

Abstract
Amyotrophic lateral sclerosis (ALS) is a debilitating and incurable disease involving the loss of motor neurons and subsequent muscle atrophy. Genetic studies have implicated deficits in autophagy and/or mitophagy in the onset of the disease. Here we review recent progress in our understanding of the pathways for autophagy and mitophagy in neurons, and how these pathways may be affected by mutations in genes including DCTN1, OPTN, TBK1, VCP, and C9ORF72. We also discuss the implications of modulating autophagy in ALS, highlighting both the potential of the approach and the concerns raised by targeting this pathway as a therapeutic strategy in neurodegenerative disease.
INTRODUCTION
Initially described in the late 19th century by Charcot and Joffroy, our scientific understanding of the devastating neurodegenerative disease Amyotrophic lateral sclerosis (ALS) has progressed significantly, particularly through the identification of causative mutations for familial forms of the disease. Genetic studies have implicated key pathways involved in pathogenesis, including autophagy and mitophagy. However, this knowledge has not yet led to the development of effective therapeutic strategies, suggesting that there are still key gaps in our understanding of the underlying mechanisms. This review summarizes progress, investigating neuronal autophagy and mitophagy, and how that knowledge contributes to our understanding of the pathobiology of ALS.
WHAT IS ALS?
ALS is a debilitating neuromuscular disease characterized by the progressive degeneration of motor neurons in the spinal cord and brain. Degeneration of these motor neurons leads to neuromuscular denervation, atrophy of voluntary skeletal muscles, and ultimately paralysis and death. The worldwide incidence of ALS is estimated at ~5/100,000 (Chio et al., 2013). Onset is usually age-dependent, although rare genetic forms may have a juvenile onset. Global estimates indicate that incidence peaks between the ages of 50 and 75 years (Chia et al., 2018). Disease progression is usually rapid, leading to death within 3–5 years of initial onset. There is currently no cure for this fatal disease, and available therapeutic options show only limited effectiveness.
Approximately 10% of ALS cases are familial in origin, with the remainder sporadic and of unknown cause. Possible causative factors for sporadic ALS include environmental factors such as infectious disease, chronic neuroinflammation, injury or trauma, and toxin exposure. However, the major shared risk factor across both sporadic and familial forms is aging.
Progress on genetic causes of ALS has been very rapid in recent years, with ~30 different genes now implicated (Chia et al., 2018). Surprisingly, these genes do not fit easily into a single cellular pathway, although some themes do emerge. For example, a number of ALS-associated genes encode RNA-binding proteins, including TDP43 and FUS, suggesting that alterations in ribostasis may contribute to pathogenesis. Alterations in proteostasis, mitochondrial function, cytoskeletal integrity and intracellular trafficking have also been implicated (Taylor et al., 2016).
Strikingly, several of the ALS-associated genes identified to date have been functionally implicated in autophagy and/or mitophagy, specifically the clearance of protein aggregates and/or damaged mitochondria. ALS genes known to function in autophagy include OPTN, TBK1, and SQSTM1. More broadly, proteins encoded by the genes C9ORF72, VCP, CHMP2B, VAPB, ALS2, and DCTN1 have all been implicated in vesicular trafficking and may affect autophagy either directly or indirectly. A central role for autophagy in ALS is well supported by the pathology of the disease, which commonly includes the accumulation of protein aggregates and swollen or dystrophic mitochondria in motor neurons of affected patients (Taylor et al., 2016).
Here we review the two best characterized pathways for autophagy in neurons, axonal autophagy and mitophagy, and discuss the available evidence linking dysfunction in these pathways to the onset and progression of ALS. We also highlight a few of the many questions that remain, such as how mutations that are ubiquitously expressed may lead to the specific loss of motor neurons, and how cell-autonomous and cell-non-autonomous mechanisms may contribute (Box 1). We also discuss the possibilities and limitations of therapeutic interventions affecting autophagy in either neurons or their support cells.
BOX 1: Outstanding questions regarding the role of autophagy in ALS.
Is autophagy necessary for motor neurons? Data from Rudnick et al. (2017) investigated the effects of Atg7 knockdown on the structure and function of the neuromuscular junction and reported relatively minor deficits? Would more severe deficits be apparent upon longer-term knockdown?
Do mutations in genes required for autophagy and/or mitophagy cause disease through cell-autonomous or cell-non-autonomous pathways? For example, is the expression of mutant OPTN or TBK1 in motor neurons sufficient to cause disease, or does disruption glial function also contribute, as has been found for mutant SOD1 (Taylor et al., 2016)?
Are all neuronal mitochondrial subject to the same quality control mechanisms? Data from Sung et al. (2016) suggest that mitophagy is rare in vivo, and is limited to the soma. Work is still needed to determine whether mitophagy is spatially constricted or occurs throughout the neuron. In addition, the contributions of other mitochondrial quality control mechanisms in neurons remains unclear.
What are the mechanistic links between deficits in mitophagy and neuroinflammation? The genes identified as functioning within the mitophagy pathway downstream of PINK1 and Parkin include OPTN and TBK1, genes which have been previously implicated in inflammatory pathways.
