

Abstract
Altered brain iron content in the striatum of premanifest and manifest Huntington disease (HD) has been reported. However, its natural history remains unclear. This study aims to investigate altered brain iron content in premanifest and early HD, and the iron deposition rate in these patients through a longitudinal one-year follow-up test, with quantitative magnetic susceptibility as an iron imaging marker. Twenty-four gene mutation carriers divided into three groups (further-from-onset, closer-to-onset and early HD) and 16 age-matched healthy controls were recruited at baseline, and of these, 14 carriers and 7 controls completed the one-year follow-up. Quantitative magnetic susceptibility and effective transverse relaxation rate (R2*) were measured at 7.0 Tesla and correlated with atrophy and available clinical and cognitive measurements. Higher susceptibility values indicating higher iron content in the striatum and globus pallidus were only observed in closer-to-onset (N = 6, p < 0.05 in caudate and p < 0.01 in putamen) and early HD (N = 9, p < 0.05 in caudate and globus pallidus and p < 0.01 in putamen). Similar results were found by R2* measurement. Such increases directly correlated with HD CAG-age product score and brain atrophy, but not with motor or cognitive scores. More importantly, significantly higher iron deposition rate (11.9%/yr in caudate and 6.1%/yr in globus pallidus) were firstly observed in closer-to-onset premanifest HD and early HD as compared to controls. These results suggest that monitoring brain iron may provide further insight into pathophysiology in HD disease progression, and may provide a biomarker for clinical trials.
Keywords: Huntington disease, Brain iron deposition, Brain atrophy, QSM, Cross-sectional study, Longitudinal study
1. Introduction
Huntington disease (HD) is a devastating inherited neurodegenerative disorder, caused by a cytosine-adenine-guanine (CAG) repeat expansion of the huntingtin (HTT) gene (Macdonald et al. 1993). The encoded polyglutamine expansions in mutant Htt protein lead to a toxic effect on neuron populations and cause neuronal dysfunction and death, resulting in progressive decline in cognitive, motor, and psychiatric functions (Ross et al. 2014; Ross and Tabrizi 2011). Early in the disease course, there is selective atrophy in basal ganglia structures, especially the corpus striatum, consisting of the caudate and putamen. The length of the CAG expansion is inversely correlated with the age of HD onset, making it possible to predict the approximate disease onset (Rosenblatt et al. 2012). A variable with the form of age times CAG repeat - the approximate threshold for disease - termed the CAG Age Product or “CAP” score has been found to be a useful way of tracking the extent of exposure to the CAG repeat expansion mutation. Motor onset of manifest HD is defined when a clinician determines that there is an extrapyramidal movement disorder indicative of HD in someone at risk. Individual mutation status can be identified by genetic testing years before clinical symptoms appear (Chan et al. 2014). There is no disease-modifying treatment currently available, though strategies for Htt protein lowering via RNA interference are being developed, using antisense oligonucleotides to be delivered intrathecally, or AAV vectors to be delivered intra-parenchymally (Wild and Tabrizi 2017). Sensitive and robust imaging biomarkers are therefore desired to monitor disease progression, and to help develop novel therapeutic approaches and new drugs for disease intervention, especially at premanifest stage.
Brain atrophy measured through structural MRI especially in the striatum is an imaging biomarker for HD that can be detected before onset of disease symptoms (Aylward et al. 2004; Ross et al. 2014), which has been demonstrated in large-scale longitudinal studies (Aylward et al. 2012; Aylward et al. 2011b; Tabrizi et al. 2012; Tabrizi et al. 2013; van den Bogaard et al. 2011). Besides brain atrophy, abnormal iron accumulation in the brain is commonly recognized as an important marker of pathological neuronal damage(Dixon and Stockwell 2014), and may predispose individuals to high risk of motor and cognitive function decline (Li et al. 2015). Increased iron accumulation in the basal ganglia and cortical regions has been associated with neurodegenerative diseases such as Parkinson disease (PD) (Du et al. 2016; He et al. 2015) and Alzheimer disease (AD) (Ayton et al. 2017; van Bergen et al. 2016b; Zecca et al. 2004). Abnormal iron deposition has long been suggested to play a role in HD pathophysiology (Bartzokis et al. 2007; Rosas et al. 2012). Since normal Htt protein is required for iron homoeostasis(Hilditch-Maguire et al. 2000), mutant HTT may lead to abnormally increased tissue iron through different mechanisms in iron metabolism (Gomez-Tortosa et al. 2001; Rivera-Mancia et al. 2010).
Different MR imaging techniques have been used to measure brain iron levels in HD. For example, increased iron levels in the basal ganglia have been indicated in manifest HD by using transverse relaxation time (T2) or T2-weight hypointensity (Jurgens et al. 2010; Vymazal et al. 2007). Bartzokis et al. performed field-dependent R2 increase (FDRI) imaging and suggested that ferritin levels in the striatum were significantly increased in HD (Bartzokis et al. 2007). Other MRI iron imaging techniques such as magnetic field correlation (MFC) (Dumas et al. 2012), effective relaxation rate (R2*) (Sanchez-Castaneda et al. 2015) and phase imaging (Rosas et al. 2012) have also been used to study brain iron levels in HD. However, known technical limitations of these imaging methods could lead to reduced sensitivity and specificity to disease related tissue iron changes (Haacke et al. 2005; Ropele and Langkammer 2017; Wang and Liu 2015). For example, R2*-based iron measures can be contaminated by the background field gradient effects, while R2 can be affected by different tissue water contents. Phase or field shift-based measures are nonlocal, i.e. affected by the magnetic susceptibility (χ) of surrounding tissue. The recent development of quantitative susceptibility mapping (QSM) overcomes these technical difficulties, providing a direct measure of bulk tissue magnetic susceptibility which has been shown to correlate linearly with tissue iron content in gray matter (Deistung et al. 2013; Langkammer et al. 2012; Lim et al. 2013), with high sensitivity and specificity to tissue iron changes (Chen et al. 2017; Deistung et al. 2017; Langkammer et al. 2018; Liu et al. 2015; Wang et al. 2017).
Using quantitative susceptibility as an imaging measure of tissue iron, recent studies have shown elevated iron levels in the striatum and globus pallidus in premanifest HD patients as compared to matched controls (Dominguez et al. 2016; van Bergen et al. 2016a). Such changes in iron levels have also been found to correlate with striatal atrophy and CAP score (Dominguez et al. 2016; van Bergen et al. 2016a). However, the natural history of abnormal iron deposition in HD is still not well understood. Namely, when extra iron starts to deposit in the striatum during the long premanifest stage, and how the iron deposition rate changes as the disease progresses has not been reported. This study therefore aims to investigate altered brain iron content (as measured indirectly through magnetic susceptibility χ) in premanifest HD at different time length to onset and early stage HD, and to assess the iron deposition rate in these patients through a longitudinal one-year follow-up test. In the present study, QSM and R2* measures were quantified in selected brain regions that have been reported to be affected in HD including striatum and globus pallidus. Other brain regions including hippocampus, substantia nigra and red nucleus that are related to movement and cognition functions (Middleton and Strick 2000; Williams et al. 2014) were also included. HD patients were divided into three disease stages: (i) more than 8 years before estimated onset (further-from-onset), (ii) less than 8 years before estimated onset (closer-to-onset), and (iii) early stage HD, and compared with healthy controls. Potential associations between iron level, structure volume and clinical assessment were tested. For the longitudinal study, closer-to-onset and early stage HD patients were pooled due to limited sample size. Differences in the changing rate of iron deposition, atrophy and clinical measures in HD patients were tested in comparison with controls using the General Linear Model (GLM).
2. Materials and Methods
2.1. Subjects
Twenty-four subjects (13 males and 11 females) carrying CAG-expanded HTT allele were recruited through the Baltimore Huntington Disease center at the Johns Hopkins University School of Medicine. The age range of these patients was from 24 to 60 years old (mean ± standard deviation of 44.8 ± 15.8 y/o). In addition, sixteen age-matched healthy controls (8 males, 8 females; 42.5 ± 12.9 y/o) were also recruited. For the longitudinal study, 14 out of the 24 CAG-expanded HTT allele carriers and 7 out of the 16 controls participated in a follow-up exam after approximately one year (1.0 ± 0.1 years). All aspects of the study were approved by the Johns Hopkins University Institutional Review Board, and all participants gave written informed consent.
To estimate the progression status of HD pathology in the patients, the CAG-age product scaled (CAPs) score as CAPs = Age × (CAG-33.66) /432.3326 was calculated based on age at study entry and CAG repeat length (Zhang et al. 2011). According to the CAPs score and predicted disease onset age, all the carriers (N = 24) were further divided into three groups, ‘> 8yr pre-HD’ (N = 9) for premanifest HD patients with estimated disease onset beyond 8 years (further-from-onset), ‘< 8yr pre-HD’ (N = 6) for premanifest HD patients with estimated onset within 8 years (closer-to-onset), and ‘early HD’ (N = 9) for participants with early-stage HD. The numbers of carriers in the >8yr pre-HD, <8yr pre-HD and early HD groups who participated in the one-year follow-up were 6, 4, 4, respectively.
