

Abstract
Chiral γ-aminobutyric acid (GABA) analogues represent abundantly prescribed drugs, which are broadly applied as anticonvulsants, as antidepressants, and for the treatment of neuropathic pain. Here we report a one-pot two-step biocatalytic cascade route for synthesis of the pharmaceutically relevant enantiomers of γ-nitrobutyric acids, starting from simple precursors (acetaldehyde and nitroalkenes), using a tailor-made highly enantioselective artificial “Michaelase” (4-oxalocrotonate tautomerase mutant L8Y/M45Y/F50A), an aldehyde dehydrogenase with a broad non-natural substrate scope, and a cofactor recycling system. We also report a three-step chemoenzymatic cascade route for the efficient chemical reduction of enzymatically prepared γ-nitrobutyric acids into GABA analogues in one pot, achieving high enantiopurity (e.r. up to 99:1) and high overall yields (up to 70%). This chemoenzymatic methodology offers a step-economic alternative route to important pharmaceutically active GABA analogues, and highlights the exciting opportunities available for combining chemocatalysts, natural enzymes, and designed artificial biocatalysts in multistep syntheses.
Keywords: systems biocatalysis, cascades, “Michaelase”, γ-aminobutyric acids, γ-nitrobutyric acids, enzyme engineering, pharmaceuticals
Introduction
Analogues of γ-aminobutyric acid (GABA, Figure 1) represent abundantly prescribed drugs, which are broadly applied as anticonvulsants, as antidepressants and for the treatment of neuropathic pain. With an increasing world population and life expectancy, the demand for GABA analogues is expected to even further increase. The efficient asymmetric synthesis of pharmaceutically active GABA analogues has therefore attracted enormous attention. With current synthesis routes often involving kinetic resolutions,1−3 there is a need to investigate alternative asymmetric synthesis routes that are potentially greener, more sustainable, and more step-economic. In this regard, the use of a systems (bio)catalysis approach4−8 in which different catalysts are combined to construct reaction cascades for efficient synthesis of GABA analogues is an attractive idea. This approach aims to minimize the number of reaction steps and improve the “pot-economy” of the process.9
Figure 1.

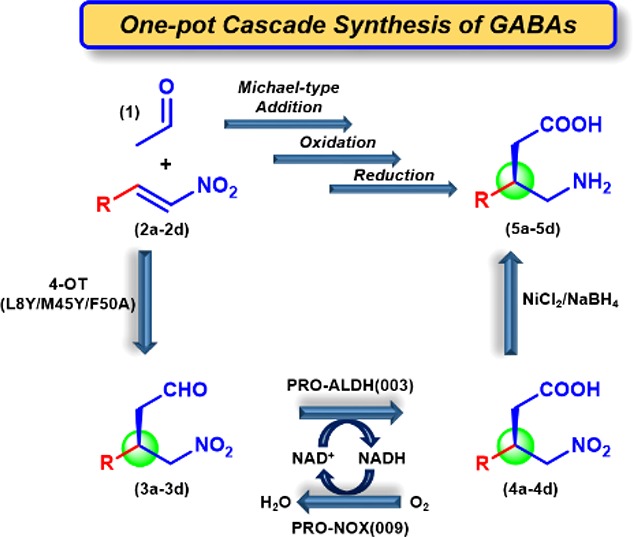
Chemoenzymatic cascade synthesis of pharmaceutically active GABA analogues. (A) Envisioned (chemo)enzymatic cascade synthesis of pharmaceutically active GABA analogues. Abbreviations: 4-OT*, newly engineered 4-OT variant that functions as a highly enantioselective artificial “Michaelase”; ALDH, aldehyde dehydrogenase; NR, nitroreductase. (B) Structures of GABA and its analogues pregabalin, phenibut, baclofen, and fluorophenibut.
We envisioned that pharmaceutically active GABA analogues, such as pregabalin, phenibut, baclofen, and fluorophenibut, could be prepared via one-pot three-step (chemo)enzymatic cascade reactions, using simple and inexpensive starting materials and avoiding (de)protecting steps and intermediate purifications (Figure 1). For establishing the required C–C bond stereochemistry, the asymmetric Michael-type addition of acetaldehyde (1) to nitroalkenes 2a–d is of high interest. This would give convenient access to chiral γ-nitroaldehydes 3a–d, which in two steps (oxidation of 3a–d into 4a–d followed by reduction of 4a–d into 5a–d) can be converted into the desired GABA analogues.
The asymmetric Michael-type addition of 1 to 2a–d is certainly not trivial. Multiple organocatalytic approaches to obtain enantioenriched γ-nitroaldehydes have been reported, mainly using small peptide- and proline-based organocatalysts.10−13 However, examples including acetaldehyde as donor substrate are scarce and a high catalyst loading in organic solvent is typically applied. Therefore, there is great interest in the development of biocatalytic procedures for the enantioselective synthesis of γ-nitroaldehydes. However, enzymes that naturally catalyze these required carbon–carbon bond-forming Michael-type additions are not known to be present in nature. In fact, only a few enzymes are known to be able to catalyze any type of C–C bond-forming Michael-type addition.14,15 Interestingly, Hilvert and co-workers published the elegant enzymatic synthesis of γ-nitroketones, but not γ-nitroaldehydes, by both acetone addition to nitroalkenes and nitroalkane addition to conjugated ketones using a highly engineered computationally designed retroaldolase.16 We have previously reported that 4-oxalocrotonate tautomerase (4-OT) can promiscuously catalyze the addition of small aldehydes, most notably the highly reactive acetaldehyde, to various aliphatic and aromatic nitroalkenes.17−20 Analogous to proline-based organocatalysts, the 4-OT enzyme utilizes an N-terminal proline (Pro-1) as key catalytic residue in promiscuous aldol condensations21,22 and Michael-type additions,19 most likely via enamine catalysis.22,23 By using mutability-landscape guided enzyme engineering,24 a mutant of 4-OT (4-OT M45Y/F50A) was generated that showed inverted enantioselectivity in acetaldehyde additions to nitroalkenes,25 allowing the enzymatic synthesis of the pharmaceutically relevant enantiomers of γ-nitroaldehydes 3a–d. However, the enantioselectivity of 4-OT M45Y/F50A is too low for biocatalytic application, providing the desired γ-nitroaldehyde products with only modest enantiomeric excess.25
Here, we report the development of a tailor-made artificial “Michaelase”, which exhibits improved enantioselectivity, activity and cosolvent stability compared to the parental enzyme 4-OT M45Y/F50A, for additions of 1 to nitroalkenes 2a–d yielding γ-nitroaldehydes 3a–d with outstanding enantiopurity. This artificial “Michaelase” was combined with a natural aldehyde dehydrogenase and a cofactor recycling NADH-oxidase to give a one-pot two-step reaction cascade for the synthesis of γ-nitrobutyric acids 4a–d in high yields and with excellent enantiomeric excess. Finally, the reaction cascade was further extended by the inclusion of nickel boride, promoting the conversion of enzymatically prepared 4a–d into the desired GABA analogues 5a–d, which resulted in a one-pot three-step chemoenzymatic reaction cascade (Figure 1A). Given that all steps were performed under aqueous conditions with high conversions, the desired GABA analogues 5a–d were obtained in good isolated yields of up to 70% and with excellent enantiomer ratio (e.r.) values of up to 99:1. This new methodology offers a step-economic alternative route to important pharmaceutically active GABA analogues, and highlights the exciting opportunities available for combining chemocatalysts, natural enzymes, and designed artificial biocatalysts in multistep syntheses of valuable chemical products.
