

Abstract
The use of subtle features as species diagnostic traits in taxa with high morphological similarity sometimes fails in discriminating intraspecific variation from interspecific differences, leading to an incorrect species delimitation. A clear assessment of species boundaries is particularly relevant in disease vector organisms in order to understand epidemiological and evolutionary processes that affect transmission capacity. Here, we assess the validity of the recently described Rhodnius taquarussuensis (da Rosa et al., 2017) using interspecific crosses and molecular markers. We did not detect differences in hatching rates in interspecific crosses between R. taquarussuensis and R. neglectus (Lent, 1954). Furthermore, genetic divergence and species delimitation analyses show that R. taquarussuensis is not an independent lineage in the R. prolixus group. These results suggest that R. taquarussuensis is a phenotypic form of R. neglectus instead of a distinct species. We would like to stress that different sources of evidence are needed to correctly delimit species. We consider this is an important step in understanding vectorial Chagas disease spread and transmission.
Introduction
The study of the speciation process requires a complete understanding of the phenotypic variation present across the range of the study taxa. This is particularly challenging in organisms where morphological differences are subtle or not obvious, and where other aspects of their biology such as reproduction, ecology, phenology and life traits are also unknown. An increasing number of studies have documented “cryptic” speciation throughout the tree of life (i.e. taxa that cannot readily be distinguished morphologically, yet evidence indicates they are on different evolutionary trajectories). However, such descriptions have been done in absence of a clear definition of what a cryptic species is, and often using alpha taxonomy as the sole approach for detecting and classifying new species [1–4]. This can lead to false species diagnosis when unreliable traits (those lacking discontinuous, nonoverlapping patterns of variation) are used [5], which is particularly important when delimiting vector species with medical relevance, as this directly impacts the control of the diseases transmitted by them.
The subfamily Triatominae has 18 genera, with Panstrongylus (Berg, 1879), Rhodnius (Stål, 1859) and Triatoma (Laporte, 1832) being the most epidemiologically important genera, since they are the main species responsible for the transmission of Trypanosoma cruzi (Chagas, 1909), the etiologic agent of Chagas disease [6, 7]. The identification of these three genera is straightforward and is based on the insertion of the antennae on the head, which is macroscopically perceptible: in Panstrongylus the antennae are inserted near the eyes, in Rhodnius these appendages are on the anterior portion of the head, and in Triatoma they are located on the middle portion of the head [8, 9]. Nonetheless, the most recent Triatominae phylogeny showed that the only monophyletic genus is Rhodnius [9–11]. Also, species delimitation within these genera remains problematic [12]. In particular, species of Rhodnius show low morphological variation and their complex identification relies on few morphological traits and/or mtDNA divergence [11, 13–16]. For example, it is difficult to differentiate between R. neglectus and R. prolixus (Stål, 1859) [17], R. robustus (Larrousse, 1827) and R. montenegrensis (da Rosa et al., 2012) [18], R. amazonicus (Almeida, Santos and Sposina, 1973) and R. pictipes (Stål, 1872) [19], R. pictipes and R. stali (Lent, Jurberg and Galvão) [20], among many other examples.
Moreover, the classic division of Rhodnius presents additional challenges. The genus is divided into three groups: prolixus, pictipes and pallescens. The first two are found east of the Andes (cis-Andean), while the third is distributed west of the Andes (trans-Andean) [21–23]. The phylogenetic relationships among these groups are still under debate, especially the position of the pictipes group that was initially considered closer to the pallescens group, but recent evidence found it as sister to the prolixus group [23–26].
Because Rhodnius has an intrinsic relation with the propagation of T. cruzi and T. rangeli (Tejera, 1920), resolving its phylogenetic relationships and accurately differentiating its species is a first step to determine the epidemiological threat associated to each species, as well as to understand their ecology and population dynamics [8, 23, 27].
Recently, a new species of the genus Rhodnius, R. taquarussuensis, was described based on phenotypic and cytogenetic traits [22]. This is the only species of the prolixus group that has dispersed heterochromatin throughout the nucleus and autosomes, and it is morphologically similar to R. neglectus [22, 28]. However, the specific status of R. taquarussuensis requires a more rigorous confirmation that implements both genetic data and tests of reproductive isolation. Here, we used six molecular markers and performed crosses between R. taquarussuensis and R. neglectus in order to address whether the former is a valid species.
Methods
Sampling and DNA extraction
Individuals of R. taquarussuensis were collected in Taquarussu, Mato Grosso do Sul, Brazil (-22.48 Lat, -53.35 Long; Table 1) and those of R. neglectus were collected in Formoso, Goiás, Brazil (-13.65 Lat, -48.88 Long; Table 1) and maintained in the Triatominae insectary of the School of Pharmaceutical Sciences, São Paulo State University (UNESP), Araraquara, São Paulo, Brazil. Rhodnius prolixus were collected in Arauca (7.08 Lat, -70.75 Long), Fortul (6.78 Lat, -71.76 Long), Puerto Rondón (6.28 Lat, -71.10 Long) and Saravena (6.95 Lat, -71.87 Long) in Colombia (Table 1). UNIVERSIDAD DEL ROSARIO provided the field permit from ANLA (Autoridad Nacional de Licensias ambientales) 63257–2014. DNA was extracted from the head, legs and intestine using the DNeasy Blood & Tissue Kit (Qiagen), following the manufacturer’s protocol. The DNA concentration was determined using a NanoDrop 1000 Spectrophotometer V3.7 (Thermo Fisher Scientific, Wilmington, DE, USA) and stored at −20°C.
Table 1. Genes, primer information and accession numbers.
| Symbol | Gene name | Rn | Rp | Rt | Primers (5'-3') | Tm (°C) | Fragment size (pb) | Accession numbers |
|---|---|---|---|---|---|---|---|---|
| CYTB | Cytochrome b** | 6 | 5 | 8 |
R: GCW CCA ATT CAR GTT ART AA F: GGA CGW GGW ATT TAT TAT GGA TC |
50 | 659 | MH704746—MH704764 |
| ND4 | NADH dehydrogenase 4** | 5 | 5 | 15 |
F: TAA TTC GTT GTC ATG GTA ATG F: TCA ACA TGA GCC CTT GGA AG |
53 | 560 | MH704765—MH704779 |
| PCB | Putative chitin binding peritrophin-a domain protein | 8 | 5 | 5 |
R: CAC TAC GGG TCG TGA AGG TT F: ACA TCC TTG GCC ACA AGA AC |
55 | 757 | MH704780—MH704797 |
| TOPO | DNA topoisomerase | 5 | 6 | 5 |
F: CAA CAC TTG TAA CCC GAG CA F: ATC ATT GGC CGC ATC TTT AG |
56 | 604 | MH704798—MH704813 |
| URO | Uroporphyrinogen decarboxylase | 11 | 6 | 6 |
R: TTA AGG GCA GCA AGA GGA GA F: AAC ACA TTT CCT GGC CAA AG |
54 | 563 | MH704814—MH704828 |
| ZNFP | Toll-like-2. Transmembrane receptor with TIR domain binding | 5 | 5 | 5 |
F: TCC TTG CGG TAA TGA TGT GA F: CTC GAA TGG TGT ACG TGG TG |
54 | 588 | MH704829—MH704852 |
Gene IDs correspond to those in the Rhodnius genome GFF file annotation.