Tests and interviews by clinical personnel were performed on the day of scanning for all gene-positive subjects: the Unified Huntington’s Disease Rating Scale (UHDRS) to determine the motor, behavior and function score, the Montreal Cognitive Assessment (Nasreddine et al. 2005) and Symbol-Digit Modalities Test (Parmenter et al. 2007) to screen for mild cognitive impairment, and the Verbal Fluency Test as an estimate of verbal fluency. Subjects in premanifest HD with known affective neuropsychiatric disorders, severe cognitive impairments or other acute medical diseases were excluded. The condition of controls as healthy was determined by interviews and scores on the Montreal Cognitive Assessment, Mini-Mental State Examination, Verbal Fluency Test, and memory tests (immediate and delayed recall).
The demographic information and clinical measurements for all the healthy controls and CAG-expand HTT allele carriers (the three HD subgroups and all HD as a whole group) are summarized in Table 1.
TABLE 1:
Demographic parameters and clinical assessment scores of controls and CAG-expanded HTT allele carriers (HD group
| Controls | All HD | > 8yr pre-HD | < 8yr pre-HD | Early HD | |
|---|---|---|---|---|---|
| Number | 16 | 24 | 9 | 6 | 9 |
| Sex (male/female) | 8:8 | 13:11 | 6:3 | 1:5 | 6:3 |
| Age (yr) | 44.8 ± 15.8 | 42.5 ± 12.9 | 34.4 ± 10.9 | 41.7 ± 11.5 | 50.8 ± 10.9 |
| Education (yr) | 16.1 ± 2.0 | 15.3 ± 2.2 | 14.7 ± 1.9 | 16.8 ± 2.6 | 14.9 ± 2.0 |
| CAG length | -- | 43.8 ± 4.3 | 41.8 ± 2.8 | 44.0 ± 4.2 | 45.6 ± 5.1 |
| CAPs score | -- | 0.9 ± 0.4 | 0.6 ± 0.2 | 1.0 ± 0.1 | 1.3 ± 0.2 |
| UHDRS Motor score | -- | 15.9 ± 17.4 | 4.3 ± 4.3 | 14.2 ± 11.8 | 28.6 ± 20.8 |
| UHDRS Function score | 24.9 ± 0.5 | 23.3 ± 3.5* | 25.0 ± 0.0 | 23.0 ± 4.0 | 21.7 ± 4.4* |
| UHDRS Behavior score | 3.5 ± 4.7 | 9.6 ± 11.2* | 7.0 ± 5.7 | 7.7 ± 4.5 | 13.6 ± 16.9 |
| Total functional capacity | 13 ± 0.0 | 11.5 ± 3.0* | 12.9 ± 0.3 | 11.0 ± 3.3 | 10.3 ± 3.9 |
| Montreal Cognitive Assessment | 29.6 s 0.7 | 28.2 ± 2.8* | 29.6 ± 0.2 | 28.2 ± 2.7 | 26.8 ± 3.6* |
| Symbol-Digit Modalities Test score | -- | 51.5 ± 22.1 | 58.1 ± 16.9 | 54.0 ± 19.8 | 42.1 ± 27.7 |
| Verbal Fluency Test score | 47.4 s 12.8 | 41.3 ± 17.4 | 50.1 ± 11.8 | 42.5 ± 16.9 | 30.5 ± 18.7* |
NOTES: CAPs score = CAG-age product scaled score; UHDRS = Unified Huntington’s Disease Rating Scale; Data are presented as means ± standard deviation
Significant difference as compared to controls with p < 0.05.
2.2. MRI Measures
Subjects were imaged on a 7T Achieva scanner (Philips Healthcare, Best, Netherlands) equipped with a quadrature transmit 32-channel receive head coil (Nova Medical, Wilmington, Massachusetts). A three-dimensional (3D) multi-echo gradient-recalled echo (GRE) sequence was used for quantitative susceptibility measurements including QSM and R2*. The acquisition parameters were: TR/TE1/∆TE = 68/4/2 ms, 8 echoes, flip angle = 9°, voxel size = 1×1×1 mm3, FOV = 220×220×100 mm3, bandwidth = 1530 Hz/voxel, SENSE factor of 2.5×2, transverse slab, scan time = 7 minutes 14 seconds. Both magnitude and phase images were saved for QSM reconstruction. In addition, a 3D T1-weighted MPRAGE sequence was used for anatomical referencing, automated image segmentation and for measuring brain structure volume with the following parameters: TR/TE = 4.1/1.8 ms, voxel size = 1 × 1 × 1 mm3, sagittal acquisition with FOV = 220 × 220 × 180 mm3, TFE factor of 352, SENSE factor of 2×2, scan time = 2 minutes 20 seconds. All images were examined for possible image artifacts or abnormalities.
2.3. Quantitative Susceptibility and R2* Mapping
Several processing steps were applied to reconstruct the quantitative susceptibility maps from the GRE phase images. First, phase wraps in the GRE phase images were removed by a Laplacian-based phase unwrapping method (Li et al. 2011). The GRE magnitude image at the first echo (TE = 4 ms) was skull-stripped using FSL BET (Smith 2002) and eroded 2 voxels to obtain a brain mask. Frequency shift maps (in Hz) were then obtained by dividing the phase by 2π*TE. Subsequently, the background field was removed by using the VSHARP method with a maximum kernel radius of 6 mm and a truncated svd regularization threshold of 0.05 (Schweser et al. 2011; Wu et al. 2012b). After removal of the background field, the resulting local frequency shift maps of the last five echoes (with TEs of 10 ms to 18 ms) were averaged to obtain a higher signal-to-noise ratio (SNR) as compared to single-echo reconstruction (Wu et al. 2012a). The ill-conditioned QSM dipole inversion was solved by using a modified structural feature based collaborative reconstruction algorithm (SFCR) (Bao et al. 2016) with only L1-norm based regularization i.e. λ1 = γ1 = 100 and λ2 = γ2 = 0 and an updated adaptive ADMM solver (Chan et al. 2011). The final reported susceptibility values for each subject were referenced to the mean susceptibility values of CSF regions in the frontal and body parts of the lateral ventricles. In addition, R2* maps were reconstructed by mono-exponential fitting to the square of the magnitude data of all available echoes in each voxel using the power method (Deistung et al. 2013).
2.4. Image Segmentation Protocol
The procedures for segmenting regions of interests (ROIs) in deep gray matter were as follows. First, the T1-weighted MPRAGE image was co-registered to the GRE magnitude image acquired at TE of 8 ms. The co-registered T1 image was then segmented using a multi-atlas matching approach which is developed as part of the Johns Hopkins University brain atlas (Tang et al. 2015; Zhang et al. 2014). Selected ROI masks in caudate nucleus, putamen, globus pallidus, hippocampus, substantia nigra and red nucleus were extracted. In addition, masks of the CSF regions in the lateral ventricle were also extracted to be used as QSM reference regions. Manual corrections on the main ROI masks were performed based on magnetic susceptibility contrast, while the CSF masks were corrected based on T1 contrast if deemed necessary after visual checking (by L.C.). All ROIs were then eroded by two voxels to avoid partial volume effects and used to calculate mean magnetic susceptibility and R2* values. The volume of each structure was determined by multiplying the number of voxels in specific ROIs with the voxel size. To eliminate the effect of different brain sizes across subjects, individual structural volume was normalized by the following approach: Corrected Structure Volume = Original Structure Volume × (Group Mean Intracranial Volume/Subject Intracranial Volume).
2.5. Statistical analysis
To investigate the differences in brain iron content and structure volume between the CAG-expanded HTT allele carriers and controls, 1-way MANCOVA was performed, with the mean magnetic susceptibility, R2* and corrected volume of each selected brain region as the outcome variables, while controlling for age and gender. Such tests were conducted for all the carriers as a whole group (All HD) and for each HD subgroup, i.e. > 8yr pre-HD, < 8yr pre-HD and early HD group in comparison to controls. The p value in multiple tests after Benjamini-Hochberg correction was considered significant at p ≤ 0.05.
To examine the associations of MRI measures with the HD gene mutation burden, Pearson partial correlation analyses was conducted with magnetic susceptibility, R2*, or corrected volume of each ROI as the outcome variable, and CAPs score as predictor while controlling for gender. Spearman’s rank partial correlation was also performed between MRI measurements, i.e. magnetic susceptibility, R2*, or corrected volume of each ROI, and ranking based clinical assessments such as UHDRS motor score, UHDRS function score, UHDRS behavior score, total functional capacity or Montreal Cognitive Assessment score while controlling for age and gender.