Experimental Methods
Library Screening
After transformation with 4-OT I2X/L8X/M45Y/F50A library DNA, individual E. coli BL21(DE3) colonies were used to inoculate 2× 1.25 mL LB supplemented with 100 μg/mL ampicillin and 100 μM isopropyl β-d-1-thiogalactopyranoside (IPTG) in 96-deep well plates (Greiner Bio-one, 96-well Masterblock). The plates were sealed with sterile gas-permeable seals (Greiner Bio-one, BREATHseal) and incubated at 37 °C, with overnight shaking at 250 rpm. After the incubation, the plates were centrifuged (2232g, 8 min). The supernatant was discarded, and the individual pellets were lysed by resuspension in 350 μL of BugBuster (Novagen) supplemented with 25 U/mL benzonase (Novagen). The lysis was continued for 20 min at room temperature under vigorous shaking. The lysates were cleared by centrifugation (2232g, 55 min, 4 °C) after which the Cell Free Extract (CFE) was obtained. The final reaction mixture for monitoring the addition of 1 to 2a consisted of the following: CFE (40% v/v), 150 mM 1, 5 mM 2a, DMSO (5% v/v) in 20 mM sodium phosphate buffer pH 6.5, 500 μL final volume. The reactions were performed in 96-deep well plates sealed by ultraviolet transparent plate seals (VIEWseal, Greiner Bio-One) at room temperature. After 50 min the reaction was stopped by extraction with 300 μL toluene, which caused the proteins to precipitate. The organic layer was separated from the water layer by centrifugation (2232g, 20 min). The plates containing the water and organic layer were incubated at −80 °C for 30 min to freeze the water layer and hence preventing accidental uptake of part of the water layer. An aliquot of 50 μL from the organic layer was transferred to a GC vial by a robotic pipetting station. For analysis of the amount and enantiopurity of the enzymatic product 3a, 8 μL of the organic layer was injected on a gas chromatograph using an Astec CHIRALDEX G-TA column, isocratic 125 °C (carrier gas He, 1.69 mL/min). Flame ionization detection; retention time S-3a: 25.6 min, retention time R-3a 26.9 min. The assignment of the absolute configuration of product 3a was based on earlier reported data.17,25
Semipreparative Scale Experiments
All semipreparative scale experiments were based on previously reported reaction conditions.17,25 Instead of DMSO, ethanol was used as a cosolvent, since the desired products 3a–d were obtained at the highest enantiopurity using 4-OT L8Y/M45Y/F50A in combination with ethanol as the cosolvent. For the enzymatic synthesis of 3a, the reaction mixture consisted of the following: 5.0 mg 2a (3 mM), 6.5 mg 4-OT L8Y/M45Y/F50A (75 μM, 2.5 mol % compared to 2a), and 2.56 mL ethanol (20% v/v) in 20 mM sodium phosphate buffer pH 6.5 to a final volume of 12.8 mL. The reaction was performed in a 25 mL Erlenmeyer flask sealed with a rubber stopper. The reaction was initiated by the addition of 108 μL of 1 (150 mM) and incubated at room temperature. At timely intervals, a sample was withdrawn from the reaction mixture and the reaction progress was monitored by following the depletion in absorbance at 249 nm corresponding to the concentration of substrate 2a.17 After the measurement, the sample was combined with the reaction mixture. After 50 min the reaction was finished and the reaction mixture was extracted 3× with 10 mL toluene. The organic layers were combined, washed with brine, and dried with anhydrous Na2SO4. The dried organic layer was concentrated in vacuo, yielding product 3a without any further purification (4.2 mg, 63% isolated yield). The enantiopurity and absolute configuration was determined by GC with a chiral stationary phase (see library screening).
For the enzymatic synthesis of 3b–d, the reaction mixture consisted of the following: nitroalkene (17.9 mg of 2b, 2 mM; 14.3 mg of 2c, 1.3 mM; or 20.1 mg of 2d, 2 mM), 11.5 mg of 4-OT L8Y/M45Y/F50A (28 μM, 1.4 mol % compared to 2b and 2d, 2.15 mol % compared to 2c), ethanol (12 mL, 20% (v/v) for 2b and 2d and 18 mL, 30% (v/v) for 2c) in 20 mM sodium phosphate buffer pH 6.5 to a final volume of 60 mL. The reaction was performed in a 100 mL Erlenmeyer flask sealed with a rubber stopper. The reaction was initiated by the addition of 169 μL 1 (50 mM) and incubated at room temperature. At timely intervals a sample was withdrawn from the reaction mixture and the reaction progress was monitored by following the depletion in absorbance (λmax of 2b and 2c at 320 nm, λmax of 2d at 322 nm). After the measurement, the sample was combined with the reaction mixture. When the reaction was finished, the reaction mixture was extracted with 3× 40 mL ethyl acetate. The organic layers were combined, washed with brine and dried with anhydrous Na2SO4. The dried organic layer was concentrated in vacuo, yielding 3b–d without any further purification (3b 20.1 mg, 87% yield; 3c 17.2 mg, 97% yield; and 3d 24.0 mg, 94% yield). The aldehyde functionality of 3b and 3c was derivatized to a cyclic acetal (see supporting methods). The enantiopurity of 3b–d was determined by reverse phase HPLC using a chiralpak AD-RH column (150 mm × 4.6 mm, Daicel) (MeCN/water 70:30 for 3b, MeCN/water 62:38 for 3c and MeCN/water 30:70 for 3d, 25 °C, 0.5 mL/min flow rate). Detection at 220 nm, retention time R-3b: 8.3 min, S-3b 10.8 min; R-3c: 29 min, S-3c 37 min; R-3d: 39 min, and S-3d: 41 min. The absolute configuration was determined by literature comparison.17
Two-Step Enzymatic Cascade Synthesis
The reaction mixture contained 2a (22 mg, 3 mM), 4-OT L8Y/M45Y/F50A (5 mol % compared to 2a), 5 mL of ethanol (10% v/v) in 0.1 M sodium phosphate buffer (pH 7.3) to a final volume of 50 mL. The reaction was initiated by the addition of 50 mM of 1 (2.5 mL from a freshly prepared 1 M stock solution) and the reaction mixture was incubated at room temperature. At timely intervals, a sample was withdrawn from the reaction mixture and the reaction progress was monitored by following the depletion in absorbance at 249 nm corresponding to the concentration of substrate 2a.17 After the measurement, the sample was combined with the reaction mixture. After 20 min, the reaction was finished (full conversion of 2a into 3a) and then to this PRO-ALDH(003) (0.5 mg/mL), PRO-NOX(009) (1 mg/mL), and NAD+ (8 mM) were added. We used this stepwise cascade approach because PRO-ALDH(003) also showed activity toward starting substrate 1. Favorably, the enzyme showed ∼3-fold higher activity toward substrate 3a (at 3 mM) than toward 1 (at 50 mM). Using this methodology, complete oxidation of 3a into 4a was achieved with the formation of less than 10 mM acetate as byproduct from 1. After the PRO-ALDH(003)-catalyzed oxidation reaction was completed, the reaction was stopped by acidifying the reaction mixture to pH 5. The reaction mixture was extracted with 3× 50 mL ethyl acetate. The organic layers were combined, washed with brine, and dried with anhydrous Na2SO4. The dried organic layer was concentrated in vacuo and the product was purified by silica gel column chromatography (hexane/ethyl acetate 4:1) to obtain 4a (18 mg, 64% yield). The acid functionality of 4a was derivatized to the corresponding methyl ester (see supporting methods). The same reaction procedures were followed for the synthesis of 4b–d. The enantiopurity of derivatized 4a was determined by GC using a chiral Astec CHIRALDEX G-TA column, 100 to 140 °C at a rate of 4 °C/min, then 140 to 160 °C at a rate of 2 °C/min and finally held at 160 °C. Flame ionization detection, retention time R-4a: 16 min, S-4a: 17 min. The enantiopurity of derivatized 4b–d was analyzed by reverse phase HPLC using a chiral column (Chiralpak-ID, 150 mm × 4.6 mm, Daicel) (MeCN/water 30:70, 25 °C, 1 mL/min flow rate). Detection at 210 nm, retention time R-4b: 22.6 min, S-4b: 24.5 min; R-4c: 19.4 min, S-4c: 24.4 min; R-4d: 23.5 min, S-4d: 26.6 min.
