

Abstract
Background
Neutrophil dysfunction plays an important role in inflammation-induced tissue injury. Previously, we identified Protein Kinase C-delta (PKCδ) as a critical controller of neutrophil activation and trafficking but how PKCδ is regulated in inflammation hasn’t been delineated. PKCδ activity is regulated by tyrosine phosphorylation on multiple sites. Tyrosine155 is a key regulator of apoptosis and gene expression, but its role in proinflammatory signaling is not known.
Methods
In vitro studies: superoxide anion (O2−) and neutrophil extracellular traps (NETs) were measured in bone marrow neutrophils (BMN) isolated from wild type (WT) and PKCδY155F knock-in mice (PKCδ tyrosine 155 → phenylalanine). Our novel 3D biomimetic microfluidic assay (bMFA) was used to delineate PKCδ-mediated regulation of individual steps in neutrophil adhesion and migration using WT and PKCδY155F BMN and lung microvascular endothelial cells (MLMVEC). In vivo studies: WT and PKCδY155F knock-in mice underwent sham or cecal ligation and puncture (CLP) surgery and the lungs harvested 24 hrs post-surgery.
Results
In vitro: PKCδY155F BMN had significantly reduced O2− and NETS release compared to WT. WT BMN, but not PKCδY155F BMN, demonstrated significant adhesion and migration across TNF-activated MLMVEC in bMFA. PKCδ inhibition significantly reduced WT BMN adhesion and migration under low shear and near bifurcations, but had no effect on PKCδY155F BMN. In vivo: mutation of PKCδ tyrosine 155 significantly decreased neutrophil migration into the lungs of septic mice.
Conclusions
PKCδ tyrosine 155 is a key phosphorylation site controlling proinflammatory signaling and neutrophil-endothelial cell interactions. These studies provide mechanistic insights into PKCδ regulation during inflammation.
Keywords: PKCδ, tyrosine phosphorylation, superoxide anion, NETS, neutrophil adhesion and migration
Introduction
Neutrophil dysfunction plays an important role in inflammatory diseases. Key to inflammation-induced tissue damage is the excessive migration of activated neutrophils across the vascular endothelium (1). While neutrophils are critical to host defense against pathogens, neutrophil dysregulation has a critical role in the early course of organ damage through release of proteases, oxygen radicals and neutrophil extracellular traps (NETs) that can damage the vascular endothelium (1, 2). During inflammation, the release of PAMPS (pathogen-associated molecular patterns) or DAMPS (damage-associated molecular patterns) activate immune cells to release cytokines/chemokines and other proinflammatory mediators that lead to increased adhesion molecule expression on neutrophils and endothelial cells resulting in enhanced neutrophil-endothelial cell interaction, vascular endothelial damage, and organ dysfunction. Neutrophil recruitment is a multi-step cascade that requires crosstalk between neutrophils and endothelial cells and is composed of a series of interactions between receptors and ligands which orchestrate rolling, adhesion, and transmigration (3, 4). Ultimately, arrested neutrophils extravasate to inflamed tissues across endothelial cells via a multi-step process controlled by concurrent chemoattractant-dependent signals, adhesive events, and hemodynamic shear forces (3, 4).
The inflammatory response is composed of multiple overlapping and redundant mechanisms, and recent research has shifted the focus to common control signaling points which are activated by diverse signals. We identified Protein Kinase C-delta (PKCδ) as a critical regulator of the inflammatory response (5–8). PKCδ is a member of the PKC family, a phospholipid-dependent family of serine/threonine kinases, and is expressed in multiple cell types and is an important regulator of neutrophil and endothelial proinflammatory signaling (5, 7, 9). In neutrophils, PKCδ regulates inflammatory signaling, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation, secretion of cytokines/chemokine, and reactive oxygen species (ROS) production (5, 9). In endothelial cells, PKCδ is involved in NF-κB activation, adhesion molecule expression, and the release of inflammatory mediators important in neutrophil transmigration (7, 10). PKCδ is activated by inflammatory mediators involved in the inflammatory response including lipopolysaccharide (LPS), tumor necrosis factor (TNF) and interleukin-1 (IL-1) (11, 12). Studies with PKCδ−/− mice and PKCδ inhibitors indicate a role for PKCδ in regulating neutrophil trafficking to the lung in response to inflammation triggered by stroke/reperfusion injury, LPS or pancreatitis (13–15). In recent studies, we demonstrated that PKCδ is activated in the lungs of septic animals, and PKCδ inhibition reduced neutrophil influx and was organ protective (6, 7, 10, 16). In vitro, PKCδ inhibition reduced human neutrophil migration across endothelial cells (7, 10). While these studies indicate a role for PKCδ in regulating neutrophil flux into the lung, they do not address specific mechanisms.
PKCδ activation requires multi-phosphorylation steps which triggers translocation from the cell cytosol to different subcellular compartments (17). PKCδ, in contrast to other PKC isotypes, is regulated by tyrosine phosphorylation patterns on multiple sites that determine activation, localization and substrate specificity (17–19). Thus, discrete cellular functions can be regulated by a single kinase through specific phosphorylation patterns. Phosphorylation of PKCδ tyrosine 155 in the regulatory domain regulates apoptosis and gene expression (19). However, the role of tyrosine 155 on pro-inflammatory signaling has not been studied. Our recent studies in a rodent model of sepsis (cecal ligation and puncture) demonstrated that sepsis triggered PKCδ activation and tyrosine 155 phosphorylation in lung endothelium suggesting a role for PKCδ tyrosine 155 phosphorylation in neutrophil-endothelial interaction (6, 16).
In this study, we used our novel 3D biomimetic microfluidic assay (bMFA) to investigate the role of PKCδ and PKCδ tyrosine 155 phosphorylation in neutrophil activation and neutrophil-endothelial cell interaction using pharmacologic (PKCδ-TAT peptide inhibitor) and genetic (PKCδ knock-in mice where PKCδ tyrosine 155 was mutated to phenylalanine: PKCδY155F KI mice) approaches. We then investigated the impact of mutation of PKCδ tyrosine 155 on neutrophil migration into the lungs of septic mice. We tested the hypothesis that PKCδ is an important regulator of neutrophil activation and migration and that PKCδ tyrosine 155 is a critical phosphorylation site.
Materials and Methods
Materials and Reagents
Mouse fibronectin (FN) was obtained from BD Biosciences (San Jose, CA). Mouse lung microvascular endothelial cells (MLMVEC) and mouse microvascular endothelial Growth Medium (EGM) were purchased from Cell Biologics (Chicago, IL). Carboxyfluorescein diacetate succinimidyl ester (CFDA/SE) and SYTOX green probes from Molecular Probes (Carlsbad, CA), Hanks’ Balanced Salt Solution (HBSS), Trypsin/EDTA, Formalin, Triton X-100, Draq5, 40kDa Texas Red conjugated dextran, and Hoechst 33342 from Thermofisher Scientific (Rockford, IL), and Alexa Fluor® 488 Phalloindin from Life Technologies Corporation (Carlsbad, CA). Recombinant mouse TNF-α was purchased from EMD Millipore (Burlington, MA). Phorbol myristate acetate (PMA), N-Formylmethionyl-leucyl-phenylalanine (fMLP), cytochalasin B, and cytochrome c were purchased from Sigma-Aldrich (St. Louis, MO).