Longitudinal changes in MRI and clinical measures were analyzed using General Linear Model (GLM) framework. Due to the limited number of patients completing the one-year follow-up, all HD patients were first grouped together (n=14) assuming no significant difference between the iron-deposition rate among all HD patients. Longitudinal changes in mean susceptibility, R2*, corrected volume of each selected ROI, UHDRS function score, UHDRS behavior score, Total functional capacity, Montreal Cognitive Assessment score and Verbal Fluency Test score were then all analyzed by means of GLM model, i.e. each measure was tested as an outcome variable of a GLM model, for repeated measures with time (2 levels) as a repeated measures factor, group (control [n = 7] vs. HD group [n = 14]) as a between subject factor, and age and gender were set as potential confounders. The GLM model can be specified as x1 = time + time×group + time×age + time×gender + error term. When a significant group × time interaction effect was found, post-hoc analyses were performed within groups using a separate GLM with time as a repeated measures factor which can be written as x2 = time + time×age + time×gender + error term. Since it has been demonstrated that atrophy rate in the striatum and the decline rate of various clinical and cognitive measures increase steadily during the progression of the Huntington disease and may even accelerate when closer to or after disease onset (Ross et al. 2014; Tabrizi et al. 2013), it is also reasonable to assume that HD patients at different disease stages may have different iron-deposition rate. Thus, similar GLM analysis was conducted with <8yr pre-HD and early HD patients pooled together as a group (n=8) to be tested against the control group. Subsequently, changes in clinical and MRI-based measurements over time were analyzed by using GLMs for repeated measures with time (2 levels) as a repeated measures factor, group (control [n = 7] vs. HD-2 group (defined as <8yr pre-HD group plus early HD group) [n = 8]) as a between subject factor, and age and gender added as covariates. When a significant group × time interaction effect was found, post-hoc analyses were performed within groups using a separate GLM with time as a repeated measures factor.
3. Results
As a whole group, all HD patients displayed no differences as compared to controls in terms of gender, age, education and Verbal Fluency Test score, but showed higher UHDRS behavior score and lower UHDRS function score, total functional capacity and Montreal cognitive assessment than the controls (p < 0.05). Compared with healthy controls, the early HD group exhibited lower UHDRS function score, Montreal cognitive assessment and Verbal Fluency Test score (p < 0.05). Typical quantitative susceptibility maps of a 61-year-old female healthy control and a 60-year-old female early stage HD patient are shown in Fig.1. Higher magnetic susceptibility (represented by higher image intensity in QSM) in deep gray matter, especially in the caudate and putamen (marked by black arrows), can be seen in the early HD patient (Fig. 1B) as compared to the age-matched control (Fig. 1A).
Figure 1:
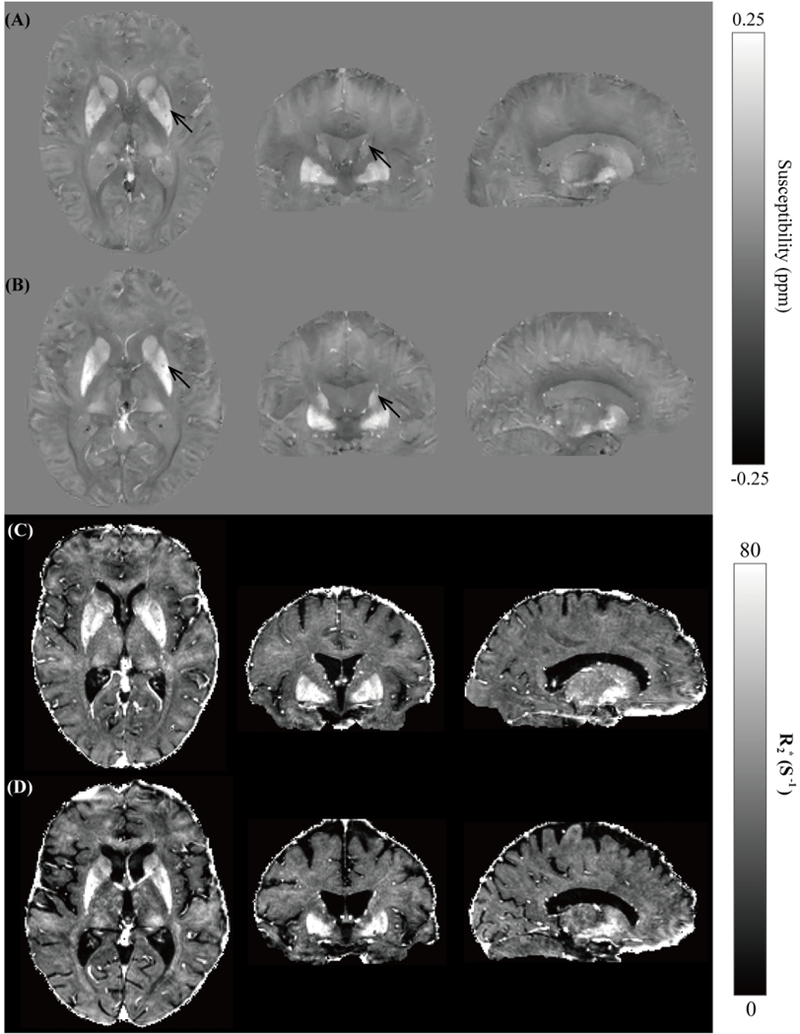
Susceptibility maps in three orthogonal views showing the striatum and globus pallidus of a 61-year-old female healthy control (A) and a 60-year-old female early stage HD patient (B). The corresponding R2* maps at similar anatomical locations are shown in (C) and (D) for the control and HD patient, respectively.
As shown in Table 2, group comparison between controls and all HD patients showed a significant susceptibility increase in the caudate, globus pallidus (p < 0.05), and putamen (p < 0.001). Increases in the caudate and putamen (p < 0.05) were also observed in R2* measurements. In addition to susceptibility and R2* changes, a significant volume decrease was found in caudate (p < 0.001), putamen, globus pallidus and substantia nigra (p < 0.01). Age and gender serve as covariates to control for their effects in all above analyses.
TABLE 2:
Mean susceptibility (χ), mean R2*, and corrected volume of ROI in controls and all HD patients.
| χ (ppm) |
R2*(s−1) |
Corrected Volume (mL) |
||||
|---|---|---|---|---|---|---|
| Control | All HD | Control | All HD | Control | All HD | |
| CN | 0.041 ± 0.004 | 0.056 ± 0.004* | 45.26 ± 1.51 | 50.86 ± 2.47* | 8.0 ± 0.2 | 6.5 ± 0.3*** |
| PT | 0.061 ± 0.006 | 0.084 ± 0.007*** | 54.41 ± 2.51 | 59.51 ± 2.32* | 9.0 ± 0.2 | 7.3 ± 0.5** |
| GP | 0.132 ± 0.004 | 0.155 ± 0.008* | 82.86 ± 3.30 | 88.76 ± 3.73 | 4.3 ± 0.2 | 3.5 ± 0.2** |
| HIPP | −0.005 ± 0.002 | −0.003 ± 0.002 | 31.94 ± 1.20 | 29.51 ± 1.14 | 6.8 ± 0.2 | 6.8 ± 0.1 |
| SN | 0.113 ± 0.008 | 0.115 ± 0.008 | 81.97 ± 5.32 | 84.32 ± 3.05 | 1.5 ± 0.1 | 1.3 ± 0.1** |
| RN | 0.081 ± 0.011 | 0.083 ± 0.006 | 62.82 ± 4.02 | 69.47 ± 3.80 | 0.6 ± 0.1 | 0.6 ± 0.1 |
NOTES: CN = caudate nuclei, PT = putamen, GP = globus pallidus, HIPP = hippocampus, SN = substantia nigra, RN = red nucleus; Data are presented as means ± standard error (SE)
p < 0.05
p < 0. 01
Significant difference as compared to controls with p < 0.001.
The multiple comparisons between HD subgroups, i.e. >8yr pre-HD, <8yr pre-HD and early HD, versus controls are shown in Figs. 2. (A)–(C). Regarding the magnetic susceptibility, the <8yr pre HD group showed significant increases in caudate (p < 0.05) and putamen (p < 0.01), and the early HD group showed significant increases in caudate, globus pallidus (p < 0.05), and putamen (p < 0.01) in comparison with the controls, with age and gender as covariates. (Fig. 2A). Similarly, significant increases in R2* measurements were observed in caudate (p < 0.001) and putamen (p < 0.05) of <8yr pre-HD group, and in caudate, putamen and globus pallidus (p < 0.05) of early HD group (Fig. 2B). Moreover, significant decreases in tissue volume, i.e. atrophy, are found in caudate and putamen (p < 0.01), globus pallidus and substantia nigra (p < 0.05) of <8yr pre-HD group as compared to controls; and in caudate, putamen, globus pallidus (p < 0.001) and substantia nigra (p < 0.01) of early HD group (Fig. 2C). With regard to the partial correlation analysis, significant inverse correlations in caudate (r = −0.586, p = 0.004), putamen (r = −0.425, p = 0.049) and globus pallidus (r = −0.730, p = 0.0001) were found between the magnetic susceptibility values and corrected ROI volumes (Fig. 2D). Similar inverse correlations were observed only in caudate (r = −0.466, p = 0.029) and globus pallidus (r = −0.582, p = 0.004) between the R2* values and corrected ROI volumes (Fig. 2E).