To what extent do defects in autophagy/mitophagy pathways contribute to the pathogenesis of sporadic ALS? Genetic studies suggests that the pathogensis of ALS involves multiple pathways, including proteostasis, ribostasis, mitochondrial health, and neuroinflammation. To what extent these pathways contribute to sporadic ALS remains to be determined.
Will therapeutic approaches that enhance autophagy have a helpful or harmful effect on motor neuron health? Data from multiple model systems suggest that activation of autophagy may have differential effects on disease onset and progression.
AUTOPHAGY IN NEURONS
Autophagy is a critical pathway to maintain homeostasis and to respond to cellular stress. The molecular components of the autophagy pathway have been well characterized over several decades of research in model systems, such as yeast. Studies in mammalian cells indicate that the overall pathway is highly conserved. More recently, however, there has been a growing appreciation that autophagy is specifically tuned in highly differentiated cells, with distinct, tissue-specific differences apparent in regulation of autophagy, such as cellular responses to stress. For example, Mizushima et al. (2004) used a transgenic mouse model expressing the autophagosome marker GFP-LC3B to demonstrate that upon starvation autophagy is significantly upregulated in tissues such as liver, but not in brain (Mizushima et al., 2004).
Differentiated and highly polarized cells, such as neurons, exhibit spatially segregated pathways for autophagy and mitophagy (Maday and Holzbaur, 2014; Sung et al., 2016). Direct imaging of autophagy in neurons expressing GFP-labeled Atg8 (LC3B in mammals) has demonstrated a robust, constitutive pathway for autophagosome formation in the distal axon both in vitro and in vivo (Maday and Holzbaur, 2014; Maday et al., 2012; Neisch et al., 2017). Autophagosomes form in a stepwise manner in the growth cone of neurons in culture and at synaptic sites such as the neuromuscular junction in vivo (Neisch et al., 2017; Stavoe et al., 2016). Once formed, distal autophagosomes are rapidly transported toward the soma via the microtubule-based motor cytoplasmic dynein and its activator dynactin (Figure 1; (Maday et al., 2012; Neisch et al., 2017)). Of note, a G59S mutation in the dynactin subunit p150Glued, encoded by the DCTN1 gene, has been identified as the cause of the late-onset and slowly progressive motor neuron disease HMN7B (Puls et al., 2003). Thus, mutations in DCTN1 can be considered as a very rare cause of ALS (Chia et al., 2018).
FIGURE 1: Autophagy and mitophagy in the neuron.
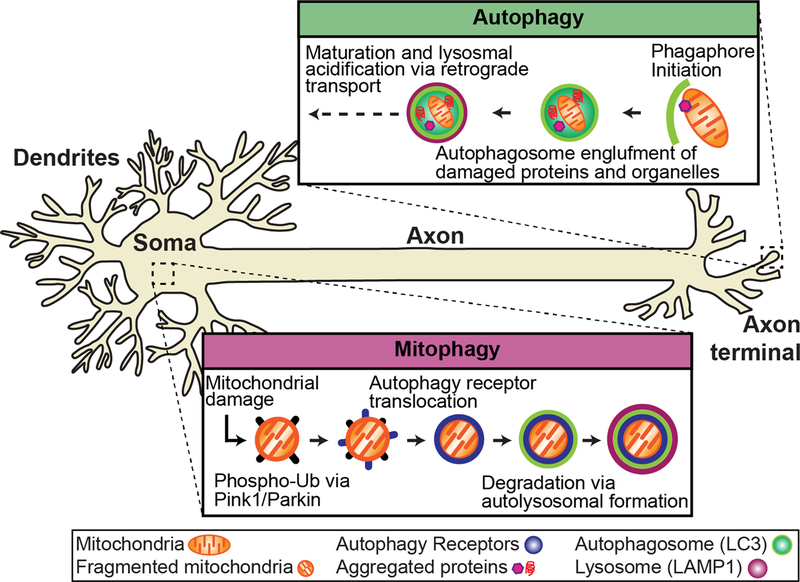
Autophagy and mitophagy are constitutive mechanisms important to in the maintenance of neuronal homeostasis that have been implicated in ALS. Autophagy occurs at the distal tip of the axon, where autophagosomes engulf damaged/aged mitochondria and protein aggregates. Through retrograde transport, the autophagosome matures and fuses with lysosomes to form a mature autolysosome that is degraded in the soma. Evidence suggests that mitophagy is spatially distinct from autophagy and occurs primarily in the soma. Damaged mitochondria are marked for degradation via phospho-ubiquitination of outer mitochondrial membrane proteins via PINK1/Parkin. Autophagy receptors (i.e. OPTN) are recruited to mitochondria and initiate the translocation of the autophagy machinery to degrade damaged mitochondria via lysosomal fusion.