**Published before. Rn: R. neglectus; Rp: R. prolixus; Rt: R. taquarussuensis
Loci amplification and sequencing
We amplified and sequenced two mitochondrial gene fragments, Cytochrome b (CYTB) and Mitochondrially Encoded NADH Dehydrogenase 4 (ND4) using the conditions reported elsewhere [29]. We also designed primers to develop new coding nuclear markers in Rhodnius. In order to do this, we used the R. prolixus genome available in VectorBase (https://www.vectorbase.org/organisms/rhodnius-prolixus) and, from the GFF file, we selected four large exon markers (≥700 bp) using a custom script. We then used BLASTn to compare these exons to the R. prolixus transcriptome and thus confirm they were single copy markers. Then, we verified the identity of the selected exons in Uniprot with the ID codes registered in the genome. Finally, we designed primers for these loci using Primer 3 [30]. The resulting nuclear markers are Putative chitin binding peritrophin-a (PCB), DNA topoisomerase (TOPO), Uroporphyrinogen decarboxylase (URO) and Toll-Like-2. Transmembrane receptor with TIR domain binding (ZNFP) (Table 1 and Table 2).
Table 2. Nuclear markers (single copy exons) designed in this study.
| Gen | Annotation in the R. prolixus genome | Region amplified | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene ID | Scaffold | Strand | Start | End | Size (bp) | Location | Start | End | ||
| ZNFP | RPRC009262-RA | Tl-like-2: Toll-like-2. Transmembrane receptor with TIR domain binding | KQ034161 | + | 481476 | 486977 | 5501 | Exon 1 | 481599 | 482146 |
| URO | RPRC013534-RA | UROD: Uroporphyrinogen decarboxylase | KQ034105 | - | 970351 | 971418 | 1067 | Exon 1 | 970699 | 971261 |
| TOPO | RPRC012703-RA | DNA topoisomerase | KQ034259 | + | 391034 | 406927 | 15893 | Exon 3 | 404730 | 405333 |
| PCB | RPRC001863-RA | Putative chitin binding peritrophin-a | KQ034056 | + | 8334541 | 8342490 | 7949 | Exon 3 | 8335296 | 8336052 |
Gene IDs correspond to those in the Rhodnius genome GFF file annotation.
PCR reactions had a final volume of 25 μl, consisting of 12.5 μl of GoTaq Green Master Mix (Promega, Madison, WI, USA), 1.25 μL (10 μM) of each primer and, 5.0 μl of DNA (20 ng) and 5μL of H2O. Amplification was conducted in a Thermal Cycler 4000 (Bio-Rad La-boratories, Inc., Hercules, CA, USA). The following PCR cycling conditions were used: 94°C for 5 min; 40 cycles of 94°C for 1 min, 50–56°C for 1 min (Table 1), and a final extension at 72°C for 10 min. PCR success was verified by electrophoresis on agarose gel stained with Fast SYBR Green (Applied Biosystems, Foster City, CA, USA) and a molecular weight marker (Promega) adding 2μl of each PCR product. The samples were purified using the PCR kit ExoSAP-IT Product Cleanup (Affymetrix, Santa Clara, CA, USA) and sequenced at Macrogen Inc. (Seoul, Korea).
Sequence analyses
Gene sequences were read, edited and aligned with CLC Main Workbench (Qiagen). For nuclear loci, haplotype inference for heterozygous calls was conducted using the PHASE algorithm implemented in DnaSP v5 [31], accepting haplotypes with a confidence higher than 90% after running 5,000 interactions per simulation. Then, we created alignments for each locus using MUSCLE [32] with the default parameters. These alignments were visualized and corrected by hand in MEGA X [33]. Finally, we translated the sequences to proteins in order to verify for stop codons using MESQUITE 3.04 [34].
Molecular phylogenetics and species delimitation
In order to assess the position of R. taquarussuensis within the group prolixus, we downloaded from the Genbank all CYTB sequences available for this group and one from Triatoma infestans (outgroup; S1 Table) using the following Entrez line: “esearch -db nucleotide -query "<organism> CYTB" | efetch -format fasta” [35]. We combined these data with our sequences and estimated a phylogenetic tree for the group prolixus using a Maximum likelihood (ML) optimization in IQ-TREE [36]. The substitution model for CYTB was established in the same software, selecting the model with the lowest BIC score. Node support was calculated with 1,000 ultrafast bootstrap replicates.
We also explored the phylogenetic relationships between R. prolixus, R. neglectus and R. taquarussuensis, concatenating all loci (nuclear and mitochondrial; 3731 bp long alignment) in Mesquite 3.04 [34] and estimating a ML phylogenetic tree with in IQ-TREE [36]. We allowed each locus to have its own substitution model, and node support was accessed as above. We also conducted a Bayesian analysis independently for each locus using BEAST 2.5, implementing linked and unlinked tree models [37]. We inferred the nucleotide substitution model, range of the rate of heterogeneity, and proportion of invariant positions during the MCMC analysis with the bModelTest package [38], with transition-transversion split option and empirical frequencies. We ran 10’000,000 generations sampling every 1,000 generations and used TRACER [39] to confirm the coverage of the chain (i.e. effective sample size >200). TreeAnnotator [37] was used to construct a consensus tree per locus and the initial 10% trees were discarded as burn-in. We superimposed and plotted consensus gene trees constructing a Multiphylo object with the densiTree function in R [40].
As the resulting ML and Bayesian topologies were identical, we used the ML tree as input for a species delimitation analysis intended to determine the species boundaries between R. taquarussuensis, R. neglectus and R. prolixus. This analysis was carried out under a phylogenetic species concept using the Bayesian and ML version of PTP with 500,000 MCMC generations, thinning = 100 and burn-in = 0.1 [41]. PTP implements a non-ultrametric phylogeny to model speciation rate as the number of substitutions reflected as branch lengths, assuming that the number of substitutions between species are significantly higher than the number of substitutions within species.
Genetic differentiation analysis and haplotype networks
We calculated segregating sites (SS), nucleotide diversity (π), haplotype diversity (Hd), number of synonymous and non-synonymous substitutions, singletons and Tajima’s D with DnaSP v5 [31]. We did not calculate relative genetic differentiation (FST) as it has been shown to be overestimated when low nucleotide diversities are obtained [42], as in our dataset (Table 3). Instead, we calculated an absolute divergence measure (DXY) and its nucleotide diversity corrected version (Da) with DnaSP v5. DXY was visualized as a heatmap drawn with the R package “fields”. We also calculated Kimura 2 parameter distance (K2P) which has been previously used in triatomines to validate different species [43].