Three-Step Chemoenzymatic Cascade Synthesis
The reaction mixture contained nitoalkene (3 mM of 2a and 4 mM of 2b–d), 4-OT L8Y/M45Y/F50A (28–32 mg; 5 mol % compared to 2a–d), and ethanol (10% v/v) in 0.1 M NaH2PO4 buffer (pH 7.3) to a final volume of 25 mL. The reaction was initiated by the addition of 50 mM 1 (1.25 mL from a freshly prepared 1 M stock solution in 0.1 M NaH2PO4 buffer pH 7.3) and the reaction mixture was incubated at room temperature. At timely intervals, a sample was withdrawn from the reaction mixture and the reaction progress was monitored by following the depletion in absorbance corresponding to the concentration of nitroalkene (see above). After the measurement the sample was combined with the reaction mixture. After the completion of the reaction, PRO-ALDH(003) (0.5 mg/mL), PRO-NOX(009) (1 mg/mL) and NAD+ (0.5 mM) were added to the reaction mixture. After 30 min, the reaction was quenched by acidifying the reaction mixture using 5 M HCl until the pH dropped to 3. To this mixture, NiCl2·6H2O (40 mM) and NaBH4 (40 mM) were added at 0 °C, and stirring was continued for 24 h at room temperature. The reaction mixture was filtered through a Celite pad and the filtrate concentrated in vacuo, after which the resulting concentrated mixture was acidified to pH 3–4. The acidified reaction mixture was loaded on a column packed with cation exchange resin (5 g of Dowex 50WX8 hydrogen form). After washing with deionized water (4 column volumes), the product was eluted out with 0.5–1 M ammonia solution (4 column volumes). The ninhydrin-positive fractions were collected, combined, and lyophilized to yield the desired products 5a–d. Products 5a–d were derivatized to diastereomers using sodium 2,4-dinitro-5-fluorophenyl-l-valine amide (see the Supporting Information) and their enantiopurity was determined using reverse phase HPLC on a C18 column (Kinetex 5u EVO C18 100A, 150 mm × 4.6 mm, Phenomenex) at 25 °C (1 mL/min flow rate). The mobile phase was a 60:40 (v/v) mixture of aqueous buffer (0.2% triethylamine, pH adjusted to 3.5 with dilute orthophosphoric acid) and MeCN. Detection at 340 nm, retention time S-5a: 16.1 min, R-5a: 21.4 min; R-5b: 10.9 min, S-5b: 12.1 min; R-5c: 17.2 min, S-5c: 20.2 min; R-5d: 12.3 min, S-5d: 13.6 min.
Results
Effect of Cosolvents on the Enzymatic Michael-Type Addition Reaction
The previously reported 4-OT mutant M45Y/F50A25 exhibits inverted enantioselectivity when compared to wild-type 4-OT, allowing the enzymatic synthesis of the pharmaceutically relevant enantiomers of γ-nitroaldehydes 3a–d (Figure 1). However, the enantioselectivity of 4-OT M45Y/F50A is too low for biocatalytic application, with for instance the production of 3a with an e.r. of 90:10 and 3c with an e.r. of only 62:38.25 To improve the enantioselectivity of the 4-OT M45Y/F50A catalyzed Michael-type additions, we initially focused our efforts on solvent engineering. Indeed, the use of different solvent systems has been reported to have a substantial effect on the enantioselectivity of different enzyme catalyzed reactions.26,27 We selected the 4-OT catalyzed addition of 1 to 2a to test the effect of cosolvents on enantioselectivity, because the optical purity of product 3a can be straightforwardly analyzed by gas chromatography. Although the enzyme was still active at concentrations of up to 40% (v/v) DMSO, increasing the DMSO concentration had a detrimental effect on the enantioselectivity of the 4-OT M45Y/F50A-catalyzed reaction (Figure 2). A similar loss of enantioselectivity was observed when short-chain alcohols such as ethanol or methanol were used as cosolvent. Notably, ethanol and methanol concentrations of >20% (v/v) led to enzyme precipitation, which indicates a destabilizing effect of the cosolvent on the biocatalyst. Using aqueous solutions of glyceline and ethaline (deep-eutectic solvents) led to inactivation of the enzyme at all tested concentrations (10–60%, v/v). Diols, however, could be used in high concentrations without observing inactivation and with a positive effect on the enantioselectivity (Figure 2). For example, using up to 60% (v/v) ethylene glycol, 1,3-propanediol or 1,4-butanediol did not affect the enzymatic activity and the e.r. of product 3a increased up to 95:5.
Figure 2.
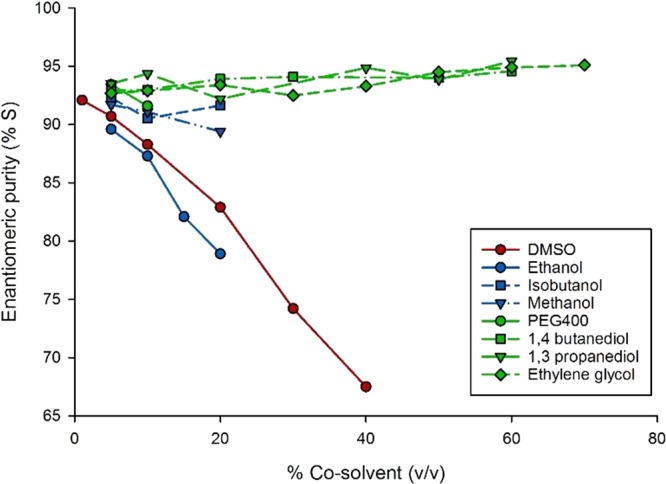
Effect of cosolvents on the enantioselectivity of the enzymatic Michael-type addition reaction. Enantiopurity of 3a (as the percentage of the S-enantiomer) in the 4-OT M45Y/F50A catalyzed addition of 1 to 2a performed in the presence of increasing concentrations of different cosolvents.