Generation of PKCδY155F knock-in mice
In PKCδ, tyrosine 155 is located in the C1 domain of the regulatory motif and is a conserved site expressed in humans and mice. PKCδ knock-in mice were generated at the University of Connecticut as outlined in Figure 1 where PKCδ tyrosine 155 was mutated to phenylalanine. Murine PKCδ is a 674-amino acid protein consisting of 18 exons and tyrosine 155 is located on exon-5 of PKCδ. The Y155F mutation was introduced into exon-5 of PRKCD locus in mice embryonic stem cells by homologous recombination (Figure 1). The clones positive for Y155F mutation were confirmed by dual selection using G418 and Gancyclovir along with PCR and sequencing. The resulting chimeras were bred with transgenic mice expressing Cre recombinase to remove the PGKneo cassette. The PKCδY155F mice were identified by PCR containing a copy of LoxP in intron-6 which is 271 bp product compared to 181 bp wildtype littermate control (Figure 1B). The mutation in the PCR product was confirmed by DNA sequence analysis (not shown). The Y155F mutation was further confirmed by another PCR using oligonucleotides that recognize the mutant allele (Figure 1C). PKCδY155F mice are viable and follow predicted mendelian ratios. Age matched male and female C57BL6 mice (Jackson Laboratories) and C57BL6/jX129sv mice (in house breeding) were used as wild type (WT) controls. There were no significant differences in neutrophil activity (O2− production and NETs release) or endothelial cell activity between the two strains.
Figure 1. Generation of PKCδY155F knock-in model.
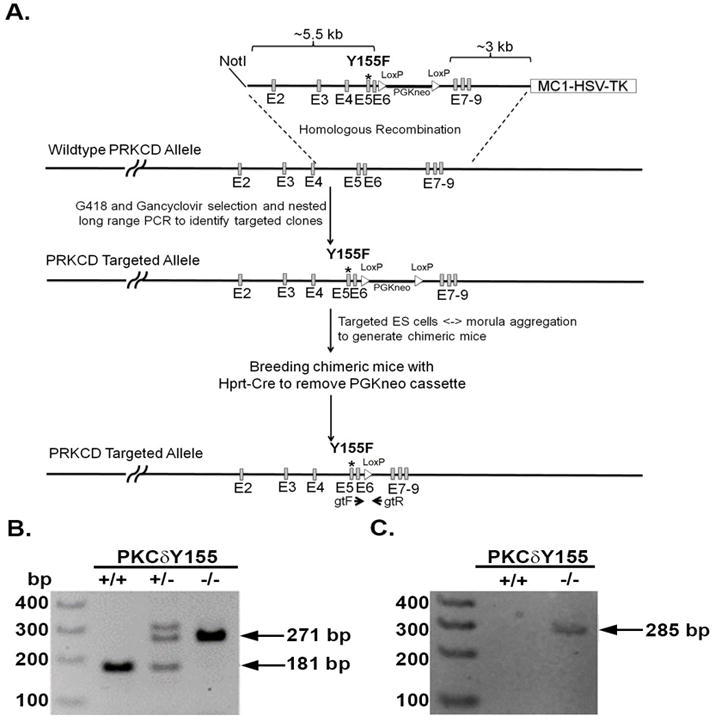
(A.) Schematic representation of targeted generation of PKCδY155F knock-in mice. Exons 2–9 are represented as grey vertical lines. PKCδ tyrosine 155 is located on Exon 5 and is indicated with an asterisk. Identification of wildtype and PKCδY155F knock-in mice using (B.) gtF/gtR primer pair (indicated in panel A) and (C.) primers that specifically recognize PKCδY155F site by PCR.
Inhibitor Peptide Synthesis
As described previously (5–7, 9, 10, 16), PKCδ activity was selectively inhibited by a peptide antagonist that consisted of a peptide derived from the first unique region (V1) of PKCδ (SFNSYELGSL: amino acids 8–17) coupled via an N-terminal Cys-Cys bond to a membrane permeant peptide sequence in the HIV TAT gene product (YGRKKRRQRRR: amino acids 47–57 of TAT) (20). Extensive in vitro and in vivo studies demonstrate that, when taken up by cells, the PKCδ TAT peptide produces a unique dominant-negative phenotype that effectively inhibits activation of PKCδ but not other PKC isotypes (5, 20). Further studies demonstrate that the TAT peptide alone is nontoxic and does not alter PKCδ activity (5, 9). The peptide was synthesized by Mimotopes (Melbourne, Australia) by 9-fluorenylmethoxycarbonyl solid-phase chemistry. Peptides were purified to >95% by preparative reverse-phase HPLC.
Animal Protocols
Animal procedures and handling were conducted in accordance to National Institutes of Health standards and were approved by the Institutional Animal Care and Use Committee at the Lewis Katz School of Medicine at Temple University (Philadelphia, PA, USA). Male and female mice (25–30g) were housed in a climate controlled facility and given free access to food and water.
CLP Model
Sepsis was induced by cecal ligation and puncture (CLP) as we described previously (6, 7, 16, 21). Sham surgery animals are subjected to sham laparotomy without cecal ligation or puncture. Briefly for studies using septic mice, under isoflurane anesthesia, a midline laparotomy was performed, the cecum identified, the mesentary trimmed, and the stalk joining the cecum to the large intestine was ligated. The cecum was punctured with a 21 gauge needle, stool expressed, the cecum returned to the abdomen, and the incision was closed in two layers. Mice were fluid resuscitated with sterile saline administered subcutaneously. At 24hrs post-surgery, the mice were anesthetized and the lungs perfused with sterile PBS, harvested and stored at −70°C.
Mouse Bone Marrow Neutrophil Isolation
To obtain mouse bone marrow neutrophils, PKCδY155 and WT mice were euthanized and the femur and tibias from both hind legs were harvested. The distal tip of each bone was cut off, bones were rinsed using HBSS, and cell clumps were dispersed. Neutrophils were isolated using a Percoll gradient sedimentation, followed by hypotonic lysis to remove erythrocytes.
Mouse lung endothelial cell isolation
To obtain mouse pulmonary endothelial cells, PKCδY155 and WT mice were anesthetized with isoflurane, the lungs removed aseptically and the animals euthanized. The lungs were minced, digested with collagenase/dispase, and mechanically dispersed to produce a single cell solution (22). The pulmonary endothelial cells are isolated by positive selection using a PECAM-1 antibody conjugated to magnetic beads. The isolated cells were cultured on gelatin coated flasks until confluent and then further purified using ICAM-2 antibody conjugated to magnetic beads. The purified endothelial cells were maintained in culture until use.
Superoxide Anion Generation
Superoxide anion (O2−) generation was measured spectrophotometrically as superoxide-dismutase (SOD)-inhibitable cytochrome c reduction. Bone marrow neutrophils (2 X 106) were activated with PMA (1μg/ml) or fMLP(10−8M) in the presence of 5 μg/ml cytochalasin B and the generation of O2− monitored over a 10 min time period (9). For experiments employing TNF as a stimulus, 96 well plates were coated with fibronectin, and cells were allowed to adhere for 30 minutes prior to addition of TNF (50ng/ml) and O2− production measured over a 120 min time period. To examine the effects of complete PKCδ inhibition, WT neutrophils were pretreated with buffer or the PKCδ-TAT peptide inhibitor (PKCδ-i) (5μM) as described previously (9).