Figure 2:
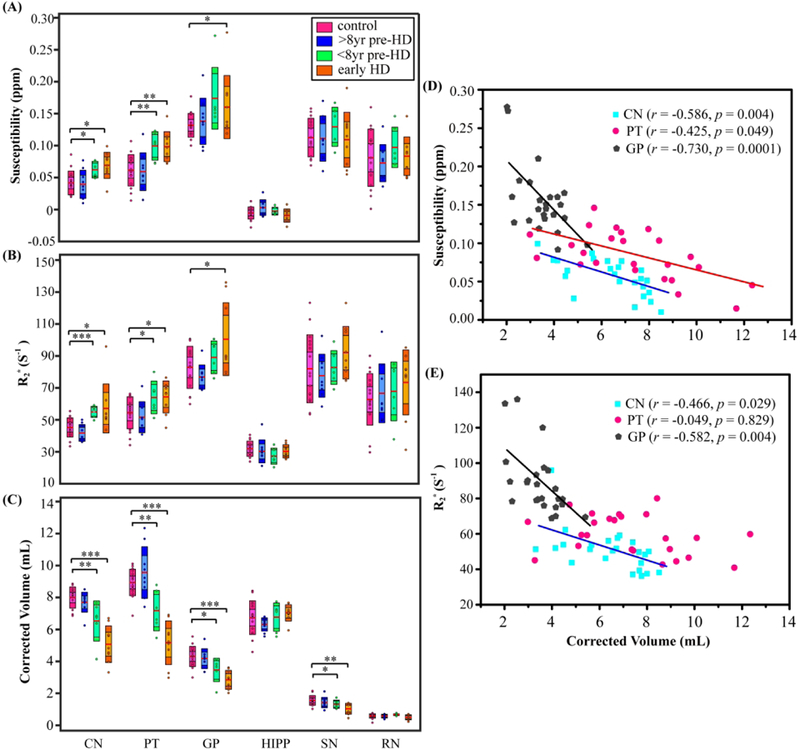
Mean magnetic susceptibility (A), R2* (B) and corrected volume (C) in each ROI of the control, >8yr pre-HD, <8yr pre-HD, and early HD groups. The boxes shaded with lighter colors denote the 95% CI and the darker colors denote ±1 SD. The red lines represent the mean. *, ** and *** indicate significant differences with p < 0.05, p < 0.01 and p < 0.001 controlling for age and gender. Correlation analysis of magnetic susceptibility (D) and R2* (E) with corrected structure volumes while controlling for age and gender in all HD patients. CN: caudate nucleus, PT: putamen, GP: globus pallidus, HIPP: hippocampus, SN: substantia nigra, RN: red nucleus.
Partial correlation analysis, with the predictor set as the UHDRS motor, function and behavior scores, total functional capacity score or Montreal Cognitive Assessment score, did not lead to significant correlations between these clinical metrics and susceptibility or R2* values in any selected brain region. However, as shown in Fig.3, the CAPs score showed positive correlations with magnetic susceptibility values in caudate (r = 0.622, p = 0.002) and putamen (r = 0.557, p = 0.006) in all HD patients, with gender as covariate. Such positive correlations were also found between R2* values and CAPs in caudate (r = 0.670, p = 0.0004), but only showed a positive trend in putamen (r = 0.355, p = 0.096). Moreover, we observed strong negative correlation between CAPs score and corrected ROI volumes in caudate (r = − 0.806, p < 0.001), putamen (r = −0.879, p < 0.001) and globus pallidus (r = −0.736, p < 0.001) with gender as covariate.
Figure 3:
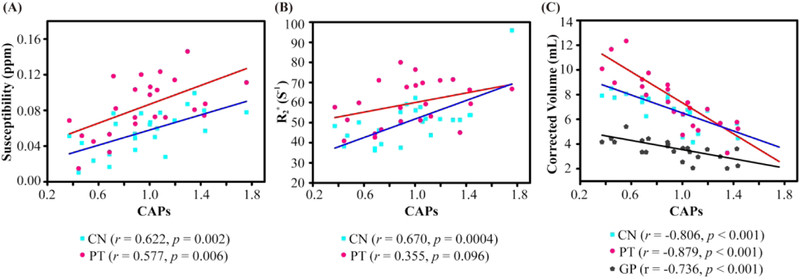
Correlation analysis of magnetic susceptibility (A), R2* (B) and corrected volume (C) with CAPs while controlling for gender in all HD patients. CN: caudate nucleus, PT: putamen, GP: globus pallidus.
In the longitudinal study, no significant group × time interaction effects in clinical measurement, magnetic susceptibility, R2* and corrected volume were found between controls and the whole HD group from the baseline to the one-year follow up by means of GLMs (Table 3). Therefore, post-hoc analyses were not implemented. When considering the HD-2 group (<8yr pre-HD plus early HD patients), GLM analysis for repeated measures showed a group × time interaction effect for the susceptibility of caudate [F-statistic(repeated time point-1, error degree of freedom) as F(1,13) = 4.94; p = 0.045], the susceptibility of globus pallidus [F(1,13) = 5.03; p = 0.043] and the corrected volume of caudate [F(1,13) = 9.77; p = 0.008]. Post-hoc analyses for the control and >8yr pre-HD group showed no significant changes of these measurements over one-year, whereas the combined closer-to-onset and early HD patients (HD-2 group) revealed an increase in susceptibility of caudate [F(1,7) = 23.35; p = 0.002] and globus pallidus [F(1,7) = 5.70; p = 0.048], as well as a decrease in volume of caudate [F(1,7) = 5.55; p = 0.050] over one-year time (Fig. 4). The changes in tissue magnetic susceptibility (indicating iron deposition rate) in the HD-2 group were 11.9%/yr in caudate and 6.1 %/yr in globus pallidus. The loss of tissue volume (indicating atrophy rate) was 4.6%/yr in caudate.
TABLE 3:
Clinical assessment scores and MRI measurements in selected ROIs of controls, HD group and HD-2 group over one-year time.