Engineering of a Highly Enantioselective Artificial “Michaelase”
Although solvent engineering slightly improved the enantioselectivity of the addition of 1 to 2a catalyzed by 4-OT M45Y/F50A, at best a product e.r. of 95:5 was achieved. Therefore, protein engineering was required to further enhance the enantioselectivity of this reaction. To guide our engineering efforts, we used the available crystal structure of 4-OT M45Y/F50A in complex with nitrostyrene 2b (PDB: 5CLO).25 This structure suggests that the substrate may bind in two distinct orientations relative to Pro-1, differing by a rotation of ∼180 deg around the longitudinal axis of the substrate molecule. One substrate orientation is consistent with the formation of the R enantiomer of the product, whereas the other is consistent with formation of the S enantiomer. We reasoned that by reducing the rotational freedom of the substrate in the active site and/or repositioning of Pro-1 relative to the substrate, the enantioselectivity of M45Y/F50A may be improved. Therefore, we selected position Leu-8, the side chain of which is involved in formation of the binding pocket for the substrate’s R-group (Figure 1), and position Ile-2, which is located next to Pro-1, for site-saturation mutagenesis.28 Accordingly, a focused library was constructed by randomizing positions Ile-2 and Leu-8 using NNK degeneracy, yielding library I2X/L8X/M45Y/F50A. As the model reaction for activity and enantioselectivity screening, the addition of 1 to 2a was chosen because the quantity and optical purity of product 3a can be easily analyzed by gas chromatography. We reasoned that if mutants displaying enhanced activity and enantioselectivity were to be found, then they could be tested for their ability to facilitate the enantioselective addition of acetaldehyde to aromatic nitroalkenes as well.
The library was transformed into Escherichia coli cells, and screened by evaluating 960 transformants. This resulted in the identification of two quadruple mutants (I2W/L8Y/M45Y/F50A and I2M/L8Y/M45Y/F50A) with significantly improved activity and enantioselectivity. These mutant enzymes were purified to homogeneity and assayed for their ability to catalyze the addition of 1 to 2a. Mutants I2W/L8Y/M45Y/F50A and I2M/L8Y/M45Y/F50A both displayed excellent enantioselectivity, providing product S-3a with an e.r. of 99:1 (Table S1), and enhanced activity (4- and 5-fold, respectively) when compared to that of M45Y/F50A (Figure 3a). Since both quadruple mutants contain the L8Y mutation, the triple mutant L8Y/M45Y/F50A was constructed to test the importance of the mutation at position Ile-2. In addition, the triple mutant L8F/M45Y/F50A was constructed to test if a structurally similar residue at position Leu-8 might result in better catalytic properties. Interestingly, mutants L8Y/M45Y/F50A and L8F/M45Y/F50A exhibited excellent enantioselectivity in the addition reaction yielding S-3a with an e.r. of 99:1 (Table S1). To our delight, the catalytic activity of mutant L8Y/M45Y/F50A for the addition of 1 to 2a was considerably improved (6-fold) compared to that of the starting mutant M45Y/F50A and higher than those of the quadruple mutants I2M/L8Y/M45Y/F50A and I2W/L8Y/M45Y/F50A (Figure 3a). The activity of mutant L8F/M45Y/F50A was slightly lower than that of L8Y/M45Y/F50A. Satisfyingly, mutant L8Y/M45Y/F50A also displayed higher activity (3.5-fold) for the addition of 1 to the aromatic nitroalkene 2c when compared to that of mutant M45Y/F50A (Figure 3b).
Figure 3.

Progress curves of Michael-type additions catalyzed by different 4-OT variants. (a) Addition of 1 (150 mM) to 2a (5 mM) in 20 mM sodium phosphate buffer (pH 6.5) and 5% (v/v) DMSO. Different 4-OT mutants were used as a catalyst at a concentration of 100 μM. YA: 4-OT M45Y/F50A, FYA: 4-OT L8F/M45Y/F50A, WYYA: 4-OT I2W/L8Y/M45Y/F50A, MYYA: 4-OT I2M/L8Y/M45Y/F50A, YYA: 4-OT L8Y/M45Y/F50A. (b) Addition of 1 (65 mM) to 2c (1.3 mM) in 20 mM sodium phosphate buffer (pH 6.5) and 45% (v/v) DMSO. Different 4-OT mutants were used as a catalyst at a concentration of 18 μM. YA: 4-OT M45Y/F50A, YYA: 4-OT L8Y/M45Y/F50A.
Coincidentally, when compared to mutant M45Y/F50A, mutant L8Y/M45Y/F50A also proved to be much more stable in the presence of the desirable cosolvent ethanol, tolerating concentrations of up to 30% (v/v) without losing activity or enantioselectivity. In fact, the enantioselectivity of the 4-OT L8Y/M45Y/F50A catalyzed addition of 1 to 2a in the presence of 25% (v/v) ethanol was exceptional, yielding optically pure S-3a (the pregabalin precursor) with an e.r. of >99:1 (Table S1). On the other hand, mutant L8F/M45Y/F50A was rather unstable in the presence of ethanol, with significantly reduced activity already at an ethanol concentration of 10% (v/v). Because the L8Y/M45Y/F50A mutant showed the highest catalytic activity and enantioselectivity, and because it tolerates relatively high concentrations of the cosolvent ethanol without loss of performance, it was selected for further study.
The synthetic usefulness of mutant L8Y/M45Y/F50A was further investigated in semipreparative scale experiments with nitroalkene acceptors 2a–d (Table 1). Ethanol was used as a cosolvent in all reactions because in 20–30% (v/v) ethanol excellent enantioselectivity was observed for the enzymatic additions. Under optimized conditions, products 3a–d could be synthesized with good isolated yields (63–97%) and excellent enantioselectivity (e.r. up to >99:1). In all cases, the pharmaceutically relevant enantiomer of the γ-nitroaldehyde was synthesized [(S)-3a and (R)-3b–d]. These results demonstrate the potential of this newly engineered artificial “Michaelase”, which displays enhanced activity, improved enantioselectivity and better ethanol stability compared to the parent enzyme, for the asymmetric synthesis of optically pure γ-nitroaldehydes, which are important precursors for the valuable pharmaceuticals pregabalin, phenibut, baclofen and fluorophenibut.
Table 1. 4-OT L8Y/M45Y/F50A Catalyzed Michael-Type Additions of 1 to 2a–d Yielding 3a–da.


Assay conditions: reaction mixtures consisted of 150 mM 1 (synthesis of 3a) or 50 mM 1 (synthesis of 3b–d), 3 mM 2a, 1.3 mM 2c or 2 mM 2b and 2d in 20 mM sodium phosphate buffer, pH 6.5. The reaction volume was 12.8 mL (synthesis of 3a) or 60 mL (synthesis of 3b–d). 1.4 mol % of 4-OT (compared to concentration nitroalkene) was used, except for entry 1 (2.5 mol %) and entry 3 (2.15 mol %).