NETs formation
NETs production was measured fluorometrically using an excitation wavelength of 492 nm and an emission wavelength of 530nm by monitoring DNA release from WT and 155KI bone marrow neutrophils. 2 X 105 neutrophils/well were seeded in 96 well black plates with 0.5% FBS and the membrane-impermeable DNA binding dye SYTOX green (Molecular Probes, Invitrogen) (5uM). Neutrophils were incubated with buffer, TNF (50 ng/ml), PMA (30 nM) or IL-1 (10 U/ml). WT neutrophil NETs production was measured ± δ-PKC TAT inhibitor (5uM). The plates were incubated at 37°C, and DNA release from bone marrow neutrophils was monitored by Sytox Green fluorescence at 0, 1, 2, 3, and 4 hours. Specificity of the reaction was determined by treatment with DNase (200U/ml).
Lung Myeloperoxidase Activity
Myeloperoxidase enzymatic activity in lung tissue was measured as we described previously (21). Lung tissue was homogenized and sonicated. Homogenates were cleared by centrifugation, and MPO levels were determined using a MPO assay kit according to the manufacturer’s instructions (Cayman, USA).
Design and Fabrication of the Microfluidic Assay
The methods for design and fabrication of the novel microfluidic assay and its in vivo validation have been previously published (10, 23). This microfluidics system used in our studies (bMFA) is one of the few devices that realistically reproduces the entire leukocyte adhesion cascade in a single assay encompassing circulation, rolling, adhesion and migration of leukocytes in a physiologically realistic three-dimensional environment under physiologically-relevant flow conditions. Briefly, a modified Geographic Information System (GIS) approach was used to digitize the in vivo microvascular networks which were lithographically patterned on polydimethylsiloxane (PDMS). To mimic the in vivo conditions, this novel microfluidics system consists of vascular channels in which cultured endothelial cells form a continuous lumen that is in communication with a tissue compartment filled with chemoattractants (e.g. fMLP). Microfabricated pillars (10 μm diameter) were used to fabricate the 3×100 μm pores resulting in a network of vascular channels connected to a tissue compartment via a 3 μm porous barrier, which is the optimum size for neutrophil migration. Leukocytes circulate in the vascular channels and interact with the endothelial cells under physiologic shear flow conditions. In agreement with the flow parameters measured in the vessels of original microvascular network in vivo (23), shear rates used in the vascular channels of bMFA ranged from 0–150 sec−1. The vascular channels form a realistic microvascular network with realistic geometry (including bifurcating capillaries) reproduced from microvascular networks observed in vivo (23). We have extensively validated this system and demonstrated that the adhesion pattern of neutrophils in bMFA is very similar to those observed in vivo by intravital microscopy (23).
Seeding of Endothelial Cells in the biomimetic MicroFluidic Assay (bMFA)
Mouse lung microvascular endothelial cells isolated from WT and PKCδY155 mice were cultured in EGM and used between passages 3–6. The bMFA were coated with fibronectin and endothelial cells were cultured at 37 °C and 5% CO2 under shear flow (inlet flow rate of 0.1 μl/min) for 48h (23). Consistent with our published data (10, 24, 25), endothelial cells in bMFA form a confluent lumen and aligned in the direction of flow (Figure 2). Formation of the 3D lumen in vascular channels under physiological conditions was confirmed using confocal microscopy (Figure 2) (10, 24). In agreement with the observed complexity of in vivo flow conditions, shear stress is different in different vessels of bMFA due to flow distribution in the complex in vivo-like geometry. However, by keeping inlet flow conditions constant, shear stress in a given vessel is the same across different measurements. Assays in which neutrophils freely entered the tissue compartment without attachment were discarded.
Figure 2. The bMFA mimics a physiologically relevant microvascular environment.

Microvascular network maps obtained in vivo are reproduced on PDMS to assemble the biomimetic microfluidic assay (scale bar 1 cm) (A). Microvascular endothelial cells, WT (scale bar 50 μm) (B) and KI155 (scale bar 50 μm) (C), formed a complete lumen in the vascular channel of bMFA (green indicates F-actin; red indicates cell nuclei).
A Nikon TE200 fluorescence microscope equipped with an automated stage was used for performing experiments. Images were acquired using an ORCA Flash 4 camera (Hamamatsu Corp., USA). PhD Ultra Syringe pump (Harvard Apparatus) was used for injecting media, permeability dye or neutrophil suspension to the bMFA with high precision. A stage warmer was used to keep the bMFA at 37°C. NIS Elements software (Nikon Instruments Inc., Melville, NY) was used to control the microscope stage and the camera.
Adhesion and Migration Experiments
Isolated neutrophils were labeled in suspension using CFDA/SE probe for 10 min at room temperature. Neutrophils were treated with either TNF-α (10U/mL) or TNF-α (10U/mL) + PKCδ-TAT peptide inhibitor (5uM, PKCδ-i) for 10 min before injection into bMFA. Neutrophils were introduced in vascular channels at an inlet flow rate of 1 μl/min. WT or PKCδY155F KI lung endothelial cells were treated with TNF (10U/ml) for 4 hrs. For PKCδ-i treatment, a solution of TNF-α and PKCδ-i (5uM) was injected into the network. At 4 hours post TNF-α with or without PKCδ-i treatment, the tissue compartment was filled with buffer (control) or fMLP (1 μM) before injecting neutrophils in the vascular compartment. Fluorescently labeled BMN isolated from WT or PKCδY155F KI mice were introduced in the vascular compartment (5 X 106 cells/ml). Cells that did not move for 30 s were considered adherent. Adhesion level of neutrophils to endothelial cells reached steady state after 10 min of flow and was quantified by scanning the entire network. The number of migrated neutrophils was quantified using time-lapse imaging every 3 min for 60 min. Nikon Elements and Fiji software were used to collect and analyze the data (10).
Permeability Measurements
The vascular compartment of the bMFA was connected to a Hamilton gas tight syringe filled with Texas Red 40 kDa dextran (25 μM in EGM) mounted on a syringe pump. Permeability was measured by imaging the bMFA every minute for 2h while the dextran solution flowed through the vascular channels (flow rate 1 μl/min). Using our previously published method (24), the following equation was used to calculate permeability (P) of dextran across the endothelium in bMFA:
| (1) |
where It is the average intensity in the tissue compartment, Iv0 is the maximum fluorescence intensity of the vascular channel and V/S is the ratio of vascular channel volume to its surface area.
Statistical Analysis
Data are presented as Mean ± SEM. All numerical data passed the Shapiro-Wilk normality test. Statistical significance was determined by one-way or two-way analysis of variance (ANOVA) with Holm-Šidák method post-hoc using SigmaPlot software for multiple group comparisons. For comparison of 2 groups, a Student t-test was employed. Differences were considered statistically significant if p<0.05.