| Clinical and MRI measures | Control (n = 7) |
HDgroup(n=14) |
HD-2 group (n = 8) |
|||
|---|---|---|---|---|---|---|
| Baseline | Follow-up | Baseline | Follow-up | Baseline | Follow-up | |
| UHDRS Function score | 25.0 ± 0.0 | 25.0 ± 0.0 | 24.7 ± 0.2 | 24.8 ± 0.2 | 24.5 ± 0.3 | 24.6 ± 0.3 |
| UHDRS Behavior score | 2.3 ± 1.2 | 1.1 ± 0.6 | 10.1 ± 3.5 | 11.2 ± 3.2 | 12.5 ± 5.9 | 12.4 ± 5.1 |
| Total Functional Capacity | 13.0 ± 0.0 | 13.0 ± 0 | 12.6 ± 0.3 | 12.6 ± 0.3 | 12.5 ± 0.5 | 12.3 ± 0.5 |
| Montreal Cognitive Assessment | 29.3 ± 0.4 | 29.8 ± 0.1 | 28.9 ± 0.4 | 28.9 ± 0.4 | 28.4 ± 0.5 | 28.4 ± 0.6 |
| Verbal Fluency Test score | 41.6 ± 4.0 | 49.8 ± 5.6 | 45.7 ± 3.8 | 48.3 ± 3.8 | 43.6 ± 5.3 | 43.0 ± 4.5 |
| Magnetic Susceptibility (ppm) | ||||||
| CN | 0.043 ± 0.006 | 0.039 ± 0.005 | 0.052 ± 0.005 | 0.055 ± 0.005 | 0.059 ± 0.003 | 0.066 ± 0.003** |
| PT | 0.060 ± 0.007 | 0.062 ± 0.007 | 0.081 ± 0.007 | 0.079 ± 0.008 | 0.094 ± 0.006 | 0.092 ± 0.005 |
| GP | 0.137 ± 0.008 | 0.135 ± 0.009 | 0.147 ± 0.008 | 0.149 ± 0.008 | 0.148 ± 0.008 | 0.157 ± 0.007* |
| HIPP | −0.004 ± 0.004 | −0.006 ± 0.004 | −0.003 ± 0.004 | −0.006 ± 0.003 | −0.009 ± 0.003 | −0.009 ± 0.002 |
| SN | 0.109 ± 0.011 | 0.117 ± 0.010 | 0.116 ± 0.009 | 0.120 ± 0.008 | 0.120 ± 0.010 | 0.128 ± 0.008 |
| RN | 0.078 ± 0.012 | 0.082 ± 0.010 | 0.090 ± 0.008 | 0.087 ± 0.010 | 0.098 ± 0.009 | 0.102 ± 0.011 |
| R2* value (S−1) | ||||||
| CN | 44.41 ± 2.21 | 44.43 ± 0.95 | 49.25 ± 2.11 | 51.22 ± 3.34 | 54.65 ± 1.46 | 55.93 ± 3.94 |
| PT | 54.24 ± 3.71 | 52.14 ± 3.29 | 60.68 ± 3.28 | 62.66 ± 4.60 | 66.70 ± 3.50 | 68.04 ± 4.08 |
| GP | 82.21 ± 5.22 | 82.24 ± 5.96 | 86.85 ± 4.47 | 92.83 ± 4.43 | 93.50 ± 6.58 | 99.59 ± 5.08 |
| HIPP | 30.60 ± 2.04 | 26.74 ± 1.21 | 29.70 ± 0.87 | 29.44 ± 1.11 | 30.56 ± 1.33 | 28.64 ± 1.26 |
| SN | 80.74 ± 9.29 | 86.46 ± 9.01 | 85.73 ± 4.15 | 89.84 ± 5.64 | 93.92 ± 5.34 | 99.15 ± 7.39 |
| RN | 61.87 ± 6.59 | 73.11 ± 6.36 | 72.97 ± 3.87 | 78.78 ± 4.96 | 80.11 ± 3.76 | 88.59 ± 6.78 |
| Corrected volume (mL) | ||||||
| CN | 8.1 ± 0.3 | 8.5 ± 0.3 | 6.8 ± 0.3 | 6.9 ± 0.4 | 6.3 ± 0.4 | 6.0 ± 0.5* |
| PT | 9.0 ± 0.3 | 9.3 ± 0.4 | 8.0 ± 0.5 | 8.5 ± 0.5 | 6.8 ± 0.5 | 7.3 ± 0.5 |
| GP | 4.5 ± 0.3 | 4.6 ± 0.2 | 3.8 ± 0.2 | 4.0 ± 0.2 | 3.4 ± 0.2 | 3.6 ± 0.2 |
| HIPP | 6.9 ± 0.4 | 6.5 ± 0.6 | 6.7 ± 0.2 | 7.0 ± 0.4 | 7.0 ± 0.2 | 7.4 ± 0.2 |
| SN | 1.7 ± 0.1 | 1.4 ± 0.1 | 1.4 ± 0.1 | 1.3 ± 0.1 | 1.4 ± 0.1 | 1.3 ± 0.1 |
| RN | 0.7 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 0.1 |
NOTE: CN = caudate nuclei, PT = putamen, GP = globus pallidus, HIPP = hippocampus, SN = substantia nigra, RN = red nucleus Data are presented as means ± standard error (SE).
HD group includes all HD patients participated in the one-year follow-up; HD-2 group contains the <8 pre-HD and early HD patients participated in the one-year follow-up.
p < 0.05
Significant difference over one-year duration with p < 0.01.
Figure 4:
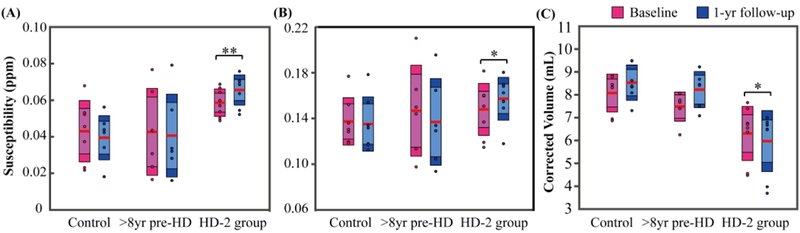
Longitudinal changes of magnetic susceptibility in caudate nuclei (A) and globus pallidus (B) and changes of corrected volumes in caudate nuclei (C) in the one-year follow-up exam. The boxes shaded with lighter colors denote the 95% CI and the darker colors denote ±1 SD. The red lines represent the mean. * and ** indicate significant difference over one-year with p < 0.05 and p < 0.01. HD-2 group contains the <8yr pre-HD and early HD patients.
4. Discussion
Two major findings emerge from our study. First, higher magnetic susceptibility values as measured by QSM indicating higher iron content in the striatum and globus pallidus were observed in closer-to-onset premanifest HD and early HD patients, but not in the further-from-onset premanifest HD group (>8yr, an average of 20 years in this study) as compared to controls. Though limited by the small sample size, this study to the best of our knowledge is the first to investigate altered brain iron content in premanifest HD at different time length to onset. Second, significantly higher iron deposition rates (11.9%/yr in caudate and 6.1%/yr in globus pallidus) were observed in closer-to-onset premanifest HD and early HD as compared to controls from our longitudinal study over one-year time period. Such findings, not reported before, help us gain more knowledge and insight on the natural history of abnormal iron deposition in HD, i.e. when extra iron starts to deposit in the striatum during the long premanifest stage, and how the iron deposition rate changes as the disease progresses.
With regard to the elevated iron deposition in caudate and putamen in premanifest HD as measured indirectly using QSM, the present study agrees in general with previous studies by us and others (Dominguez et al. 2016; van Bergen et al. 2016a). It was noticed that the premanifest HD populations in previous studies resemble the <8yr premanifest (closer-to-onset) HD group more in this study in terms of disease stage, i.e. with a mean CAPs score of 0.9 (van Bergen et al. 2016a) or 0.85 (Dominguez et al. 2016). Such elevated iron deposition was observed to be not significant in the further-from-onset stage, i.e. in the >8yr premanifest HD group in this study (20.3 ± 10.9 estimated years to onset). Such non-significance in brain iron changes at very early stage of the disease could of course be due to the small sample size used in this study. Such elevated iron deposition becomes more obvious at closer-to-onset stage (3.8 ± 2.5 estimated years to onset). Elevated iron content in globus pallidus was only found significant in the early HD patients in the present study (using either QSM or R2*). This is discrepant with the two previous QSM studies showing elevated tissue susceptibility in globus pallidus in premanifest HD. Such discrepancy is attributed partly to the limited sample size in the <8yr pre-HD group (n = 6) in the present study.
In terms of brain atrophy, our study results are consistent with existing literature suggesting HD-related neuronal damage to begin from the striatum (more significant volume loss), whereas a secondary neurodegenerative process may affect the globus pallidus and substantia nigra (Ross and Tabrizi 2011; van Bergen et al. 2016a). Controversies still exist considering the atrophy in globus pallidus in the premanifest stages. Our previous study reported no significant decrease in the volume of globus pallidus in the premanifest HD even though with observed susceptibility changes (van Bergen et al. 2016a), while several other studies reported the opposite (Jurgens et al. 2008; van den Bogaard et al. 2011). The present study further suggests that the volume loss in globus pallidus is distinguishable in the <8yr pre-HD group, but not in >8yr pre-HD group agreeing with the general understanding that brain atrophy becomes more severe as the disease progresses. In addition, the atrophy in caudate and putamen, which happens in a decade or more before predicted disease onset (Aylward et al. 1996; van den Bogaard et al. 2011), was not found in the >8yr pre-HD group in our study. This is most likely due to the small sample size and relatively long time to disease onset of this group of patients (20 years to onset on average) thus relatively small effect size of atrophy. Furthermore, the corrected volume in some regions, e.g. in the putamen of the HD and HD-2 group over one-year time showed some increase (Table 3), though not statistically significant. The reason of such increase may be partly due to the inaccuracy of the anatomical parcellation, i.e. in the ROI delineation procedure. Note that since we defined these ROIs mainly based on susceptibility contrast, it may be possible that the boundaries of these deep gray matter regions expand visually on QSM maps due to extra iron deposition or demyelination in surrounding white matter area.
The significant correlations between corrected ROI volumes and iron levels in caudate, putamen and globus pallidus demonstrate that atrophy and iron deposition in the striatum and pallidum observed in HD are two processes that are linked in time, which agrees with previous studies using R2* and QSM (Dominguez et al. 2016; van Bergen et al. 2016a). It should be noted that our current study cannot rule out the possibility that the observed increase of tissue iron level in striatum simply reflect the effect of atrophy assuming iron remains in place while structural loss happens, i.e. iron concentration increases without increase of total iron content. However, this is less likely the case when considering previous studies showing unmatched tissue iron changes and atrophy (Dumas et al. 2012; van Bergen et al. 2016a) in addition to the suggested involvement of Htt in the regulation of brain iron homeostasis (Dietrich et al. 2017; Muller and Leavitt 2014).