For 3a, the e.r. was determined by GC with a chiral stationary phase. For 3b and 3c, the enzymatic product was first converted into the corresponding ethylene glycol acetal, of which the e.r. was determined by HPLC with a chiral stationary phase. For 3d, the e.r. was directly determined by HPLC with a chiral stationary phase. The absolute configuration was determined by comparison to literature data.17,25
Structural Basis for the Enhanced Enantioselectivity
To obtain insight into how the active site of 4-OT L8Y/M45Y/F50A is remodelled to facilitate the highly enantioselective Michael-type addition reactions, we determined its crystal structure at 1.9 Å resolution (Table S2). The overall hexameric structure of 4-OT L8Y/M45Y/F50A is very similar to that of wild-type 4-OT (PDB entry: 4X19)23 and of mutant M45Y/F50A (PDB entry: 5CLN),25 with Cα-backbone root-mean-square-deviations of 0.38 and 0.69 Å, respectively. A more detailed structural comparison of 4-OT L8Y/M45Y/F50A and the parental enzyme 4-OT M45Y/F50A with bound nitrostyrene (PDB entry: 5CLO)25 shows that the extra mutation (L8Y) in the triple mutant results in a slight shift of residues 5–17, comprising the last residues of strand β1, the following loop, and the N-terminal residues of helix α1 (maximum shift is 1.4 Å). In addition, residues 46–48 comprising the loop/310-helix after strand β2 show a maximum shift of 2.2 Å. Residues Tyr-45 and Ala-50 line the active site pocket, their side chains having a conformation very similar to that observed in the M45Y/F50A mutant. Notably, two conformations of Tyr-45 were observed. In one conformation, the side chain of Tyr-45 is rotated toward the substrate binding pocket. Overlaying this conformation with the structure of M45Y/F50A in complex with nitrostyrene (2b) would result in a clash between the aromatic ring of 2b and Tyr-45, suggesting that this is not the active conformation. In the second conformation, the side chain of Tyr-45 is flipped to the back of the active site, making H-bond interactions with His-6, similar as is observed in the structure of M45Y/F50A.
Most importantly, at the interface of each dimer of the L8Y/M45Y/F50A mutant, the Tyr-8 side chain lies “above” the hydrophobic pocket that binds the aromatic ring of 2b in the M45Y/F50A mutant (Figure 4a). While the pocket has become narrower due to the extra L8Y mutation, the comparison with the M45Y/F50A structure indicates that it remains capable to accommodate 2b, with the aromatic rings of Tyr-8 and the substrate forming a parallel displaced π–π stacking interaction (Figure 4b). The narrower substrate binding pocket is expected to be less tolerant toward accommodating the alternate binding mode of 2b, which would give the opposite enantiomer of the corresponding product, because the substrate has little freedom to adjust its conformation and orientation to minimize unfavorable close contacts. These results indicate that the enhanced enantioselectivity of the L8Y/M45Y/F50A mutant is not due to major structural changes but is related to the replacement of Leu-8 by a larger tyrosine residue, resulting in a narrower substrate binding pocket that prevents the substrate to bind in two distinct orientations relative to Pro-1. Thus, the reduction in size of the substrate binding pocket explains the excellent enantioselectivity of the 4-OT L8Y/M45Y/F50A mutant.
Figure 4.

Crystal structure of the newly engineered 4-OT L8Y/M45Y/F50A mutant. (a) Overlay of the crystal structures of mutant M45Y/F50A with bound nitrostyrene (2b, in light gray color, PDB entry: 5CLO) and mutant L8Y/M45Y/F50A (in orange color, this work). The extra L8Y mutation of the triple mutant has the tyrosine side chain lying above a hydrophobic pocket where nitrostyrene (2b) is bound in the double mutant, close to the catalytic proline residue (Pro-1). (b) Close-up view at a similar orientation, with the surface representation of the L8Y/M45Y/F50A mutant added, showing that nitrostyrene (2b) would snugly fit in the hydrophobic pocket.
One-Pot Two-Step Enzymatic Cascade Synthesis of γ-Nitrobutyric Acids
Having constructed an artificial “Michaelase” that is highly enantioselective for the addition of acetaldehyde (1) to nitroalkenes 2a–d, yielding γ-nitroaldehydes (S)-3a and (R)-3b–d (Table 1), we focused our attention on the development of a two-step enzymatic cascade process for the synthesis of γ-nitrobutyric acids 4a–d, which are more stable and practical GABA analogue precursors than the corresponding γ-nitroaldehydes. The synthesis of these compounds requires the coupling of the newly developed “Michaelase” with an appropriate aldehyde oxidase or aldehyde dehydrogenase (Table 2). Notably, using an oxidase would result in the formation of hydrogen peroxide as a byproduct, which could unfavorably react with the aldehyde substrates and enzyme catalysts, necessitating an additional step for its removal. Therefore, an aldehyde dehydrogenase with a broad non-natural substrate scope, PRO-ALDH(003) available from Prozomix Ltd., was used. Initial experiments showed that PRO-ALDH(003) was dependent on either NAD+ or NADP+ and could accept both (R)-3a and (S)-3a as substrates. For cofactor regeneration, we used an NADH oxidase, PRO-NOX(009) available from Prozomix Ltd., which converts NADH to NAD+ using O2 and producing H2O. A water-forming NOX enzyme is particularly suited to include in the envisioned cascade synthesis, as it does not employ substrates or generate products that complicate subsequent workup, resulting in increased purity and yield of 4a–d. Using our newly engineered artificial “Michaelase” 4-OT L8Y/M45Y/F50A in combination with PRO-ALDH(003) and PRO-NOX(009), we developed a one-pot two-step biocatalytic cascade to synthesize the desired product (S)-4a in 64% isolated yield and with an excellent e.r. of 99:1 (Table 2, entry 1). Using similar conditions, also (R)-4b–d could be synthesized in good isolated yield (67–70%) and with excellent e.r. values of up to 99:1 (Table 2, entries 2–4).
Table 2. One-Pot Two-Step Biocatalytic Enantioselective Synthesis of γ-Nitrobutyric Acids 4a–da.


Michael-type addition of 1 to 2a–d catalyzed by 4-OT L8Y/M45Y/F50A or 4-OT A33D to form either S-3a and R-3b–d or R-3a and S-3b–d respectively, followed by aldehyde oxidation catalyzed by PRO-ALDH(003), using PRO-NOX(009) for cofactor recycling, to form either S-4a and R-4b–d or R-4a and S-4b–d. The reaction mixtures consisted of 50 mM 1, 3 mM 2a or 4 mM 2b–d in 100 mM sodium phosphate buffer pH 7.3 and 10% (v/v) ethanol. Five mol % of 4-OT (compared to concentration nitroalkene) was used, except for entry 6 (1.5 mol %) and entry 7 (3 mol %). PRO-ALDH(003) was added to a final concentration of 0.5 mg/mL, PRO-NOX(009) was added to a final concentration of 1 mg/mL. The concentration NAD+ was 8 mM (4a) or 10 mM (4b–d). The amounts of applied cofactor were adjusted such that short reaction times and high conversions were achieved.
Monitored by UV spectroscopy.
Monitored by HPLC.
Products 4a–d were esterified and the e.r. was determined by GC or HPLC with a chiral stationary phase.
Interestingly, the modularity of this enzymatic cascade approach and the ability of PRO-ALDH(003) to accept both enantiomers of 3a–d as substrates, also allowed for the synthesis of the opposite enantiomers of γ-nitrobutyric acids 4a–d. To this end, we replaced 4-OT L8Y/M45Y/F50A by the previously engineered enantiocomplementary 4-OT variant A33D, which produces the opposite enantiomers of 3a–d.25 Under optimized conditions and using the appropriate combination of enzymes in one pot, the desired products (R)-4a and (S)-4b–d were obtained with excellent e.r. values (99:1) and in 62–74% isolated yield (Table 2, entries 5–8).