Table 3. Summary statistics for each locus.
| Species | Gene | Pi (π) | SS | Tajima's D* | Hd | Synonymous sites | Non- synonymous sites | Singletons |
|---|---|---|---|---|---|---|---|---|
| R. neglectus | CYTB | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ND4 | 0.00089 | 1 | -0.61 | 0.5 | 0 | 1 | 1 | |
| PCB | 0.0012 | 2 | 1.085 | 0.49 | 1 | 1 | 0 | |
| TOPO | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| URO | 0.00015 | 1 | -1.15 | 0.083 | 0 | 1 | 1 | |
| ZNFP | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| R. taquarussuensis | CYTB | 1.00E-07 | 1 | -1.05 | 0.25 | 1 | 0 | 1 |
| ND4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| PCB | 0.00074 | 1 | 1.38 | 0.53 | 0 | 1 | 0 | |
| TOPO | 0.00353 | 6 | 0.02 | 0.62 | 3 | 3 | 0 | |
| URO | 0.00143 | 4 | -1.38 | 0.56 | 0 | 4 | 3 | |
| ZNFP | 0.00091 | 1 | 0.85 | 0.81 | 0 | 1 | 0 | |
| R. prolixus | CYTB | 0.00965 | 13 | -1.1 | 1 | 2 | 12 | 12 |
| ND4 | 0.00714 | 4 | 0 | 1 | 1 | 3 | 4 | |
| PCB | 0.00141 | 3 | 0.021 | 0.35 | 1 | 2 | 0 | |
| TOPO | 0.00028 | 1 | -1.14 | 0.17 | 1 | 0 | 1 | |
| URO | 0.00328 | 4 | 1.39 | 0.77 | 1 | 3 | 0 | |
| ZNFP | 0.00181 | 2 | 1.031 | 0.53 | 0 | 2 | 0 |
*None of the Tajima’s D were significant.
Genetic clustering between R. neglectus and R. taquarussuensis was validated with a discriminant analysis of principal components (DAPC) performed with both nDNA and mtDNA using the ‘adegenet’ R package [44]. We did this by transforming fasta sequences into a genind object that contains individual genotypes and loading it into ‘adegenet’ [44]. We performed a principal component analysis (PCA) on these data and retained the first two components (that accounted for >90% of the total variation in both mtDNA and nDNA). We then applied a discriminant analysis using the dapc function and assuming two prior groups (i.e. two species). This produced a single canonical function that summarizes the individual genetic variability, which was then visualized with a density plot. Finally, we constructed haplotype median-joining networks per locus with POPART [45].
Interspecific crosses
As a first attempt to determine the presence of reproductive isolation between R. taquarussuensis and R. neglectus, we performed interspecific (direct and reciprocal) and conspecific crosses. These were conducted in the Triatominae insectary of the School of Pharmaceutical Sciences, São Paulo State University (UNESP), Araraquara, São Paulo, Brazil, following the methodology established by Costa et al. [46] and Mendonça et al. [47]. Each cross was replicated three times for a total of 12 matings. First, insects were sexed as 5th instar nymphs [48], and males and females were kept separately until they reached the adult stage [49]. Then, a virgin female was placed with a male inside a plastic box (5cm diameter × 10cm height) for a maximum period of 120 days and kept at room temperature. The success or failure of mating was recorded by direct observation. After seven days, we collected the eggs of each cross weekly throughout the females’ oviposition period (120 days). The eggs collected were placed inside a plastic box (5cm diameter × 10 cm height) and their hatching was recorded weekly.
We calculated hatching success of the interspecific crosses as a measure of egg viability relative to conspecific crosses. A likelihood approximation was implemented in Betabino 1.1 [50] to analyze these data. Because using a binomial model alone does not account for the variation in hatching rate among families in each type of cross, Betabino fits a beta-binomial distribution to count data (in our case, number of eggs that hatched), thus solving this issue. Four alternative models that contrast the number of parameters in the data (i.e. mean and variance in the hatching rate) were tested. For details see http://www.ucl.ac.uk/~ucbhdjm/bin/betabino/betabino.pdf and the appendix section in [50].
Results
Molecular phylogenetics and species delimitation
All sequences obtained for this study were deposited in the Genbank and their accession numbers are found in Table 1. Our dataset for the CYTB gene consisted of 162 sequences corresponding to six species and confirmed the phylogenetic relationships previously shown by Monteiro et al. [11]. Briefly, the ML topology obtained with this gene (evolution model TN+F+I; BIC score 4339.957) revealed that the prolixus group is subdivided into two clades, one exclusively formed by R. barreti (Abad-Franch, Palomeque and Monteiro, 2013), and the second consisting of R. robustus, R. montenegrensis, R. prolixus, R. neglectus, R. nasutus (Stål, 1859), and R. taquarussuensis. The relations within this latter clade are complicated. For example, we recovered the four groups previously described for R. robustus [11], where R. robustus-I falls inside the R. prolixus clade, and R. montenegrensis is part of R. robustus-II (Fig 1 and S1 Fig). Additionally, the species R. neglectus is recovered as sister to R. prolixus and contains all individuals from the newly described species R. taquarussuensis, which although monophyletic, has virtually no differentiation from R. neglectus (Fig 1 and S1 Fig).
Fig 1. Maximum Likelihood tree for Rhodnius based on CYTB.

Numbers on the nodes are bootstrap supports. The vertical bar on the right highlight the prolixus group. The focal species, namely, R. taquarussuensis and R. neglectus, are highlighted in the green square. Green branches and the collapsed clade (green triangle) correspond to the sequences obtained here for R. taquarussuensis and R. neglectus respectively.
To better explore this unexpected pattern, we constructed haplotype networks of the gene fragments studied with R. neglectus, R. taquarussuensis and R. prolixus (Fig 2). In the case of CYTB, we found R. prolixus separated from the other two species by 15 mutational steps. In contrast, R. taquarussuensis haplotypes were less distant to R. neglectus (only two mutational steps). In fact, the divergence of R. taquarussuensis from R. neglectus (H.1 and H.2) is less than the divergence between such haplotypes and others from the same species (i.e. H.3 to H.8). Consistently, nucleotide diversity of R. prolixus and R. neglectus is higher than that of R. taquarussuensis (Table 3).
Fig 2. Haplotype networks.

(a) CYTB; (b) ND4; (c) PCB; (d) TOPO; (e) URO; (f) ZNFP. Ticks on branches indicate mutational steps between haplotypes. Circle size is proportional to the number of individuals having a haplotype.