It is widely known that CAG repeats play an important role in HD disease toxicity, and CAG repeat length and age at study entry (i.e. CAPs score) account for a determinant of clinical progression (Zhang et al. 2011). Higher CAPs score is associated with faster clinical progression and faster striatal atrophy (Aylward et al. 2011a). Our results provide further support to this showing that higher CAPs score is indeed associated with greater atrophy in caudate, putamen and globus pallidus. In addition, our results further confirm that the elevated brain iron in the striatum is associated with CAPs or disease severity found in previous studies (Dominguez et al. 2016; van Bergen et al. 2016a). It still remains unclear whether the iron deposition in the basal ganglia is a primary cause of tissue damage, or is a secondary effect of other neuronal damages. However, considering its correlation with CAPs, iron content may serve as a potential alternative marker for monitoring therapy for disease intervention that would lead to detectable brain iron changes.
The increased iron contents in the basal ganglia in the premanifest and manifest HD groups are further confirmed by the increased R2* values in caudate and putamen, consistent with our previous study (van Bergen et al. 2016a). Another R2* study however only showed higher R2* in pre-symptomatic HD in left caudate (Sanchez-Castaneda et al. 2015). The absence of a R2* difference in globus pallidus, smaller relative changes between all carriers and controls, and the missing correlation between R2* and corrected ROI volume in putamen suggests a lower detection sensitivity and accuracy of R2* maps to tissue iron levels in comparison with QSM, which was also concluded in previous studies (Chen et al. 2017; Dominguez et al. 2016; van Bergen et al. 2016a). This is partly because R2* contrast can be easily contaminated by field inhomogeneity effects unrelated to tissue iron content (Barbosa et al. 2015; Wang and Liu 2015).
Due to the small sample size of patients who completed the one-year follow up, we tried to pool them together into a whole HD group for our first longitudinal analysis. Group differences in longitudinal changes for this HD group were however not statistically significant for magnetic susceptibility, R2*values, structure volume as well as cognitive function measurements as compared to controls. However, subsequent analysis showing significantly higher one-year iron-deposition rate in caudate and globus pallidus, and higher volume-loss rate in caudate in the HD-2 group (combined closer-to-onset and early HD) compared to the controls and no significant difference in the whole HD group compared to the controls is consistent with the reported steadily increased atrophy rate in the striatum during the progression of the HD from further-from-onset to closer-to-onset and afterward (Ross et al. 2014; Tabrizi et al. 2013). The non-significant structural volume loss in the whole HD-group and the significant atrophy found only in caudate in the HD-2 group is somewhat diverse to the previous literature reporting early atrophy in HD and is likely caused by the rather small sample size. Nevertheless, this study suggests for the first time that the iron deposition rate in the striatum may follow a similar pattern of acceleration of striatal atrophy in HD, i.e. faster iron deposition rate at closer-to-onset and early HD than further-from-onset stage.
The present results are consistent with the gain-of function model for HD pathogenesis, i.e. loss of Htt expression in adulthood has been reported to cause decreased rather than increased iron (Dietrich et al. 2017). One consequence of the increased delineation of basal ganglia structures especially the striatum using QSM, might be to facilitate mapping of these structures in patients for neurosurgical interventions (Wild and Tabrizi 2017).
The present study does have limitations. Other potential sources such as calcium and other trace metal elements may influence the iron assessment by magnetic susceptibility. However, such effects should be limited (Drayer et al. 1986) and the increase of magnetic susceptibility observed in our study should reflect primarily increased tissue iron content, consistent with other QSM studies (Ayton et al. 2017; Bilgic et al. 2012; Du et al. 2016; Ropele and Langkammer 2017; Schweser et al. 2011; Shmueli et al. 2009). The second limitation is the small sample size and relatively short follow-up period (one year) employed in this study. The sample size is especially limited for the closer-to-onset and early HD patients who completed the follow-up (n = 4 in each subgroup), which made it impossible to test them as a separate group in the longitudinal analysis. Further studies with larger sample size and longer follow-up period are needed to further validate the present findings.
5. Conclusions
The present study confirms that increased iron deposition in striatum can be measured by quantitative susceptibility mapping in premanifest and manifest Huntington disease and thus has potential to serve as a marker to monitor disease progression. The altered iron content in striatum is indicated to be more obvious in the closer-to-onset stage and less so in further-from-onset stage. The inverse correlations between structural volumes and iron levels in striatum and globus pallidus indicate volume loss and iron accumulation are two time locked processes. Both iron deposition and volume loss are related to disease progression as indicated by their correlations with CAPs score. Furthermore, we have shown through a one-year longitudinal study that iron deposition rate in caudate and globus pallidus is also higher in the closer-to-onset and early stage HD patients than controls.
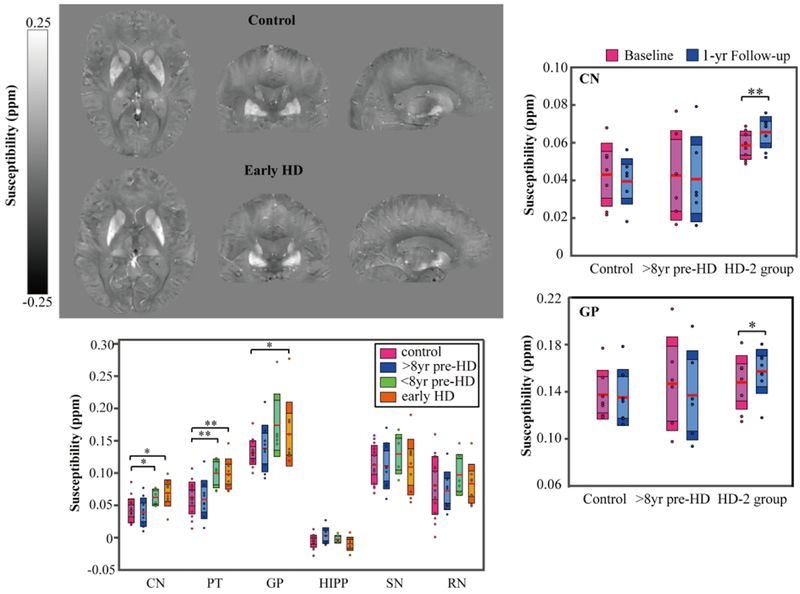
Quantitative susceptibility mapping (QSM) was used to investigate altered brain iron content and iron deposition rate in Huntington disease (HD). Left top panel shows example QSM maps of a 61-yr-old female control as compared to example QSM maps of a 60-yr-old female early HD patients. Higher iron content in the striatum and pallidum were observed in the closer-to-onset premanifest HD and early-HD patients as compared to the control group, but not in the further-from-onset group (left bottom panel). In addition, significantly higher iron deposition rates in caudate and pallidum were firstly observed in closer-to-onset premanifest HD and early HD (HD-2 group) as compared to controls over one-year
Significance.
This study tried to investigate the natural history of altered brain iron content at different stages of premanifest and manifest Huntington disease (HD). Two significant findings are shown. First, higher magnetic susceptibility values as measured by QSM indicating higher iron content in the striatum and globus pallidus were observed in closer-to-onset premanifest HD and early HD patients, but not in the further-from-onset premanifest HD group as compared to controls. Second, significantly higher iron deposition rates (11.9%/yr in caudate and 6.1%/yr in globus pallidus) were observed in closer-to-onset premanifest HD and early HD as compared to controls over one-year time period.
Acknowledgement
The authors would like to thank Mr. Joseph Gillen, Ms. Terri Brawner, Ms. Kathleen Kahl, Ms. Ivana Kusevic, for their assistance with data acquisition.
Funding sources: National Center for Research Resources and the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health (P41 EB015909); Dana Foundation and the Huntington’s Disease Society of America (HDSA); Chinese Scholarship Council (201706310087 to Lin Chen); National Natural Science Foundation of China (11775184).
Footnotes
Conflict of Interest Statement
Dr. Peter van Zijl is a paid lecturer for Philips Healthcare and is the inventor of technology that is licensed to Philips. This arrangement has been approved by The Johns Hopkins University in accordance with its Conflict of Interest policies.
Data Accessibility
The data sets generated and analyzed during the current study are available from the corresponding author based on reasonable requests.