One-Pot Three-Step Chemoenzymatic Cascade Synthesis of γ-Aminobutyric Acids
Having developed a modular one-pot two-step enzymatic cascade for the production of both enantiomers of γ-nitrobutyric acids 4a–d, we focused our attention on the extension of this cascade by including a third step, converting 4a–d into the desired pharmaceutically active γ-aminobutyric acids 5a–d. To the best of our knowledge, no enzymes have been reported that can catalyze the full reduction of aliphatic nitro groups, such as those present in 4a–d, to the corresponding amines.29 While several procedures for the chemical reduction of nitro groups have been reported, most of these use organic solvents and are not suitable to be executed in aqueous solutions. To prevent the necessity of intermediate purification of 4a–d, we selected nickel boride as catalyst (NiCl2·6H2O in combination with NaBH4), because this system has been reported to catalyze the reduction of nitro groups in aqueous solutions.30,31 Pleasingly, when using 10 equiv of nickel boride (NaBH4/NiCl2·6H2O), the complete reduction of 4a–d into 5a–d in aqueous buffer at pH 3 was achieved.
Having established that nickel boride can be used to set up a three-step chemoenzymatic cascade reaction, 4-OT L8Y/M45Y/F50A, PRO-ALDH(003), PRO-NOX(009), and NaBH4/NiCl2·6H2O were combined in one pot. This chemoenzymatic cascade proved to be efficient, converting the simple starting materials 1 and 2a into the desired product (S)-5a with an isolated yield of 55% and an excellent e.r. of 98:2 (Table 3, entry 1). Similarly, this one-pot three-step reaction cascade could also be used to efficiently synthesize (R)-5b–d in good isolated yields (65–70%) and with excellent e.r. values of 99:1 (Table 3, entries 2–4). Thus, access to the pharmaceuticals pregabalin, phenibut, baclofen and fluorophenibut has been achieved using this newly developed one-pot, three-step chemoenzymatic cascade process.
Table 3. One-Pot Three-Step Chemoenzymatic Cascade Synthesis of Pharmaceuticals 5a–da.


Michael-type addition of 1 to 2a–d catalyzed by 4-OT L8Y/M45Y/F50A to form S-3a and R-3b–d, followed by aldehyde oxidation catalyzed by PRO-ALDH(003), using PRO-NOX(009) for cofactor recycling, to form S-4a and R-4b–d, followed by nitro reduction catalyzed by nickel boride to form S-5a and R-5b–d. The reaction mixtures consisted of 50 mM 1, 3 mM 2a or 4 mM 2b–d in 100 mM sodium phosphate buffer pH 7.3 and 10% (v/v) ethanol. Five mol % of 4-OT (compared to concentration of nitroalkene) was used; PRO-ALDH(003) was added to a final concentration of 0.5 mg/mL; PRO-NOX(009) was added to a final concentration of 1 mg/mL; 0.5 mM of NAD+ was added; 40 mM of NiCl2 and 40 mM of NaBH4 were used. Because of the poor stability of nickel boride in aqueous buffer, a 10-fold excess of this reagent (40 mM versus 3–4 mM 2a–d) is required to achieve high conversion.
Monitored by UV spectroscopy.
Monitored by HPLC.
Monitored by TLC.
Purified by cation exchange chromatography.
Products 5a–d were derivatized using Nα-(2,4-dinitro-5-fluorophenyl)-l-valinamide, and the e.r. of the corresponding diastereomers was determined by HPLC with an achiral stationary phase. Notably, the e.r. of product 5a is likely to be 99:1; however, because a minor contaminant from the derivatizing agent has the same retention time as the R enantiomer of product 5a, the e.r. for product 5a is cautiously reported as 98:2, consistent with the observed peaks in the HPLC chromatogram.
Discussion
The use of different enzymes to build reaction cascades for efficient synthesis of valuable chemical products is an emerging concept in biocatalysis.4−7 This approach aims to minimize the number of reaction steps, prevent the necessity of isolation of intermediate products, and improve the “pot-economy” of the process.9 Using such a systems biocatalysis strategy, we have successfully demonstrated a one-pot two-step biocatalytic cascade reaction for the synthesis of the pharmaceutically relevant enantiomers of γ-nitrobutyric acids starting from simple achiral precursors (acetaldehyde and nitroalkenes). This synthetic route highlights a highly enantioselective carbon–carbon bond-forming step catalyzed by a newly engineered artificial “Michaelase”, 4-OT L8Y/M45Y/F50A, which has no known biological counterpart. This tailor-made enzyme shows non-natural substrate promiscuity, accepting acetaldehyde for enantioselective addition to various nitroalkenes. The modularity of this two-step cascade reaction was demonstrated by using a previously engineered enantiocomplementary 4-OT mutant, 4-OT A33D,25 to synthesize the opposite enantiomers of the γ-nitrobutyric acids. We also developed a three-step chemoenzymatic cascade route for the conversion of enzymatically prepared γ-nitrobutyric acids by chemical reduction into the corresponding γ-aminobutyric acids in one pot. Given that the efficient large-scale process for the manufacture of pregabalin (5a) by Pfizer is based on a lipase-catalyzed kinetic resolution with ex situ enantiomer recycling,32 our one-pot asymmetric chemoenzymatic cascade synthesis offers an alternative route to pharmaceutically active γ-aminobutyric acid products, as exemplified by the enantioselective synthesis of pregabalin (3 steps, 55% yield), phenibut (3 steps, 70% yield), baclofen (3 steps, 65% yield), and fluorophenibut (3 steps, 68% yield). This shows the exciting opportunities available for combining chemocatalysts, natural enzymes, and designed artificial biocatalysts in multistep syntheses.5 It is anticipated that such a rapid synthesis strategy, which avoids (de)protecting steps and intermediate purifications, could be flexibly extended to various other targets, for example by using aldehyde donors with different chain length or by starting from structurally distinct nitroalkenes.
The key step of the developed cascade synthesis is the enantioselective Michael addition of acetaldehyde to a nitroalkene catalyzed by an engineered 4-OT variant (mutant L8Y/M45Y/F50A). The development of this highly enantioselective 4-OT variant was achieved by using an engineering strategy that involves simultaneous randomization at two sites (Ile-2 and Leu-8, Figure 4) lining the substrate binding pocket of 4-OT M45Y/F50A, a double mutant previously designed for inverting the stereoselectivity to that required for GABA precursor synthesis.25 The major benefit of this structure-guided engineering strategy lies in the reduction of the sequence space that has to be covered in order to identify valuable mutants.28 Screening of 960 transformants, which is only a relatively small fraction of clones with respect to statistical requirements for significant coverage of possible combinations in a double-site SSM library with NNK codons, yielded two quadruple mutants (I2W/L8Y/M45Y/F50A and I2M/L8Y/M45Y/F50A) with significantly improved activity and the desired high enantioselectivity. The best performing triple mutant, L8Y/M45Y/F50A, was deductively constructed from these quadruple mutants. Notably, this triple mutant must have been included in the double-site SSM library, but apparently was not identified due to insufficient screening. From this result it can be concluded that a single-site SSM strategy at both positions would have been an even more efficient engineering strategy, with only the need for screening of ∼200 transformants with acceptable statistical coverage of possible mutants. In summary, we provide evidence that three amino acid substitutions (L8Y/M45Y/F50A) are sufficient to invert and dramatically enhance the enantioselectivity of 4-OT for the Michael addition of acetaldehyde to various nitroalkenes.
A structural comparison of 4-OT M45Y/F50A complexed with a nitroalkene substrate25 and unliganded 4-OT L8Y/M45Y/F50A suggested that the new triple mutant has a narrower binding pocket for the nitroalkene substrate (Figure 4). This reduction in size of the substrate binding pocket may prevent the nitroalkene substrate to bind in two distinct orientations relative to Pro-1, potentially explaining the enhanced enantioselectivity. However, the possibility that the observed nitroalkene substrate orientation is not relevant for the catalytic bond-forming event cannot be excluded, because the covalent enzyme-bound enamine intermediate, which is formed between Pro-1 and acetaldehyde,23 may influence the binding orientation of the nitroalkene substrate and the trajectory of the nucleophilic addition.