We recovered the same multilocus phylogeny for R. prolixus, R. neglectus and R. taquarussuensis with ML and Bayesian approaches (ML substitution models were CYTB: HKY+F+I; ND4: HKY+F; PCB: F81+I; TOPO: F81+I; URO: HKY+F; ZNFP: TPM2+F+I). The three species were monophyletic and all of them with posterior probabilities of 100 (Fig 3A) Bootstrap support values were > 90 for R. prolixus and R. neglectus, while R. taquarussuensis has a bootstrap support of 78. Also, the branch length of R. taquarussuensis is less than one in a thousand changes. The unlinked and superimposed Bayesian gene trees consistently recovered two main clades: one exclusively composed of R. prolixus, and the second where R. neglectus and R. taquarussuensis show incomplete coalescence (Fig 3B). Consistently, in the analysis of species delimitation (PTP), both the Maximum Likelihood and Bayesian inference found two species as the most probable partition (Fig 4). These two partitions correspond to R. prolixus and R. neglectus. All other internal nodes had probabilities lower than 0.1 (Fig 4).
Fig 3. Phylogenetic trees for R. prolixus, R. neglectus and R. taquarussuensis based on all molecular markers.

A. Multilocus phylogeny where node support is indicated on each branch: bootstrap (above) and posterior probability (below). B. Bayesian superimposed gene trees: red (CYTB), blue (ND4), green (TOPO), yellow (URO), orange (PCB) and black (ZNFP). The alignment consisted of 3731 bp.
Fig 4. Species delimitation based on the Poisson Tree Process (PTP).
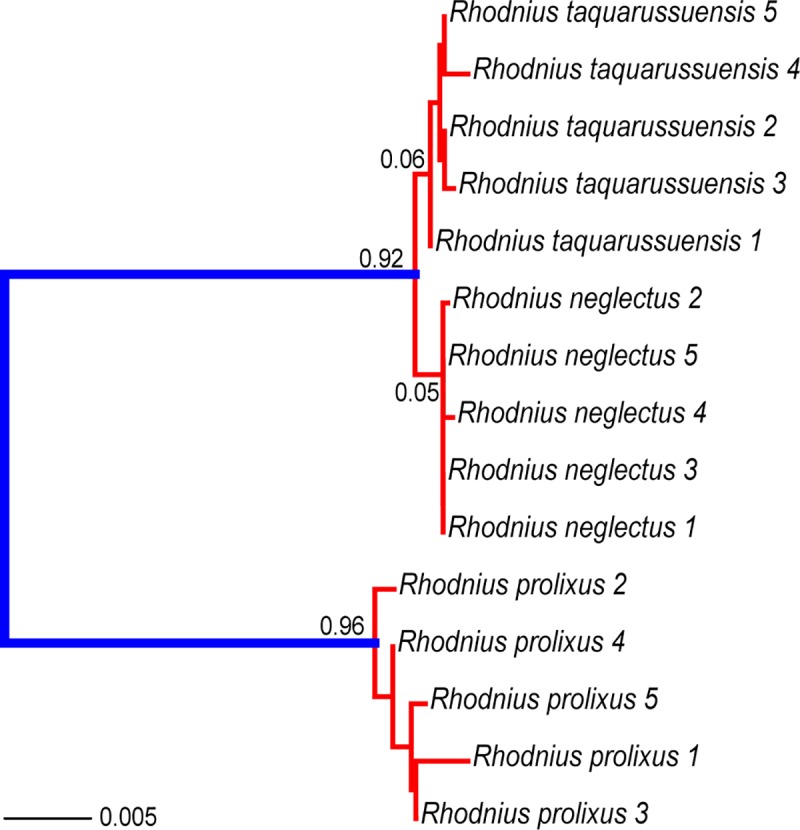
Maximum Likelihood and Bayesian inference yielded identical results. Numbers on each node are posterior probabilities of the inner taxa forming one species. Thus, red branches indicate taxa that should be considered as part of the same lineage.
Genetic differentiation
Overall, all markers showed low genetic diversity for the three taxa, R. prolixus, R. neglectus and R. taquarussuensis. In particular, the loci PCB and ND4 showed the same pattern as CYTB, where R. taquarussuensis is less diverse than the other two species (Table 4). The remaining loci showed R. taquarussuensis less diverse than R. prolixus and the diversity of R. neglectus was zero. This is consistent with the low number of haplotypes observed in the haplotype networks (Fig 2), where R. prolixus has private haplotypes that clearly differentiate it from the other two species (Fig 2B–2F), while R. taquarussuensis and R. neglectus exhibit substantial haplotype sharing (Fig 2).
Table 4. Absolute genetic divergence corrected by nucleotide diversity (Da) and Kimura 2 Parameter distance (K2P) between R. prolixus, R. taquarussuensis and R. neglectus.
| Gene | Species pair | Da | K2P |
|---|---|---|---|
| CYTB | R. neglectus–R. taquarussuensis | 0.003 | 0.003 |
| R. neglectus–R. prolixus | 0.06639 | 0.082 | |
| R. taquarussuensis–R. prolixus | 0.06939 | 0.086 | |
| ND4 | R. neglectus–R. taquarussuensis | 0 | 0 |
| R. neglectus–R. prolixus | 0.0625 | 0.075 | |
| R. taquarussuensis–R. prolixus | 0.0625 | 0.075 | |
| PCB | R. neglectus–R. taquarussuensis | 0.00037 | 0.001 |
| R. neglectus–R. prolixus | 0.0359 | 0.038 | |
| R. taquarussuensis–R.prolixus | 0.0359 | 0.039 | |
| TOPO | R. neglectus–R. taquarussuensis | 0.00221 | 0.004 |
| R. neglectus–R. prolixus | 0.01325 | 0.014 | |
| R.taquarussuensis–R. prolixus | 0.01545 | 0.018 | |
| URO | R. neglectus–R. taquarussuensis | 0.00476 | 0.005 |
| R. neglectus–R. prolixus | 0.01701 | 0.017 | |
| R. taquarussuensis–R. prolixus | 0.01701 | 0.012 | |
| ZNFP | R. neglectus–R. taquarussuensis | 0.00635 | 0.007 |
| R. neglectus–R. prolixus | 0.02234 | 0.024 | |
| R. taquarussuensis–R. prolixus | 0.02585 | 0.028 |
Consistent with these findings, DXY shows R. prolixus highly differentiated from R. neglectus and R. taquarussuensis in all loci whilst the latter two taxa do not differentiate between them (S2 Fig). When correcting for the nucleotide diversity, the same pattern is observed (Table 4). The genetic distance (K2P) between R. neglectus and R. taquarussuensis in all loci was less than 7.5%, a value previously used to define species in triatomines using CYTB [43]. Also, the discriminant analysis = of genetic variation for both mtDNA and nDNA fails to separate the taxa R. neglectus and R. taquarussuensis, which is reflected by the overlap of their densities on the canonical function (S3 Fig).
Interspecific crosses
All interspecific matings attempted were successful (n = 6), suggesting that there are no mechanical and/or gametic mechanisms that act against hybridization between R. neglectus and R. taquarussuensis. When we tested homogeneity across categories in the hatching rate, we did not observe differences between interspecific crosses (direct or reciprocal) and controls (Table 5; G6 = 7.06, P = 0.3152). Models that have multiple means (G3 = 1.243, P = 0.7428) or variances (G3 = 2.097, P = 0.5525) for the hatching rate were not supported by the data, indicating the absence of maternal or cytoplasmic effects.