References
- Aylward E, Mills J, Liu D, Nopoulos P, Ross CA, Pierson R, Paulsen JS. 2011a. Association between age and striatal volume stratified by CAG repeat length in prodromal huntington disease. PLoS currents 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylward EH, Codori AM, Barta PE, Pearlson GD, Harris GJ, Brandt J. 1996. Basal ganglia volume and proximity to onset in presymptomatic Huntington disease. Archives of Neurology 53(12):1293–1296. [DOI] [PubMed] [Google Scholar]
- Aylward EH, Liu DW, Nopoulos PC, Ross CA, Pierson RK, Mills JA, Long JD, Paulsen JS, Grp HS. 2012. Striatal volume contributes to the prediction of onset of Huntington disease in incident cases. Biol Psychiat 71(9):822–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylward EH, Nopoulos PC, Ross CA, Langbehn DR, Pierson RK, Mills JA, Johnson HJ, Magnotta VA, Juhl AR, Paulsen JS, Investigators P-H, Coordinators of Huntington Study G. 2011b. Longitudinal change in regional brain volumes in prodromal Huntington disease. Journal of Neurology, Neurosurgery, and Psychiatry 82(4):405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylward EH, Sparks BF, Field KM, Yallapragada V, Shpritz BD, Rosenblatt A, Brandt J, Gourley LM, Liang K, Zhou H, Margolis RL, Ross CA. 2004. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology 63(1):66–72. [DOI] [PubMed] [Google Scholar]
- Ayton S, Fazlollahi A, Bourgeat P, Raniga P, Ng A, Lim YY, Diouf I, Farquharson S, Fripp J, Ames D, Doecke J, Desmond P, Ordidge R, Masters CL, Rowe CC, Maruff P, Villemagne VL, Salvado O, Bush AI, Life AIB. 2017. Cerebral quantitative susceptibility mapping predicts amyloid-beta-related cognitive decline. Brain 140:2112–2119. [DOI] [PubMed] [Google Scholar]
- Bao LJ, Li X, Cai CB, Chen Z, van Zijl PCM. 2016. Quantitative susceptibility mapping using Structural Feature based Collaborative Reconstruction (SFCR) in the human brain. Ieee T Med Imaging 35(9):2040–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa JH, Santos AC, Tumas V, Liu M, Zheng W, Haacke EM, Salmon CE. 2015. Quantifying brain iron deposition in patients with Parkinson’s disease using quantitative susceptibility mapping, R2 and R2*. Magnetic Resonance Imaging 33(5):559–565. [DOI] [PubMed] [Google Scholar]
- Bartzokis G, Lu PH, Tishler TA, Fong SM, Oluwadara B, Finn JP, Huang D, Bordelon Y, Mintz J, Perlman S. 2007. Myelin breakdown and iron changes in Huntington’s disease: pathogenesis and treatment implications. Neurochemical Research 32(10):1655–1664. [DOI] [PubMed] [Google Scholar]
- Bilgic B, Pfefferbaum A, Rohlfing T, Sullivan EV, Adalsteinsson E. 2012. MRI estimates of brain iron concentration in normal aging using quantitative susceptibility mapping. NeuroImage 59(3):2625–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AW, Xu Y, Jiang J, Rahim T, Zhao D, Kocerha J, Chi T, Moran S, Engelhardt H, Larkin K, Neumann A, Cheng H, Li C, Nelson K, Banta H, Zola SM, Villinger F, Yang J, Testa CM, Mao H, Zhang X, Bachevalier J. 2014. A two years longitudinal study of a transgenic Huntington disease monkey. BMC Neuroscience 15:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SH, Khoshabeh R, Gibson KB, Gill PE, Nguyen TQ. 2011. An augmented Lagrangian method for total variation video restoration. IEEE Trans Image Process 20(11):3097–3111. [DOI] [PubMed] [Google Scholar]
- Chen L, Cai C, Yang T, Lin J, Cai S, Zhang J, Chen Z. 2017. Changes in brain iron concentration after exposure to high-altitude hypoxia measured by quantitative susceptibility mapping. Neuroimage 147:488–499. [DOI] [PubMed] [Google Scholar]
- Deistung A, Schafer A, Schweser F, Biedermann U, Turner R, Reichenbach JR. 2013. Toward in vivo histology: a comparison of quantitative susceptibility mapping (QSM) with magnitude-, phase-, and R2*-imaging at ultra-high magnetic field strength. Neuroimage 65:299–314. [DOI] [PubMed] [Google Scholar]
- Deistung A, Schweser F, Reichenbach JR. 2017. Overview of quantitative susceptibility mapping. NMR in Biomedicine 30(4). [DOI] [PubMed] [Google Scholar]
- Dietrich P, Johnson IM, Alli S, Dragatsis I. 2017. Elimination of huntingtin in the adult mouse leads to progressive behavioral deficits, bilateral thalamic calcification, and altered brain iron homeostasis. Plos Genet 13(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Stockwell BR. 2014. The role of iron and reactive oxygen species in cell death. Nat Chem Biol 10(1):9–17. [DOI] [PubMed] [Google Scholar]
- Dominguez JF, Ng AC, Poudel G, Stout JC, Churchyard A, Chua P, Egan GF, Georgiou-Karistianis N. 2016. Iron accumulation in the basal ganglia in Huntington’s disease: cross-sectional data from the IMAGE-HD study. Journal of Neurology, Neurosurgery, and Psychiatry 87(5):545–549. [DOI] [PubMed] [Google Scholar]
- Drayer B, Burger P, Darwin R, Riederer S, Herfkens R, Johnson GA. 1986. Magnetic resonance imaging of brain iron. Am J Neuroradiol 7(3):373–380. [Google Scholar]
- Du G, Liu T, Lewis MM, Kong L, Wang Y, Connor J, Mailman RB, Huang X. 2016. Quantitative susceptibility mapping of the midbrain in Parkinson’s disease. Movement Disord 31(3):317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas EM, Versluis MJ, van den Bogaard SJA, van Osch MJP, Hart EP, van Roon-Mom WMC, van Buchem MA, Webb AG, van der Grond J, Roos RAC. 2012. Elevated brain iron is independent from atrophy in Huntington’s Disease. Neuroimage 61(3):558–564. [DOI] [PubMed] [Google Scholar]
- Gomez-Tortosa E, MacDonald ME, Friend JC, Taylor SA, Weiler LJ, Cupples LA, Srinidhi J, Gusella JF, Bird ED, Vonsattel JP, Myers RH. 2001. Quantitative neuropathological changes in presymptomatic Huntington’s disease. Ann Neurol 49(1):29–34. [PubMed] [Google Scholar]
- Haacke EM, Chengb NYC, House MJ, Liu Q, Neelavalli J, Ogg RJ, Khan A, Ayaz M, Kirsch W, Obenaus A. 2005. Imaging iron stores in the brain using magnetic resonance imaging. Magnetic Resonance Imaging 23(1):1–25. [DOI] [PubMed] [Google Scholar]
- He NY, Ling HW, Ding B, Huang J, Zhang Y, Zhang ZP, Liu CL, Chen KM, Yan FH. 2015. Region-specific disturbed iron distribution in early idiopathic Parkinson’s disease measured by quantitative susceptibility mapping. Human Brain Mapping 36(11):4407–4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilditch-Maguire P, Trettel F, Passani LA, Auerbach A, Persichetti F, MacDonald ME. 2000. Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum Mol Genet 9(19):2789–2797. [DOI] [PubMed] [Google Scholar]
- Jurgens CK, Jasinschi R, Ekin A, Witjes-Ane MN, Middelkoop H, van der Grond J, Roos RA. 2010. MRI T2 Hypointensities in basal ganglia of premanifest Huntington’s disease. PLOS Currents 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgens CK, van de Wiel L, van Es AC, Grimbergen YM, Witjes-Ane MN, van der Grond J, Middelkoop HA, Roos RA. 2008. Basal ganglia volume and clinical correlates in ‘preclinical’ Huntington’s disease. Journal of Neurology 255(11):1785–1791. [DOI] [PubMed] [Google Scholar]
- Langkammer C, Schweser F, Krebs N, Deistung A, Goessler W, Scheurer E, Sommer K, Reishofer G, Yen K, Fazekas F, Ropele S, Reichenbach JR. 2012. Quantitative susceptibility mapping (QSM) as a means to measure brain iron? A post mortem validation study. Neuroimage 62(3):1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langkammer C, Schweser F, Shmueli K, Kames C, Li X, Guo L, Milovic C, Kim J, Wei H, Bredies K, Buch S, Guo Y, Liu Z, Meineke J, Rauscher A, Marques JP, Bilgic B. 2018. Quantitative susceptibility mapping: Report from the 2016 reconstruction challenge. Magn Reson Med 79(3):1661–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Langkammer C, Chou YH, Petrovic K, Schmidt R, Song AW, Madden DJ, Ropele S, Liu CL. 2015. Association between increased magnetic susceptibility of deep gray matter nuclei and decreased motor function in healthy adults. Neuroimage 105:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Wu B, Liu CL. 2011. Quantitative susceptibility mapping of human brain reflects spatial variation in tissue composition. Neuroimage 55(4):1645–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim IA, Faria AV, Li X, Hsu JT, Airan RD, Mori S, van Zijl PC. 2013. Human brain atlas for automated region of interest selection in quantitative susceptibility mapping: application to determine iron content in deep gray matter structures. Neuroimage 82:449–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li W, Tong KA, Yeom KW, Kuzminski S. 2015. Susceptibility-weighted imaging and quantitative susceptibility mapping in the brain. J Magn Reson Imaging 42(1):23–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, Groot N, Macfarlane H, Jenkins B, Anderson MA, Wexler NS, Gusella JF, Bates GP, Baxendale S, Hummerich H, Kirby S, North M, Youngman S, Mott R, Zehetner G, Sedlacek Z, Poustka A, Frischauf AM, Lehrach H, Buckler AJ, Church D, Doucettestamm L, Odonovan MC, Ribaramirez L, Shah M, Stanton VP, Strobel SA, Draths KM, Wales JL, Dervan P, Housman DE, Altherr M, Shiang R, Thompson L, Fielder T, Wasmuth JJ, Tagle D, Valdes J, Elmer L, Allard M, Castilla L, Swaroop M, Blanchard K, Collins FS, Snell R, Holloway T, Gillespie K, Datson N, Shaw D, Harper PS. 1993. A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntingtons-Disease Chromosomes. Cell 72(6):971–983. [DOI] [PubMed] [Google Scholar]