Results
Impact of PKCδ Tyr155 phosphorylation on neutrophil function
The role of PKCδ Tyr155 phosphorylation in neutrophil activation has not been delineated. In these studies, we examined the role of PKCδ and PKCδ Tyr155 phosphorylation in superoxide anion (O2−) generation in response to PMA (1ug/ml), fMLP (10−8M) and TNF (50ng/ml). O2− production was measured as SOD-inhibitable cytochrome c reduction in bone marrow neutrophils isolated from WT and PKCδY155F KI mice. As shown in Figures 3 and 4, mouse bone marrow neutrophils generated significant quantities of O2− in response to PMA, fMLP and TNF. In the absence of PKCδ Tyr155 phosphorylation, fMLP-stimulated O2− production was inhibited by 42% as compared to WT O2− production but had no significant effect on PMA, an activator of multiple PKC isotypes, mediated O2− generation (Figure 3).
Figure 3. PMA- and fMLP-stimulated Superoxide anion generation (O2−).
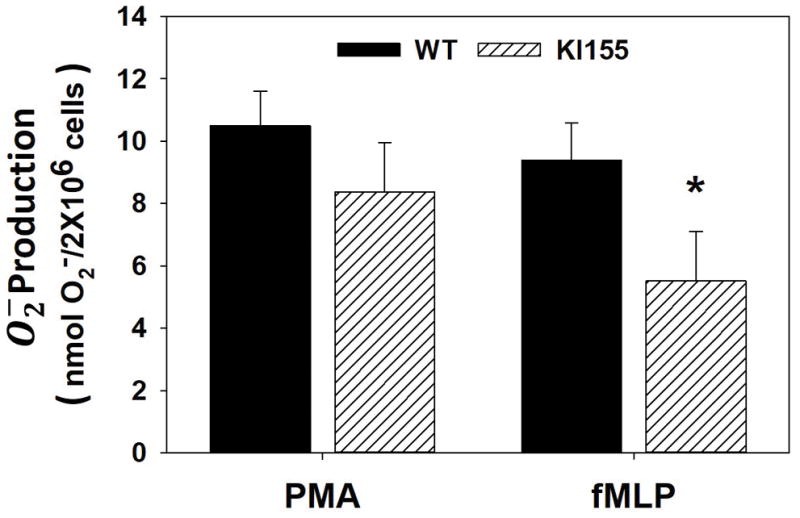
Superoxide onion generation by WT BMN and KI 155 BMN in response to PMA or fMLP was measured as SOD-inhibitable cytochrome c reduction. The measurements indicate significant quantities of superoxide anion generation in response to PMA and fMLP. While WT BMN O2− generation in response to either PMA or fMLP was similar, lack of PKCδ Tyr 155 phosphorylation in KI155 BMN decreased O2− generation significantly in response to fMLP, but not PMA. (N=11; Mean±SEM; * p<0.05 student t-test)
Figure 4. TNF-stimulated O2− Generation.

Superoxide anion generation in WT BMN with or without PKCδ-TAT peptide inhibitor (PKCδ-i) treatment and KI 155 BMN in response to TNF-α measured as SOD-inhibitable cytochrome c reduction. TNF-α stimulated O2− generation was significantly decreased in KI155 BMN and in WT BMN treated with the PKCδ-i (A). Vmax of the reaction was also significantly reduced with KI155 BMN and WT BMN treated with the PKCδ-i as compared to WT BMN (B). There was a significant increase in the lag time to O2− production in both the KI155 BMN and WT BMN treated with the PKCδ inhibitor as compared to WT BMN (C). (N=24 for WT group, N=9 for KI155 group, N=5 for WT+PKCδ-i group; Mean±SEM; * p<0.05, ** p<0.01 one-way ANOVA)
Full activation of neutrophils by proinflammatory mediators, such as TNF, requires adherence and ligation of integrins (9). Adherence of human neutrophils to extracellular matrix (ECM) proteins such as FN produces significant alterations in the kinetics of oxygen radical production in response to soluble mediators. There is a significant delay of approximately 60 min, followed by O2− generation, which is enhanced significantly as compared with non-adherent neutrophil O2− generation. In WT BMN, PKCδ inhibition significantly reduced O2− production in response to TNF (Figure 4A, p<0.01). In PKCδY155F KI BMN, O2− production was also significantly decreased in response to TNF (Figure 4A, p<0.05). The Vmax of the reaction was also significantly reduced in PKCδY155F KI BMN and WT BMN treated with the PKCδ inhibitor as compared to WT BMN (Figure 4B, p<0.01). Further analysis of the kinetics of O2− production demonstrated a significant increase in the lag time to O2− production in both the PKCδY155F KI BMN and WT BMN treated with the PKCδ inhibitor as compared to WT BMN suggesting a role for PKCδ in the assembly of the activated NADPH oxidase (Figure 4C, p<0.05).
NET formation was measured fluorometrically in BMN by monitoring DNA release. WT BMN produced NETs in response to PMA, IL-1 and TNF. PKCδ inhibition in WT BMN significantly decreased NETs in response to TNF (p<0.05) and IL-1 (p<0.05), but not PMA (Figure 5A and 5B). NET formation was also significantly attenuated in response to IL-1 (P<0.05) and TNF (P<0.05) in PKCδY155F KI BMN as compared to WT BMN (Figure 5C). Thus, PKCδ is an important regulator of O2− and NETs release, key components of neutrophil-mediated damage of vascular endothelium during inflammation. Furthermore, PKCδ tyrosine155 is a key phosphorylation site controlling neutrophil proinflammatory signaling.
Figure 5. Role of PKCδ in Neutrophil Extracellular Trap (NET) Formation.

NET formation was measured fluorometrically in BMN by monitoring DNA release. NET formation in WT BMN in response to IL-1 (A) (N=5) and fMLP (B) (N=5) increases over 4 hrs. Inhibition of PKCδ with the inhibitor decreased NET formation in WT BMN significantly over the course of 4hrs ((A) and (B)). NET formation was also significantly attenuated in response to IL-1 and TNF-α in KI155 BMN as compared to WT BMN (C) (N=5 for WT group, N=4 for WT+PKCδ-i and KI155 groups). (Mean±SEM; * p<0.05 two-way ANOVA)
Impact of PKCδ Tyr155 phosphorylation on endothelial cell function
PKCδ is also expressed in endothelial cells and plays an important role in cell function, but the role of PKCδ tyrosine155 phosphorylation in endothelial cell activation is not known. We next examined the role of PKCδ tyrosine155 phosphorylation on endothelial cell structure and function. We utilized our novel bMFA to determine the role of PKCδ and Tyr155 phosphorylation on endothelial cell permeability in response to inflammation. The expression of f-actin filaments in lung microvascular endothelial cells from WT and PKCδY155F KI mice under physiologically relevant flow condition was investigated using immunofluorescence staining and confocal microscopy. Under flow conditions, WT and PKCδY155F KI endothelial cells completely covered the vascular channels, formed a 3D lumen and aligned in the direction of flow (Figure 2).
The integrity of the endothelial cell barrier was directly assessed in the bMFA by measuring 40 kDa dextran permeation from the vascular channels to the tissue compartment. Permeability rates across the endothelial barrier were measured in WT and PKCδY155F KI endothelial cells under the following conditions: no treatment, TNF-α treatment (4h activation) and TNF-α +PKCδ-TAT peptide inhibitor (PKCδ-i) treatment (4 h activation). Permeability was similar between PKCδY155F KI endothelial cells and WT endothelial cells under control conditions (No Treatment) (Figure 6). TNF-α treatment significantly increased dextran permeability by 84% in WT but not to PKCδY155F KI endothelial cells (Figure 6). Treatment of cells with the PKCδ-i significantly reduced the permeability rates in WT endothelial cells to control levels (No Treatment) from 7.9×10−7 to 4.7×10−7 cm/s in WT cells but had no significant effect on permeability of PKCδY155F KI cells (Figure 6).
