1.61 ± 0.11 | < −4 |
| 1022 | GCE/FePc | −2453± 260 | 2.7± 0.3 | −1.73 ± 0.11 | −1.89 ± 0.12 | < −4 |
| 1022 | GCE/CoPc | −2026± 226 | 2.3± 0.3 | −1.67 ± 0.12 | −1.78 ± 0.07 | < −4 |
| 1022 | GCE/NiPc | −2556± 380 | 2.9± 0.4 | −1.75 ± 0.15 | −1.96 ± 0.04 | < −4 |
| 1022 | GCE/ZnPc | −1902± 398 | 2.1± 0.4 | −1.57 ± 0.05 | −1.84 ± 0.21 | < −4 |
| 840 | GCE/FePc | −1024± 66 | 1.1± 0.1 | −1.79 ± 0.18 | −1.57 ± 0.01 | < −4 |
| 880 | GCE/CoPc | −840 ± 173 | 0.9± 0.2 | −1.75 ± 0.01 | −1.41 ± 0.16 | < −4 |
| 814 | GCE/NiPc | −370 ± 151 | 0.4± 0.2 | −1.57 ± 0.12 | −1.09 ± 0.14 | < −4 |
| 811 | GCE/ZnPc | −812 ± 299 | 0.9± 0.3 | −1.73 ± 0.07 | −1.43 ± 0.10 | < −4 |
While reducing the CPA applied potential benefitted or maintained selectivity against nitrite, the same could not be said for the selectivity versus AA. Even though the applied potential was reduced to the +800-900 mV range with MPc modification, these potentials still represent vast overpotentials to AA oxidation (Ea,AA = 100-300 mV), precluding differential signal reduction over NO. As shown in Table 3, the selectivity coefficients against AA were diminished as a result. As with DPV measurements, no activity versus CO was observed, regardless of MPc modification or applied potential.
4. Conclusions
Metallophthalocyanines, and electrocatalysts more generally, exert their catalytic effects through two primary mechanisms: signal sensitivity amplification and reduction in the required overpotential. How these effects translate to NO sensor selectivity enhancements was explored using DPV and CPA. Signal sensitivity amplification (~1.5×) was perfectly specific to NO over AA for all MPc complexes studied herein, but only perfectly selective over nitrite in the case of FePc. The MPc catalysts greatly enhanced the NO signal under CPA (2-3×), but only a portion of that amplification was due to specific catalytic effects. The claim that MPc-enabled lowering of the applied potential improves selectivity is only true in the case of high potential interferents such as nitrite. The signal contributions of low potential interferents such as AA (and inferably others, including acetaminophen, dopamine, and uric acid) remain largely unaffected and continue to pose a significant challenge to selective, continuous detection of NO. Therefore, signal sensitivity amplification (as opposed to peak shifting) is the only route by which MPc complexes may meaningfully improve NO selectivity. Compared to permselective membranes capable of mitigating interferent transport >1000-fold, the suggested selectivity benefits of MPc incorporation are real, but ultimately limited.
Supplementary Material
Acknowledgments
This work was performed in part at the Chapel Hill Analytical and Nanofabrication Laboratory, CHANL, a member of the North Carolina Research Triangle Nanotechnology Network, TRNN, which is supported by the National Science Foundation, Grant ECCS-1542015, as part of the National Nanotechnology Coordinated Infrastructure, NNCI. Specific thanks is given to Carrie Donley, PhD for her assistance with XPS spectra collection and interpretation.
Funding
This work was supported by the National Institutes of Health (AI112064).
Associated Content
Supporting information associated with this work is available online. Chemical structures and surface analysis of MPc-modified glassy carbon via XPS are included (PDF).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Notes
The corresponding author declares the following competing financial interest: Mark Schoenfich is a founder of and maintains financial interest in Clinical Sensors, Inc. Clinical Sensors is an early-stage medical device company developing NO sensors for use in hospital settings.