Table 5. Results for interspecific and conspecific crosses.
R denotates replicate number for each cross. SE = standard error.
| Type of cross | Laid eggs (hatched) | Proportion of viable eggs (SE) | Variance (SE) | ||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | Total | ||||
| Interspecific | R. taquarussuensis ♀ x R. neglectus ♂ | 230 (198) | 86 (80) | 230 (193) | 510 (471) | 0.83 (0.03) | 0.0016 (0.002) |
| R. neglectus ♀ x R. taquarussuensis ♂ | 300 (275) | 181 (105) | 256 (244) | 708 (624) | 0.88 (0.02) | 0.0006 (0.0007) | |
| Conspecific | R. neglectus ♀ x R. neglectus ♂ | 337 (308) | 409 (346) | 174 (155) | 901 (809) | 0.86 (0.02) | 0.0001 (0.0016) |
| R. taquarussuensis ♀ x R. taquarussuensis ♂ | 151 (127) | 168 (150) | 201 (156) | 501 (433) | 0.78 (0.14) | 0.034 (0.046) | |
Discussion
Rhodnius exhibits morphological traits that facilitate its identification at the genus level [18, 51], but the low morphological variation within the genus precludes an easy species identification based on morphology alone [23]. This has led to suggest the existence of cryptic species in Rhodnius, where multiple look-alike lineages should be considered as different species based on their genetic differentiation [11, 16, 23, 51]. However, morphological species identification in Rhodnius relies on intraspecifically variable traits, which can lead to over-estimate the number of species [5]. Therefore, it is necessary to validate the status of the currently described species in the genus implementing a comprehensive approach that uses morphology, genetics, and measures of reproductive isolation.
R. taquarussuensis is the most recently described species in Rhodnius, based on morphological, morphometric and cytogenetic evidence [22]. However, the description of this species lacked other crucial evidence. Here, we tested the species status of R. taquarussuensis sequencing six molecular markers and performing interspecific crosses. Our results suggest that, despite the morphological differences between R. taquarussuensis and R. neglectus [22], these taxa constitute a single species.
Firstly, the known distribution range of R. taquarussuensis overlaps that of R. neglectus (Fig 5). Thus, for them to be different species it would be necessary to evolve strong intrinsic and/or extrinsic isolation barriers that restrict gene flow. In contrast, we found that R. taquarussuensis and R. neglectus successfully cross and there are no maternal or cytoplasmic effects that affect offspring viability, as reflected by the high hatching rates we obtained. This also suggests the absence of mechanical or gametic mechanisms acting against their hybridization. Although we did not test the fertility of the “hybrid” offspring, the egg viability observed in our crosses is higher than that reported for other interspecific crosses between different species in the subfamily Triatominae, where hybrid disfunction has been detected [47, 52–54]. However, the role of other pre-zygotic barriers such as temporal asynchrony, mate choice and/or habitat differences, among others, remains to be tested.
Fig 5. Geographical distribution of R. neglectus (blue) and R. taquarussuensis (red).
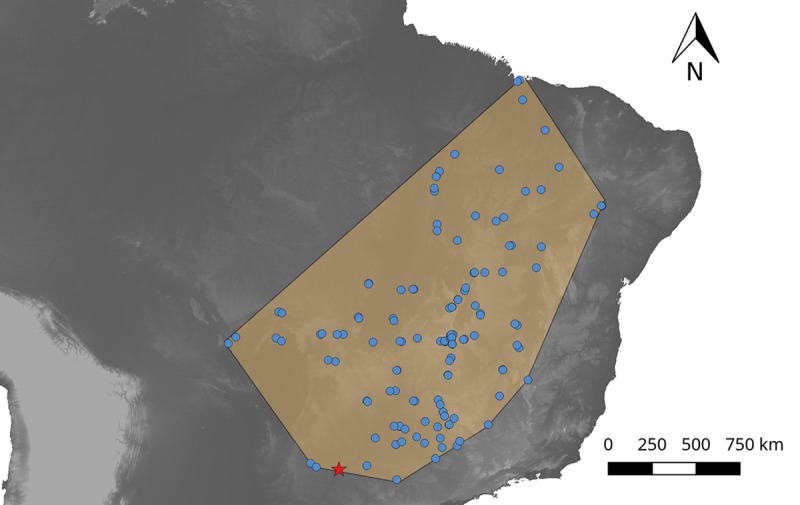
Distribution of R. neglectus is based on records available on DataTri [55] whilst that of R. taquarussuensis is based on collections made by the authors.
Secondly, our phylogenies and haplotype networks showed R. taquarussuensis nested within R. neglectus, with no differentiation from this species. Consequently, the species delimitation analysis collapsed these two taxa as a single one. Additionally, genetic differentiation measures as well as the discriminant analysis failed to show genetic structure between these lineages. Recent genomic analysis in animals have established that ‘good-species’ usually have a genetic divergence (Da) > 2%, although there is a “grey zone” of speciation (in which taxonomy is often controversial), that spans from 0.5% to 2% of Da. However, any Da < 0.5% undoubtedly corresponds to populations of the same species [56]. Therefore, our Da values are consistent with a scenario of R. taquarussuensis being R. neglectus rather than a different species. Furthermore, our genetic distance (K2P) estimates between R. neglectus and R. taquarussuensis were lower than those between R. neglectus and R. prolixus, and between R. taquarussuensis and R. prolixus. This genetic similarity between R. taquarussuensis and R. neglectus in all our analyses contrast with the clear differentiation observed between R. neglectus and R. prolixus, which are known to be distinct yet closely related species. In agreement with these findings, recent studies have suggested that R. milesi (Carcavalho et al., 2001), another species described based on cytogenetic differences [57, 58], shows high genetic similarity with R. neglectus thus questioning its validity as a true species [11]. This further suggests that R. neglectus may be a species that shows important polymorphism in cytogenetic patterns, which should not be used for species diagnosis.