Figure 6. Role of PKCδ Tyr155 phosphorylation on permeability of MLMVEC.
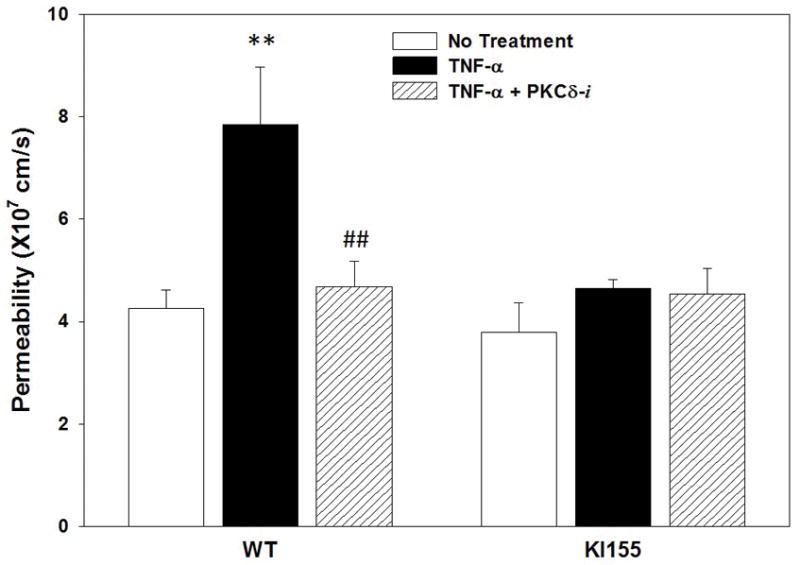
WT MLMVEC permeability increased significantly after TNF-α activation. Pharmacological inhibition of PKCδ in WT MLMVEC using PKCδ-i reduced the permeability to No Treatment levels. In the absence of PKCδ Tyr 155 phosphorylation in KI155 MLMVEC, permeability was not altered in response to TNF-α activation or after treatment with PKCδ-i. (N=3, Mean±SEM, ** p<0.01 compared to No Treatment; ## p<0.01 compared to TNF-α, two-way ANOVA)
PKCδ Tyr155 Phosphorylation differentially impacts adhesion and migration of neutrophils to endothelial cells in bMFA
To delineate the mechanism by which PKCδ regulates individual steps in neutrophil-endothelial cell interaction during inflammation, we utilized the bMFA to ascertain the role of PKCδ and PKCδ Tyr155 phosphorylation on the spatial distribution of adhering/migrating neutrophils. As shown in Figure 7A, there was significant migration of WT BMN across TNF-activated WT endothelial cells in response to the chemoattractant fMLP as compared to No Treatment (Figure 7A). Incubation with the PKCδ TAT peptide inhibitor resulted in a significant (p<0.001 at 60 minutes) decrease in WT BMN migration. In contrast, while baseline (No Treatment) migration of PKCδY155F BMN across WT lung endothelial cells (Figure 7B) was similar to baseline (No Treatment) migration of WT BMN across WT endothelial cells (Figure 7A), migration of PKCδY155F BMN across WT lung endothelial cells was not increased in response to TNF/fMLP activation (Figure 7B). Treatment with the PKCδ inhibitor did not result in further inhibition of migration of PKCδY155F BMN across WT lung endothelial cells. Furthermore, migration of PKCδY155F BMN across PKCδY155F KI lung microvascular endothelial cells was also significantly reduced as compared to WT BMN and was not increased in response to TNF/fMLP stimulation (Figure 7C). Again, treatment with the PKCδ inhibitor did not result in further inhibition of migration. The migration patterns of PKCδY155F BMN across WT endothelial cells (Figure 7B) were not significantly different from their migration pattern across PKCδY155F KI endothelial cells (Figure 7C) supporting the hypothesis that in neutrophils, and not endothelial cells, PKCδ Tyr155 phosphorylation is the dominant regulatory mechanism regulating migration across endothelial cells.
Figure 7. Migration of WT and KI155 BMN across MLMVEC in bMFA.

There was a significant increase in migration of WT BMN across TNF-activated WT MLMVEC in response to fMLP. Pharmacological inhibition with the PKCδ-TAT inhibitor (PKCδ-i) significantly decreased the number of migrated WT neutrophils across MLMVEC (A). Migration of TNF-activated KI155 BMN across WT MLMVEC in response to fMLP was not significantly different as compared to No Treatment levels. PKCδ inhibition did not change migration of KI155 BMN significantly (B). Migration of KI155 BMN across TNF-activated KI155 MLMVEC in response to fMLP did not increase compared to No treatment levels. Treatment with PKCδ-i did not significantly impact KI155 BMN migration across KI155 MLMVEC (C). (N=3; Mean±SEM; *** p<0.001 compared to No Treatment; ## p<0.01, ### p<0.001 compared TNF-α, one-way ANOVA)
Neutrophil adhesion to endothelium under shear flow precedes its migration and is a determinant of its efficiency. Activation with TNF-α resulted in a significant increase in WT BMN adhesion to WT lung endothelial cells. As shown in Figure 8, BMN preferentially adhered to activated endothelial cells near bifurcations and in regions of low shear with minimal adhesion in high shear regions (shear rate > 30 s−1), indicating that flow conditions strongly influence cell adhesion in these microvascular networks (10). PKCδ inhibition resulted in a significant reduction in BMN adherence (Figure 8A). In contrast, TNF-activation or treatment with the PKCδ-i did not impact adhesion of PKCδY155F KI BMN to WT endothelial cells (Figure 8B). Similarly, TNF-activation did not increase adhesion of PKCδY155F KI BMN to PKCδY155F KI lung endothelial cells (Figure 8C). Furthermore, treatment with the PKCδ inhibitor did not significantly alter PKCδY155F KI BMN adhesion to PKCδY155F KI lung microvascular endothelial cells (Figure 8C). Moreover, the adhesion level of PKCδY155F BMN to PKCδY155F endothelial cells (Figure 8C) was significantly different from its adhesion level of WT BMN to WT endothelial cells (Figure 8A), but not significantly different from adhesion level of PKCδY155F BMN to WT endothelial cells (Figure 8B), supporting the hypothesis that both neutrophil and endothelial PKCδ Tyr155 phosphorylation play a role in regulating adhesion of neutrophils to endothelial cells.
Figure 8. Adhesion of WT and KI155 BMN to MLMVEC.