The currently employed amounts (1.4–5.0 mol %) of 4-OT L8Y/M45Y/F50A are lower and reactions times of ≤30 min are generally shorter than in the few conventional organocatalytic methodologies for identical type of Michael reactions.12,33,34 Moreover, we take advantage of the water solubility of the enzyme to achieve the reactions in environmentally friendly aqueous buffer and not in organic solvent. Nevertheless, the relatively high amounts of the 4-OT L8Y/M45Y/F50A enzyme employed, required to ensure short reaction times to outcompete nonenzymatic water addition to nitroalkene substrates 2a–d, might pose a possible constraint on the use of our developed chemoenzymatic cascade synthesis for large scale transformations. To address this issue, we currently run protein engineering studies with the aim to further enhance the unnatural “Michaelase” activities of 4 OT L8Y/M45Y/F50A, which would allow lower catalyst loadings.
While all three steps of the cascade reaction were performed under aqueous conditions with high conversions, the chemical reduction step requires 10 equiv of nickel boride and has to be performed at acidic pH, which is not directly compatible with the enzymatic steps that require neutral pH. It will be interesting to investigate whether the chemical reduction step can be replaced by an enzymatic step, converting the γ-nitrobutyric acids 4a–d into GABA analogues 5a–d. However, such an entirely enzymatic cascade route for the straightforward synthesis of pharmaceutically active GABA analogues awaits the discovery or engineering of an appropriate nitroreductase capable of completely reducing the aliphatic nitro group of 4a–d to the amine group [at the expense of NAD(P)H] in a controlled and efficient fashion, without formation of unstable, reactive intermediates that lower the yield of the desired amino acid product. Although increasingly investigated, reported nitroreductases prefer nitroaromatic substrates and often catalyze only partial reduction of nitro groups to the reactive nitroso or hydroxylamino products.29 We have initiated studies aimed at exploring a large collection of putative nitroreductases, as well as engineered variants thereof, for their ability to efficiently reduce γ-nitrobutyric acids 4a–d into γ-aminobutyric acids 5a–d. As such, our work sets the stage for further development of “artificial metabolisms” for the greener, more sustainable and step-economic synthesis of an important class of pharmaceuticals.
Acknowledgments
We acknowledge financial support from The Netherlands Organization of Scientific Research (VICI grant 724.016.002), the European Research Council (PoC grant 713483), and the European Union’s Horizon 2020 Research and Innovation Programme under grant agreement No 635595 (CarbaZymes). We thank M.H. de Vries and M. Jeronimus-Stratingh for their expert assistance in acquiring ESI-MS data, and Yan Ni for insightful discussions. Parts of this research were carried out at PETRA III at DESY, a member of the Helmholtz Association (HGF). We would like to thank E. Reddem and A. Burkhardt for assistance in using the P11 beamline.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.8b04299.
Additional experimental procedures and characterizations of compounds (PDF)
Author Contributions
⊥ These authors contributed equally: L.B. and T.S.
The authors declare no competing financial interest.
Notes
All data are available from the corresponding author upon reasonable request.
Supplementary Material
References
- Winkler C. K.; Clay D.; Davies S.; O’Neill P.; McDaid P.; Debarge S.; Steflik J.; Karmilowicz M.; Wong J. W.; Faber K. Chemoenzymatic Asymmetric Synthesis of Pregabalin Precursors via Asymmetric Bioreduction of β-Cyanoacrylate Esters Using Ene-Reductases. J. Org. Chem. 2013, 78, 1525–1533. 10.1021/jo302484p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debarge S.; McDaid P.; O’Neill P.; Frahill J.; Wong J. W.; Carr D.; Burrell A.; Davies S.; Karmilowicz M.; Steflik J. Evaluation of Several Routes to Advanced Pregabalin Intermediates: Synthesis and Enantioselective Enzymatic Reduction Using Ene-Reductases. Org. Process Res. Dev. 2014, 18, 109–121. 10.1021/op4002774. [DOI] [Google Scholar]
- Hoekstra M. S.; Sobieray D. M.; Schwindt M. A.; Mulhern T. A.; Grote T. M.; Huckabee B. K.; Hendrickson V. S.; Franklin L. C.; Granger E. J.; Karrick G. L. Chemical Development of CI-1008, an Enantiomerically Pure Anticonvulsant. Org. Process Res. Dev. 1997, 1, 26–38. 10.1021/op9600320. [DOI] [Google Scholar]
- Muschiol J.; Peters C.; Oberleitner N.; Mihovilovic M. D.; Bornscheuer U. T.; Rudroff F. Cascade Catalysis – Strategies and Challenges En Route to Preparative Synthetic Biology. Chem. Commun. 2015, 51, 5798–5811. 10.1039/C4CC08752F. [DOI] [PubMed] [Google Scholar]
- Rudroff F.; Mihovilovic M. D.; Gröger H.; Snajdrova R.; Iding H.; Bornscheuer U. T. Opportunities and Challenges for Combining Chemo- and Biocatalysis. Nat. Catal. 2018, 1, 12–22. 10.1038/s41929-017-0010-4. [DOI] [Google Scholar]
- Fessner W. Systems Biocatalysis: Development and Engineering of Cell-Free “Artificial Metabolisms” for Preparative Multi-Enzymatic Synthesis. New Biotechnol. 2015, 32, 658–664. 10.1016/j.nbt.2014.11.007. [DOI] [PubMed] [Google Scholar]
- Faber K.; Fessner W.; Turner N. J. Engineering Biocatalysts for Synthesis Including Cascade Processes. Adv. Synth. Catal. 2015, 357, 1565–1566. 10.1002/adsc.201500450. [DOI] [Google Scholar]
- Payer S. E.; Pollak H.; Schmidbauer B.; Hamm F.; Juričić F.; Faber K.; Glueck S. M. Multienzyme One-Pot Cascade for the Stereoselective Hydroxyethyl Functionalization of Substituted Phenols. Org. Lett. 2018, 20, 5139–5143. 10.1021/acs.orglett.8b02058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y. Pot Economy and One-Pot Synthesis. Chem. Sci. 2016, 7, 866–880. 10.1039/C5SC02913A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner M.; Upert G.; Angelici G.; Wennemers H. Enamine Catalysis with Low Catalyst Loadings - High Efficiency via Kinetic Studies. J. Am. Chem. Soc. 2010, 132, 6–7. 10.1021/ja9068112. [DOI] [PubMed] [Google Scholar]