Axonal autophagosomes form constitutively (Maday and Holzbaur, 2014; Maday and Holzbaur, 2016; Maday et al., 2012; Neisch et al., 2017; Stavoe et al., 2016), and cargo-loading appears to be nonselective (Maday et al., 2012). Engulfed cargos found within axonal autophagosomes include mitochondrial fragments and aggregated proteins (Figure 1), and may include synaptic vesicles under some circumstances (Maday et al., 2012; Okerlund et al., 2017; Wong and Holzbaur, 2014b). Both in vitro and in vivo, autophagosome formation is most prominent in the distal axon (Maday and Holzbaur, 2014; Maday and Holzbaur, 2016; Neisch et al., 2017; Soukup et al., 2016; Stavoe et al., 2016; Vanhauwaert et al., 2017).
During transport toward the soma, autophagosomes fuse with late endosomes and lysosomes to form a more mature, acidified organelle known as an autolysosome. In contrast to newly formed autophagosomes which are primarily found in the distal axon, autolysosomes are found along the length of the axon and accumulated within the soma (Maday and Holzbaur, 2016). The autolysosome contains degradative proteases such as cathepsins and efficiently degrades internalized cargos so that the components can be released and recycled. As effective autophagy depends on fusion of nascent autophagosomes with functional lysosomes to form a proteolytically active compartment, conditions that affect lysosomal integrity, such as mutations in the ALS-linked gene VCP (Papadopoulos et al., 2017) or extracellular deposits of β-amyloid (Gowrishankar et al., 2015) affect both lysosomal and autophagosomal processing in neurons.
ALS-LINKED GENES IMPLICATED IN AUTOPHAGY
In addition to DCTN1, which encodes an essential component of the microtubule-based motor complex that moves autophagosomes from the axon toward the soma, the protein products of multiple ALS-linked genes have been implicated in the dynamics or regulation of autophagy. These include p62/SQSTM1 (Gal et al., 2009), a ubiquitin-binding protein that associates with both protein aggregates (for example, see (Rudnick et al., 2017)) and damaged mitochondria (Wong and Holzbaur, 2014a). Optineurin/OPTN is another ubiquitin-binding protein that is also implicated in the autophagic clearance of aggregated proteins (Korac et al., 2013) and depolarized or damaged mitochondria (Wong and Holzbaur, 2014a). As noted below, both p62/SQSTM1 and OPTN are recruited to ubiquitinated substrates, and induce their sequestration by an autophagosome via their LC3-binding motifs. TBK1 was identified as an ALS-linked gene by exome sequencing, and is a kinase known to phosphorylate both p62 and OPTN (Cirulli et al., 2015). While cellular assays have clearly established roles for p62, OPTN, and TBK1 in the autophagic clearance of protein aggregates as well as mitophagy (discussed below), these three proteins have also been implicated in innate immunity pathways, and thus may cause disease by mechanisms unrelated to autophagy/mitophagy. Further work is required to establish the critical pathogenic mechanism or mechanisms involved.
Other ALS-associated genes have also been linked to autophagy, although their specific roles remain to be determined. These include VCP, implicated in the autophagy of stress granules (Buchan et al., 2013) and the maintenance of lysosomal integrity (Papadopoulos et al., 2017), and VAPB, an ER-tethering protein (Dong et al., 2016). Expansion of hexanucleotide repeats within the noncoding sequence of the C9ORF72 gene are the most frequent cause of familial ALS (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Recent data implicate the protein product of the C9ORF72 gene in the regulation of ULK1, and thus in the initiation of autophagy; however, C9ORF72 has also been proposed to function in the regulation of lysosomal fusion or function (Amick et al., 2016; Jung et al., 2017; Yang et al., 2016). However, it remains unclear if decreased C9ORF72 expression contributes to the onset of neurodegeneration, as mouse knockouts do not show a neuron-specific phenotype (O’Rourke et al., 2016); other mechanisms such as RAN translation or the formation of RNA foci may instead drive pathogenesis (Ash et al., 2013; Mori et al., 2013; Zu et al., 2011; Zu et al., 2013)
AUTOPHAGIC CLEARANCE OF PROTEIN AGGREGATES IN ALS
In vitro studies of neurons expressing the ALS-linked mutation SOD1G93A demonstrated the uptake of mutant SOD1 into axonal autophagosomes (Maday et al., 2012). Surprisingly, over-expression of the mutant protein did not result in upregulation of autophagosome formation or flux, suggesting that the axonal pathway for autophagosome biogenesis has a limited ability to respond to proteotoxic stress. Similar studies in neurons expressing mutant Huntingtin support this conclusion, as Huntingtin aggregates were readily taken up by axonal autophagosomes, but no increase in biogenesis or flux was induced by the expression of this aggregation-prone protein (Wong and Holzbaur, 2014b). This study demonstrated a detrimental effect of mutant Huntingtin on the ability of autophagosomes to degrade their cargo, suggesting that while autophagosome biogenesis is not effectively upregulated, turnover may be inhibited leading to the accumulation of autophagosomes within neurons exposed to proteotoxic stress.