The original description of R. taquarussuensis reported differences in the constitutive heterochromatin pattern and nanocomposition of TA and CG rich DNA base pairs between R. taquarussuensis and R. neglectus, mainly because R. taquarussuensis shows more heterochromatic blocks in the autosomes and the Y chromosome compared to the other Rhodnius species. Although gain and/or loss of constitutive heterochromatin has been previously used as evidence of species differentiation in the R. pallescens group [59], the T. sordida subcomplex [60, 61], and T. dimidiata (Latreille, 1811) [62], such heterochromatin differences between R. neglectus and R. taquarussuensis are likely just intraspecific polymorphism of R. neglectus. The presence of intraspecific heterochromatin variation with no apparent consequences on speciation is not new in Triatominae and has been observed in T. infestans (Klug, 1834) [63–65], P. geniculatus (Latreille, 1811) [66], and R. pallescens [67]. Therefore, although cytogenetics is a valuable methodology for taxonomic studies [68], heterochromatin variation between populations (i.e. the existence of cytotypes) is not a reliable trait to delimit species when evaluated alone. This agrees with the fact that cytogenetics is known to have a 20% failure rate in delimiting arthropods’ species [69]. In conclusion, after performing a comprehensive analysis using mitochondrial and newly developed nuclear markers, as well as crosses between R. taquarussuensis and R. neglectus, we can confidently suggest that R. taquarussuensis is not a separate species and must be considered a synonym of R. neglectus. Our study highlights the importance of revising carefully the current taxonomy of Rhodnius, because only a confident species delimitation will permit to study the processes and mechanisms involved in their diversification, as well as to unveil vector/parasite associations with epidemiological relevance.
Supporting information
(DOCX)
(DOCX)
(a) CYTB; (b) ND4; (c) PCB; (d) TOPO; (e) URO; (f) ZNFP. Note that DXY scale for all genes is not the same.
(DOCX)
Discriminant analysis based on mtDNA (a) and nDNA (b). Densities for a single discriminant function are shown, with red being R. taquarussuensis and blue being R. neglectus.
(DOCX)
Data Availability
All relevant data are within the manuscript.
Funding Statement
This word was funded by DIRECCION DE INVESTIGACION UNIVERSIDAD DEL ROSARIO. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Bickford D, Lohman DJ, Sodhi NS, Ng PKL, Meier R, Winker K, et al. Cryptic species as a window on diversity and conservation. Trends in Ecology & Evolution. 2007;22(3):148–55. 10.1016/j.tree.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Minard G, Tran Van V, Tran FH, Melaun C, Klimpel S, Koch LK, et al. Identification of sympatric cryptic species of Aedes albopictus subgroup in Vietnam: new perspectives in phylosymbiosis of insect vector. Parasites & Vectors. 2017;10(1):276 10.1186/s13071-017-2202-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noireau F, Gutierrez T, Zegarra M, Flores R, Brenière F, Cardozo L, et al. Cryptic speciation in Triatoma sordida (Hemiptera: Reduviidae) from the Bolivian Chaco. Tropical medicine & international health: TM & IH. 1998;3(5):364–72. [DOI] [PubMed] [Google Scholar]
- 4.Skoracka A, Magalhães S, Rector BG, Kuczyński L. Cryptic speciation in the Acari: a function of species lifestyles or our ability to separate species? Experimental and Applied Acarology. 2015;67(2):165–82. 10.1007/s10493-015-9954-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zapata F, Jimenez I. Species delimitation: inferring gaps in morphology across geography. Systematic biology. 2012;61(2):179–94. Epub 2011/08/16. 10.1093/sysbio/syr084 . [DOI] [PubMed] [Google Scholar]
- 6.Galvão C. Vetores da doença de chagas no Brasil: SciELO-Sociedade Brasileira de Zoologia; 2014. [Google Scholar]
- 7.WHO. Chagas Disease (American trypanosomiasis). Fact sheet 340. 2018;http://www.who.int/mediacentre/factsheets/fs340/en/ 2018(cited 2018 October 3).
- 8.Lent H, Wygodzinsky P. Revision of the Triatominae (Hemiptera, Reduviidae), and their significance as vectors of Chagas' disease. Bulletin of the American museum of Natural History. 1979;163(3):123–520. [Google Scholar]
- 9.Pinto C. Valor do rostro e antenas na caracterização dos gêneros de Triatomídeos. Hemiptera, Reduvidioidea. Bol Biol. 1931;19:45–136. [Google Scholar]
- 10.Justi SA, Galvão C, Schrago CG. Geological changes of the Americas and their influence on the diversification of the Neotropical kissing bugs (Hemiptera: Reduviidae: Triatominae). PLoS neglected tropical diseases. 2016;10(4):e0004527 10.1371/journal.pntd.0004527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monteiro FA, Weirauch C, Felix M, Lazoski C, Abad-Franch F. Evolution, Systematics, and Biogeography of the Triatominae, Vectors of Chagas Disease. Advances in parasitology. 2018;99:265–344. 10.1016/bs.apar.2017.12.002 [DOI] [PubMed] [Google Scholar]
- 12.Monteiro FA, Barrett TV, Fitzpatrick S, Cordon‐Rosales C, Feliciangeli D, Beard CB. Molecular phylogeography of the Amazonian Chagas disease vectors Rhodnius prolixus and R. robustus. Molecular Ecology. 2003;12(4):997–1006. [DOI] [PubMed] [Google Scholar]
- 13.Rosa JA, Mendonça VJ, Gardim S, de Carvalho DB, de Oliveira J, Nascimento JD, et al. Study of the external female genitalia of 14 Rhodnius species (Hemiptera, Reduviidae, Triatominae) using scanning electron microscopy. Parasites & vectors. 2014;7(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neiva A, Pinto C. Estado actual dos conhecimentos sobre o gênero Rhodnius Stål, com a descrição de uma nova espécie. Bras Med. 1923;37:20–4. [Google Scholar]
- 15.Pavan M, Monteiro F. A multiplex PCR assay that separates Rhodnius prolixus from members of the Rhodnius robustus cryptic species complex (Hemiptera: Reduviidae). Tropical Medicine & International Health. 2007;12(6):751–8. [DOI] [PubMed] [Google Scholar]
- 16.Pavan MG, Mesquita RD, Lawrence GG, Lazoski C, Dotson EM, Abubucker S, et al. A nuclear single-nucleotide polymorphism (SNP) potentially useful for the separation of Rhodnius prolixus from members of the Rhodnius robustus cryptic species complex (Hemiptera: Reduviidae). Infection, Genetics and Evolution. 2013;14:426–33. 10.1016/j.meegid.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barata JMS. Aspectos morfológicos de ovos de triatominae: II-Características macroscópicas e exocoriais de dez espécies do gênero Rhodnius Stal, 1859 (Hemiptera-Reduviidae). Revista de Saúde pública. 1981;15:490–542. [DOI] [PubMed] [Google Scholar]