References
- 1.Fang FC. J Clin Invest. 1997;99:2818. doi: 10.1172/JCI119473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luo J-d, Chen AF. Acta Pharmacol Sin. 2005;26:259. doi: 10.1111/j.1745-7254.2005.00058.x. [DOI] [PubMed] [Google Scholar]
- 3.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Proc Natl Acad Sci U S A. 1987;84:9265. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garthwaite J, Boulton CL. Annu Rev Physiol. 1995;57:683. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- 5.Hunter RA, Storm WL, Coneski PN, Schoenfisch MH. Anal Chem. 2013;85:1957. doi: 10.1021/ac303787p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Archer S. FASEB J. 1993;7:349. doi: 10.1096/fasebj.7.2.8440411. [DOI] [PubMed] [Google Scholar]
- 7.Hurst RD, Clark JB. Sensors. 2003;3:321. [Google Scholar]
- 8.Privett BJ, Shin JH, Schoenfisch MH. Chem Soc Rev. 2010;39:1925. doi: 10.1039/b701906h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coneski PN, Schoenfisch MH. Chem Soc Rev. 2012;41:3753. doi: 10.1039/c2cs15271a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trouillon R. Biol Chem. 2013;394:17. doi: 10.1515/hsz-2012-0245. [DOI] [PubMed] [Google Scholar]
- 11.Davies IR, Zheng X. Nitric Oxide Selective Electrodes. In: Colowick SP, Kaplan NO, editors. Methods in Enzymology. Vol. 436. Academic Press; New York: NY: 2008. pp. 63–95. [DOI] [PubMed] [Google Scholar]
- 12.Nyokong T, Vilakazi S. Talanta. 2003;61:27. doi: 10.1016/S0039-9140(03)00356-4. [DOI] [PubMed] [Google Scholar]
- 13.Bedioui F, Griveau S. Electroanalysis. 2013;25:587. [Google Scholar]
- 14.Brown MD, Schoenfisch MH. ACS Sensors. 2016;1:1453. doi: 10.1021/acssensors.6b00596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu T, Scafa N, Xu L-p, Su L, Li C, Zhou S, Liu Y, Zhang X. Electroanalysis. 2014;26:449. [Google Scholar]
- 16.Zagal JH, Griveau S, Silva JF, Nyokong T, Bedioui F. Coord Chem Rev. 2010;254:2755. [Google Scholar]
- 17.Kim IK, Chung HT, Oh GS, Bae HO, Kim SH, Chun HJ. Microchem J. 2005;80:219. [Google Scholar]
- 18.Caro CA, Zagal JH, Bedioui F. J Electrochem Soc. 2003;150:E95. [Google Scholar]
- 19.Nguyen TQ, Padama AAB, Escano MCS, Kasai H. ECS Trans. 2013;45:91. [Google Scholar]
- 20.Nguyen TQ, Escano MCS, Kasai H. J Phys Chem B. 2010;114:10017. doi: 10.1021/jp1035426. [DOI] [PubMed] [Google Scholar]
- 21.Liao M-S, Scheiner S. J Chem Phys. 2001;114:9780. [Google Scholar]
- 22.Vilakazi SL, Nyokong T. J Electroanal Chem. 2001;512:56. [Google Scholar]
- 23.Cardenas-Jiron GI, Gonzalez C, Benavides J. J Phys Chem C. 2012;116:16979. [Google Scholar]
- 24.Kim-Shapiro DB, Schechter AN, Gladwin MT. Arterioscler, Thromb, Vasc Biol. 2006;26:697. doi: 10.1161/01.ATV.0000204350.44226.9a. [DOI] [PubMed] [Google Scholar]
- 25.Pelletier MM, Kleinbongard P, Ringwood L, Hito R, Hunter CJ, Schechter AN, Gladwin MT, Dejam A. Free Radic Biol Med. 2006;41:541. doi: 10.1016/j.freeradbiomed.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 26.Reiber H, Ruff M, Uhr M. Clinica Chimica Acta. 1993;217:163. doi: 10.1016/0009-8981(93)90162-w. [DOI] [PubMed] [Google Scholar]
- 27.Lee Y, Kim J. Anal Chem. 2007;79:7669. doi: 10.1021/ac070814z. [DOI] [PubMed] [Google Scholar]
- 28.Park SS, Kim J, Lee Y. Anal Chem. 2012;84:1792. doi: 10.1021/ac2031628. [DOI] [PubMed] [Google Scholar]
- 29.Mho S-i, Ortiz B, Park S-M, Ingersoll D, Doddapaneni N. J Electrochem Soc. 1995;142:1436. [Google Scholar]
- 30.Zagal JH, Griveau S, Santander-Nelli M, Granados SG, Bedioui F. J Porphyrins Phthalocyanines. 2012;16:713. [Google Scholar]
- 31.Francisco Silva J, Griveau S, Richard C, Zagal JH, Bedioui F. Electrochem Commun. 2007;9:1629. [Google Scholar]
- 32.Raveh O, Peleg N, Bettleheim A, Silberman I, Rishpon J. Bioelectrochem Bioenerg. 1997;43:19. [Google Scholar]
- 33.Jin J, Miwa T, Mao L, Tu H, Jin L. Talanta. 1999;48:1005. doi: 10.1016/s0039-9140(98)00308-7. [DOI] [PubMed] [Google Scholar]
- 34.Bedioui F, Trevin S, Devynck J, Lantoine F, Brunet A, Devynck MA. Biosens Bioelectron. 1997;12:205. doi: 10.1016/s0956-5663(97)85338-9. [DOI] [PubMed] [Google Scholar]
- 35.Yu F, Niu G-x, Huang Y, Huang Y-f, Cheng D-s. J Fudan Univ Nat Sci. 2006;45:335. [Google Scholar]
- 36.Caro CA, Bedioui F, Zagal JH. Electrochim Acta. 2002;47:1489. [Google Scholar]
- 37.Pereira-Rodrigues N, Albin V, Koudelka-Hep M, Auger V, Pailleret A, Bedioui F. Electrochem Commun. 2002;4:922. [Google Scholar]
- 38.Nyokong T. Curr Top Electrochem. 2003;9:197. [Google Scholar]
- 39.Tran NL, Kummel AC. J Chem Phys. 2007;127:214701/1. doi: 10.1063/1.2799988. [DOI] [PubMed] [Google Scholar]
- 40.Isvoranu C, Wang B, Ataman E, Knudsen J, Schulte K, Andersen JN, Bocquet M-L, Schnadt J. J Phys Chem C. 2011;115:24718. [Google Scholar]
- 41.Zagal JH, Koper MTM. Angew Chem, Int Ed. 2016;55:14510. doi: 10.1002/anie.201604311. [DOI] [PubMed] [Google Scholar]
- 42.Gilman S. J Phys Chem. 1964;68:70. [Google Scholar]
- 43.Pallini M, Curulli A, Amine A, Palleschi G. Electroanalysis. 1998;10:1010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.