Rudnick et al. (2017) examined autophagy in motor neurons in a transgenic mouse model expressing SOD1G93A, and identified the formation of large ubiquitinated protein aggregates positive for p62, the product of the ALS-associated gene SQSTM1. Both large autophagosomes positive for Atg8 homologs, ~3 μm in diameter and termed round-bodies, and skein-like inclusions were observed. The SOD1G93A transgenic mouse has a stereotypical time course of disease onset, progression and death, allowing the investigators to compare autophagosome accumulation to disease progression. Prior to frank onset of disease, round bodies were relatively abundant while the skein-like inclusions became more prevalent with disease progression. Potentially, the accumulation of the skein-like inclusions may reflect a deficit in the ability of the motor neuron to effectively clear protein aggregates via autophagy, due to either an aging-dependent decline in autophagy or the high burden of proteotoxic stress. While the formation of p62-positive aggregates was most prominent in motor neurons early in disease progression, similar structures were observed in interneurons at later time points, indicating that while the observed pathology induced by over-expression of mutant SOD1G93A preferentially affected motor neurons, it was not motor neuron-specific. Together, these studies suggest that overexpression of aggregation-prone proteins readily overwhelms the ability of the neuron to effectively clear aggregates by autophagy.
MITOPHAGY IN NEURONS
Damaged or aged mitochondria are selectively sequestered and eliminated through the autophagic process of mitophagy (Tal et al., 2007). This pathway is important for neuronal homeostasis and is most commonly linked to Parkinson’s disease, due to the discovery of two proteins (PINK1 and Parkin) linked to the familial form of the disease (Kitada et al., 1998; Valente et al., 2004; Youle and Narendra, 2011). However, many neurodegenerative diseases overlap on the subcellular level with defects in neuronal mitophagy. In the case of ALS, the accumulation of damaged or dysfunctional mitochondria are thought to be a contributing factor to the disease. Despite our knowledge of this pathway and disease links between PINK-1/Parkin and OPTN/TBK1 to Parkinson’s disease and ALS, respectively, it remains unclear as to the significance of this overlap (Cirulli et al., 2015; Freischmidt et al., 2015; Kitada et al., 1998; Maruyama and Kawakami, 2013; Valente et al., 2004).
The mitophagy pathway has been worked out in detail in immortalized cell lines, identifying a highly regulated process that involves the stepwise recruitment of multiple proteins (Lazarou et al., 2015; Moore and Holzbaur, 2016; Wong and Holzbaur, 2014a). Upon damage, PINK1 is stabilized on the mitochondrial surface which leads to the recruitment of an E3 ubiquitin ligase, Parkin (for reviews of PINK1/Parkin mitophagy see (Nguyen et al., 2016; Pickrell and Youle, 2015; Youle and Narendra, 2011)). Phospho-ubiquitination of outer mitochondrial membrane proteins initiates a feed-forward cascade that marks mitochondria for degradation (Kane et al., 2014; Kazlauskaite et al., 2014; Matsuda et al., 2010; Narendra et al., 2008; Narendra et al., 2010). Specificity for the autophagosome engulfment of damaged mitochondria relies on OPTN, an autophagy receptor, and its kinase TBK1 (Lazarou et al., 2015; Moore and Holzbaur, 2016; Wong and Holzbaur, 2014a). Translocation of OPTN to damaged mitochondria leads to LC3B recruitment and degradation via lysosomal fusion (Figure 1; (Moore and Holzbaur, 2016; Rogov et al., 2014; Wong and Holzbaur, 2014a)). It should be noted that p62 is also recruited to damage mitochondria, but is independent of OPTN and does not lead to recruitment of LC3B (Wong and Holzbaur, 2014a).
In immortalized cell lines, OPTN translocation was shown to initiate within tens of minutes of mitochondrial damage via a mitochondrial KillerRed (mt-KR) construct, where light-induced activation of mt-KR causes local damage to mitochondria vis ROS production (Bulina et al., 2006), or CCCP treatment, an inhibitor of oxidative phosphorylation; robust OPTN recruitment and stabilization was observed in ~40 minutes (Moore and Holzbaur, 2016; Wong and Holzbaur, 2014a). The use of an ALS-associated E478G ubiquitin binding-deficient mutant of OPTN significantly decreased translocation to damaged mitochondria, as the mutant failed to stably associate with the mitochondrial surface (Lazarou et al., 2015; Wong and Holzbaur, 2014a). Moreover, the use of an ALS-linked TBK1 mutant significantly reduced OPTN and LC3B recruitment to damaged mitochondria; this was also observed by silencing TBK1 or using a potent TBK1 inhibitor (Lazarou et al., 2015; Moore and Holzbaur, 2016). Therefore, loss of function of OPTN or TBK1 results in impaired mitophagy and accumulation of damaged mitochondria. Thus, inefficient turnover of mitochondria via ALS-associated mutants could be a contributing factor leading to mitochondrial dysfunction and accumulation, a prevalent feature in the motor neurons of ALS patients.
Surprisingly, substantially less is known about the regulation of mitophagy in neurons. As neurons are highly polarized and require long-range transport, it remains controversial whether distal mitochondria are subject to the same quality control mechanisms as those near the cell body. Since prompt removal of damaged mitochondria is critical for cell viability and the energy demands of neuronal mitochondria are substantial, it raises the question of how/where Pink1/Parkin-mediated mitophagy occur in neurons? Using mt-KR or global damage via antimycin a treatment, Ashrafi et. al (2014) showed that damage to axonal mitochondria triggered translocation of the mitophagy machinery within tens of minutes. In neurons lacking PINK1 or Parkin, recruitment was not visualized. It should be noted that despite the implications of PINK1 and Parkin in neurodegenerative disease, knockout mice fail to develop significant neurological defects (Whitworth and Pallanck, 2017). The authors concluded that while autophagosome biogenesis is a constitutive pathway that occurs distally in neurons, autophagosome induced formation around damaged mitochondria is a distinct pathway that can occur anywhere along the axon (Ashrafi et al., 2014).