- 18.Rosa JA, Rocha CS, Gardim S, Pinto MC, Mendonca VJ, Ferreira Filho J, et al. Description of Rhodnius montenegrensis n. sp.(Hemiptera: Reduviidae: Triatominae) from the state of Rondônia, Brazil. Zootaxa. 2012;3478:62–76. [Google Scholar]
- 19.Almeida FBd, Santos EI, Sposina G. Triatomíneos da Amazônia III.(). Acta Amazônica. 1973;3(2):43–6. [Google Scholar]
- 20.Lent H, Jurberg J, Galvão C. Rhodnius stali n. sp. related to Rhodnius pictipes Stal, 1872 (Hemiptera, Reduviidae, Triatominae). Memórias do Instituto Oswaldo Cruz. 1993;88(4):605–14. [Google Scholar]
- 21.Abad-Franch F, Monteiro FA, Jaramillo N, Gurgel-Gonçalves R, Dias FBS, Diotaiuti L. Ecology, evolution, and the long-term surveillance of vector-borne Chagas disease: a multi-scale appraisal of the tribe Rhodniini (Triatominae). Acta Tropica. 2009;110(2–3):159–77. 10.1016/j.actatropica.2008.06.005 [DOI] [PubMed] [Google Scholar]
- 22.Rosa JA, Justino HHG, Nascimento JD, Mendonça VJ, Rocha CS, de Carvalho DB, et al. A new species of Rhodnius from Brazil (Hemiptera, Reduviidae, Triatominae). ZooKeys. 2017;(675):1 10.3897/zookeys.675.12024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Justi SA, Galvão C. The evolutionary origin of diversity in Chagas disease vectors. Trends in parasitology. 2017;33(1):42–52. 10.1016/j.pt.2016.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hypša V, Tietz DF, Zrzavý J, Rego RO, Galvao C, Jurberg J. Phylogeny and biogeography of Triatominae (Hemiptera: Reduviidae): molecular evidence of a New World origin of the Asiatic clade. Molecular phylogenetics and evolution. 2002;23(3):447–57. [DOI] [PubMed] [Google Scholar]
- 25.Justi SA, Russo CA, dos Santos Mallet JR, Obara MT, Galvão C. Molecular phylogeny of Triatomini (Hemiptera: Reduviidae: Triatominae). Parasites & vectors. 2014;7(1):149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyman DF, Monteiro FA, Escalante AA, Cordon-Rosales C, Wesson DM, Dujardin J-P, et al. Mitochondrial DNA sequence variation among triatomine vectors of Chagas' disease. The American journal of tropical medicine and hygiene. 1999;60(3):377–86. [DOI] [PubMed] [Google Scholar]
- 27.Meneguetti DUdO Soares EB, Campaner M Camargo LMA. First report of Rhodnius montenegrensis (Hemiptera: Reduviidae: Triatominae) infection by Trypanosoma rangeli. Revista da Sociedade Brasileira de Medicina Tropical. 2014;47(3):374–6. [DOI] [PubMed] [Google Scholar]
- 28.Ravazi A, Alevi K, Oliveira J, Rosa J, Azeredo-Oliveira M. Cytogenetic analysis in different populations of Rhodnius prolixus and R. nasutus from different countries of South America. Brazilian Journal of Biology. 2018;78(1):183–5. [DOI] [PubMed] [Google Scholar]
- 29.Díaz S, Panzera F, Jaramillo-O N, Pérez R, Fernández R, Vallejo G, et al. Genetic, cytogenetic and morphological trends in the evolution of the Rhodnius (Triatominae: Rhodniini) trans-Andean group. PLoS One. 2014;9(2):e87493 10.1371/journal.pone.0087493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics (Oxford, England). 2007;23(10):1289–91. Epub 2007/03/24. 10.1093/bioinformatics/btm091 . [DOI] [PubMed] [Google Scholar]
- 31.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinforma. 2009;25 10.1093/bioinformatics/btp187 [DOI] [PubMed] [Google Scholar]
- 32.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar V, Reinartz W. Customer relationship management: Concept, strategy, and tools: Springer; 2018. [Google Scholar]
- 34.Maddison WPMaddison D. Mesquite: a modular system for evolutionary analysis. Version 3.04. 2015.
- 35.Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic acids research. 2010;39(suppl_1):D52–D7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molecular biology and evolution. 2014;32(1):268–74. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bouckaert R, Vaughan TG, Barido-Sottani J, Duchene S, Fourment M, Gavryushkina A, et al. BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. bioRxiv. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bouckaert RR, Drummond AJ. bModelTest: Bayesian phylogenetic site model averaging and model comparison. BMC Evolutionary Biology. 2017;17(1):42 10.1186/s12862-017-0890-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Systematic biology. 2018;67(5):901–4. 10.1093/sysbio/syy032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schliep KP. phangorn: phylogenetic analysis in R. Bioinformatics (Oxford, England). 2011;27(4):592–3. Epub 2010/12/21. 10.1093/bioinformatics/btq706 ; PubMed Central PMCID: PMCPmc3035803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang J, Kapli P, Pavlidis P, Stamatakis A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics (Oxford, England). 2013;29(22):2869–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merilä J, Crnokrak P. Comparison of genetic differentiation at marker loci and quantitative traits. Journal of Evolutionary Biology. 2001;14(6):892–903. 10.1046/j.1420-9101.2001.00348.x [DOI] [Google Scholar]
- 43.Monteiro FA, Donnelly MJ, Beard CB, Costa J. Nested clade and phylogeographic analyses of the Chagas disease vector Triatoma brasiliensis in Northeast Brazil. Mol Phylogenet Evol. 2004;32(1):46–56. Epub 2004/06/10. 10.1016/j.ympev.2003.12.011 . [DOI] [PubMed] [Google Scholar]
- 44.Jombart T, Ahmed I. adegenet 1.3–1: new tools for the analysis of genome-wide SNP data. Bioinformatics (Oxford, England). 2011;27(21):3070–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leigh JW, Bryant D. popart: full‐feature software for haplotype network construction. Methods in Ecology and Evolution. 2015;6(9):1110–6. [Google Scholar]
- 46.Costa J, Almeida CE, Dujardin JP, Beard CB. Crossing experiments detect genetic incompatibility among populations of Triatoma brasiliensis Neiva, 1911 (Heteroptera, Reduviidae, Triatominae). Memórias do Instituto Oswaldo Cruz. 2003;98(5):637–9. [DOI] [PubMed] [Google Scholar]
- 47.Mendonça VJ, Alevi KCC, de Oliveira Medeiros LM, Nascimento JD, de Azeredo-Oliveira MTV, da Rosa JA. Cytogenetic and morphologic approaches of hybrids from experimental crosses between Triatoma lenti Sherlock & Serafim, 1967 and T. sherlocki Papa et al., 2002 (Hemiptera: Reduviidae). Infection, Genetics and Evolution. 2014;26:123–31. 10.1016/j.meegid.2014.05.015 [DOI] [PubMed] [Google Scholar]