There was a significant increase in adhesion of WT BMN to TNF-α activated WT MLMVEC in the presence of fMLP, especially at low shear rates and near bifurcations. Pharmacological inhibition with the PKCδ-TAT inhibitor (PKCδ-i) significantly reduced the adhesion level of WT BMN to WT MLMVEC to No Treatment levels (A). TNF-α activation of KI155 BMN did not increase adhesion to WT MLMVEC above the No Treatment levels. PKCδ-i treatment did not significantly change the adhesion level of KI155 BMN to WT MLMVEC (B). Adhesion of KI155 BMN to TNF-activated KI155 MLMVEC was not significantly different from No Treatment. Treatment with PKCδ-i did not significantly alter the adhesion level of KI155 BMN to KI155 MLMVEC (C) (N=3; Mean±SEM; * p<0.05, ** p<0.01, *** p<0.001, compared to No Treatment; # p<0.05, ## p<0.01, compared to TNF-α, one-way ANOVA)
Impact of PKCδ Tyr155 phosphorylation on neutrophil recruitment to the lung in sepsis
Using our bMFA in vitro model system, we demonstrated that PKCδ is a critical regulator of murine neutrophil adhesion and migration through PMVEC in response to inflammation, and the Tyr155 phosphorylation site is critical to neutrophil adhesion and migration through pulmonary endothelium. We next sought to confirm that PKCδ Tyr155 phosphorylation had an important role regulating neutrophil migration into the lung in vivo in response to sepsis induced by CLP. As shown in Figure 9, lung MPO activity, a marker of neutrophil infiltration, increased significantly in WT mice 24 hrs post CLP surgery as compared to WT sham mice. When sepsis was induced in PKCδY155F KI mice, MPO activity was reduced 66% as compared to WT mice. Thus, we have demonstrated both in vitro and in vivo that this tyrosine phosphorylation site has an important role in regulating neutrophil activation and migration through pulmonary endothelium.
Figure 9. Role of PKCδ Tyr155 phosphorylation on Pulmonary MPO Activity in Sepsis.
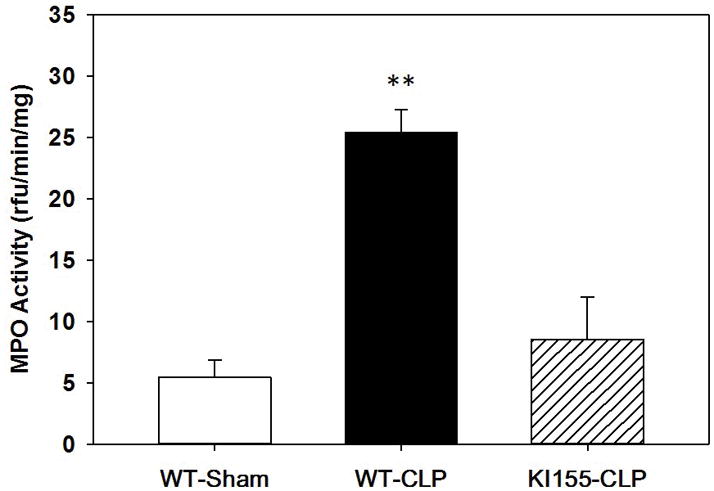
MPO analysis was performed in mouse lung samples harvested 24h post-surgery. There was a significant increase in MPO activity in the lungs from WT septic mice (WT-CLP, n=4) as compared to sham surgery WT mice (WT-sham, n=4). MPO activity was significantly lower in lungs from KI155 septic mice (KI155, n=3) as compared to WT septic mice (WT-CLP). Values are expressed as MEAN±SEM in relative fluorescence units (RFU)/min/mg. (** p<0.01, WT-CLP vs. WT-sham and WT-CLP vs. KI155-CLP, one-way ANOVA)
Discussion
Previous studies from our group (5–7, 9, 10, 16) have demonstrated a key role for PKCδ in the regulation of proinflammatory signaling. How PKCδ is activated under proinflammatory conditions has not been fully delineated but site-specific tyrosine phosphorylation may be a critical step. The results of the present study demonstrate for the first time that PKCδ tyrosine 155 phosphorylation plays an important regulatory role in neutrophil activation and migration. We demonstrate that this phosphorylation site on PKCδ is critical for O2-generation in response to TNF and the bacterial peptide fMLP but is not required for PMA-stimulated O2-production. We further demonstrate that PKCδ tyrosine 155 phosphorylation is also required for NETs formation and the extracellular release of DNA. The regulatory role of 155-tyrosine phosphorylation is not limited to neutrophils but also has a key role in the regulation of endothelial cell permeability, neutrophil adherence and transmigration. Lastly, we show that this phosphorylation site has a key role regulating neutrophil recruitment to the lung in sepsis induced by CLP. Thus, we have demonstrated both in vitro and in vivo that this tyrosine phosphorylation site has an important role in regulating neutrophil activation and migration through pulmonary endothelium. These studies provide mechanistic insights into the regulation of PKCδ during inflammation.
PKCδ, in contrast to other members of the PKC family, is regulated by tyrosine phosphorylation on multiple sites including phosphorylation sites in PKCδ regulatory domain (Tyr52, Tyr155, and Tyr187), catalytic domain (Tyr512 and Tyr523), and hinge region (Tyr311 and Tyr332) (26). These tyrosine phosphorylation patterns determine activation, localization and substrate specificity (17–19). Tyrosine phosphorylation in the catalytic domain of PKCδ increases kinase activity, whereas tyrosine phosphorylation in the regulatory domain mediates cellular actions without influencing kinase activity (17). Furthermore, tyrosine phosphorylation may also regulate protein-protein interactions and serve as docking sites for other proteins (26). Thus, discrete cellular functions are regulated by a single kinase through specific phosphorylation patterns and can be positive or negative regulators of cell function. In support of this concept, we previously demonstrated that PKCδ differentially regulates dense granule secretion in human platelets (27), and specific PKCδ tyrosine phosphorylations were stimulus-dependent. These results indicate that PKCδ tyrosine residues are differentially phosphorylated, and phosphorylation can regulate discrete cellular events that are cell type and stimulus dependent (17).
Our previous studies demonstrated an important role for PKCδ in TNF-mediated O2− production in systemic human neutrophils through PKCδ association and phosphorylation of the p47phox component of the NADPH oxidase enzyme complex (9). In this study, we establish that PKCδ is also a key regulator of murine O2− production in bone marrow neutrophils and that tyrosine 155 phosphorylation is a key phosphorylation site. Treatment of WT bone marrow neutrophils with the PKCδ-TAT peptide inhibitor resulted in reduced O2− production in response to TNF and fMLP, but not to PMA. A similar decrease in O2− generation in response to TNF was reported in bone marrow neutrophils isolated from PKCδ−/− mice (13). In the current study, we demonstrate for the first time that PKCδ tyrosine 155 is a critical phosphorylation site in regulating O2− generation stimulated by TNF and fMLP, but not PMA. The finding that the response to PMA was unchanged in response to the PKCδ inhibitor or in the PKCδY155F cells indicate that the requirement for PKCδ activity and tyrosine 155 phosphorylation is stimulus dependent.
During inflammation, activated neutrophils can release NETS composed of chromatin filaments studded with histones and granular proteins such as MPO, elastase and MMP9. While NETs are important in pathogen sequestration and killing, NETS can also damage the vascular endothelium and have an important role in the pathophysiology of sepsis-induced tissue damage (2). NETS formation can be triggered by inflammatory stimuli such as IL-1 and TNF (2), but the signaling pathways that regulate NET formation have yet to be fully elucidated. NET formation requires NADPH oxidase activity and activation of the MEK-ERK pathway (2, 28). PMA, an activator of multiple PKC isoforms including PKCδ, is a potent stimulus of NET formation suggesting that PKCδ may have a role in regulating NET formation. PKCδ regulates TNF-mediated ERK activation and the NADPH oxidase in neutrophils (this study and (5, 9). In this study, we found that PKCδ inhibition significantly decreased NET formation in response to TNF and IL-1, but not PMA. Furthermore, neutrophil PKCδ tyrosine 155 phosphorylation is required for TNF and IL-1 triggered NET formation but not PMA-induced NET formation indicating that PMA-induced NETs is PKCδ independent. In support of this concept, a role for PKCβ, but not PKCδ, in PMA-induced NETs was previously reported (29).