This idea was recently called into question by studies in Drosophila which suggested that the soma is the primary focus of PINK1/Parkin-mediated mitophagy in neurons. Devireddy and colleagues showed that while loss of PINK1 reduced mitochondrial membrane potential, it failed to lead to an accumulation of mitochondria or alter mitochondrial length in axons. However, irregular mitochondrial morphology was observed in the soma (Devireddy et al., 2015). Consistent with this notion, Parkin-deficient flies had normal motility and morphology in the axons of motor neurons, but an abnormal mitochondrial network in the cell body (Sung et al., 2016). The authors argued that in motor neurons in vivo mitophagy is a rare event due to an additional quality control step that allows only healthy mitochondria to exit the soma and enter the axon.
Much of the work thus far to study neuronal mitophagy has focused on a PINK1/Parkin-mediated process. The use of chemical uncouplers has shown that mitophagy in neurons is remarkably slow compared to the time-course that was observed in immortalized cell lines. Additionally, neuronal mitochondria are much more resistant to mitophagy initiation than organelles in cell lines, which could be due to the fact that they are terminally differentiated. Thus, it is plausible that neurons have developed additional quality control mechanisms, prior to mitophagy, as preventative measures to decrease the number of stressed or aged organelles likely to enter the mitophagy pathway (Sugiura et al., 2014). For example, recent work from Lin et al. (2017) has provided new evidence to suggest that stressed neuronal mitochondria are removed from the axon via mitochondria-derived cargos and that this pathway is independent of Parkin, Drp1, and autophagy. The authors observed an alteration in mitochondrial membrane potential when synatphilin (SNPH), a mitochondrial anchoring protein, was overexpressed suggesting that axonal transport of mitochondria is required to maintain integrity and health. Additionally, stressed mitochondria release SNPH containing vesicles as a mechanism to shift from a stationary position to actively transport damaged mitochondria from the axon for degradation (Lin et al., 2017). This finding is in line with a recent report showing mature mitochondria are tethered near presynaptic terminals in cortical neurons (Lewis et al., 2016). Together these results suggest that aged mitochondria are more susceptible to damage due to their immobilization.
It is plausible that these discrepancies in mitophagy location are due to the types of treatments (i.e. global vs. local damage), as well as the severity of the chemical uncouplers (i.e. CCCP or antimycin a) used to induce mitochondrial damage. Additionally, the rates of mitophagy could be heavily influenced by in vitro versus in vivo model systems (Sung et al., 2016). While Pink1/Parkin-mediated mitophagy in immortalized cell lines appeared to be straightforward, it has become apparent that this mechanism in neurons is complex and work is still needed to fully address where and how this process occurs. For example, the interplay of mitophagy and neuroinflammation is now receiving attention, as neuroinflammation is a hallmark of ALS and increasing evidence suggests that mitophagy and neuroinflammation are linked (Komine and Yamanaka, 2015; Oakes et al., 2017).
AUTOPHAGY: FRIEND OR FOE FOR THE TREATMENT OF ALS?
Autophagy and mitophagy are considered to be essential homeostatic pathways in neurons, as genetic ablation of key autophagy genes is sufficient to induce the degeneration of neurons (Hara et al., 2006; Komatsu et al., 2006). Further, the identification of mutations in proteins thought to function within the autophagy/mitophagy pathways (SQSTM1, C9ORF72, PINK1, Parkin, OPTN, TBK1) in both Parkinson’s disease and ALS further highlights the potential importance of these cellular mechanisms.
In cellular models of ALS, there have been reports that pathology can be rescued by the induction of autophagy. For example, Marrone et al. (2018) developed iPSC lines expressing wild type or mutant FUS-eGFP, and observed the recruitment of both proteins to stress granules upon oxidative stress in both iPSCs and an induced mixed population of neuronal cells containing motor neurons. Dynamics of stress granule formation were altered in mutant FUS-eGFP iPSCs and could be rescued by treating the cells with mTOR inhibitors that can induce autophagy (Marrone et al., 2018). Of note, the drug-induced induction of autophagy in iPSC-derived neurons was much less than that observed in the parent iPSC lines, consistent with observations that mTOR inhibition does not effectively upregulate autophagy in neurons (Maday and Holzbaur, 2016).