- 48.Rosa JA, Barata JMS, Barelli N, Santos JLF, Neto B, Miguel F. Sexual distinction between 5th instar nymphs of six species of Triatominae (Hemiptera, Reduviidae). Memórias do Instituto Oswaldo Cruz. 1992;87(2):257–64. [Google Scholar]
- 49.Martínez-Ibarra J, Grant-Guillén Y, Delgadillo-Aceves I, Zumaya-Estrada F, Rocha-Chávez G, Salazar-Schettino P, et al. Biological and genetic aspects of crosses between phylogenetically close species of mexican triatomines (Hemiptera: Reduviidae). Journal of medical entomology. 2011;48(3):705–7. [DOI] [PubMed] [Google Scholar]
- 50.Jiggins CD, Linares M, Naisbit RE, Salazar C, Yang ZH, Mallet J. Sex-linked hybrid sterility in a butterfly. Evolution; international journal of organic evolution. 2001;55(8):1631–8. Epub 2001/10/03. . [DOI] [PubMed] [Google Scholar]
- 51.Abad-Franch F, Pavan MG, Jaramillo-O N, Palomeque FS, Dale C, Chaverra D, et al. Rhodnius barretti, a new species of Triatominae (Hemiptera: Reduviidae) from western Amazonia. Memórias do Instituto Oswaldo Cruz. 2013;108:92–9. 10.1590/0074-0276130434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heitzmann-Fontenelle T. Bionomia comparativa de triatomíneos. VI-Híbridos de Triatoma brasiliensis Neiva, 1911 X Triatoma lenti, Sherlocki & Serafim, 1967 (Hemiptera, Reduviidade). Memórias do Instituto Butantan. 1984;47:175–81. [Google Scholar]
- 53.Mendonca VJ, Alevi KCC, Pinotti H, Gurgel-Goncalves R, Pita S, Guerra AL, et al. Revalidation of Triatoma bahiensis Sherlock & Serafim, 1967 (Hemiptera: Reduviidae) and phylogeny of the T. brasiliensis species complex. Zootaxa. 2016;4107(2):239–54. 10.11646/zootaxa.4107.2.6 [DOI] [PubMed] [Google Scholar]
- 54.Perez R, Hernández M, Quintero O, Scvortzoff E, Canale D, Mendez L, et al. Cytogenetic analysis of experimental hybrids in species of Triatominae (Hemiptera-Reduviidae). Genetica. 2005;125(2–3):261–70. 10.1007/s10709-005-0369-z [DOI] [PubMed] [Google Scholar]
- 55.Ceccarelli S, Balsalobre A, Medone P, Cano ME, Gonçalves RG, Feliciangeli D, et al. DataTri, a database of American triatomine species occurrence. Scientific data. 2018;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roux C, Fraisse C, Romiguier J, Anciaux Y, Galtier N, Bierne N. Shedding light on the grey zone of speciation along a continuum of genomic divergence. PLoS biology. 2016;14(12):e2000234 10.1371/journal.pbio.2000234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pita S, Panzera F, Ferrandis I, Galvão C, Gómez-Palacio A, Panzera Y. Chromosomal divergence and evolutionary inferences in Rhodniini based on the chromosomal location of ribosomal genes. Memórias do Instituto Oswaldo Cruz. 2013;108(3):376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Valente VdC, Valente SAdS, Carcavallo RU, Rocha DdS, Galvão C, Jurberg J. Considerações sobre uma nova espécie do gênero Rhodnius stal, do Estado do Pará, Brasil (Hemiptera, Reduviidae, Triatominae. Entomol vectores. 2001;8(1):65–80. [Google Scholar]
- 59.Alevi K, Ravazi A, Franco-Bernardes M, Rosa J, Azeredo-Oliveira M. Chromosomal evolution in the pallescens group (Hemiptera, Triatominae). Genetics and Molecular Research. 2015;14(4):12654–9. 10.4238/2015.October.19.9 [DOI] [PubMed] [Google Scholar]
- 60.Bardella VB, Pita S, Vanzela ALL, Galvão C, Panzera F. Heterochromatin base pair composition and diversification in holocentric chromosomes of kissing bugs (Hemiptera, Reduviidae). Memórias do Instituto Oswaldo Cruz. 2016;111(10):614–24. 10.1590/0074-02760160044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Panzera F, Hornos S, Pereira J, Cestau R, Canale D, Diotaiuti L, et al. Genetic variability and geographic differentiation among three species of triatomine bugs (Hemiptera-Reduviidae). The American journal of tropical medicine and hygiene. 1997;57(6):732–9. [DOI] [PubMed] [Google Scholar]
- 62.Panzera F, Ferrandis I, Ramsey J, Ordonez R, Salazar‐Schettino P, Cabrera M, et al. Chromosomal variation and genome size support existence of cryptic species of Triatoma dimidiata with different epidemiological importance as Chagas disease vectors. Tropical Medicine & International Health. 2006;11(7):1092–103. [DOI] [PubMed] [Google Scholar]
- 63.Panzera F, Dujardin JP, Nicolini P, Caraccio MN, Rose V, Tellez T, et al. Genomic changes of Chagas disease vector, South America. Emerging Infectious Diseases. 2004;10(3):438 10.3201/eid1003.020812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Panzera F, Ferreiro MJ, Pita S, Calleros L, Pérez R, Basmadjián Y, et al. Evolutionary and dispersal history of Triatoma infestans, main vector of Chagas disease, by chromosomal markers. Infection, Genetics and Evolution. 2014;27:105–13. 10.1016/j.meegid.2014.07.006 [DOI] [PubMed] [Google Scholar]
- 65.Panzera Y, Pita S, Ferreiro M, Ferrandis I, Lages C, Pérez R, et al. High dynamics of rDNA cluster location in kissing bug holocentric chromosomes (Triatominae, Heteroptera). Cytogenetic and Genome Research. 2012;138(1):56–67. 10.1159/000341888 [DOI] [PubMed] [Google Scholar]
- 66.Crossa RP, Hernández M, Caraccio MN, Rose V, Valente SAS, da Costa Valente V, et al. Chromosomal evolution trends of the genus Panstrongylus (Hemiptera, Reduviidae), vectors of Chagas disease. Infection, Genetics and Evolution. 2002;2(1):47–56. [DOI] [PubMed] [Google Scholar]
- 67.Gómez-Palacio A, Jaramillo-Ocampo N, Triana-Chávez O, Saldaña A, Calzada J, Pérez R, et al. Chromosome variability in the Chagas disease vector Rhodnius pallescens (Hemiptera, Reduviidae, Rhodniini). Memórias do Instituto Oswaldo Cruz. 2008;103(2):160–4. [DOI] [PubMed] [Google Scholar]
- 68.Ueshima N. Cytotaxonomy of the triatominae (Reduviidae: Hemiptera). Chromosoma. 1966;18(1):97–122. [Google Scholar]
- 69.Schlick-Steiner BC, Steiner FM, Seifert B, Stauffer C, Christian E, Crozier RH. Integrative taxonomy: a multisource approach to exploring biodiversity. Annual review of entomology. 2010;55:421–38. Epub 2009/09/10. 10.1146/annurev-ento-112408-085432 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
(DOCX)
(a) CYTB; (b) ND4; (c) PCB; (d) TOPO; (e) URO; (f) ZNFP. Note that DXY scale for all genes is not the same.
(DOCX)
Discriminant analysis based on mtDNA (a) and nDNA (b). Densities for a single discriminant function are shown, with red being R. taquarussuensis and blue being R. neglectus.
(DOCX)
Data Availability Statement
All relevant data are within the manuscript.