- Middleton FA, Strick PL. 2000. Basal ganglia and cerebellar loops: motor and cognitive circuits. Brain Res Rev 31(2–3):236–250. [DOI] [PubMed] [Google Scholar]
- Muller M, Leavitt BR. 2014. Iron dysregulation in Huntington’s disease. J Neurochem 130(3):328–350. [DOI] [PubMed] [Google Scholar]
- Nasreddine ZS, Phillips NA, Bedirian V, Charbonneau S, Whitehead V, Collin I, Cummings JL, Chertkow H. 2005. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. Journal of the American Geriatrics Society 53(4):695–699. [DOI] [PubMed] [Google Scholar]
- Parmenter BA, Weinstock-Guttman B, Garg N, Munschauer F, Benedict RHB. 2007. Screening for cognitive impairment in multiple sclerosis using the Symbol Digit Modalities Test. Mult Scler 13(1):52–57. [DOI] [PubMed] [Google Scholar]
- Rivera-Mancia S, Perez-Neri I, Rios C, Tristan-Lopez L, Rivera-Espinosa L, Montes S. 2010. The transition metals copper and iron in neurodegenerative diseases. Chem-Biol Interact 186(2):184–199. [DOI] [PubMed] [Google Scholar]
- Ropele S, Langkammer C. 2017. Iron quantification with susceptibility. NMR in Biomedicine 30(4). [DOI] [PubMed] [Google Scholar]
- Rosas HD, Chen YI, Doros G, Salat DH, Chen NK, Kwong KK, Bush A, Fox J, Hersch SM. 2012. Alterations in brain transition metals in Huntington disease: an evolving and intricate story. Archives of Neurology 69(7):887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt A, Kumar BV, Mo A, Welsh CS, Margolis RL, Ross CA. 2012. Age, CAG repeat length, and clinical progression in Huntington’s disease. Movement Disord 27(2):272–276. [DOI] [PubMed] [Google Scholar]
- Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, Scahill RI, Leavitt BR, Stout JC, Paulsen JS, Reilmann R, Unschuld PG, Wexler A, Margolis RL, Tabrizi SJ. 2014. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nature Review Neurology 10(4):204–216. [DOI] [PubMed] [Google Scholar]
- Ross CA, Tabrizi SJ. 2011. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10(1):83–98. [DOI] [PubMed] [Google Scholar]
- Sanchez-Castaneda C, Squitieri F, Di Paola M, Dayan M, Petrollini M, Sabatini U. 2015. The role of iron in gray matter degeneration in Huntington’s disease: a magnetic resonance imaging study. Human brain mapping 36(1):50–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweser F, Deistung A, Lehr BW, Reichenbach JR. 2011. Quantitative imaging of intrinsic magnetic tissue properties using MRI signal phase: an approach to in vivo brain iron metabolism? Neuroimage 54(4):2789–2807. [DOI] [PubMed] [Google Scholar]
- Shmueli K, de Zwart JA, van Gelderen P, Li TQ, Dodd SJ, Duyn JH. 2009. Magnetic susceptibility mapping of brain tissue in vivo using MRI phase data. Magn Reson Med 62(6):1510–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM. 2002. Fast robust automated brain extraction. Human brain mapping 17(3):143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Reilmann R, Roos RAC, Durr A, Leavitt B, Owen G, Jones R, Johnson H, Craufurd D, Hicks SL, Kennard C, Landwehrmeyer B, Stout JC, Borowsky B, Scahill RI, Frost C, Langbehn DR, Investigators T-H. 2012. Potential endpoints for clinical trials in premanifest and early Huntington’s disease in the TRACK-HD study: analysis of 24 month observational data. Lancet Neurol 11(1):42–53. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Scahill RI, Owen G, Durr A, Leavitt BR, Roos RA, Borowsky B, Landwehrmeyer B, Frost C, Johnson H, Craufurd D, Reilmann R, Stout JC, Langbehn DR, Investigators T-H. 2013. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol 12(7):637–649. [DOI] [PubMed] [Google Scholar]
- Tang X, Crocetti D, Kutten K, Ceritoglu C, Albert MS, Mori S, Mostofsky SH, Miller MI. 2015. Segmentation of brain magnetic resonance images based on multi-atlas likelihood fusion: testing using data with a broad range of anatomical and photometric profiles. Frontiers in Neuroscience 9:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bergen JMG, Hua J, Unschuld PG, Lim IAL, Jones CK, Margolis RL, Ross CA, van Zijl PCM, Li X. 2016a. Quantitative susceptibility mapping suggests altered brain iron in premanifest Huntington disease. Am J Neuroradiol 37(5):789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bergen JMG, Li X, Hua J, Schreiner SJ, Steininger SC, Quevenco FC, Wyss M, Gietl AF, Treyer V, Leh SE, Buck F, Nitsch RM, Pruessmann KP, van Zijl PCM, Hock C, Unschuld PG. 2016b. Colocalization of cerebral iron with Amyloid beta in Mild Cognitive Impairment. Sci Rep-Uk 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaard SJ, Dumas EM, Acharya TP, Johnson H, Langbehn DR, Scahill RI, Tabrizi SJ, van Buchem MA, van der Grond J, Roos RA, Group T-HI. 2011. Early atrophy of pallidum and accumbens nucleus in Huntington’s disease. Journal of Neurology 258(3):412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vymazal J, Klempir J, Jech R, Zidovska J, Syka M, Ruzicka E, Roth J. 2007. MR relaxometry in Huntington’s disease: Correlation between imaging, genetic and clinical parameters. J Neurol Sci 263(1–2):20–25. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu T. 2015. Quantitative susceptibility mapping (QSM): Decoding MRI data for a tissue magnetic biomarker. Magn Reson Med 73(1):82–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Spincemaille P, Liu Z, Dimov A, Deh K, Li JQ, Zhang Y, Yao YH, Gillen KM, Wilman AH, Gupta A, Tsiouris AJ, Kovanlikaya I, Chiang GCY, Weinsaft JW, Tanenbaum L, Chen WW, Zhu WZ, Chang SX, Lou M, Kopell BH, Kaplitt MG, Devos D, Hirai T, Huang XM, Korogi Y, Shtilbans A, Jahng GH, Pelletier D, Gauthier SA, Pitt D, Bush AI, Brittenham GM, Prince MR. 2017. Clinical quantitative susceptibility mapping (QSM): Biometal imaging and its emerging roles in patient care. J Magn Reson Imaging 46(4):951–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild EJ, Tabrizi SJ. 2017. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurology 16(10):837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams PTJA, Kim S, Martin JH 2014. Postnatal maturation of the red nucleus motor map depends on rubrospinal connections with forelimb motor pools. J Neurosci 34(12):4432–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Li W, Avram AV, Gho SM, Liu CL. 2012a. Fast and tissue-optimized mapping of magnetic susceptibility and T2*with multi-echo and multi-shot spirals. Neuroimage 59(1):297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Li W, Guidon A, Liu CL. 2012b. Whole brain susceptibility mapping using compressed sensing. Magn Reson Med 67(1):137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecca L, Youdim MBH, Riederer P, Connor JR, Crichton RR. 2004. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci 5(11):863–873. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Long JD, Mills JA, Warner JH, Lu WJ, Paulsen JS. 2011. Indexing disease progression at study entry with individuals at-risk for Huntington disease. Am J Med Genet B 156b(7):751–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Zhang JY, Hsu J, Oishi K, Faria AV, Albert M, Miller MI, Mori S. 2014. Evaluation of group-specific, whole-brain atlas generation using Volume-based Template Estimation (VTE): Application to normal and Alzheimer’s populations. Neuroimage 84:406–419. [DOI] [PMC free article] [PubMed] [Google Scholar]