Surprisingly, however, Rudnick et al. (2017) reported that the inhibition of autophagy induced by a targeted knockdown of Atg7 is not sufficient to induce motor neuron cell death in mice up to 150 days old, although abnormal synaptic structure and function were observed in Atg7 conditional knockout (cKO) mice. Crossing Atg7-cKO mice to mice expressing mutant SOD1 led to some counter-intuitive findings. The deficiency in autophagy induced by depletion of Atg7 led to an earlier disease onset by some but not all measures, as onset of hind limb tremor was observed 22 days earlier but there was no difference in disease-associated weight loss. However, depletion of Atg7 was found to extend lifespan by ~22%, although motor neuron survival was not affected as compared to mice expressing mutant SOD1 in the presence of wildtype levels of Atg7 (Rudnick et al., 2017).
In a parallel study, rilmenidine was administered to induce autophagy in the SOD1G93A mouse. While autophagy was upregulated, both motor neuron degeneration and symptom progression were deleteriously affected (Perera et al., 2017). Non-cell autonomous effects of autophagy modulation must also be considered (Fabbrizio et al., 2017). For example, Staats et al. (2013) found that rapamycin treatment of SOD1G93A mice did not increase survival. However, rapamycin has an immunosuppressive effect on lymphocytes. To avoid this complication, SOD1G93A mice were crossed to RAG1−/− mice to generate animals deficient in mature lymphocytes. In this line, rapamycin treatment induced a mild extension of lifespan (6.5 days). Disease onset was not affected, instead the principal effect was on disease progression (Staats et al., 2013).
Together, results to date raise concern that therapeutic approaches aimed generally at enhancing autophagy may not be uniformly beneficial for patients with ALS. More nuanced strategies may be effective, such as enhancing lysosomal function and thus counteracting the accumulation of autophagosomes or autolysosomes without sufficient degradative ability. These strategies must be built on a better understanding of the cell biology of autophagy in neurons, and how the dynamics of autophagy are affected in both familial and sporadic ALS.
CONCLUSIONS
Recent progress is beginning to map out how and when autophagy occurs in neurons, and how neurons use this pathway to counter cellular stressors such as proteotoxic stress or damaged mitochondria. Studies at the cellular level are profiting from the recent advances in the genetics of ALS and other, related neurodegenerative diseases. We are beginning to understand how disease-associated genes can be grouped into consensus pathways, allowing an improved focus on disease-causing mechanisms involved in pathogenesis. While current data clearly implicate defects in autophagy and mitophagy in familial ALS, further research is necessary to understand the molecular basis for the defects, as well as the most effective approaches to intervene (Box 1). Hopefully, the progress made in understanding familial cases will then directly inform our understanding of the predominant, sporadic forms of ALS, and lead to the design of more effective therapeutic approaches in future.
HIGHLIGHTS.
ALS-associated genes are implicated in autophagy and mitophagy
Constitutive autophagy and induced mitophagy are distinct pathways in neurons
Therapeutic targeting of autophagy in ALS has promise but also potential concerns
ACKNOWLEDGMENTS
The authors thank Phuong Nguyen for her careful reading of the manuscript, and gratefully acknowledge funding from the HHMI Hanna H. Gray Fellowship to CSE and NIH R37 NS060698 to ELFH.
REFERENCES
- Amick J, Roczniak-Ferguson A, and Ferguson SM. 2016. C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol Biol Cell 27:3040–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW 3rd, Rademakers R, Boylan KB, Dickson DW, and Petrucelli L. 2013. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77:639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Schlehe JS, LaVoie MJ, and Schwarz TL. 2014. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. The Journal of cell biology 206:655–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan JR, Kolaitis RM, Taylor JP, and Parker R. 2013. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153:1461–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulina ME, Chudakov DM, Britanova OV, Yanushevich YG, Staroverov DB, Chepurnykh TV, Merzlyak EM, Shkrob MA, Lukyanov S, and Lukyanov KA. 2006. A genetically encoded photosensitizer. Nature biotechnology 24:95–99. [DOI] [PubMed] [Google Scholar]
- Chia R, Chio A, and Traynor BJ. 2018. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol 17:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, and White LA. 2013. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 41:118–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, Wang Q, Krueger BJ, Ren Z, Keebler J, Han Y, Levy SE, Boone BE, Wimbish JR, Waite LL, Jones AL, Carulli JP, Day-Williams AG, Staropoli JF, Xin WW, Chesi A, Raphael AR, McKenna-Yasek D, Cady J, Vianney de Jong JM, Kenna KP, Smith BN, Topp S, Miller J, Gkazi A, Consortium FS, Al-Chalabi A, van den Berg LH, Veldink J, Silani V, Ticozzi N, Shaw CE, Baloh RH, Appel S, Simpson E, Lagier-Tourenne C, Pulst SM, Gibson S, Trojanowski JQ, Elman L, McCluskey L, Grossman M, Shneider NA, Chung WK, Ravits JM, Glass JD, Sims KB, Van Deerlin VM, Maniatis T, Hayes SD, Ordureau A, Swarup S, Landers J, Baas F, Allen AS, Bedlack RS, Harper JW, Gitler AD, Rouleau GA, Brown R, Harms MB, Cooper GM, Harris T, Myers RM, and Goldstein DB. 2015. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347:1436–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, and Rademakers R. 2011. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devireddy S, Liu A, Lampe T, and Hollenbeck PJ. 2015. The Organization of Mitochondrial Quality Control and Life Cycle in the Nervous System In Vivo in the Absence of PINK1. The Journal of neuroscience : the official journal of the Society for Neuroscience 35:9391–9401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong R, Saheki Y, Swarup S, Lucast L, Harper JW, and De Camilli P. 2016. Endosome-ER Contacts Control Actin Nucleation and Retromer Function through VAP-Dependent Regulation of PI4P. Cell 166:408–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrizio P, Amadio S, Apolloni S, and Volonte C. 2017. P2X7 Receptor Activation Modulates Autophagy in SOD1-G93A Mouse Microglia. Front Cell Neurosci 11:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K, Marroquin N, Nordin F, Hubers A, Weydt P, Pinto S, Press R, Millecamps S, Molko N, Bernard E, Desnuelle C, Soriani MH, Dorst J, Graf E, Nordstrom U, Feiler MS, Putz S, Boeckers TM, Meyer T, Winkler AS, Winkelman J, de Carvalho M, Thal DR, Otto M, Brannstrom T, Volk AE, Kursula P, Danzer KM, Lichtner P, Dikic I, Meitinger T, Ludolph AC, Strom TM, Andersen PM, and Weishaupt JH. 2015. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nature neuroscience 18:631–636. [DOI] [PubMed] [Google Scholar]
- Gal J, Strom AL, Kwinter DM, Kilty R, Zhang J, Shi P, Fu W, Wooten MW, and Zhu H. 2009. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J Neurochem 111:1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar S, Yuan P, Wu Y, Schrag M, Paradise S, Grutzendler J, De Camilli P, and Ferguson SM. 2015. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proceedings of the National Academy of Sciences of the United States of America 112:E3699–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, and Mizushima N. 2006. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889. [DOI] [PubMed] [Google Scholar]
- Jung J, Nayak A, Schaeffer V, Starzetz T, Kirsch AK, Muller S, Dikic I, Mittelbronn M, and Behrends C. 2017. Multiplex image-based autophagy RNAi screening identifies SMCR8 as ULK1 kinase activity and gene expression regulator. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, and Youle RJ. 2014. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. The Journal of cell biology 205:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, and Muqit MM. 2014. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J 460:127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, and Shimizu N. 1998. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, and Tanaka K. 2006. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884. [DOI] [PubMed] [Google Scholar]
- Komine O, and Yamanaka K. 2015. Neuroinflammation in motor neuron disease. Nagoya J Med Sci 77:537–549. [PMC free article] [PubMed] [Google Scholar]
- Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, Behl C, Terzic J, and Dikic I. 2013. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci 126:580–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, and Youle RJ. 2015. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524:309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TL Jr., Turi GF, Kwon SK, Losonczy A, and Polleux F. 2016. Progressive Decrease of Mitochondrial Motility during Maturation of Cortical Axons In Vitro and In Vivo. Current biology : CB 26:2602–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MY, Cheng XT, Tammineni P, Xie Y, Zhou B, Cai Q, and Sheng ZH. 2017. Releasing Syntaphilin Removes Stressed Mitochondria from Axons Independent of Mitophagy under Pathophysiological Conditions. Neuron 94:595–610 e596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, and Holzbaur EL. 2014. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell 30:71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, and Holzbaur EL. 2016. Compartment-Specific Regulation of Autophagy in Primary Neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 36:5933–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Wallace KE, and Holzbaur EL. 2012. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. The Journal of cell biology 196:407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrone L, Poser I, Casci I, Japtok J, Reinhardt P, Janosch A, Andree C, Lee HO, Moebius C, Koerner E, Reinhardt L, Cicardi ME, Hackmann K, Klink B, Poletti A, Alberti S, Bickle M, Hermann A, Pandey UB, Hyman AA, and Sterneckert JL. 2018. Isogenic FUS-eGFP iPSC Reporter Lines Enable Quantification of FUS Stress Granule Pathology that Is Rescued by Drugs Inducing Autophagy. Stem Cell Reports 10:375–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama H, and Kawakami H. 2013. Optineurin and amyotrophic lateral sclerosis. Geriatrics & gerontology international 13:528–532. [DOI] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, and Tanaka K. 2010. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. The Journal of cell biology 189:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, and Ohsumi Y. 2004. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AS, and Holzbaur EL. 2016. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proceedings of the National Academy of Sciences of the United States of America 113:E3349–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, and Edbauer D. 2013. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339:1335–1338. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, and Youle RJ. 2008. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of cell biology 183:795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, and Youle RJ. 2010. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8:e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neisch AL, Neufeld TP, and Hays TS. 2017. A STRIPAK complex mediates axonal transport of autophagosomes and dense core vesicles through PP2A regulation. The Journal of cell biology 216:441–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TN, Padman BS, and Lazarou M. 2016. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol 26:733–744. [DOI] [PubMed] [Google Scholar]