During inflammation, mediators damage the vascular endothelium resulting in increased permeability and excessive neutrophil migration into critical organs, with the lung being an early target. The vascular endothelium is an active participant in the dynamic process of neutrophil recruitment and activation through production of chemokines/cytokines and adhesion molecule expression (1, 3). To further investigate the role of PKCδ Tyr155 phosphorylation, we have used a novel biomimetic microfluidic assay (bMFA) that reproduces the endothelial barrier function and entire neutrophil adhesion cascade in a physiologically realistic three-dimensional environment and has already been validated against in vivo data (23, 25). We have previously shown that PKCδ has a critical role in regulating human neutrophil-endothelial cells interaction (10) but its molecular mechanisms were not identified. In this study, our findings indicate that PKCδ is a critical regulator of pulmonary endothelial cells permeability and murine neutrophil-endothelial cells interaction cascade during the inflammation. Moreover, we show that the Tyr155 phosphorylation site is a critical regulator for microvascular endothelium barrier function, neutrophil adhesion to, and migration through pulmonary endothelium. Consistent with our previous findings (10, 30), PKCδ was found to play a more significant role in regulating migration of neutrophils across endothelial cells as opposed to their adhesion to endothelial cells.
Previously, we demonstrated that in CLP-induced sepsis, PKCδ is activated in the lungs, is phosphorylated on tyrosine 155, and the kinase translocated to the nucleus, a process regulated by tyrosine 155 phosphorylation (6, 16, 31, 32). In these studies, we demonstrate for the first time that PKCδ tyrosine 155 phosphorylation is an important regulator of PKCδ proinflammatory activity during sepsis regulating neutrophil influx into the lung. Our studies show that PKCδY155F mice had significantly decreased MPO activity in their lungs as compared to WT mice 24 hrs post CLP, similar to what we previously observed following treatment with the PKCδ-TAT inhibitor (6, 7, 10, 16). The finding that sepsis triggers PKCδ tyrosine155 phosphorylation and nuclear translocation is important as these cellular events are also associated with tissue injury following ischemia-reperfusion and radiation damage (33–35).
In this study we used a novel microfluidic assay to mimic many aspects of the complex physiological conditions that regulate leukocyte adhesion/migration and leukocyte-endothelial interaction. Complete microvascular networks, including post-capillary venules, are reproduced in this in vitro model resulting in complex flows and a range of shear rates similar to those observed in vivo (10, 23–25, 30). Nevertheless, as with any other in vivo or in vitro model of inflammation, bMFA does not reproduce the entirety of the clinical condition. For example, in this study isolated neutrophils were in EGM as they interacted with endothelial cells that formed the 3D vessel lumen in the bMFA. Further studies may be required to determine whether the presence of other blood components may alter the observed neutrophil-endothelial interaction patterns. Similarly, the version of bMFA used in this study has a uniform diameter of 100 μm in all vessels and additional studies may shed further light on the possible effects of diameter variability on neutrophil-endothelial interaction patterns. Furthermore, other perivascular cell types, such as smooth muscle cells, can be co-cultured in the tissue compartment of bMFA with endothelial cells to provide a more realistic environment for studying the inflammatory response. Nevertheless, given the extensive validation of this bMFA against in vivo data (23–25) and the overall agreement between the findings from various methods presented in this study, we believe that our results provide a novel approach for better understanding the inflammatory response.
These findings provide a clear indication that PKCδ Tyr155 phosphorylation is not only an important molecular regulator of endothelial cell function but also plays a critical role in regulating the interaction of neutrophils with endothelial cells at a functional level. Overall, these findings indicate that regulating the PKCδ pathway may provide novel therapeutic strategies for treating inflammation.
Footnotes
Conflict of Interest Disclosure
L.E.K. is listed as an inventor on US patent #8,470,766 entitled “Novel Protein Kinase C Therapy for the Treatment of Acute Lung Injury” which is assigned to Children’s Hospital of Philadelphia and the University of Pennsylvania.
DISCLOSURE OF FUNDINGS: F.S. is a Predoctoral Fellow of the American Heart Association (grant No. 16PRE29860006). This work was supported by the American Heart Association (grant No. 16GRNT29980001) and National Institutes of Health (grant No. GM114359, HL111552, and HL93231).
References
- 1.Brown KA, Brain SD, Pearson JD, Edgeworth JD, Lewis SM, Treacher DF. Neutrophils in development of multiple organ failure in sepsis. The Lancet. 2006;368:157–169. doi: 10.1016/S0140-6736(06)69005-3. [DOI] [PubMed] [Google Scholar]
- 2.Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. 2011;17:1381–1390. doi: 10.1038/nm.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 4.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 5.Kilpatrick LE, Sun S, Mackie D, Baik F, Li H, Korchak HM. Regulation of TNF mediated antiapoptotic signaling in human neutrophils: role of {delta}-PKC and ERK1/2. J Leuk Biol. 2006;80:1512–1521. doi: 10.1189/jlb.0406284. [DOI] [PubMed] [Google Scholar]
- 6.Kilpatrick LE, Standage SW, Li H, Raj NR, Korchak HM, Wolfson MR, Deutschman CS. Protection against sepsis-induced lung injury by selective inhibition of protein kinase C-δ (δ-PKC) J Leuk Biol. 2011;89:3–10. doi: 10.1189/jlb.0510281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mondrinos MJ, Zhang T, Sun S, Kennedy PA, King DJ, Wolfson MR, Knight LC, Scalia R, Kilpatrick LE. Pulmonary Endothelial Protein Kinase C-Delta (PKCδ) Regulates Neutrophil Migration in Acute Lung Inflammation. Amer J Pathology. 2014;184:200–213. doi: 10.1016/j.ajpath.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mondrinos MJ, Kennedy PA, Lyons M, Deutschman CS, Kilpatrick LE. Protein Kinase C and Acute Respiratory Distress Syndrome. Shock. 2013;39:467–479. doi: 10.1097/SHK.0b013e318294f85a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kilpatrick LE, Sun S, Li H, Vary TC, Korchak HM. Regulation of TNF-induced oxygen radical production in human neutrophils: role of δ-PKC. J Leuk Biol. 2010;87:153–164. doi: 10.1189/jlb.0408230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soroush F, Zhang T, King DJ, Tang Y, Deosarkar S, Prabhakarpandian B, Kilpatrick LE, Kiani MF. A novel microfluidic assay reveals a key role for protein kinase C delta in regulating human neutrophil-endothelium interaction. J Leuk Biology. 2016;100:1027–1035. doi: 10.1189/jlb.3MA0216-087R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kilpatrick LE, Lee JY, Haines KM, Campbell DE, Sullivan KE, Korchak HM. A role for PKC-delta and PI 3-kinase in TNF-alpha-mediated antiapoptotic signaling in the human neutrophil. Am J Physiol Cell Physiol. 2002;283:C48–57. doi: 10.1152/ajpcell.00385.2001. [DOI] [PubMed] [Google Scholar]