- Wiesner M.; Revell J. D.; Tonazzi S.; Wennemers H. Peptide Catalyzed Asymmetric Conjugate Addition Reactions of Aldehydes to Nitroethylene - A Convenient Entry into γ2-Amino Acids. J. Am. Chem. Soc. 2008, 130, 5610–5611. 10.1021/ja801027s. [DOI] [PubMed] [Google Scholar]
- Wiesner M.; Revell J. D.; Wennemers H. Tripeptides as Efficient Asymmetric Catalysts for 1,4-Addition Reactions of Aldehydes to Nitroolefins–A Rational Approach. Angew. Chem. 2008, 120, 1897–1900. 10.1002/ange.200704972. [DOI] [PubMed] [Google Scholar]
- Wiesner M.; Neuburger M.; Wennemers H. Tripeptides of the Type H-D-Pro-Pro-Xaa-NH2 as Catalysts for Asymmetric 1,4-Addition Reactions: Structural Requirements for High Catalytic Efficiency. Chem. - Eur. J. 2009, 15, 10103–10109. 10.1002/chem.200901021. [DOI] [PubMed] [Google Scholar]
- Miao Y.; Rahimi M.; Geertsema E. M.; Poelarends G. J. Recent Developments in Enzyme Promiscuity for Carbon–Carbon Bond-Forming Reactions. Curr. Opin. Chem. Biol. 2015, 25, 115–123. 10.1016/j.cbpa.2014.12.020. [DOI] [PubMed] [Google Scholar]
- Geertsema E. M.; Poelarends G. J.. Enzymatic Carbon–Carbon Bond-Forming Michael-Type Additions. In Science of Synthesis: Biocatalysis in Organic Synthesis 2; Faber K., Fessner W. D., Turner N., Eds.; Thieme: Stuttgart, Germany, 2014; Vol. 2, pp 313–333. [Google Scholar]
- Garrabou X.; Verez R.; Hilvert D. Enantiocomplementary Synthesis of γ-Nitroketones Using Designed and Evolved Carboligases. J. Am. Chem. Soc. 2017, 139, 103–106. 10.1021/jacs.6b11928. [DOI] [PubMed] [Google Scholar]
- Geertsema E. M.; Miao Y.; Tepper P. G.; de Haan P.; Zandvoort E.; Poelarends G. J. Biocatalytic Michael-Type Additions of Acetaldehyde to Nitroolefins with the Proline Based Enzyme 4-Oxalocrotonate Tautomerase Yielding Enantioenriched γ-Nitroaldehydes. Chem. - Eur. J. 2013, 19, 14407–14410. 10.1002/chem.201302351. [DOI] [PubMed] [Google Scholar]
- Miao Y.; Tepper P. G.; Geertsema E. M.; Poelarends G. J. Stereochemical Control of Enzymatic Carbon–Carbon Bond-Forming Michael-Type Additions by “Substrate Engineering. Eur. J. Org. Chem. 2016, 2016, 5350–5354. 10.1002/ejoc.201601126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandvoort E.; Geertsema E. M.; Baas B.; Quax W. J.; Poelarends G. J. Bridging Between Organocatalysis and Biocatalysis: Asymmetric Addition of Acetaldehyde to β-Nitrostyrenes Catalyzed by a Promiscuous Proline-Based Tautomerase. Angew. Chem. 2012, 124, 1266–1269. 10.1002/ange.201107404. [DOI] [PubMed] [Google Scholar]
- Miao Y.; Geertsema E. M.; Tepper P. G.; Zandvoort E.; Poelarends G. J. Promiscuous Catalysis of Asymmetric Michael-Type Additions of Linear Aldehydes to β-Nitrostyrene by the Proline-Based Enzyme 4-Oxalocrotonate Tautomerase. ChemBioChem 2013, 14, 191–194. 10.1002/cbic.201200676. [DOI] [PubMed] [Google Scholar]
- Zandvoort E.; Geertsema E. M.; Quax W. J.; Poelarends G. J. Enhancement of the Promiscuous Aldolase and Dehydration Activities of 4-Oxalocrotonate Tautomerase by Protein Engineering. ChemBioChem 2012, 13, 1274–1277. 10.1002/cbic.201200225. [DOI] [PubMed] [Google Scholar]
- Zandvoort E.; Baas B.; Quax W. J.; Poelarends G. J. Systematic Screening for Catalytic Promiscuity in 4-Oxalocrotonate Tautomerase: Enamine Formation and Aldolase Activity. ChemBioChem 2011, 12, 602–609. 10.1002/cbic.201000633. [DOI] [PubMed] [Google Scholar]
- Poddar H.; Rahimi M.; Geertsema E. M.; Thunnissen A. M. W. H.; Poelarends G. J. Evidence for the Formation of an Enamine Species During Aldol and Michael-Type Addition Reactions Promiscuously Catalyzed by 4-Oxalocrotonate Tautomerase. ChemBioChem 2015, 16, 738–741. 10.1002/cbic.201402687. [DOI] [PubMed] [Google Scholar]
- van der Meer J.; Biewenga L.; Poelarends G. J. The Generation and Exploitation of Protein Mutability Landscapes for Enzyme Engineering. ChemBioChem 2016, 17, 1792–1799. 10.1002/cbic.201600382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer J.; Poddar H.; Baas B.; Miao Y.; Rahimi M.; Kunzendorf A.; van Merkerk R.; Tepper P. G.; Geertsema E. M.; Thunnissen A. M. W. H.; Quax W. J.; Poelarends G. J. Using Mutability Landscapes of a Promiscuous Tautomerase to Guide the Engineering of Enantioselective Michaelases. Nat. Commun. 2016, 7, 10911. 10.1038/ncomms10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrea G.; Ottolina G.; Riva S. Role of Solvents in the Control of Enzyme Selectivity in Organic Media. Trends Biotechnol. 1995, 13, 63–70. 10.1016/S0167-7799(00)88907-6. [DOI] [Google Scholar]
- Klibanov A. M. Enzymatic Catalysis in Anhydrous Organic Solvents. Trends Biochem. Sci. 1989, 14, 141–144. 10.1016/0968-0004(89)90146-1. [DOI] [PubMed] [Google Scholar]
- Reetz M. T.; Bocola M.; Carballeira J. D.; Zha D.; Vogel A. Expanding the Range of Substrate Acceptance of Enzymes: Combinatorial Active-Site Saturation Test. Angew. Chem., Int. Ed. 2005, 44, 4192–4196. 10.1002/anie.200500767. [DOI] [PubMed] [Google Scholar]
- Durchschein K.; Hall M.; Faber K. Unusual Reactions Mediated by FMN-Dependent Ene- and Nitro-Reductases. Green Chem. 2013, 15, 1764–1772. 10.1039/c3gc40588e. [DOI] [Google Scholar]
- Khurana J. M.; Gogia A. Synthetically Useful Reactions with Nickel Boride. A Review. Org. Prep. Proced. Int. 1997, 29, 1–32. 10.1080/00304949709355171. [DOI] [Google Scholar]
- Osby J. O.; Ganem B. Rapid and Efficient Reduction of Aliphatic Nitro Compounds to Amines. Tetrahedron Lett. 1985, 26, 6413–6416. 10.1016/S0040-4039(00)99014-2. [DOI] [Google Scholar]
- Martinez C. A.; Hu S.; Dumond Y.; Tao J.; Kelleher P.; Tully L. Development of a Chemoenzymatic Manufacturing Process for Pregabalin. Org. Process Res. Dev. 2008, 12, 392–398. 10.1021/op7002248. [DOI] [Google Scholar]
- García-García P.; Ladépêche A.; Halder R.; List B. Catalytic Asymmetric Michael Reactions of acetaldehyde. Angew. Chem., Int. Ed. 2008, 47, 4719–4721. 10.1002/anie.200800847. [DOI] [PubMed] [Google Scholar]
- Hayashi Y.; Itoh T.; Ohkubo M.; Ishikawa H. Asymmetric Michael Reaction of Acetaldehyde Catalyzed by Diphenylprolinol Silyl Ether. Angew. Chem., Int. Ed. 2008, 47, 4722–4724. 10.1002/anie.200801130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.