- O’Rourke JG, Bogdanik L, Yanez A, Lall D, Wolf AJ, Muhammad AK, Ho R, Carmona S, Vit JP, Zarrow J, Kim KJ, Bell S, Harms MB, Miller TM, Dangler CA, Underhill DM, Goodridge HS, Lutz CM, and Baloh RH. 2016. C9orf72 is required for proper macrophage and microglial function in mice. Science 351:1324–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes JA, Davies MC, and Collins MO. 2017. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain 10:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okerlund ND, Schneider K, Leal-Ortiz S, Montenegro-Venegas C, Kim SA, Garner LC, Waites CL, Gundelfinger ED, Reimer RJ, and Garner CC. 2017. Bassoon Controls Presynaptic Autophagy through Atg5. Neuron 93:897–913 e897. [DOI] [PubMed] [Google Scholar]
- Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, Poehler R, Dressler A, Fengler S, Arhzaouy K, Lux V, Ehrmann M, Weihl CC, and Meyer H. 2017. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. The EMBO journal 36:135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera ND, Sheean RK, Lau CL, Shin YS, Beart PM, Horne MK, and Turner BJ. 2017. Rilmenidine promotes MTOR-independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression. Autophagy:1–18. [DOI] [PMC free article] [PubMed]
- Pickrell AM, and Youle RJ. 2015. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85:257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH Jr., Ludlow CL, and Fischbeck KH. 2003. Mutant dynactin in motor neuron disease. Nat Genet 33:455–456. [DOI] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Consortium I, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, and Traynor BJ. 2011. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogov V, Dotsch V, Johansen T, and Kirkin V. 2014. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Molecular cell 53:167–178. [DOI] [PubMed] [Google Scholar]
- Rudnick ND, Griffey CJ, Guarnieri P, Gerbino V, Wang X, Piersaint JA, Tapia JC, Rich MM, and Maniatis T. 2017. Distinct roles for motor neuron autophagy early and late in the SOD1(G93A) mouse model of ALS. Proceedings of the National Academy of Sciences of the United States of America 114:E8294–E8303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukup SF, Kuenen S, Vanhauwaert R, Manetsberger J, Hernandez-Diaz S, Swerts J, Schoovaerts N, Vilain S, Gounko NV, Vints K, Geens A, De Strooper B, and Verstreken P. 2016. A LRRK2-Dependent EndophilinA Phosphoswitch Is Critical for Macroautophagy at Presynaptic Terminals. Neuron 92:829–844. [DOI] [PubMed] [Google Scholar]
- Staats KA, Hernandez S, Schonefeldt S, Bento-Abreu A, Dooley J, Van Damme P, Liston A, Robberecht W, and Van Den Bosch L. 2013. Rapamycin increases survival in ALS mice lacking mature lymphocytes. Molecular neurodegeneration 8:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavoe AK, Hill SE, Hall DH, and Colon-Ramos DA. 2016. KIF1A/UNC-104 Transports ATG-9 to Regulate Neurodevelopment and Autophagy at Synapses. Dev Cell 38:171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura A, McLelland GL, Fon EA, and McBride HM. 2014. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. The EMBO journal 33:2142–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung H, Tandarich LC, Nguyen K, and Hollenbeck PJ. 2016. Compartmentalized Regulation of Parkin-Mediated Mitochondrial Quality Control in the Drosophila Nervous System In Vivo. The Journal of neuroscience : the official journal of the Society for Neuroscience 36:7375–7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal R, Winter G, Ecker N, Klionsky DJ, and Abeliovich H. 2007. Aup1p, a yeast mitochondrial protein phosphatase homolog, is required for efficient stationary phase mitophagy and cell survival. The Journal of biological chemistry 282:5617–5624. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Brown RH Jr., and Cleveland DW. 2016. Decoding ALS: from genes to mechanism. Nature 539:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, and Wood NW. 2004. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–1160. [DOI] [PubMed] [Google Scholar]
- Vanhauwaert R, Kuenen S, Masius R, Bademosi A, Manetsberger J, Schoovaerts N, Bounti L, Gontcharenko S, Swerts J, Vilain S, Picillo M, Barone P, Munshi ST, de Vrij FM, Kushner SA, Gounko NV, Mandemakers W, Bonifati V, Meunier FA, Soukup SF, and Verstreken P. 2017. The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. The EMBO journal 36:1392–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth AJ, and Pallanck LJ. 2017. PINK1/Parkin mitophagy and neurodegeneration-what do we really know in vivo? Curr Opin Genet Dev 44:47–53. [DOI] [PubMed] [Google Scholar]
- Wong YC, and Holzbaur EL. 2014a. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proceedings of the National Academy of Sciences of the United States of America 111:E4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, and Holzbaur EL. 2014b. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. The Journal of neuroscience : the official journal of the Society for Neuroscience 34:1293–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Liang C, Swaminathan K, Herrlinger S, Lai F, Shiekhattar R, and Chen JF. 2016. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv 2:e1601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, and Narendra DP. 2011. Mechanisms of mitophagy. Nature reviews. Molecular cell biology 12:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, and Ranum LP. 2011. Non-ATG-initiated translation directed by microsatellite expansions. Proceedings of the National Academy of Sciences of the United States of America 108:260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, Ostrow LW, Rothstein JD, Troncoso JC, and Ranum LP. 2013. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proceedings of the National Academy of Sciences of the United States of America 110:E4968–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]