- 12.Page K, Li J, Zhou L, Iasvovskaia S, Corbit KC, Soh JW, Weinstein IB, Brasier AR, Lin A, Hershenson MB. Regulation of airway epithelial cell NF-kappa B-dependent gene expression by protein kinase C delta. J Immunol. 2003;170:5681–5689. doi: 10.4049/jimmunol.170.11.5681. [DOI] [PubMed] [Google Scholar]
- 13.Chou WH, Choi DS, Zhang H, Mu D, McMahon T, Kharazia VN, Lowell CA, Ferriero DM, Messing RO. Neutrophil protein kinase Cdelta as a mediator of stroke-reperfusion injury. J Clin Invest. 2004;114:49–56. doi: 10.1172/JCI21655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramnath R, Sun J, Bhatia M. PKC δ mediates pro-inflammatory responses in a mouse model of caerulein-induced acute pancreatitis. J Molecular Med. 2010;88:1–9. doi: 10.1007/s00109-010-0647-9. [DOI] [PubMed] [Google Scholar]
- 15.Chichger H, Grinnell KL, Casserly B, Chung CS, Braza J, Lomas-Neira J, Ayala A, Rounds S, Klinger JR, Harrington EO. Genetic disruption of protein kinase Cdelta reduces endotoxin-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2012;303:L880–888. doi: 10.1152/ajplung.00169.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mondrinos MJ, Knight LC, Kennedy PA, Wu J, Kauffman M, Baker ST, Wolfson MR, Kilpatrick LE. Biodistribution and Efficacy of Targeted Pulmonary Delivery of a Protein Kinase C-δ Inhibitory Peptide: Impact on Indirect Lung Injury. J Pharm Exper Therapeut. 2015;355:86–98. doi: 10.1124/jpet.115.224832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinberg SF. Distinctive activation mechanisms and functions for protein kinase Cdelta. Biochem J. 2004;384:449–459. doi: 10.1042/BJ20040704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kronfeld I, Kazimirsky G, Lorenzo PS, Garfield SH, Blumberg PM, Brodie C. Phosphorylation of Protein Kinase Cδ on Distinct Tyrosine Residues Regulates Specific Cellular Functions. J Biol Chem. 2000;275:35491–35498. doi: 10.1074/jbc.M005991200. [DOI] [PubMed] [Google Scholar]
- 19.Steinberg SF. Structural Basis of Protein Kinase C Isoform Function. Physiol Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen L, Hahn H, Wu G, Chen CH, Liron T, Schechtman D, Cavallaro G, Banci L, Guo Y, Bolli R, et al. Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc Natl Acad Sci U S A. 2001;98:11114–11119. doi: 10.1073/pnas.191369098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liverani E, Mondrinos MJ, Sun S, Kunapuli SP, Kilpatrick LE. Role of Protein Kinase C-delta in regulating platelet activation and platelet-leukocyte interaction during sepsis. PLOS ONE. 2018;13:e0195379. doi: 10.1371/journal.pone.0195379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sobczak M, Dargatz J, Chrzanowska-Wodnicka M. Isolation and Culture of Pulmonary Endothelial Cells from Neonatal Mice. JoVE. 2010:e2316. doi: 10.3791/2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamberti G, Prabhakarpandian B, Garson C, Smith A, Pant K, Wang B, Kiani MF. Bioinspired Microfluidic Assay for In Vitro Modeling of Leukocyte-Endothelium Interactions. Anal Chem. 2014;86:8344–8351. doi: 10.1021/ac5018716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deosarkar SP, Prabhakarpandian B, Wang B, Sheffield JB, Krynska B, Kiani MF. A Novel Dynamic Neonatal Blood-Brain Barrier on a Chip. PLoS ONE. 2015;10:e0142725. doi: 10.1371/journal.pone.0142725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang Y, Soroush F, Sheffield JB, Wang B, Prabhakarpandian B, Kiani MF. A Biomimetic Microfluidic Tumor Microenvironment Platform Mimicking the EPR Effect for Rapid Screening of Drug Delivery Systems. Sci Reports. 2017;7:9359. doi: 10.1038/s41598-017-09815-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rybin VO, Guo J, Sabri A, Elouardighi H, Schaefer E, Steinberg SF. Stimulus-specific differences in protein kinase C delta localization and activation mechanisms in cardiomyocytes. J Biol Chem. 2004;279:19350–19361. doi: 10.1074/jbc.M311096200. [DOI] [PubMed] [Google Scholar]
- 27.Chari R, Getz T, Nagy B, Bhavaraju K, Mao Y, Bynagari YS, Murugappan S, Nakayama K, Kunapuli SP. Protein Kinase Cδ Differentially Regulates Platelet Functional Responses. Arterioscler Thromb Vasc Biol. 2009;29:699–705. doi: 10.1161/ATVBAHA.109.184010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, Waldmann H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol. 2011;7:75–77. doi: 10.1038/nchembio.496. [DOI] [PubMed] [Google Scholar]
- 29.Gray R, Lucas C, MacKellar A, Li F, Hiersemenzel K, Haslett C, Davidson D, Rossi A. Activation of conventional protein kinase C (PKC) is critical in the generation of human neutrophil extracellular traps. J Inflamm. 2013;10:12. doi: 10.1186/1476-9255-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soroush F, Tang Y, Zaidi J, Sheffield L, Kilpatrick LE, Kiani MF. PKCδ inhibition as a novel medical countermeasure for radiation-induced vascular damage. FASEB J. 2018;32 doi: 10.1096/fj.201701099. [DOI] [PubMed] [Google Scholar]
- 31.Humphries MJ, Ohm AM, Schaack J, Adwan TS, Reyland ME. Tyrosine phosphorylation regulates nuclear translocation of PKC[delta] Oncogene. 2007;27:3045–3053. doi: 10.1038/sj.onc.1210967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pabla N, Dong G, Jiang M, Huang S, Kumar MV, Messing RO, Dong Z. Inhibition of PKCδ reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J Clin Invest. 2011;121:2709–2722. doi: 10.1172/JCI45586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kostyak JC, Hunter JC, Korzick DH. Acute PKCδ inhibition limits ischaemia-reperfusion injury in the aged rat heart: Role of GSK-3β. Cardiovascular Res. 2006;70:325–334. doi: 10.1016/j.cardiores.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 34.Wie SM, Adwan TS, DeGregori J, Anderson SM, Reyland ME. Inhibiting Tyrosine Phosphorylation of Protein Kinase Cδ (PKCδ) Protects the Salivary Gland from Radiation Damage. J Biol Chem. 2014;289:10900–10908. doi: 10.1074/jbc.M114.551366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimohata T, Zhao H, Sung JH, Sun G, Mochly-Rosen D, Steinberg GK. Suppression of deltaPKC activation after focal cerebral ischemia contributes to the protective effect of hypothermia. J Cereb Blood Flow Metab. 2007;27:1463–1475. doi: 10.1038/sj.jcbfm.9600450. [DOI] [PubMed] [Google Scholar]